
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-nanofibrillation strategy to an unusual combination of strength and toughness for poly(lactic acid)†
Lan Xie *,
Xin Sun,
Yaozhu Tian,
Fuping Dong,
Min He,
Yuzhu Xiong* and
Qiang Zheng
*,
Xin Sun,
Yaozhu Tian,
Fuping Dong,
Min He,
Yuzhu Xiong* and
Qiang Zheng
Department of Polymer Materials and Engineering, College of Materials and Metallurgy, Guizhou University, Guiyang 550025, China. E-mail: lancysmile@163.com; mm.lanxie@gzu.edu.cn; 932271187@qq.com
First published on 14th February 2017
Abstract
Despite various efforts made to toughen poly(lactic acid) (PLA), currently developed routes (e.g., blending and chain modification) are prominently hampered by economical concerns and strength–ductility trade-off dilemma. Here we disclose an additive-free, one-step approach to create numerous PLA nanofibrils with a high structural integrity and self-reinforcing effect. Specifically, adequately crystallized PLA was partially melted below the melting point and refined into tiny crystalline blocks by intensive extrusion. The chain networks crosslinked by shear-aligned crystals with high melt strength were stretched at drawing ratios as high as 20, conferring extremely ordered alignment in the stretching flow and straightforward nanofibrillation by PLA itself. The perfectly aligned nanofibrils featured an ultralow diameter approaching 80 nm and perfect alignment, contributing to direct formation of compact shish-kebabs with ordered chain packing and enhanced thermal stability. The unique structural features allowed self-nanofibrillar PLA to achieve an impressive combination of high strength and toughness, reaching a remarkable increase of 36%, 4077% and 3976% in tensile strength, elongation at break and toughness (63.7 MPa, 325.8% and 101.9 MJ m−3) compared to normal PLA, respectively. The profound morphology control, the simplicity of manufacturing, together with the versatility of multiple property improvements, should make the self-nanofibrillation strategy useful to guide the design of high-performance PLA with industrial feasibility.
Introduction
In the emerging bioplastics market, development of poly(lactic acid) (PLA) has been among the first to revolutionize and implement renewable materials solutions.1,2 Nevertheless, PLA generally falls into the category of “brittle materials” with impact strength below 5 kJ m−2 and elongation at break below 10%, imposing profound application restrictions for PLA.3–6 The quest to develop versatile PLA focuses increasingly on the promotion of ductility and toughness, essential to prompt broader applications ranging from biomedical scaffolds to drug and food packaging.7–10Existing efforts of improving the mechanical properties of PLA are in principle associated with incorporation of tough polymers and modification of chain flexibility,11–15 whereas the improvements made in ductility and toughness frequently lead to poor elasticity and low deformation resistance. This is exemplified by dramatic decline of tensile strength from 63 MPa for pure PLA to 30 MPa for PLA blended with 30 wt% poly(butylene succinate-co-adipate) (PBSA),16 while the tensile strength and Young's modulus of PLA witnessed over 40% and nearly 70% decrease after copolymerization with poly(ε-decalactone), respectively.17 In addition to the inferior property balance, the conventional routes usually involve the application of specific processing techniques and use of components that are high-cost or non-degradable. This brings to light four essential factors that are in special relation to the realistic development of high-performance PLA: (1) flexible technical demands for modification process with affordable production costs, (2) low environmental impact during the synthesis and fabrication, (3) readiness for scaled-up production, and (4) good balance for the strength–ductility–toughness property profiles. However, it is still challenging to simultaneously satisfy all the four requirements, despite some notable efforts made recently.18–21
As an endeavor to simultaneous promotion of strength and toughness for PLA, we conceptualize the crystal-directed nanofibrillation approach amenable to industrial production based on common processing techniques, strategically involving (1) isothermal annealing at 120 °C for 30 min to foster PLA crystals, (2) intensive extrusion at 165 °C to partially melt the less-ordered crystals, combined with strong shear flow to refine the crystalline domains, and (3) melt stretching with high drawing ratios to sufficiently orient the chains crosslinked by fine crystal blocks, readying the crystal-directed nanofibrillation along the flow field (Fig. 1a). We hypothesize that a considerable amount of ordered crystals survived during the extrusion are of critical importance to sustain high melt strength for high-speed stretching and to serve as precursors for subsequent formation of oriented new crystals during stretching (Fig. 1b). Once the stretched chain networks crosslinked by the aligned crystals are created, it is highly expected to trigger density fluctuation between the as-stretched networks and the neighboring base so as to remold a non-steady phase structure, laying down the prerequisite to generate nanofibrillar textures. This assumption is supported by the supramolecular model by Dorgan et al., pointing out the essential units of highly oriented crystals and amorphous chains for the microfibrillar structures formed in melt-spun PLA fibers.22,23
