
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis of novel N-substituted 2-carboxy-4-quinolones via lithium bis(trimethylsilyl)amide (LiHMDS)-induced in situ cyclocondensation reaction†
Phool Hasanab,
Babita Anejaa,
Mir M. Masooda,
Md. Belal Ahmadb,
Umesh Yadavac,
Constantin G. Daniliucd and
Mohammad Abid‡
 *a
*a
aMedicinal Chemistry Lab, Department of Biosciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India. E-mail: mabid@jmi.ac.in; Fax: +91-11-26980229; Tel: +91-87-50295095
bDepartment of Chemistry, TNB College, TM Bhagalpur University, Bhagalpur 812007, Bihar, India
cDepartment of Physics, Deen Dayal Upadhyay Gorakhpur University, Gorakhpur, UP 273009, India
dOrganisch-Chemisches Institut, Westfälische Wilhelm-Universität Münster, 48149, Germany
First published on 13th February 2017
Abstract
A different approach for the synthesis of N-substituted 2-carboxy-4-quinolones using direct reductive amination followed by LiHMDS-induced cyclocondensation has been developed. A range of analogues of the title compounds with broad substrate scope were obtained in moderate yields and good regioselectivity.
Introduction
4-Quinolone, a nitrogen containing heterocycle, constitutes the core structure in a wide line up of natural products, synthetic materials and pharmacologically active molecules.1 Besides constituting a vital class of marketed antibiotics (e.g. ciprofloxacin, moxifloxacin, levofloxacin, gemifloxacin, norfloxacin, ofloxacin etc.)2 quinolone derivatives also been used in various therapeutic areas as they exhibit antimitotic,3 anticancer,3b,4 antimalarial,5 antimicrobial,6 antidiabetic,7 antiviral,8 anti-HIV9 and anti-inflammatory activities.10 Moreover, this scaffold exhibits the potential to act as an inhibitor of various vital enzymes such as topoisomerase I,11 topoisomerase II and IV gyrase,12 farnesyltransferase,13 tubulin,14 P-glycoprotein15 and casein kinase 2 (CK2).16 Such interesting biological applications have fascinated medicinal chemists worldwide, thus this scaffold is the foremost synthetic target, and chemists are seeking to develop new synthetic protocols to access quinolone bearing pharmacophores (Fig. 1).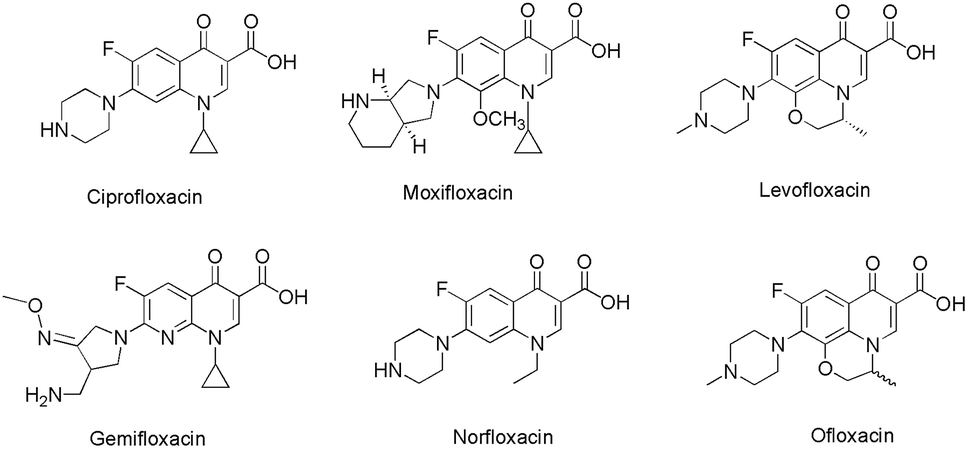 | ||
| Fig. 1 Few examples of antibiotics bearing quinolone core. | ||
Most commonly adopted methods for the construction of 4-quinolones such as Conard–Limach, Gould–Jacobs, Niementowski, Grohe–Heitzer and Camp cyclizations require the condensation of aniline and carboxylate derivatives followed by cyclisation.17 Some other methodologies were further developed with certain modifications including cycloacylation of aza-Michael adducts in the presence of Eaton's reagent,18 diphenyl ether,19 PPA,20 dowtherm,21 Pd-catalysed N-arylation of Z-enamines,22 and alkoxide promoted rearrangement of isatinacetamides23 (Scheme 1). Although these reactions were efficient in introducing ester functionality at the 2-position of 4-quinolones, most of them require harsh conditions such as the use of metal catalysts,22 strong acids and high temperatures.18–21 These extremely harsh conditions make them complicated in terms of synthesis and purification of end products and thus result in unsatisfactory yields, narrow substrate scope and poor regioselectivities. Moreover, N-aryl substitution requires a multi-step procedure which further added to its disadvantages.24 So it is necessary to design simple and cost effective procedures for the synthesis of N-aryl-2-carboxy substituted 4-quinolone compounds.
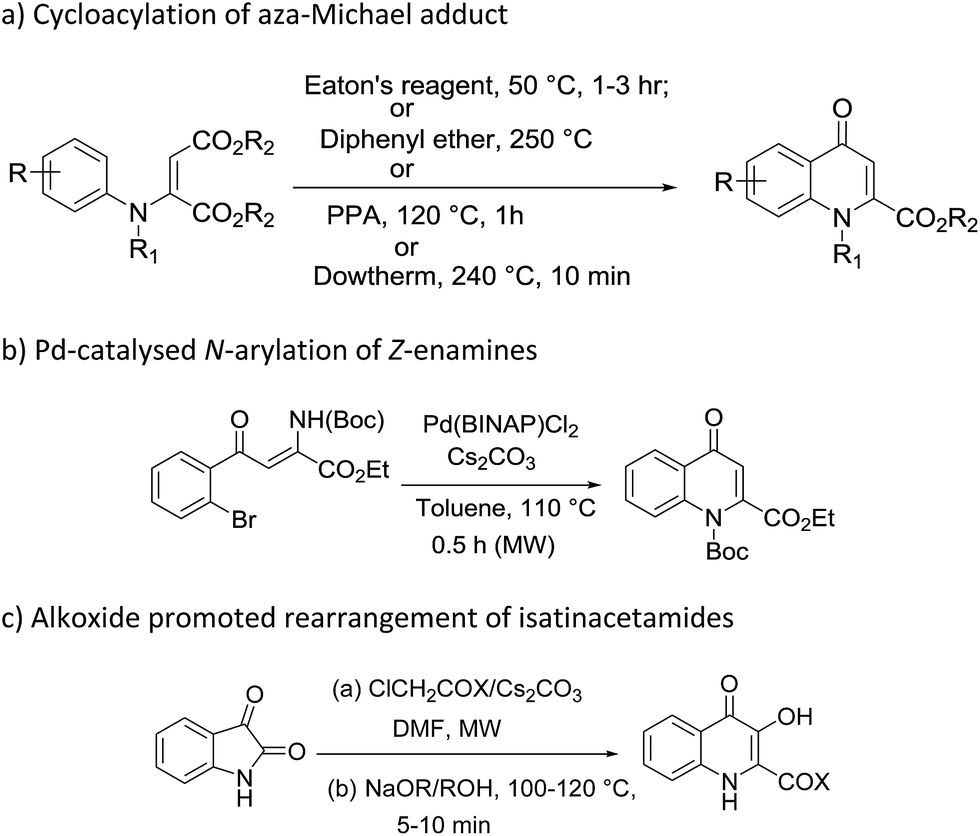 | ||
| Scheme 1 Methods for the construction of 4-quinolones. | ||
Earlier Haesslein et al. synthesised 1,3-disubstituted-2-carboxy quinolones via reductive amination followed by condensation in the presence of a mild base. Although this effective and practical methodology provides an easy route for the synthesis of N-aryl-2-carboxy substituted 4-quinolone compounds, its two-step procedure and incompatibility of reaction conditions to introduce different heterocyclic scaffolds at the 1-position add to its disadvantages.25 So in order to overcome these drawbacks, we have devised a LiHMDS induced in situ cyclocondensation reaction to give N-aryl-2-carboxy substituted 4-quinolones from readily available aldehydes and 2′-amino acetophenone. To the best of our knowledge, this method is the first example of LiHMDS catalyzed direct C–N bond formation in one step via in situ cyclocondensation to furnish N-aryl-2-carboxy substituted 4-quinolone compounds (Scheme 2).
