
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From seleno-mediated radical polymerization to seleno-containing branched polymers and dynamic hydrogel†
Weihong Lu,
Xiaowei An,
Jian Zhu,
Nianchen Zhou,
Zhengbiao Zhang,
Xiangqiang Pan * and
Xiulin Zhu*
* and
Xiulin Zhu*
State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: panxq@suda.edu.cn; xlzhu@suda.edu.cn
First published on 2nd February 2017
Abstract
Diselenocarbonate-end capped hyperbranched copolymers and diselenide containing dynamic hydrogel were synthesized on the basis of a new RDRP methodology mediated by diselenocarbonates. Copolymerization of O-(4-methoxyphenyl)Se-(4-vinylbenzyl)carbonodiselenoate with vinyl monomers such as styrene, methyl acrylate, n-butyl acrylate, methyl methacrylate, and N-isopropylacrylamide afforded hyperbranched copolymers with variable branch length and degree of branching. A dynamic diselenide containing hydrogel was obtained from a hyperbranched PNIPAM via aminolysis and spontaneous oxidation reaction. This hydrogel showed redox and temperature responses.
Introduction
Selenium-containing polymers have drawn considerable attention in a wide range of fundamental and applied contexts due to their unique redox responsive properties. These polymers show potential applications in physiological condition as responsive drug delivery vehicles,1 artificial enzymes2 and self-healing materials.3 Until now, diverse topologies of the polymers such as main chain selenium-containing block copolymers, covalent or noncovalent side-chain selenium-containing polymers, selenium-containing dendrimers and hyperbranched polymers, have been prepared. Amongst these structures, selenium-containing branched polymers have unique properties in comparison to their linear analogues, such as high branching density and high amount of terminal groups, which make them attractive for many applications such as drug release.4 However, there are rare reports about selenium-containing branched polymers to date. Zhang et al. reported a series of dendrimers with a diselenide core by the reaction of Na2Se2 with dendron bromide.5 But the production of dendrimers requires multistep syntheses, with time consuming purification. Hyperbranched polymers are more attractive architectures as they are easier to produce with less perfect branching structures than dendrimers.6 The creation and development of hyperbranched selenium-containing polymer via one-step processes has been poorly documented in the literature.4,7 They can be synthesized by step-growth polymerization via polycondensation or addition polymerization of multifunctional monomers. Yan and co-workers reported a combination of A2 + B3 monomers to synthesize novel hyperbranched polyselenides, which consist of alternative hydrophobic diselenide and hydrophilic phosphate groups in the backbone framework.4a Smet and co-workers used sodium hydrogen selenide as A2 monomer to synthesize the hyperbranched polyselenides, which show high glutathione peroxidase-like activities.7bGenerally, hyperbranched polymers can also be produced by chain growth polymerization using branching monomers, which act as both monomers and transfer agents or as monomers and initiators,8 due to their many advantages such as relatively mild reaction conditions, wide range of monomers, and tolerance of various functionalities.9 Among them, the combination of reversible addition–fragmentation chain-transfer polymerization and self-condensing vinyl polymerization (RAFT-SCVP) has proved to be a useful technique for the synthesis of hyperbranched polymers with well-defined architectures.10 Our group developed recently, a new reversible-deactivation radical polymerization (RDRP) technique mediated by diselenocarbonates (Se-RAFT).11 Some novel topological structures of selenium-containing polymer, such as linear and cyclic copolymers, have been constructed based on this method.12 To our best knowledge, there are few reports available on the preparation of selenium capped branched/hyperbranched copolymer which could present excellent properties for their particular structure.13 In this article, we investigate, for the first time, the use of Se-RAFT-SCVP to produce hyperbranched copolymers in a one-pot reaction. The synthesized polymers were characterized by NMR, UV-vis, size-exclusion chromatography coupled with multiangle laser light scattering (SEC-MALLS), and thermal analyses. Finally, the Se-RAFT process was used to generate novel architectures, namely redox and temperature responsive diselenide containing hyperbranched-core hydrogel. The synthetic route for the formation of selenium-containing hyperbranched copolymers is shown in Scheme 1.