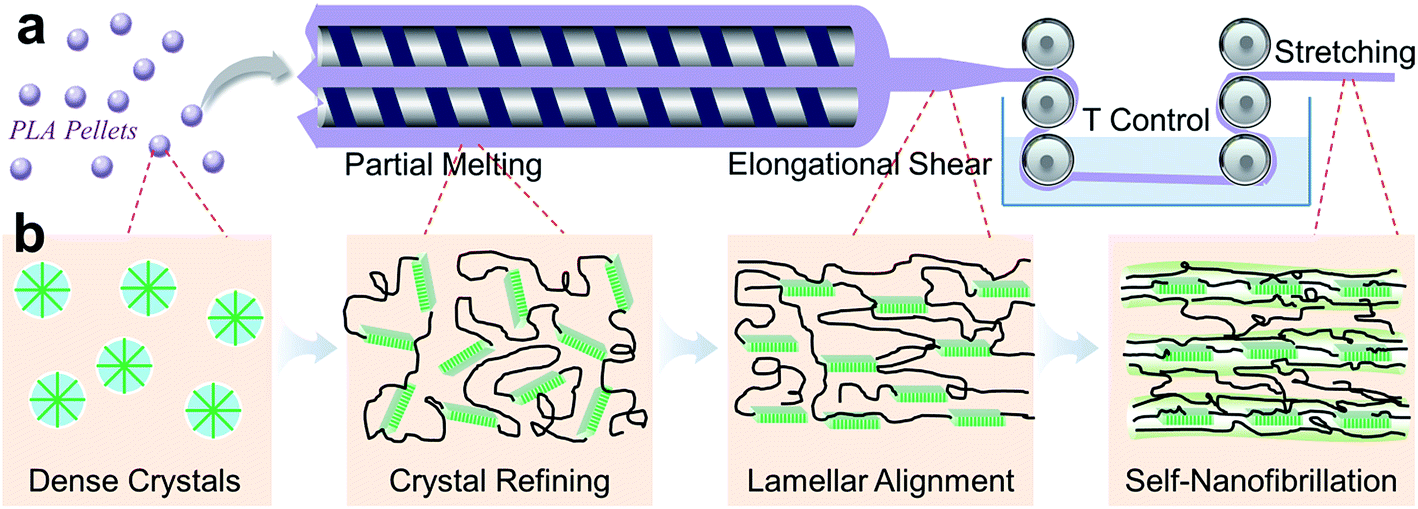 | ||
| Fig. 1 Synthetic roadmap to produce self-nanofibrillar PLA. (a) Annealed PLA was partially melted and intensively sheared in a twin-screw extruder featuring relatively low temperatures, followed by high-speed stretching to align PLA lamellae and chains and rapid quenching to freeze the as-formed nanofibrils. (b) Schematic illustration of structural evolution during the proposed self-nanofibrillation process. The shear-refined PLA crystals, together with the PLA chains crosslinked by neighboring lamella blocks, are aligned in the stretching flow, leading to the formation of nanofibrils comprising oriented chain bundles and lamellae. | ||
Results and discussion
The intensity gradient of melt stretching was controlled by fixing the drawing ratios at 1, 10 and 20 to produce normal PLA, moderately drawn PLA (MD PLA) and highly drawn PLA (HD PLA), respectively. Our hypothesis on the structural transformation of PLA by self-nanofibrillation strategy was examined by scanning electronic microscopy (SEM) observation (Fig. 2). It is of interest to observe that nanosized fibrils of extremely high density were perfectly aligned along the stretching flow in both MD PLA and HD PLA. The weak dependence on drawing ratio was essentially attributed to the robust execution of nanofibrillation process by the proposed approach, readying the industrial feasibility for scaled-up fabrication of nanofibrillar structures for PLA. The nanofibrils were characterized by ultralow diameter, arriving at 95 ± 12 nm and 82 ± 5 nm for MD PLA and HD PLA, respectively. The variations of diameter and its distribution with drawing ratios manifested that the nanofibrillar textures could be directly controlled in the process of nanofibrillation. The ultralow size likely conferred PLA nanofibrils with large specific surface area and high surface activity, encouraging the creation of strong interactions with neighboring nanofibrils and non-fibrillar base.24,25 It explains the generation of tight ligaments that interlocked the adjacent nanofibrils, probably acting as important contributors to the structural integrity of self-nanofibrillar PLA. Aligning within the non-fibrillar base, nanochannels with uniform spatial distribution were elongated along the stretching flow to space the adjacent nanofibrils, which were of high potential to function as energy-dispersive elements.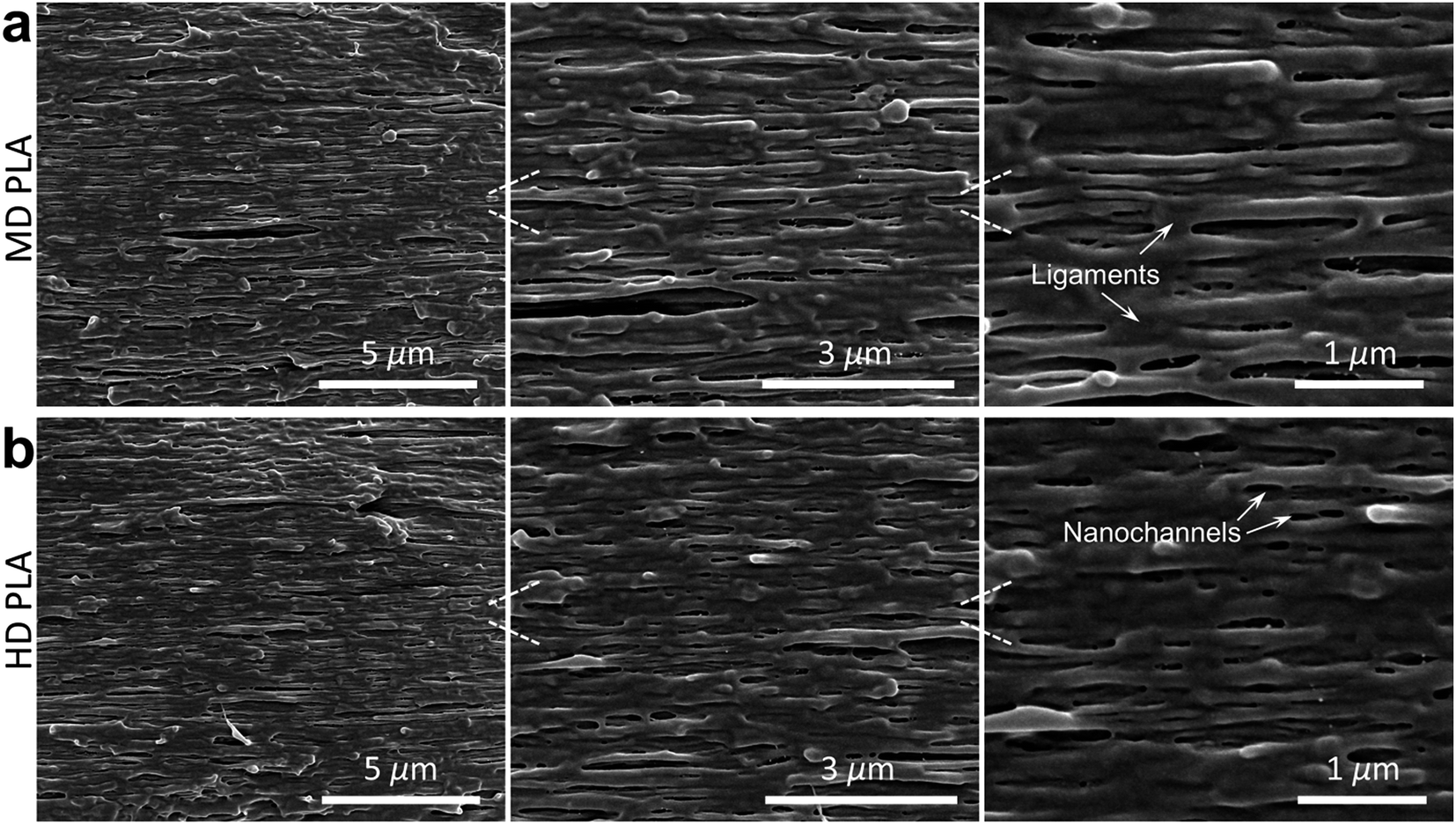 | ||
| Fig. 2 Morphological features of PLA nanofibrils. SEM images of cryogenically fractured surfaces examining the formation of compact and perfectly ordered nanofibrils spaced by fine nanochannels in (a) MD PLA and (b) HD PLA. The stretching flow is horizontal. | ||
In the pursuit of high-strength polymeric materials, generation of fibrillar extended structures is of significance to mechanical property improvements, yet challenged by the lack of available systems and accessible processing routes.26–28 The quest to develop nanofibrillar structures focuses mainly on two- and multi-component polymer blends with poor miscibility, e.g., polypropylene/poly(ethylene terephthalate) blends.29–31 The flexibility of suitable dispersed phase is further restricted when attempting to develop nanofibrillar reinforcements for PLA. Recently, poly(butylene succinate) (PBS),25,32 polyamide 6 (PA6)33 and poly(butylene adipate-co-terephatalate) (PBAT)34 have been converted to microsized or even nanosized fibrils in PLA matrix. It is pertinent to point out that the currently accessible routes are usually related to complex processing scenarios and specific technical demands. As an example, extrusion compounding followed by cold drawing and annealing treatment was used to create PBAT microfibrils in PLA matrix,34 while hot stretching and compression molding of extruded PLA/PA6 blends were used to produce microfibrils.33 The self-nanofibrillation strategy proposed here hold distinct advantages that are unattainable with conventional methods: (1) ease of processing by one-step nanofibrillation, (2) no use of other components that are non-degradable or high-cost, (3) facile control of the nanofibrillar texture. The strategy is therefore of significance to prompt high flexibility for the design of nanofibrillar structures in PLA materials.