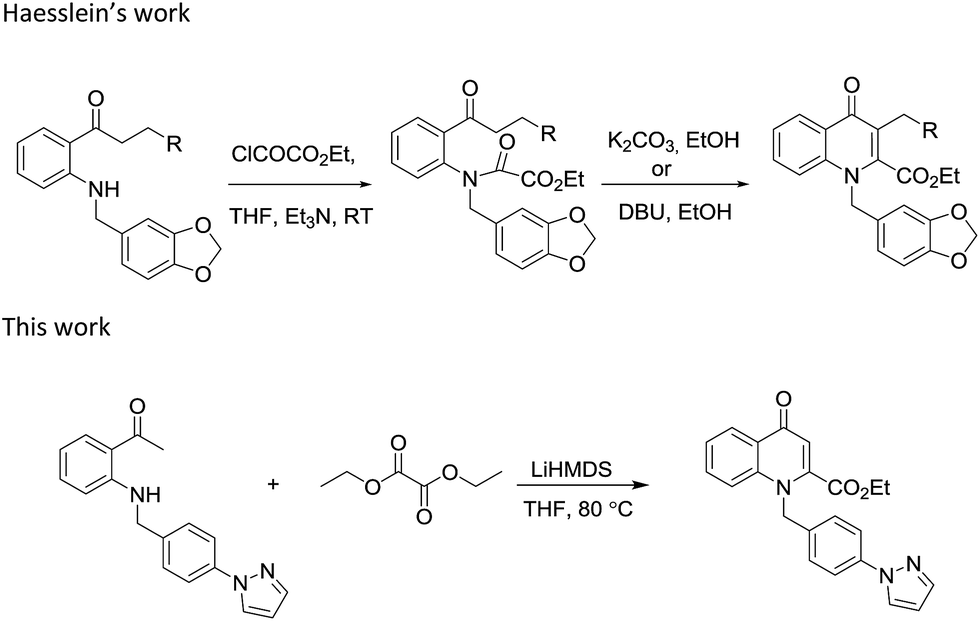 | ||
| Scheme 2 Theme of this work. | ||
Results and discussion
Our preliminary studies focused on the reductive amination approach between commercially available aldehydes and 2′-aminoacetophenone in the presence of hydride reducing agents such as sodium triacetoxyborohydride (NaBH(OAc)3),26 sodium cyanoborohydride (NaBH3CN),27 phenylsilane (PhSiH3)/dibutyltin dichloride (Bu2SnCl2)28 (Scheme 3). Among all three methods, reductive amination with PhSiH3/Bu2SnCl2 gave excellent yields of all the products in the range of 84–95% while the other two methods gave low to moderate yields (Table 1). The structures of compounds 3c and 3e were also confirmed by single crystal X-ray diffraction. ORTEP diagrams of these compounds with ellipsoids drawn at the 30% probability level along with their atomic numbering schemes are shown in Fig. 2.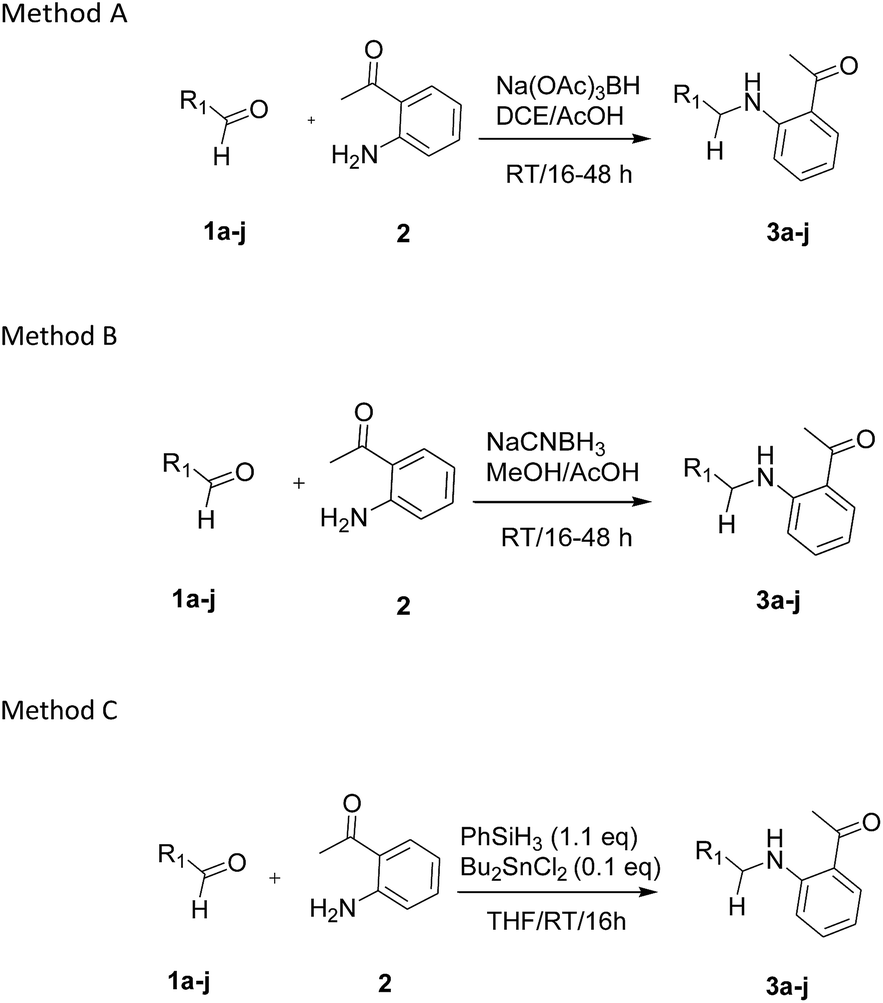 | ||
| Scheme 3 Methods used for reductive amination in this study. | ||