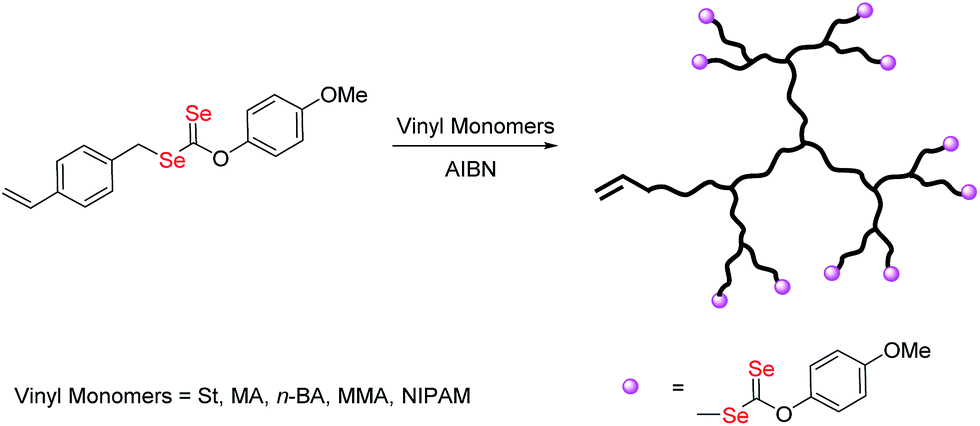 | ||
| Scheme 1 Synthesis of branched copolymers by RAFT copolymerization of O-(4-methoxyphenyl)Se-(4-vinylbenzyl)carbonodiselenoate (VBDSe, M1) with vinyl monomers (M2). | ||
Experimental
Materials
Monomers were purchased from Sinopharm Chemical Reagent Co., Ltd and purified before use. Styrene (St), methyl acrylate (MA), methyl methacrylate (MMA) and n-butyl acrylate (n-BA) were passed through a column filled with basic alumina oxide and kept in a refrigerator at 0 °C. N-Isopropyl acrylamide (NIPAM) was recrystallized in hexane and kept in a desiccator. 2,2′-Azoisobutyronitrile (AIBN, 98%) was recrystallized in ethanol for three times and stored in a refrigerator at 4 °C. p-Methoxyphenol was recrystallized from petroleum ether. The organoselenium compound, O-(4-methoxyphenyl)Se-(4-vinylbenzyl)carbonodiselenoate (VBDSe), was synthesized according to a previously reported method.12a All other chemicals (analytical grade) used in this study were purchased from Shanghai Chemical Reagents and used as received.Synthesis of O-(4-methoxyphenyl)Se-(4-vinylbenzyl)carbonodiselenoate (VBDSe)
A 100 mL round flask was filled with p-methoxyphenol (2.48 g, 20 mmol) dissolved in N,N-dimethylformamide (20 mL). Sodium hydroxide (0.88 g, 22 mmol) was dissolved in 0.5 mL water, then added to the p-methoxyphenol solution at −2 °C and well-mixed. Carbon diselenide (4.08 g, 24 mmol) dissolved in 6 mL DMF was added dropwise to the solution and stirred for 30 min at −2 °C. 4-Vinylbenzyl chloride (3.05 g, 20 mmol) was dissolved in 6 mL DMF, and then added dropwise to the mixture. The reaction was then stirred at room temperature for 4 h. Afterwards, 100 mL of water was added to the product mixture, extracted with petroleum ether for three times, and then dried with anhydrous magnesium sulfate. The residue was purified by column chromatography (petroleum ether/dichloromethane (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (v/v)) to afford the desired compound as a yellow solid (4.51 g, 55% yield).
:1) (v/v)) to afford the desired compound as a yellow solid (4.51 g, 55% yield).
1H NMR (300 MHz, CDCl3) δ 7.41–7.30 (m, 4H), 7.06 (d, J = 7.5 Hz, 2H), 6.94 (d, J = 7.8 Hz, 2H), 6.70 (dd, J = 17.6, 11.0 Hz, 1H), 5.75 (d, J = 17.6 Hz, 1H), 5.26 (d, J = 10.9 Hz, 1H), 4.53 (s, 2H), 3.83 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 221.84, 158.66, 150.86, 137.58, 136.89, 136.27, 130.11, 127.18, 123.36, 115.24, 114.81, 56.21, 39.96. 77Se NMR (115 MHz, CDCl3) δ 971.48, 739.20. HR-ESI-MS: calculated m/z C17H16O2Se2 [M + H+]: 412.9553; experimental m/z [M + H+]: 412.9513.