Fig. 3 offers insights into the profound modification of crystalline morphology for self-nanofibrillar PLA. Due to the intrinsic poor crystallization ability of PLA, normal PLA showed nearly amorphous state without any trace of crystals (Fig. S3†), in agreement with the common scenarios documented for PLA-based materials.35,36 Of particular interest was the formation of compact crystalline domains of high structural regularity in MD PLA and HD PLA (Fig. 3a and b). Numerous thread-like crystal fibrils with length as high as several micrometers were perfectly extended along the stretching flow, perpendicular to which the oriented lamellae were orderly induced.37 It is worth stressing that the lamellae featuring extraordinarily high density were tightly embedded into adjacent crystal fibrils, making it difficult to distinguish individual entities from each other. The structure elucidation allows us to assume that classic shish-kebab superstructure was induced by PLA nanofibrils. This assumption is supported by structural determination by two-dimensional wide-angle X-ray diffraction (2D-WAXD) and two-dimensional small-angle X-ray scattering (2D-SAXS) in Fig. 3c–e. Several pairs of symmetric arc-like diffraction patterns in 2D-WAXD images indicated that a large amount of oriented crystals were created in MD PLA and HD PLA,38 in sharp contrast to the homogeneous halo reflecting low crystallinity and orientation degree for normal PLA (Fig. 3c and S6†). It is well in line the 2D-SAXS patterns (Fig. 3d), displaying distinct lobes for self-nanofibrillar PLA especially HD PLA, as a result of oriented crystalline domains.39 The combination of SEM, 2D-WAXD and 2D-SAXS results thus evidenced that the formation of classic shish-kebabs were induced in self-nanofibrillar PLA. Fig. 3e gives some interesting results for the structural determination of PLA shish-kebabs. Prominent peaks centered at q = 0.259 nm−1 and 0.273 nm−1 were recorded exclusively for the 1D-SAXS intensity curves of MD PLA and HD PLA, respectively, yielding the values of 24.3 nm and 23.0 nm for the long period, respectively.40 The decrease of 1.3 nm in the long period as increasing the drawing ratio was presumably due to the chain packing of higher order under the intensified stretching flow,39,41 in line with the SEM observation (Fig. 3a and b). Differential scanning calorimetry (DSC) heating traces in Fig. 3f delineate that the thermal properties of PLA were significantly promoted with the existence of nanofibril-induced shish-kebabs. The glass transition temperature (Tg) witnessed an increase of 1.4 °C for MD PLA and 2.1 °C for HD PLA compared to normal PLA (59.0 °C), accompanied by substantial increase of nearly 7 °C in melting point (Tm) for self-nanofibrillar PLA (around 171.7 °C). The rise of Tg and Tm was primarily attributed to the extended alignment of chains and crystalline blocks in self-nanofibrillar PLA, giving rise to constrained chain mobility and enhanced thermal stability.42–45
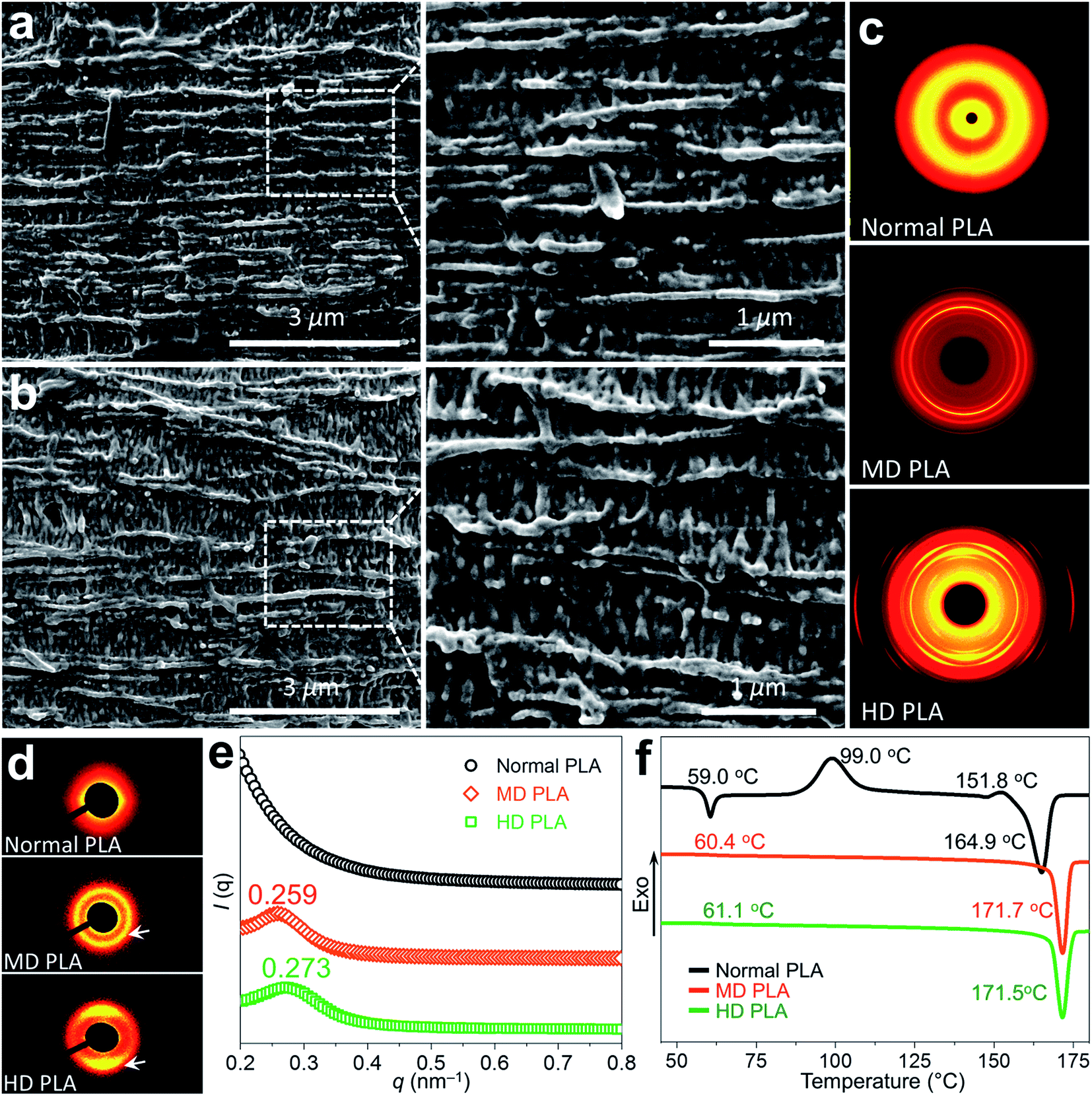 | ||
| Fig. 3 Crystalline morphology in self-nanofibrillar PLA. SEM images of (a) MD PLA and (b) HD PLA showing the formation of highly ordered shish-kebabs. (c) 2D-WAXD patterns demonstrating amorphous state for normal PLA and dense oriented crystals in self-nanofibrillar PLA. (d) 2D-SAXS images and (e) 1D-SAXS intensity profiles of normal PLA and self-nanofibrillar PLA. (f) DSC heating curves indicating the decreased chain mobility and increased crystallinity and melting points of PLA after self-nanofibrillation. | ||