| S. no. | Aldehyde | Product | Yield (method A) | Yield (method B) | Yield (method C) |
|---|---|---|---|---|---|
| 1 | 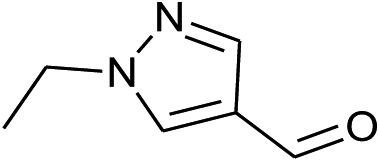 |
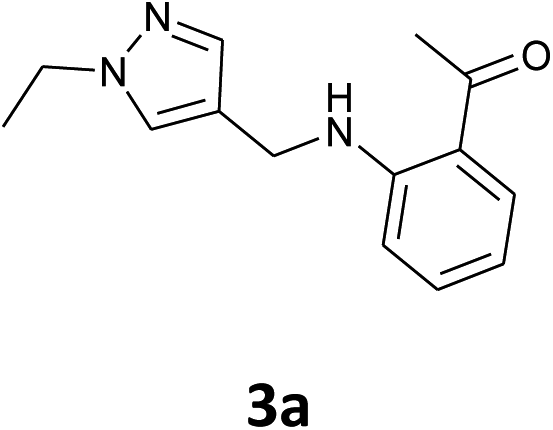 |
45% | 43% | 91% |
| 2 | 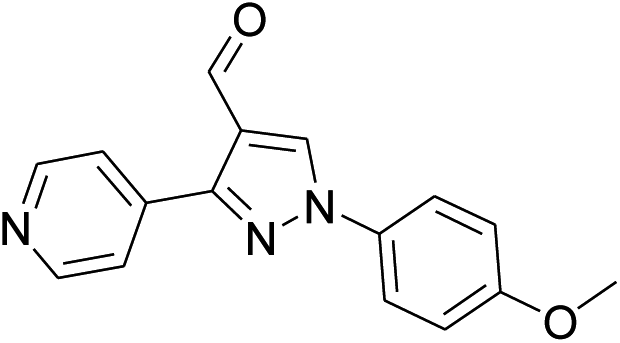 |
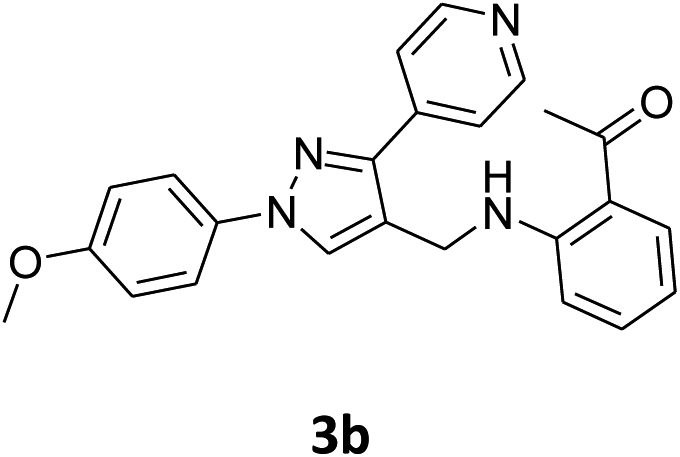 |
48% | 45% | 95% |
| 3 | 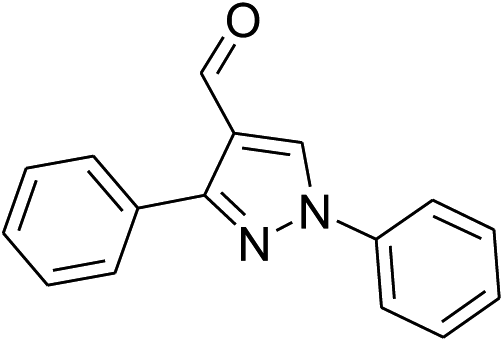 |
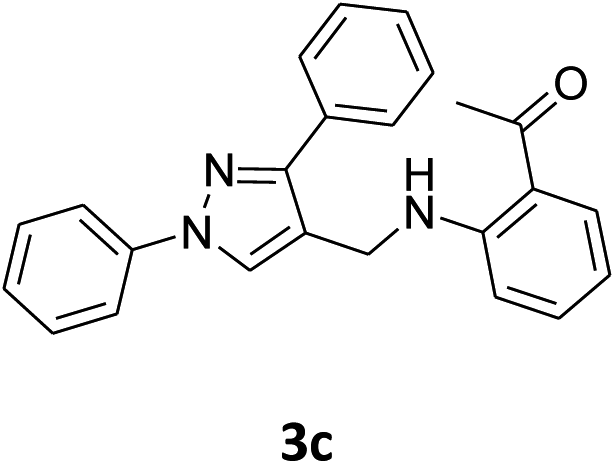 |
54% | 51% | 95% |
| 4 | 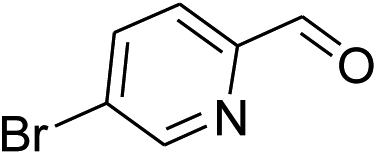 |
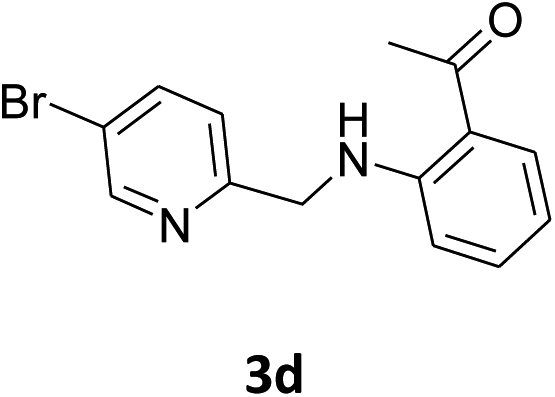 |
40% | 43% | 84% |
| 5 | 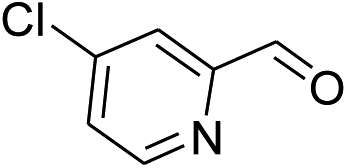 |
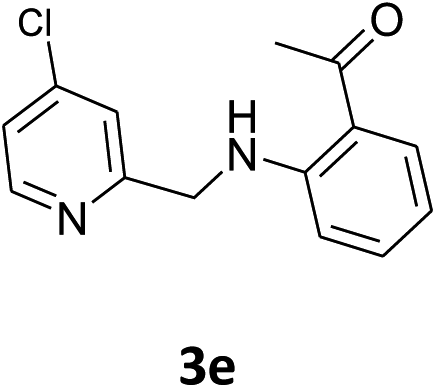 |
50% | 47% | 92% |
| 6 | 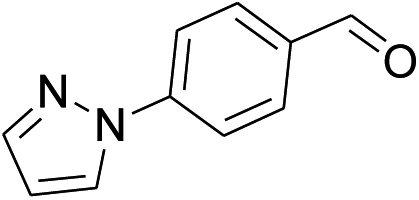 |
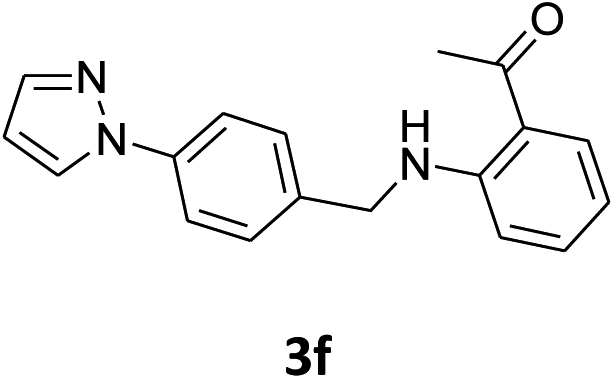 |
53% | 60% | 88% |
| 7 | 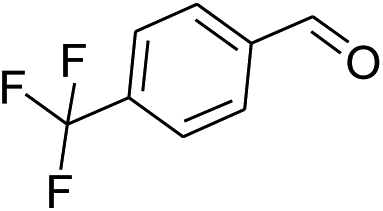 |
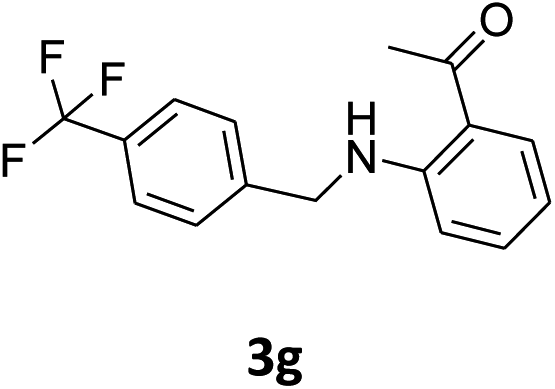 |
40% | 45% | 91% |
| 8 | 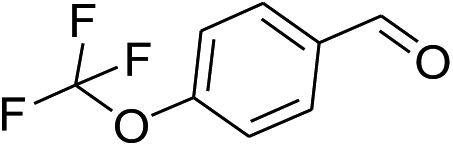 |
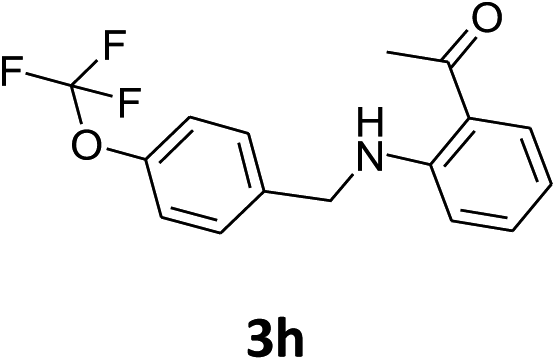 |
42% | 37% | 91% |
| 9 |  |
 |
34% | 41% | 85% |
| 10 |  |
 |
30% | 40% | 90% |