Se-RAFT-SCVP of vinyl monomers
The typical procedure for the Se-RAFT-SCVP of styrene is summarized as followed: a 10 mL-ampule was filled with styrene (17.8 mmol, 1.8539 g), VBDSe (0.178 mmol, 0.0730 g), AIBN (0.089 mmol, 0.146 g) and toluene (4 mL). The solution was deoxygenated by three standard freeze–pump–thaw cycles. The ampule was flame sealed and placed into an oil bath at 60 °C. After the decided time interval, the ampule was immersed into an ice water and then opened. The content was dissolved in 2 mL THF and precipitated in 200 mL methanol three times. The polymer was obtained by filtration and dried at room temperature under vacuum to constant weight.Other polymers were synthesized according to a similar approach and obtained by precipitation into cold hexane (for branched poly(VBDSe-co-MA), poly(VBDSe-co-MMA)) or methanol (for branched poly(VBDSe-co-St) and poly(VBDSe-co-n-BA)).
Poly(VBDSe-co-NIPAM) was synthesized in DMF using the following conditions: a 10 mL-ampule was filled with NIPAM (20 mmol, 2.2636 g), VBDSe (0.5 mmol, 0.2051 g), AIBN (0.25 mmol, 0.0411 g) and DMF (4 mL). The solution was deoxygenated by three standard freeze–pump–thaw cycles. The ampule was flame sealed and placed into an oil bath at 70 °C. After reaction, the ampule was immersed in an ice water and then opened. The content was dissolved in chloroform and precipitated three times in cold diethyl ether. The polymer was obtained by filtration and dried at room temperature under vacuum.
Linear PNIPAM was synthesized in a 5 mL-ampule filled with NIPAM (20 mmol, 2.2636 g), AIBN (0.1 mmol, 0.0164 g) and isopropanol (4 mL). The solution was deoxygenated by three standard freeze–pump–thaw cycles. The ampule was flame sealed and placed into an oil bath at 70 °C. At the end of the reaction, the ampule was immersed in an ice water and then opened. The content was dissolved in chloroform and precipitated into cold diethyl ether for three times. The polymer was obtained by filtration and dried at room temperature under vacuum.
The monomer conversions (Conv., %) were determined by 1H NMR spectra of the crude polymerization mixture by comparing the integrated areas of characteristic signals of monomer and polymer using the following equations: CSt = 1 − (5 × I5.27–5.23)/(3 × I7.43–7.12), CMA = 1 − 3 × I6.14–6.05/I3.95–3.55, CMMA = I3.59/I3.85–3.45, and Cn-BA = 1 − 3 × I5.80–5.76/I0.93, where Ia–b means the integrated areas from a to b ppm in 1H NMR spectra.
For the polystyrene derivative, the signals at δ 5.27–5.23 ppm (integration = I5.27–5.23) were attributed to the alkene protons of St, and the signals at δ 7.43–7.12 ppm (integration = I7.43–7.12) were ascribed to all aryl protons of the St and polystyrene. In the case of the polymerization of the methyl acrylate, the signals at δ 6.14–6.05 ppm (integration = I6.14–6.05) were ascribed to the alkene protons of the MA, and the signals at about δ 3.95–3.55 ppm (integration = I3.95–3.55) were attributed to all methyl protons of the MA and PMA. The 1H NMR characterization of PMMA showed signals at δ 3.59 ppm (integration = I3.59) which were ascribed to the methyl protons of the PMMA, and signals at about δ 3.85–3.45 ppm (integration = I3.85–3.45) which were ascribed to all methyl protons of MMA and PMMA. On the 1H NMR spectrum of the crude PBA mixture, the signals at δ 5.80–5.76 ppm (integration = I5.80–5.76) were attributed to the alkene protons of the n-BA, and the signals at about δ 0.93 ppm (integration = I0.93) were ascribed to all methyl protons of the n-BA and PBA.
Chain extension
The procedure of the chain extension experiment was similar to the polymerization described above except that VBDSe was substituted by the poly(VBDSe-co-St) (Mn = 5800 g mol−1).Typical aminolysis oxidation procedure of hyperbranched copolymer from Se-RAFT-SCVP
A solution of poly(N-isopropyl acrylamide) (PNIPAM) (2.45 g, Mn,SEC = 37000 g mol−1) and n-hexamine (0.66 mL) in THF was stirred for 15 min at room temperature. After oxidation in the air, the gel was formed and dialyzed against THF/H2O (1:1) (v/v) for three days and against H2O for two more days. The polymer was obtained by freeze-drying.