Long been perceived as excellent self-reinforcements of polymeric materials, shish-kebab superstructures have been particularly attractive to both the technical community and the academic researchers.46–49 Generally shish-kebabs are created in traditional polyolefins like polyethylene and polypropylene, normally involving the application of flow fields by shearing, stretching and spinning the polymer melts or solutions.50–55 The structural manipulation is in favor of mechanical property improvements for PLA-based materials, yet quite challenging to achieve, mainly ascribed to two reasons: (1) relatively rigid chain backbone undesirable for molecular motion and orientation along the flow fields, (2) insufficiently high chain length to suppress the relaxation and disengagement of oriented precursors.56 It appears that, when targeting self-reinforcing merit, the property improvements of PLA are normally restricted to limited shish-kebab formation or chain degradation under prohibitively intense fields.56–58 By one-step nanofibrillation, extended nanofibrils of high length and density rendered the formation of numerous shish-kebabs with high structural integrity, opening up new possibilities for facile fabrication of PLA shish-kebabs as self-reinforcing elements.
Large improvements in mechanical properties of self-nanofibrillar PLA arising from controlled self-nanofibrillation are illustrated in Fig. 4. It is apparent from the stress–strain curves that normal PLA was directly fractured near the yield point after limited extension of below 8% (Fig. 4a and b), following the typical brittle deformation mode of PLA-based materials.12,59 In sharp contrast, self-nanofibrillation in PLA triggered the brittle-to-ductile transition, displaying extremely large plastic deformation with elongation approaching 150% for MD PLA and over 300% for HD PLA. It is moreover unexpected to observe the strain hardening for HD PLA before catastrophic fracture (in the range of 300–325% strain), which indicated the varied mechanism to strengthen the resistance capability to tensile deformation, probably involving the alignment of nanofibrils, orientation of recrystallized lamellae, distortion of crystal lattices and extension of chain conformations. While strain hardening behavior is generally documented for polyolefins with rubber elasticity like polyethylene and polycarbonate,60,61 the unusual occurrence in self-nanofibrillar PLA may provide unique structural database for theoretical understanding on the strain-hardening mechanisms of glassy polymers.
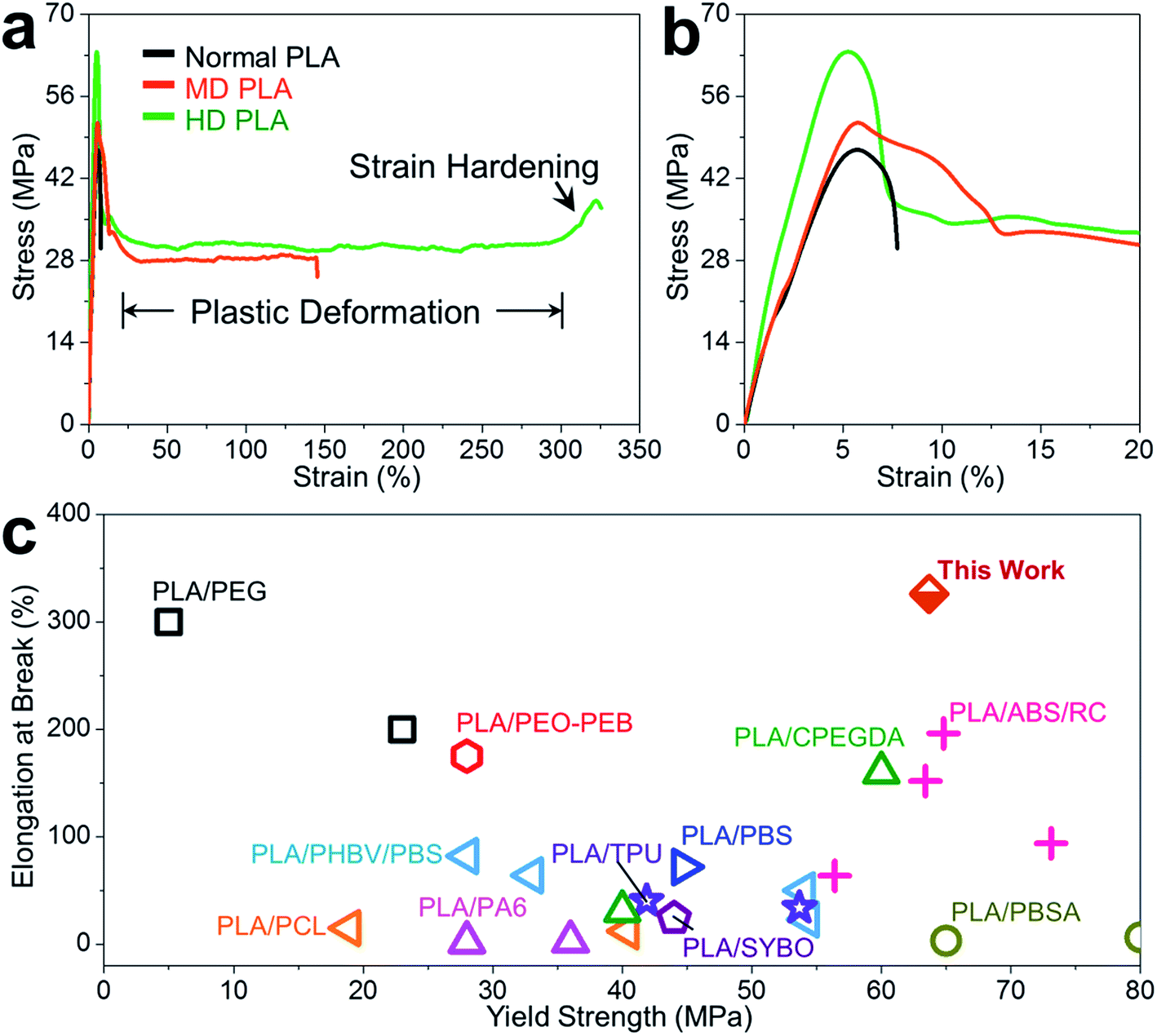 | ||
| Fig. 4 Combination of strength and ductility for self-nanofibrillar PLA. Typical stress–strain curves of self-nanofibrillar PLA: (a) in full range, (b) within 20% strain. (c) Property space for tensile ductility versus yield strength for PLA toughened by our nanofibrillation strategy and blending with PEG,62 poly(ethylene oxide)-b-poly(butylene oxide) (PEO-PEB),59 acrylonitrile–butadiene–styrene copolymer (ABS) compatibilized by reactive comb (RC) polymers,11 crosslinked PEG diacylate (CPEGDA),68 PHBV/PBS,5 PCL,63 PA6,33 soybean oil (SYBO),69 PBS,70 thermoplastic polyurethane (TPU)71 and PBSA.64 | ||
Optimization of the strength–ductility–toughness property profile of PLA by self-nanofibrillation is well illustrated by the key tensile property criteria (Table S1†). The improvement in the extensibility of self-nanofibrillar PLA was particularly striking: a 41-fold increase of elongation at break was achieved for HD PLA (325.8%) from the lowest point of normal PLA (7.8%). The ductility distinction was in line with the astounding increase of 3976% in the toughness for HD PLA (101.9 MJ m−3), as calculated by the area under the measured stress–strain curves that indicates energy dissipation during tensile deformation.25 Equally desirable was the simultaneous promotion of strength and stiffness, as exemplified by the substantially increased yield strength and Young's modulus for HD PLA (63.7 MPa and 2193 MPa) compared to the lowest values of normal PLA (46.9 MPa and 1316 MPa). To highlight the anomaly in the mechanical property improvements by self-nanofibrillation, we compared the ductility–strength property space for PLA toughened by our