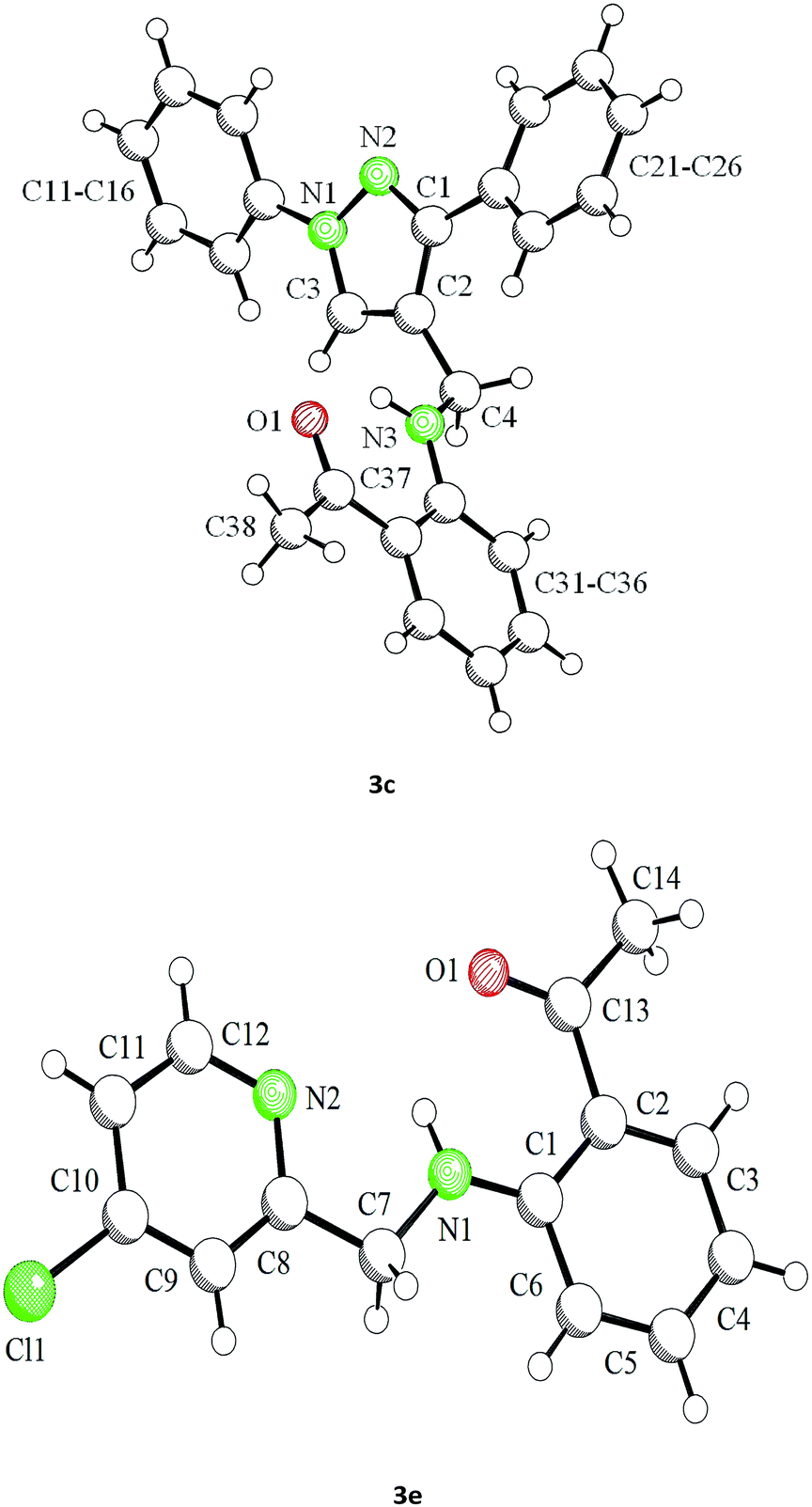 | ||
| Fig. 2 Molecular structures of 3c and 3e in the solid state (SCHAKAL plot). | ||
Further, we examined the reaction between 3g and diethyl oxalate to investigate the experimental conditions which include the optimization of base, solvent and temperature. As shown in Table 2, four different bases (entries 1–15) were examined in the presence of 2.5 eq. of diethyl oxalate where LiHMDS (entry 15) showed the highest reaction rate as compared to only 10% yield of target product (entry 12) observed in the presence of NaH. Other bases such as NaOMe and NatOBt were ineffective in producing any product. We also tested solvents in combination with different bases out of which, LiHMDS/THF combination gave the best results. The effect of temperature was also investigated in the presence of different combinations of bases and solvent and it was concluded that when temperature was raised from 0 °C to reflux temperature using LiHMDS/THF, the reaction gave the product in 56% yield.