Typical gel–sol/sol–gel procedure of poly(VBDSe-co-NIPAM) copolymer
A dry cuvette was filled with deionized water (2.5 mL) and the gel (0.1331 g) obtained from aminolysis-oxidative coupling. After deoxygenating by nitrogen flush for 15 min, DL-1,4-dithiothreitol (DTT) (0.0416 g) was added and the hydrogel exhibited gel-to-sol transition. The reversible sol-to-gel transition was exhibited after removing the bottle cap and leaving the sample for one day under air atmosphere.Characterization
The number-average molecular weight (Mn) and molecular weight distribution (Đ) of the resulting polymers were determined by SEC-MALLS using an Agilent Technologies PL-GPC 50 apparatus, equipped with triple detectors (refractive-index detector, multi angles light scattering detector and viscosity detector), using PL gel 5 μm MIXED-C column with molecular weights ranging from 200 to 2 × 106 g mol−1. Tetrahydrofuran was used as the eluent with a flow rate of 1.0 mL min−1 at 40 °C. SEC samples were injected manually. The molecular weights were calibrated with PS standards. The 1H NMR and 13C NMR of spectra were recorded on a Bruker 300 MHz nuclear magnetic resonance instrument using CDCl3 as the solvent and tetramethylsilane (TMS) as an internal standard. The 77Se NMR spectrum of the VBDSe was recorded on a Bruker 400 MHz nuclear magnetic resonance instrument using CDCl3 as the solvent and tetramethylsilane (TMS) as an internal standard. UV-vis spectra of VBDSe and polymers were recorded using a Shimadzu U-2600 spectrophotometer at room temperature. Thermogravimetric analysis (TGA) was carried out on a Discovery TGA of TA instruments with a heating rate of 10 °C min−1 from the room temperature to 800 °C under nitrogen atmosphere. Differential scanning calorimetric analysis (DSC) were performed using a Discovery Q200 from TA Instruments, and the heating rate was 10 °C min−1 under nitrogen. Lower Critical Solution Temperature (LCST) measurement was performed on a Shimadzu U-2600 spectrophotometer with S-1700 Shimadzu Temperature Controller. The relative transmission of a laser beam with a wavelength of 700 nm was recorded for each experiment. The measurements were performed at a temperature range between 15 and 40 °C and a heating rate of 1 °C min−1. Critical solution temperatures derived from these experiments were taken at 50% relative transmission.Results and discussion
In order to synthesize selenium containing hyperbranched copolymers, a novel Se-functionalized chain transfer agent (Se-CTA), O-(4-methoxyphenyl)Se-(4-vinylbenzyl)carbonodiselenoate (VBDSe) was prepared. Herein VBDSe was prepared in a relatively good yield via the one-pot reaction of 4-methoxyphenol, CSe2 and 4-vinylbenzyl chloride according to a previously reported protocol (Fig. S1†).11 The use of this styryl-diselenocarbonate allowed copolymerization with vinyl monomers to prepare the hyperbranched