self-nanofibrillation strategy and conventional blending method (Fig. 4c). As distinguished from the poor property balance in conventional PLA blends, self-nanofibrillar PLA stands out with clearly highest elongation at break and yield strength. The strength–ductility trade-off dilemma is usually encountered when attempting to develop PLA blends, as exemplified by large decrease of strength and stiffness for PLA plasticized by poly(ethylene glycol) (PEG)62 and polycaprolactone (PCL),63 as well as evident decline in ductility and toughness for PLA/PBSA64 and PLA/poly(3-hydroxybutyrateco-hydroxyvalerate) (PHBV)/PBS blends.5 In addition to the unprecedented combination of high strength and ductility for PLA, the proposed additive-free strategy provides distinct advantages related to full degradability and ease of processing, holding great potential to fashion a new paradigm for PLA modification. Beyond the traditional melt-spun PLA in the form of fibers,65–67 the self-nanofibrillar PLA marks another useful case for facilitating the industrial revolution of multiform PLA materials.
The deformation mechanisms underlying the mechanical robustness of self-nanofibrillar PLA were appraised from direct observation of fracture surfaces after tensile failure (Fig. 5a–c). In contrast to the smooth and flat topography in normal PLA arising from the intrinsic brittleness (Fig. 5a), rubber-like plastic deformation acted as the predominant fracture mode with the existence of PLA nanofibrils independent of stretching ratios (Fig. 5b and c). It is important to note that the nanofibril bundles were highly extended and evolved into sheet-like tenacious ligaments. The plastically deformed zones were featured by high density of extended ligaments and elongated nanovoids enclosing the extended nanofibrils. The slit-like nanovoids with a wide range of diameter distribution from tens to hundreds of nanometer were uniformly embedded in the highly deformed base, likely serving as numerous energy-dispersive catalysts by localizing, transferring and redistributing the penetrating stress.72,73 The remarkable microstructure features in self-nanofibrillar PLA that rendered the combination of high elasticity and extensibility are of particular significance, and the underlying mechanisms are interpreted in Fig. 5d. The aligned nanofibrils comprising amorphous chains and ordered crystals are interspaced by fine channels and inter-tangled by compact kebabs. These features allow facilitation of stress transfer through the nanofibrils during the initial elastic deformation, triggering the extensive alignment of amorphous chains and neighboring crystals.32,74 The oriented chains crosslinked by dense crystals along the nanofibrils exhibit high strength and elasticity to arrest, localize and resist the penetrating stress so as to increase the resistance to yielding.75 The additional elastic energy dissipating mechanism is triggered by the formation of nanovoids in the channel base and the anchoring interactions between adjacent nanofibrils. Upon increasing deformation after the yield point, the consumption of deformable phase is accommodated principally by structural regularization within the nanofibrils and nanochannels, encouraging a series of energy-dissipating mechanisms: (1) amorphization of excessively extended crystals followed by reorientation of newly unfolded chains,76 (2) formation and growth of uniform nanovoids to deflect crack tips and constrain crack propagation,77 (3) deflection and reorientation of kebabs protruding from the nanofibrils, (4) friction between interactional nanofibrils and channels due to the displacement incompatibility, (5) gradual evolution into longer, thinner and stronger ultrafine nanofibrils. The synergistic mechanisms occurring in self-nanofibrillar PLA contribute to the combination of strength and toughness until the entire consumption of deformable phase.