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 3g (2.04 mmol), diethyloxalate (5.1 mmol), base (2.5 eq.), solvent (20 mL), 16 h.b The yields of isolated products.c No reaction. | ||||
| 1 | NaOMe | MeOH | 0 | NRc |
| 2 | NaOMe | MeOH | RT | NRc |
| 3 | NaOMe | MeOH | 80 | NRc |
| 4 | NaOMe | THF | 0 | NRc |
| 5 | NaOMe | THF | RT | NRc |
| 6 | NaOMe | THF | 80 | NRc |
| 7 | NatOBut | THF | 0 | NRc |
| 8 | NatOBut | THF | RT | NRc |
| 9 | NatOBut | THF | 80 | NRc |
| 10 | NaH | THF | 0 | NRc |
| 11 | NaH | THF | RT | NRc |
| 12 | NaH | THF | 80 | 10 |
| 13 | LiHMDS | THF | 0 | NRc |
| 14 | LiHMDS | THF | RT | NRc |
| 15 | LiHMDS | THF | 80 | 56 |
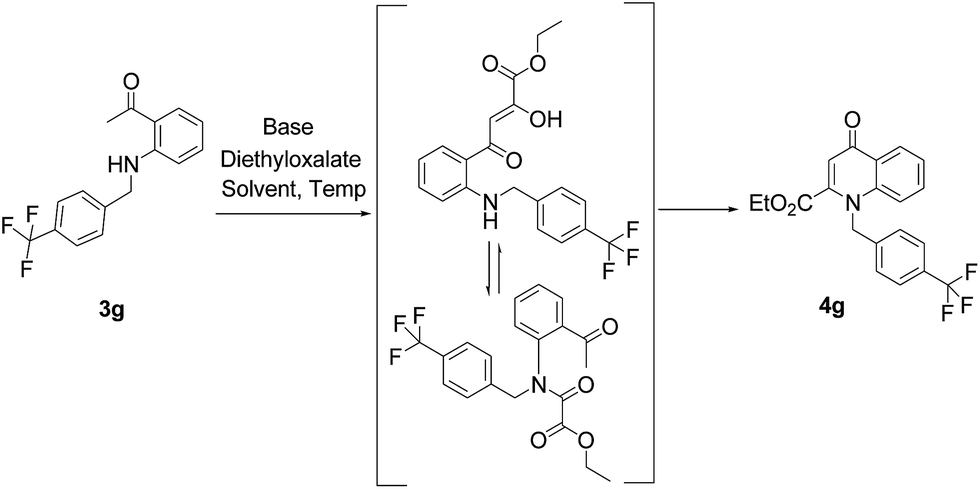
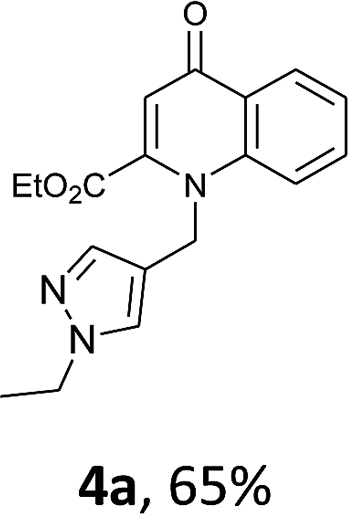
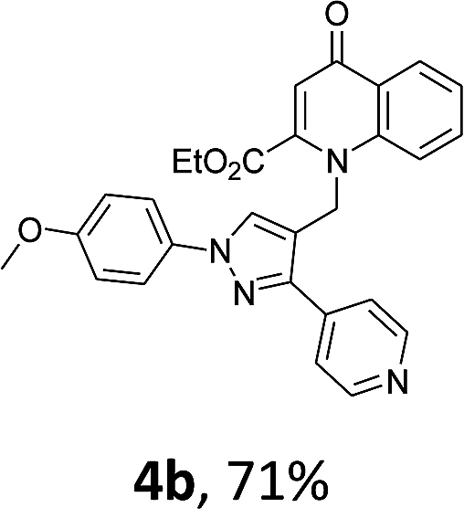
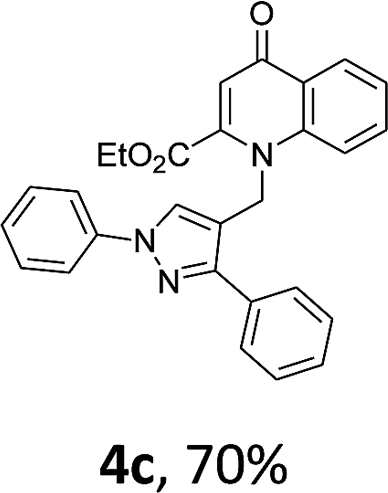
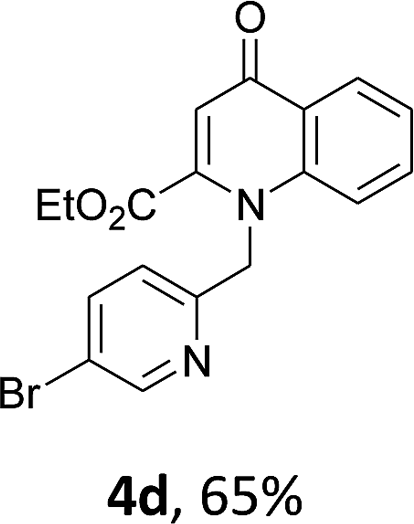
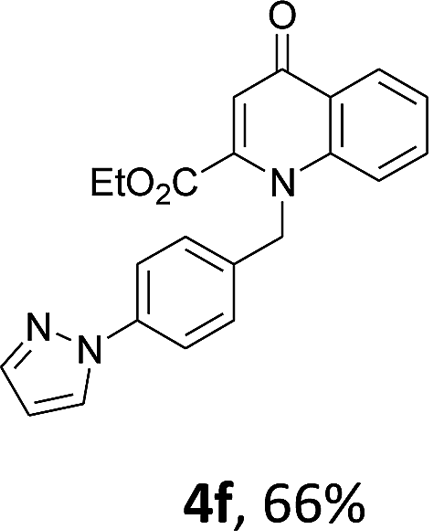
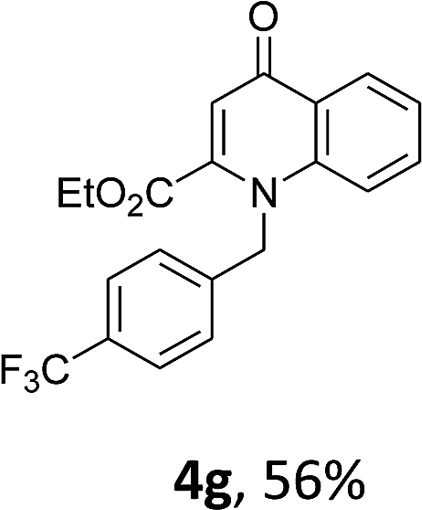
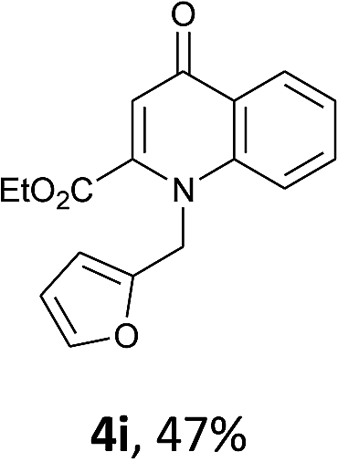
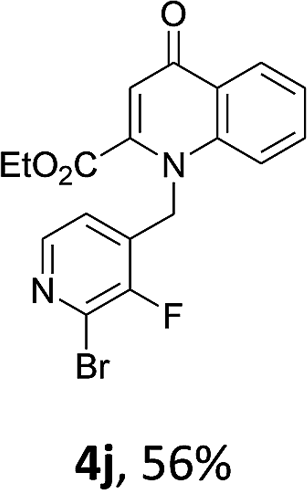
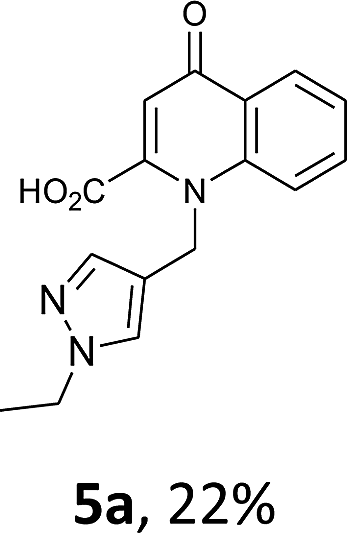
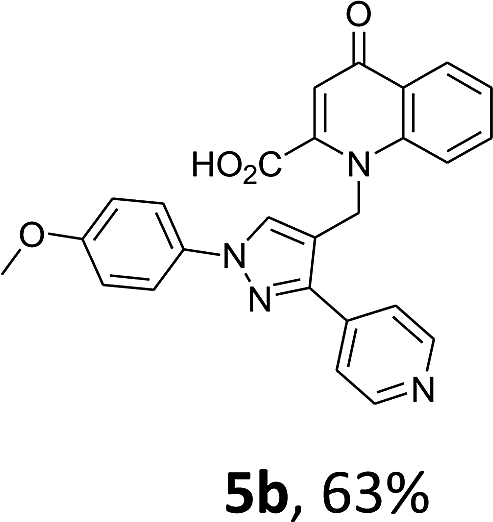
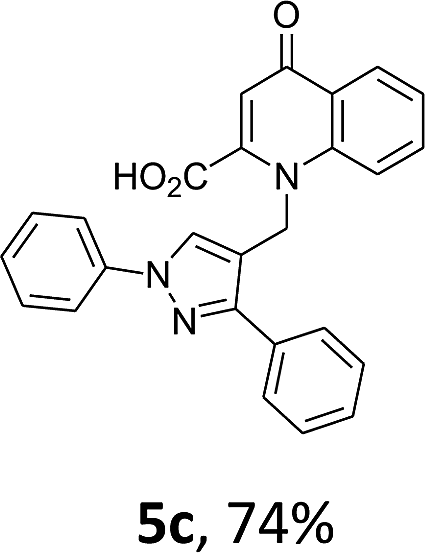
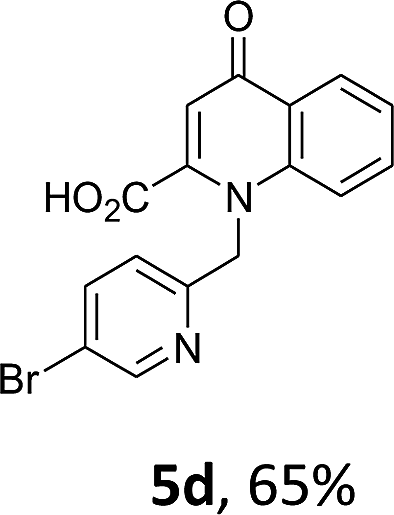
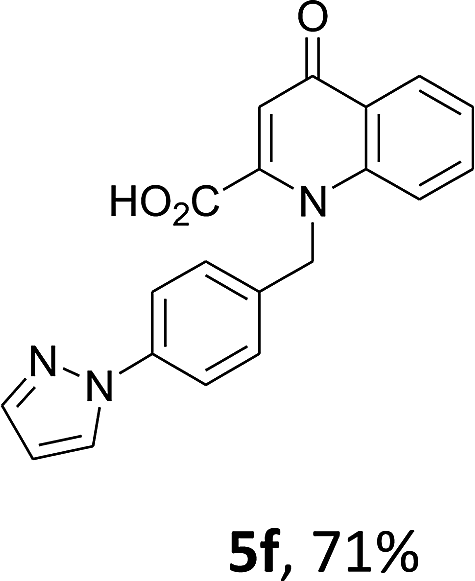
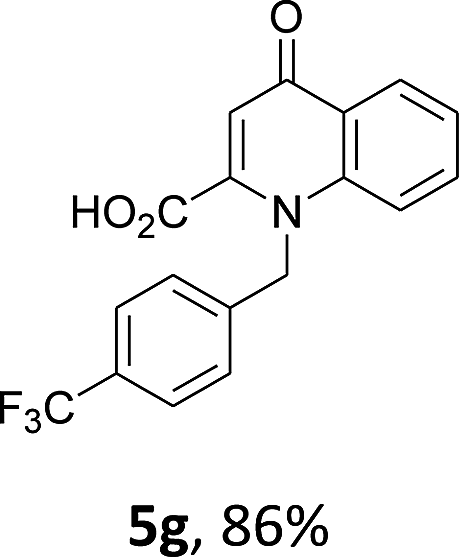
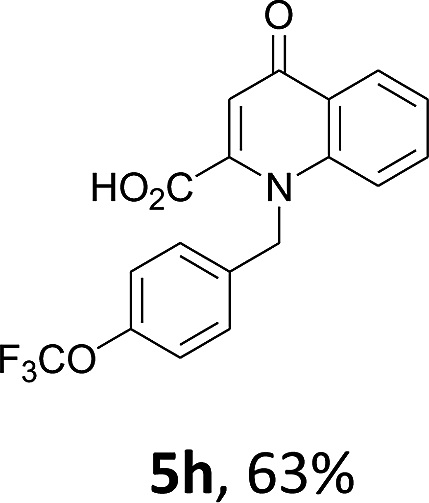
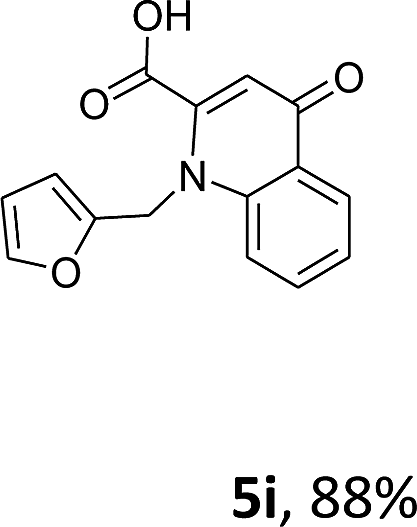
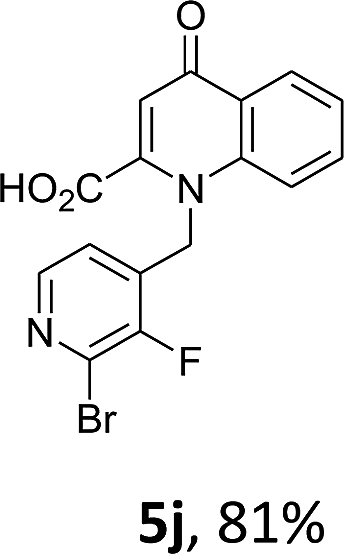
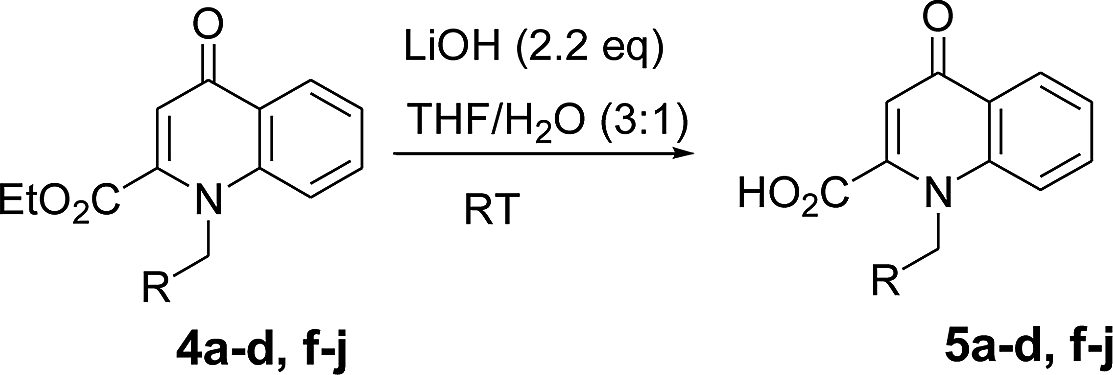
Using the optimized conditions, we next explored the scope of LiHMDS induced in situ cyclisation to yield N-substituted 4-quinolones. As shown in Table 3, the corresponding N-aryl-2-carboxy substituted 4-quinolones were obtained in moderate yield at reflux temperature with all the reductive amination products. The substrates bearing electron-withdrawing or electron-donating substituents on aryl as well as