copolymers via presence of a styryl double bond and chain branching. To investigate the effects of the reaction time on copolymerization, Se-RAFT of VBDSe with styrene (St) using a feed ratio of 100 and a total monomer concentrations of 3.0 mol L−1 were conducted in toluene at 60 °C. The monomer conversions, determined with 1H NMR by comparing the integrated areas of the signals of monomer and polymer, increased linearly with the increase of reaction time (Fig. S2†). The copolymerization results are listed in Table 1. As one can see on Table 1, the styrene conversion is gradually increasing to 79.9% after 48 h. The molecular weights and dispersity index (Đ) of poly(VBDSe-co-St) branched copolymers tend to increase with St conversion (Fig. 1). The significant increase of the amount of repeating units per branch (RB) with polymerization time in Table 1 confirmed that the branch length is increasing with St conversion.| Entry | T (h) | CStb (%) | Mnc (g mol−1) | Mwd (g mol−1) | Mpc (g mol−1) | Đ | RBe | DBf |
|---|---|---|---|---|---|---|---|---|
| a Polymerization condition: [St]0:[VBDSe]0:[AIBN]0 = 100:1:0.5, in toluene at 60 °C [M1]0 + [M2]0 = 3 mol L−1.b St conversions were determined by 1H NMR.c Determined by DRI.d Determined by MALLS.e Determined by 1H NMR, herein RB = (I1.2–2.6/3)/(I3.66–3.86/3).f Degree of branching (DB) = 1/RB. |
||||||||
| 1 | 10 | 17.2 | 5500 | 5900 | 5200 | 1.07 | 19.43 | 0.0515 |
| 2 | 12 | 23.2 | 3900 | 4500 | 3600 | 1.15 | 21.11 | 0.0474 |
| 3 | 18 | 32.7 | 5800 | 7700 | 5400 | 1.33 | 28.44 | 0.0352 |
| 4 | 24 | 36.9 | 5600 | 7100 | 5400 | 1.27 | 34.84 | 0.0287 |
| 5 | 30 | 51.7 | 7600 | 11800 |
6500 | 1.55 | 50.92 | 0.0196 |
| 6 | 48 | 79.9 | 10700 |
20000 |
8800 | 1.87 | 58.89 | 0.0170 |
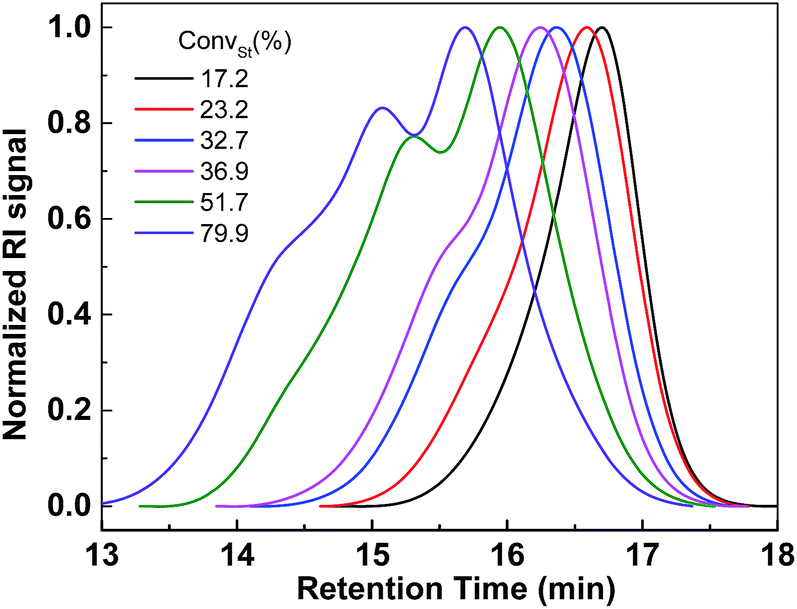 | ||
| Fig. 1 SEC traces of hyperbranched poly(VBDSe-co-St) shown in Table 1. | ||
In 1H NMR spectra of poly(VBDSe-co-St) branched copolymers, the proton signals of CH2Se in VBDSe unit with unreacted diselenocarbonate functionality appeared at 4.00–4.20 ppm (c). The signals of CHSe in terminal St and VBDSe unit appeared at 4.30–4.50 (e) and 4.50–4.70 ppm (d), respectively. The signals of CH3O in terminal diselenocarbonate functionality appeared at 3.66–3.86 ppm (f). The signals of CH in the terminal vinyl group were found around 5.18 ppm (Z-alkene H, a) and 5.67 ppm (E-alkene H, b), respectively. The signals of CH and CH2 in the chain appeared at 1.2–2.6 ppm (h) (Fig. 2).
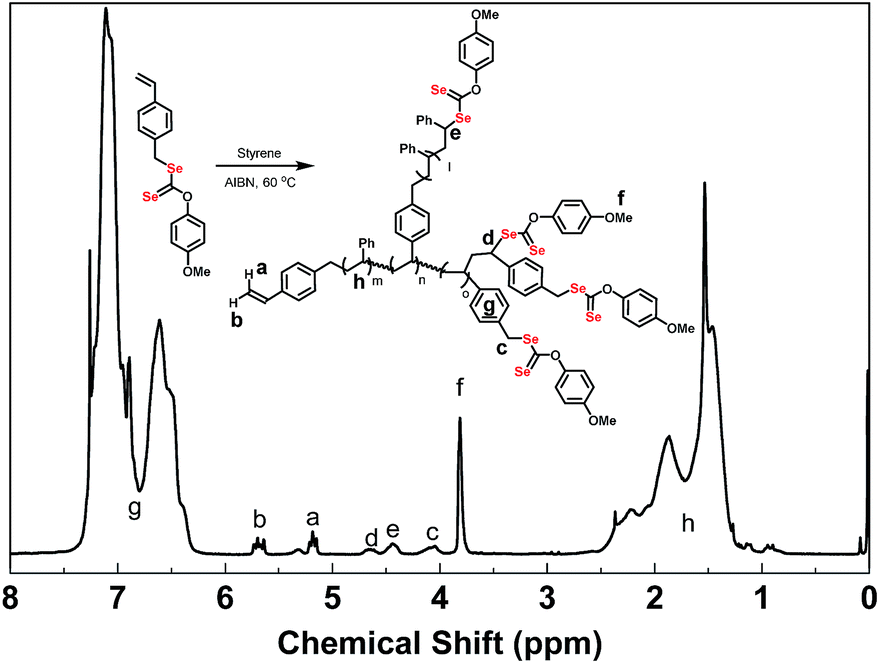 | ||
| Fig. 2 1H NMR spectra of hyperbranched poly(VBDSe-co-St) in CDCl3 corresponding to entry 1 in Table 1. | ||