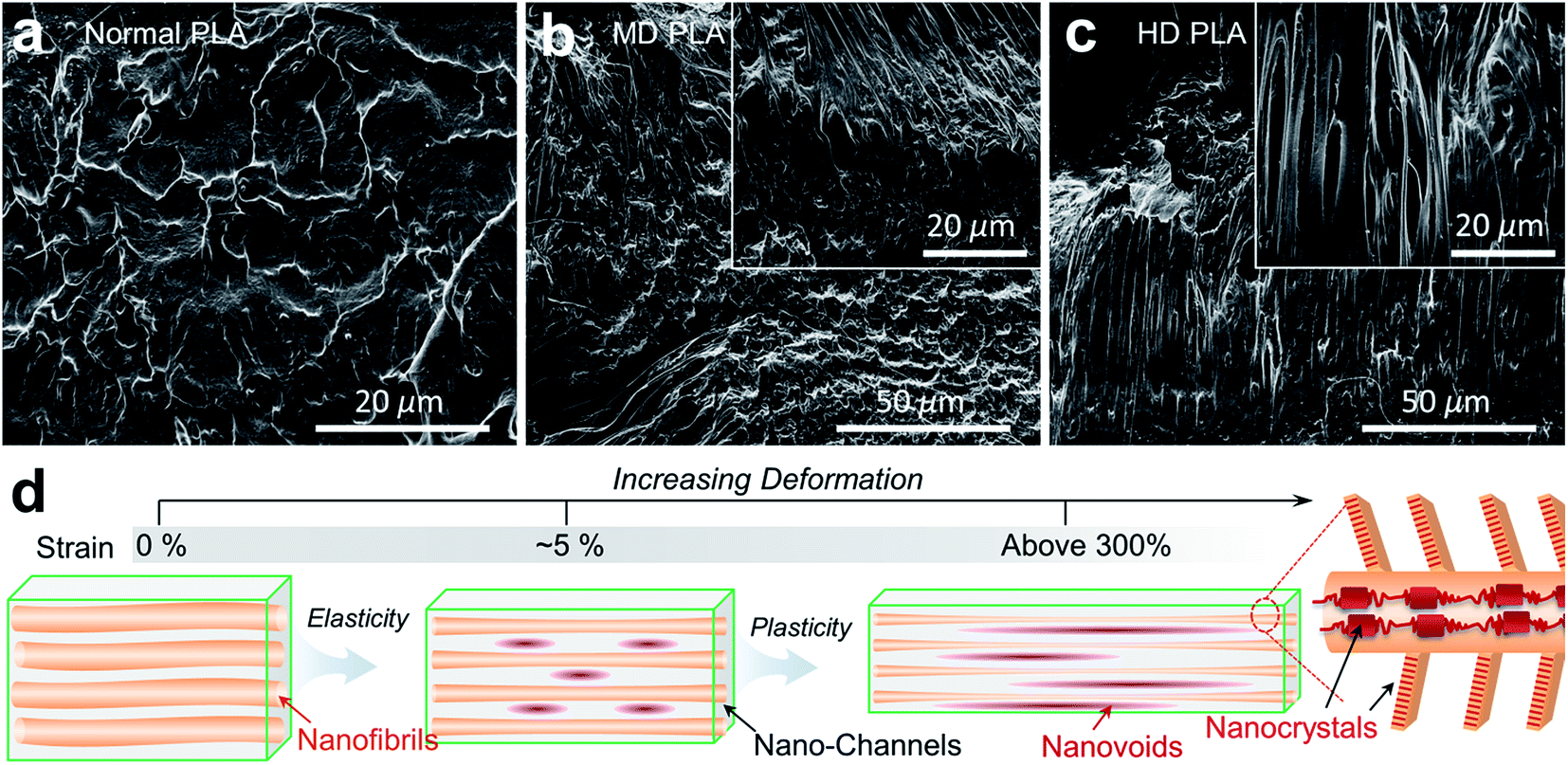 | ||
| Fig. 5 Toughening mechanisms in self-nanofibrillar PLA. (a–c) SEM images of fracture surfaces after tensile failure showing a large quantity of rubber-like plastic deformation in self-nanofibrillar PLA. (d) Schematic illustration explaining the mechanism of elasticity and plasticity for self-nanofibrillar PLA. The oriented chains and nanocrystals within the aligned nanofibrils are responsible for high elastic resistance, while a large quantity of deformation is accommodated by growth of nanovoids, amorphization of nanocrystals and chain alignment. | ||
Conclusions
Mechanically strong and tough PLA was realized by directly creating nanofibrillar textures of high density and structural integrity under industrial conditions of processing. The key element was intensive extrusion slightly below the melting point of PLA, rendering a considerable amount of shear-aligned crystal blocks to crosslink chain networks. It enabled creation of non-equilibrium phase states between the as-stretched networks and the neighboring base during the melt stretching process, which was of particular importance to direct nanofibrillation by PLA itself. The highly ordered nanofibrils featuring ultralow diameter of ∼80 nm were perfectly aligned along the stretching flow, exhibiting high surface activity to induce the formation of dense shish-kebabs with oriented alignment. The unique structural features conferred simultaneous promotion of strength, ductility and toughness for self-nanofibrillar PLA, achieving remarkable increase of 36%, 4077% and 3976% for the tensile strength, elongation at break and toughness of HD PLA (63.7 MPa, 325.8% and 101.9 MJ m−3) compared to normal PLA, respectively. This work, by strategically controlling the phase structure and crystalline morphology in self-nanofibrillar PLA, paves a facile yet effective way to high-performance PLA with well-balanced strength, ductility and toughness.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The National Natural Science Foundation of China (21604016) and National Undergraduate Innovation Training Program (201610657004) are acknowledged for the financial support. The synchrotron X-ray measurements were performed in Shanghai Synchrotron Radiation Facility (SSRF).References
- A. Höglund, K. Odelius and A.-C. Albertsson, ACS Appl. Mater. Interfaces, 2012, 4, 2788–2793 Search PubMed.
- T. Tsujimoto and H. Uyama, ACS Sustainable Chem. Eng., 2014, 2, 2057–2062 CrossRef CAS.
- T. Lebarbé, E. Grau, B. Gadenne, C. Alfos and H. Cramail, ACS Sustainable Chem. Eng., 2015, 3, 283–292 CrossRef.
- V. Ojijo, S. S. Ray and R. Sadiku, ACS Appl. Mater. Interfaces, 2013, 5, 4266–4276 CAS.