heteroaryl rings showed no marked difference in the transformation. However, introduction of furan ring resulted in the reduced yield of target compound, 4i which might be due to presence of lone pair of electron on oxygen which is making NH proton partially available for abstraction with base. Under the optimized conditions, substrate containing p-chloro pyridinyl, 3e was not tolerated as it might be due to the presence of chlorine at para position which is enhancing the electron density on the pyridine ring thus attracting the NH proton towards the nitrogen of pyridine through hydrogen bonding, therefore making it unavailable for abstraction even by the strong base. While, various other functionalities such as ethyl, methoxy, fluoro, bromo, trifluoromethoxy, trifluoromethyl were well tolerated under the standard conditions. Further hydrolysis of the ester substituted N-aryl-4-quinolones in the presence of LiOH/H2O/THF gave corresponding acid in moderate to good yield for 5b–d, 5f–j (Table 4). However, the low yield (22%) of 5a might be due to the quaternization of pyrazole ring during work up.
|
||
|---|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
 |
|
||
|---|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
 |
A plausible mechanism for in situ cyclocondensation step has been elucidated in Scheme 4. Initially, anion generated from the secondary amine in the presence of base attacks the carbonyl group of diethyloxalate to generate the N-acetylated product. Then the abstraction of α-proton from methylketone resulted in the generation of anion which in turn attack the amidic carbonyl carbon of the N-acetylated intermediate. Then under the basic conditions, cyclocondensation reaction takes place to form quinolone ring with the elimination of water molecule.
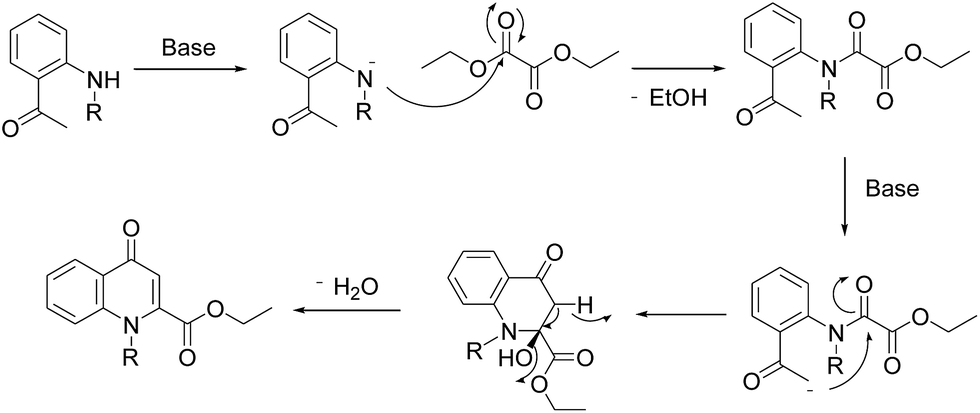 | ||
| Scheme 4 Possible mechanism for in situ cyclocondensation. | ||
Conclusions
In summary, we successfully developed a LiHMDS catalyzed protocol for C–N bond formation in one step via in situ cyclocondensation to give the title compounds. The secondary amines utilized for the formation of quinolones tolerated a wide variety of functional groups. The biological studies of these quinolones are under process and further applications of this protocol for broader substrate scope to explore the synthesis of other biologically active molecules is underway in our laboratory. We are also working on alternative methods to improve the yield as well as avoiding such harsh conditions.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Mohammad Abid gratefully acknowledges University Grant Commission (UGC), Govt. of India for RAMAN Postdoctoral Fellowship (F. No. 5-123/2016 (IC)) to work at Eppley Institute for Cancer Research, UNMC, USA. PH thanks Prof. Jyotindra Choudhary, HOD and Prof. Faiz Ahmad, P.G. Department of Chemistry, for their guidance and support. BA acknowledges BSR fellowship support from UGC, INDIA.Notes and references
- J. Huang, Y. Chen, A. O. King, M. Dilmeghani, R. D. Larsen and M. M. Faul, Org. Lett., 2008, 10, 2609–2612 CrossRef CAS PubMed.
- S. Gupta, P. Ghosh, S. Dwivedi and S. Das, RSC Adv., 2014, 4, 6254–6260 RSC.
- (a) S. C. Kuo, H. Z. Lee, J. P. Juang, Y. T. Lin, T. S. Wu, J. J. Chang, D. Lednicer, K. D. Paull and C. M. Lin, J. Med. Chem., 1993, 36, 1146–1156 CrossRef CAS PubMed; (b) M. Hadjeri, E. L. Peiller, C. Beney, N. Deka, M. A. Lawson, C. Dumontet and A. Boumendjel, J. Med. Chem., 2004, 47, 4964–4970 CrossRef CAS PubMed.
- S. Nakamura, M. Kozuka, K. F. Bastow, H. Tokuda, H. Nishino, M. Suzuki, J. Tatsuzaki, S. L. M. Natschke, S. C. Kuo and K. H. Lee, Bioorg. Med. Chem., 2005, 13, 4396–4401 CrossRef CAS PubMed.