- K. Zhang, A. K. Mohanty and M. Misra, ACS Appl. Mater. Interfaces, 2012, 4, 3091–3101 CAS.
- D. Li, H. Sun, L. Jiang, K. Zhang, W. Liu, Y. Zhu, J. Fangteng, C. Shi, L. Zhao, H. Sun and B. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 9402–9410 CAS.
- G. Li, M. Lamberti, D. Pappalardo and C. Pellecchia, Macromolecules, 2012, 45, 8614–8620 CrossRef CAS.
- A.-L. Esquirol, P. Sarazin and N. Virgilio, Macromolecules, 2014, 47, 3068–3075 CrossRef CAS.
- R.-Y. Bao, W.-R. Jiang, Z.-Y. Liu, W. Yang, B.-H. Xie and M.-B. Yang, RSC Adv., 2015, 5, 34821–34830 RSC.
- Y. Huang, R. Chang, L. Han, G. Shan, Y. Bao and P. Pan, ACS Sustainable Chem. Eng., 2015, 4, 121–128 CrossRef.
- W. Dong, M. He, H. Wang, F. Ren, J. Zhang, X. Zhao and Y. Li, ACS Sustainable Chem. Eng., 2015, 3, 2542–2550 CrossRef CAS.
- W. Dong, H. Wang, F. Ren, J. Zhang, M. He, T. Wu and Y. Li, ACS Sustainable Chem. Eng., 2016, 4, 4480–4489 CrossRef CAS.
- Z. Liu, Y. Luo, H. Bai, Q. Zhang and Q. Fu, ACS Sustainable Chem. Eng., 2015, 4, 111–120 CrossRef.
- T. Li, J. Zhang, D. K. Schneiderman, L. F. Francis and F. S. Bates, ACS Macro Lett., 2016, 5, 359–364 CrossRef CAS.
- J. Zhang, T. Li, A. M. Mannion, D. K. Schneiderman, M. A. Hillmyer and F. S. Bates, ACS Macro Lett., 2016, 5, 407–412 CrossRef CAS.
- V. Ojijo, H. Cele and S. Sinha Ray, Macromol. Mater. Eng., 2011, 296, 865–877 CrossRef CAS.
- A. M. Mannion, F. S. Bates and C. W. Macosko, Macromolecules, 2016, 49, 4587–4598 CrossRef CAS.
- H. Bai, H. Xiu, J. Gao, H. Deng, Q. Zhang, M. Yang and Q. Fu, ACS Appl. Mater. Interfaces, 2012, 4, 897–905 CAS.
- V. Arias, J. Odent, J.-M. Raquez, P. Dubois, K. Odelius and A.-C. Albertsson, ACS Sustainable Chem. Eng., 2016, 4, 3757–3765 CrossRef CAS.
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2016, 4, 2899–2916 CrossRef CAS.
- B. H. Tan, J. K. Muiruri, Z. Li and C. He, ACS Sustainable Chem. Eng., 2016, 4, 5370–5391 CAS.
- J. A. Cicero, J. R. Dorgan, J. Garrett, J. Runt and J. S. Lin, J. Appl. Polym. Sci., 2002, 86, 2839–2846 CrossRef CAS.
- J. A. Cicero, J. R. Dorgan, J. Janzen, J. Garrett, J. Runt and J. S. Lin, J. Appl. Polym. Sci., 2002, 86, 2828–2838 CrossRef CAS.
- H. Xu, X. Yang, L. Xie and M. Hakkarainen, Biomacromolecules, 2016, 17, 985–995 CrossRef CAS PubMed.
- L. Xie, H. Xu, L.-B. Li, B. S. Hsiao, G.-J. Zhong and Z.-M. Li, Sci. Rep., 2016, 6, 34572 CrossRef CAS PubMed.
- M. Evstatiev and S. Fakirov, Polymer, 1992, 33, 877–880 CrossRef CAS.
- R. J. Shields, D. Bhattacharyya and S. Fakirov, J. Mater. Sci., 2008, 43, 6758–6770 CrossRef CAS.
- S. Fakirov, Macromol. Mater. Eng., 2013, 298, 9–32 CrossRef CAS.
- M. Evstatiev, S. Fakirov, J. Schultz and K. Friedrich, Polym. Eng. Sci., 2001, 41, 192–204 CAS.
- S. Fakirov, Compos. Sci. Technol., 2013, 89, 211–225 CrossRef CAS.
- K. Friedrich, M. Evstatiev, S. Fakirov, O. Evstatiev, M. Ishii and M. Harrass, Compos. Sci. Technol., 2005, 65, 107–116 CrossRef CAS.
- L. Xie, H. Xu, B. Niu, X. Ji, J. Chen, Z.-M. Li, B. S. Hsiao and G.-J. Zhong, Biomacromolecules, 2014, 15, 4054–4064 CrossRef CAS PubMed.
- A. R. Kakroodi, Y. Kazemi, W. Ding, A. Ameli and C. B. Park, Biomacromolecules, 2015, 16, 3925–3935 CrossRef CAS PubMed.
- M. Evstatiev, S. Simeonova, K. Friedrich, X.-Q. Pei and P. Formanek, J. Mater. Sci., 2013, 48, 6312–6330 CrossRef CAS.
- N. Rahman, T. Kawai, G. Matsuba, K. Nishida, T. Kanaya, H. Watanabe, H. Okamoto, M. Kato, A. Usuki, M. Matsuda, K. Nakajima and N. Honma, Macromolecules, 2009, 42, 4739–4745 CrossRef CAS.
- H.-Y. Yin, X.-F. Wei, R.-Y. Bao, Q.-X. Dong, Z.-Y. Liu, W. Yang, B.-H. Xie and M.-B. Yang, ACS Sustainable Chem. Eng., 2015, 3, 654–661 CrossRef CAS.
- A. Mahendrasingama, D. J. Blundella, M. Partona, A. K. Wrighta, J. Rasburnb, T. Narayananc and W. Fullera, Polymer, 2005, 46, 6009–6015 CrossRef.
- Y. Furuhashi, Y. Kimura, N. Yoshie and H. Yamane, Polymer, 2006, 47, 5965–5972 CrossRef CAS.
- Y. Shinohara, K. Yamazoe, T. Sakurai, S. Kimata, T. Maruyama and Y. Amemiya, Macromolecules, 2012, 45, 1398–1407 CrossRef CAS.
- L. Xie, H. Xu, J.-B. Chen, Z.-J. Zhang, B. S. Hsiao, G.-J. Zhong, J. Chen and Z.-M. Li, ACS Appl. Mater. Interfaces, 2015, 7, 8023–8032 CAS.