- (a) N. Mahmoudi, L. Ciceron, J. F. Franetich, K. Farhati, O. Silvie, W. Eling, R. Sauerwein, M. Danis, D. Mazier and F. Derouin, Antimicrob. Agents Chemother., 2003, 47, 2636–2639 CrossRef CAS PubMed; (b) R. M. Cross, A. Monastyrskyi, T. S. Mutka, J. N. Burrows, D. E. Kyle and R. Manetsch, J. Med. Chem., 2010, 53, 7076–7094 CrossRef CAS PubMed.
- (a) D. T. Chung, C. Y. Tsai, S. J. Chen, L. W. Chang, C. H. R. King, C. H. Hsu, K. M. Chiu, H. C. Tan, Y. T. Chang and M. C. Hsu, Antimicrob. Agents Chemother., 2010, 54, 411–417 CrossRef CAS PubMed; (b) E. Meyer, F. Schwab, P. Gastmeier, H. Ruden and A. Heininger, J. Antimicrob. Chemother., 2007, 60, 619–624 CrossRef CAS PubMed.
- D. Edmont, R. Rocher, C. Plisson and J. Chenault, Bioorg. Med. Chem. Lett., 2000, 10, 1831–1834 CrossRef CAS PubMed.
- (a) B. A. Lucero, C. R. B. Gomes, I. C. P. P. Frugulhetti, L. V. Faro, L. Alvarenga, M. C. B. V. de-Souza, T. M. L. de-Souza and V. F. Ferreiraa, Bioorg. Med. Chem. Lett., 2006, 16, 1010–1013 CrossRef CAS PubMed; (b) M. Llinas-Brunet, M. D. Bailey, E. Ghiro, V. Gorys, T. Halmos, M. Poirier, J. Rancourt and N. Goudreau, J. Med. Chem., 2004, 47, 6584–6594 CrossRef CAS PubMed.
- (a) F. Santos, P. Abreu, H. Castro, I. Paixão, C. Cirne-Santos, V. Giongo, J. Barbosa, B. Simonetti, V. Garrido, D. Bou-Habib, D. Silva, P. Batalha, J. Temerozo, T. Souza, C. Nogueira, A. Cunha, C. Rodrigues, V. Ferreira and M. de Souza, Bioorg. Med. Chem., 2009, 17, 5476–5481 CrossRef CAS PubMed; (b) M. Sato, T. Motomura, H. Aramaki, T. Matsuda, M. Yamashita, Y. Ito, H. Kawakami, Y. Matsuzaki, W. Watanabe, K. Yamataka, S. Ikeda, E. Kodama, M. Matsuoka and H. Shinkai, J. Med. Chem., 2006, 49, 1506–1508 CrossRef CAS PubMed.
- J. Regan, T. W. Lee, R. M. Zindell, Y. Bekkali, J. Bentzien, T. Gilmore, A. Hammach, T. M. Kirrane, A. J. Kukulka, D. Kuzmich, R. M. Nelson, J. R. Proudfoot, M. Ralph, J. Pelletier, D. Souza, L. Zuvela-Jelaska, G. Nabozny and D. S. Thomson, J. Med. Chem., 2006, 49, 7887–7896 CrossRef CAS PubMed.
- Q. D. You, Z. Y. Li, C. H. Huang, Q. Yang, X. J. Wang, Q. L. Guo, X. G. Chen, X. G. He, T. K. Li and J. W. Chern, J. Med. Chem., 2009, 52, 5649–5661 CrossRef CAS PubMed.
- K. Drlica and X. Zhao, Microbiol. Mol. Biol. Rev., 1997, 3, 377–392 Search PubMed.
- Q. Li, A. Claiborne, T. Li, L. Hasvold, V. S. Stoll, S. Muchmore, C. G. Jakob, W. Gu, J. Cohen, C. Hutchins and D. Frost, Bioorg. Med. Chem. Lett., 2004, 14, 5367–5370 CrossRef CAS PubMed.
- Y. H. Chang, M. H. Hsu, S. H. Wang, L. J. Huang, K. Qian, S. L. Morris-Natschke, E. Hamel, S. C. Kuo and K. H. Lee, J. Med. Chem., 2009, 52, 4883–4891 CrossRef CAS PubMed.
- B. Medeiros, H. Landau, M. Morrow, R. Lockerbie, T. Pitts and S. Eckhardt, Leukemia, 2007, 21, 739–746 CAS.
- A. Golub, O. Yakovenko, V. Bdzhola, V. Sapelkin, P. Zien and S. Yarmoluk, J. Med. Chem., 2006, 49, 6443–6450 CrossRef CAS PubMed.
- (a) R. M. Cross and R. Manetsch, J. Org. Chem., 2010, 75, 8654–8657 CrossRef CAS PubMed; (b) C. P. Jones, K. W. Anderson and S. L. Buchwald, J. Org. Chem., 2007, 72, 7968–7973 CrossRef CAS PubMed.
- D. Zewge, C. Y. Chen, C. Deer, P. G. Dormer and D. L. Hughes, J. Org. Chem., 2007, 72, 4276–4279 CrossRef CAS PubMed.
- A. K. Pandey, R. Sharma, R. Shivahare, A. Arora, N. Rastogi, S. Gupta and P. M. S. Chauhan, J. Org. Chem., 2013, 78, 1534–1546 CrossRef CAS PubMed.
- F. Salvaggio, J. T. Hodgkinson, L. Carro, S. M. Geddis, W. R. J. D. Galloway, M. Welch and D. R. Spring, Eur. J. Org. Chem., 2016, 434–437 CrossRef CAS.
- I. Borza, S. Kolok, K. Galgóczy, A. Gere, C. Horváth, S. Farkas, I. Greiner and G. Domány, Bioorg. Med. Chem. Lett., 2007, 17, 406–409 CrossRef CAS PubMed.
- K. Y. Park, J. Lee, S. J. Park, J. N. Heo and H. J. Lim, Adv. Synth. Catal., 2015, 357, 3917–3926 CrossRef CAS.
- M. S. Shmidt, I. A. Perillo, A. Camelli, M. A. Fernández and M. M. Blanco, Tetrahedron Lett., 2016, 57, 1022–1026 CrossRef CAS.
- X. Chen, J. Klöckner, J. Holze, C. Zimmermann, W. K. Seemann, R. Schrage, A. Bock, K. Mohr, C. Tränkle, U. Holzgrabe and M. Decker, J. Med. Chem., 2014, 58, 560–576 CrossRef PubMed.
- J. L. Haesslein, I. Baholet, M. Fortin, A. Iltis, J. Khider, A. M. Periers, C. Pierre and J. P. Vevert, Bioorg. Med. Chem. Lett., 2000, 10, 1487–1490 CrossRef CAS PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897–2904 CrossRef CAS.
- R. Apodaca and W. Xiao, Org. Lett., 2001, 3, 1745–1748 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1470319 (3c) and 1470320 (3e). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28631c |
| ‡ Present address: Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 68198-6805, USA. |
| This journal is © The Royal Society of Chemistry 2017 |