- L. Balzano, S. Rastogi and G. Peters, Macromolecules, 2011, 44, 2926–2933 CrossRef CAS.
- F. Zuo, J. K. Keum, L. Yang, R. H. Somani and B. S. Hsiao, Macromolecules, 2006, 39, 2209–2218 CrossRef CAS.
- J. Bai, J. Wang, W. Wang, H. Fang, Z. Xu, X. Chen and Z. Wang, ACS Sustainable Chem. Eng., 2016, 4, 273–283 CrossRef CAS.
- L. Xie, H. Xu, Z. M. Li and M. Hakkarainen, Macromol. Rapid Commun., 2016, 37, 745–751 CrossRef CAS PubMed.
- D. Cavallo, F. Azzurri, L. Balzano, S. S. Funari and G. C. Alfonso, Macromolecules, 2010, 43, 9394–9400 CrossRef CAS.
- G. Kalay and C. R. Kalay, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1828–1834 CrossRef CAS.
- C. Gao, L. Yu, H. Liu and L. Chen, Prog. Polym. Sci., 2012, 37, 767–780 CrossRef CAS.
- H. Fukushima, Y. Ogino, G. Matsuba, K. Nishida and T. Kanaya, Polymer, 2005, 46, 1878–1885 CrossRef CAS.
- O. O. Mykhaylyk, P. Chambon, C. Impradice, J. P. A. Fairclough, N. J. Terrill and A. J. Ryan, Macromolecules, 2010, 43, 2389–2405 CrossRef CAS.
- Y. Wang, K. Meng, S. Hong, X. Xie, C. Zhang and C. C. Han, Polymer, 2009, 50, 636–644 CrossRef CAS.
- K. Wang, F. Chen, Q. Zhang and Q. Fu, Polymer, 2008, 49, 4745–4755 CrossRef CAS.
- Y. Hayashi, G. Matsuba, Y. Zhao, K. Nishida and T. Kanaya, Polymer, 2009, 50, 2095–2103 CrossRef CAS.
- T. Yan, B. Zhao, Y. Cong, Y. Fang, S. Cheng, L. Li, G. Pan, Z. Wang, X. Li and F. Bian, Macromolecules, 2010, 43, 602–605 CrossRef CAS.
- N. Tian, W. Zhou, K. Cui, Y. Liu, Y. Fang, X. Wang, L. Liu and L. Li, Macromolecules, 2011, 44, 7704–7712 CrossRef CAS.
- H. Murase, Y. Ohta and T. Hashimoto, Macromolecules, 2011, 44, 7335–7350 CrossRef CAS.
- H. Xu, G.-J. Zhong, Q. Fu, J. Lei, W. Jiang, B. S. Hsiao and Z.-M. Li, ACS Appl. Mater. Interfaces, 2012, 4, 6774–6784 CAS.
- S. Yamazaki, M. Itoh, T. Oka and K. Kimura, Eur. Polym. J., 2010, 46, 58–68 CrossRef CAS.
- H. Xu, L. Xie and M. Hakkarainen, ACS Sustainable Chem. Eng., 2015, 3, 1443–1452 CrossRef CAS.
- T. Li, J. Zhang, D. K. Schneiderman, L. F. Francis and F. S. Bates, ACS Macro Lett., 2016, 5, 359–364 CrossRef CAS.
- R. N. Haward, Macromolecules, 1993, 26, 5860–5869 CrossRef CAS.
- H. G. H. van Melick, L. E. Govaert and H. E. H. Meijer, Polymer, 2003, 44, 2493–2502 CrossRef CAS.
- Y. Hu, M. Rogunova, V. Topolkaraev, A. Hiltner and E. Baer, Polymer, 2003, 44, 5701–5710 CrossRef CAS.
- C.-C. Chen, J.-Y. Chueh, H.-M. Huang and S.-Y. Lee, Biomaterials, 2003, 24, 1167–1173 CrossRef CAS PubMed.
- V. Ojijo, S. Sinha Ray and R. Sadiku, ACS Appl. Mater. Interfaces, 2012, 4, 6690–6701 CAS.
- J. A. Cicero and J. R. Dorgan, J. Polym. Environ., 2001, 9, 1–10 CrossRef CAS.
- L. Fambri, A. Pegoretti, R. Fenner, S. D. Incardona and C. Migliaresi, Polymer, 1997, 38, 79–85 CrossRef CAS.
- X. Yuan, A. F. T. Mak, K. W. Kwok, B. K. O. Yung and K. Yao, J. Appl. Polym. Sci., 2001, 81, 251–260 CrossRef CAS.
- H. Fang, F. Jiang, Q. Wu, Y. Ding and Z. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13552–13563 CAS.
- S. C. Mauck, S. Wang, W. Ding, B. J. Rohde, C. K. Fortune, G. Yang, S.-K. Ahn and M. L. Robertson, Macromolecules, 2016, 49, 1605–1615 CrossRef CAS.
- G.-X. Chen, H.-S. Kim, E.-S. Kim and J.-S. Yoon, Polymer, 2005, 46, 11829–11836 CrossRef CAS.
- E. Oliaei, B. Kaffashi and S. Davoodi, J. Appl. Polym. Sci., 2016, 133, 3560–3572 CrossRef.
- L. Daelemans, S. van der Heijden, I. De Baere, H. Rahier, W. Van Paepegem and K. De Clerck, ACS Appl. Mater. Interfaces, 2016, 8, 11806–11818 CAS.
- X. Zhang, K. Schneider, G. Liu, J. Chen, K. Brüning, D. Wang and M. Stamm, Polymer, 2011, 52, 4141–4149 CrossRef CAS.
- N. D. Wanasekara, L. E. Matolyak and L. T. J. Korley, ACS Appl. Mater. Interfaces, 2015, 7, 22970–22979 CAS.
- L. Daelemans, S. van der Heijden, I. De Baere, H. Rahier, W. Van Paepegem and K. De Clerck, ACS Appl. Mater. Interfaces, 2016, 8, 11806–11818 CAS.
- J. Mohanraj, D. C. Barton and I. M. Ward, Plast., Rubber Compos., 2008, 37, 13–22 CrossRef CAS.
- H. Dong, Y. R. Sliozberg, J. F. Snyder, J. Steele, T. L. Chantawansri, J. A. Orlicki, S. D. Walck, R. S. Reiner and A. W. Rudie, ACS Appl. Mater. Interfaces, 2015, 7, 25464–25472 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27643a |
| This journal is © The Royal Society of Chemistry 2017 |