Se-RAFT tolerates a wide range of monomers,11 so it is possible to generate various kinds of branched copolymers via copolymerization of VBDSe with different monomers. To confirm the versatility and generality of this method, Se-RAFT copolymerization of VBDSe and other vinyl monomers such as methyl methacrylate, methyl acrylate and n-butyl acrylate were performed, and the results are listed in Table 2. This convenient procedure allowed approximate quantitative monomer conversion in 24 h and yielded high branched copolymers (Entries 3, 5–8 in Table 2). On 1H NMR spectra of poly(VBDSe-co-M2) (M2 = MA, n-BA, and MMA) branched copolymers, the proton signals of CH3O in terminal diselenocarbonate functionality appeared at 3.81–3.83 ppm, the signals of CHSe appeared at 4.35–4.55 ppm, and the signals of CH in styryl unit were found at 6.75–7.32 ppm (Fig. S3†). On the basis of the integration values, proportions of terminal diselenocarbonate, M2 unit and styryl unit originated from reacted VBDSe in hyperbranched copolymers could be quantitatively determined. SEC curves of branched copolymers shown on Fig. 3, evidenced that higher ratios of monomer/VBDSe result in increased molecular weight, which is in good agreement with previous reports.10d It should be noted that a copolymer with a dispersity of 1.27 was obtained at conversion of styrene around 37% using a feed ratio of 100 at 60 °C for 24 h. The SEC trace of this copolymer exhibited almost unimodal distribution with the presence of a slight shoulder at high molecular weight indicating the resultant copolymer was composed of linear as well as branched polymers (Fig. 3). This phenomenon could be ascribed to relatively low rate of polymerization of styrene and to the fact that in the initial stage, VBDSe behave as a chain transfer agent rather than a cross-linker. Thus, an increase of the dispersity values of the obtained polymers with an increase of monomer conversion was observed (Fig. 1). On the other hand, the copolymerization of other monomers with VBDSe gave broad dispersity values and the MW is higher using the ratio of 100:1 than using the ratio of 10:1 because of the high conversion of monomers after 24 h.
| Entry | M2 | x | CM2b (%) | Mnc (g mol−1) | Mwd (g mol−1) | Đc | RBe | DBe | Tg |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization condition: [M2]0:[VBDSe]0:[AIBN]0 = x:1:0.5, [M1]0 + [M2]0 = 3 mol L−1, in toluene at 60 °C for 24 h.b VBDSe (CM1) and M2 (CM2) conversions were determined by 1H NMR.c Determined by SEC-MALLS.d Determined by MALLS.e Determined by 1H NMR. |
|||||||||
| 1 | St | 10 | 76.0 | 6500 | 13300 |
2.05 | 9.61 | 0.1041 | 88.9 |
| 2 | St | 100 | 36.9 | 5600 | 7100 | 1.27 | 34.84 | 0.0287 | 91.1 |
| 3 | MMA | 10 | 99.1 | 10900 |
77400 |
7.08 | 10.10 | 0.0990 | 106.0 |
| 4 | MMA | 100 | 93.7 | 35100 |
133100 |
3.80 | 83.48 | 0.0121 | 124.6 |
| 5 | MA | 10 | 99.1 | 9700 | 33600 |
3.47 | 10.10 | 0.0990 | 28.3 |
| 6 | MA | 100 | 98.2 | 18900 |
43900 |
2.33 | 93.42 | 0.0107 | 13.5 |
| 7 | n-BA | 10 | 98.7 | 12200 |
35800 |
2.95 | 11.73 | 0.0853 | −30.0 |
| 8 | n-BA | 100 | 98.9 | 37100 |
85200 |
2.30 | 102.39 | 0.0098 | −44.2 |
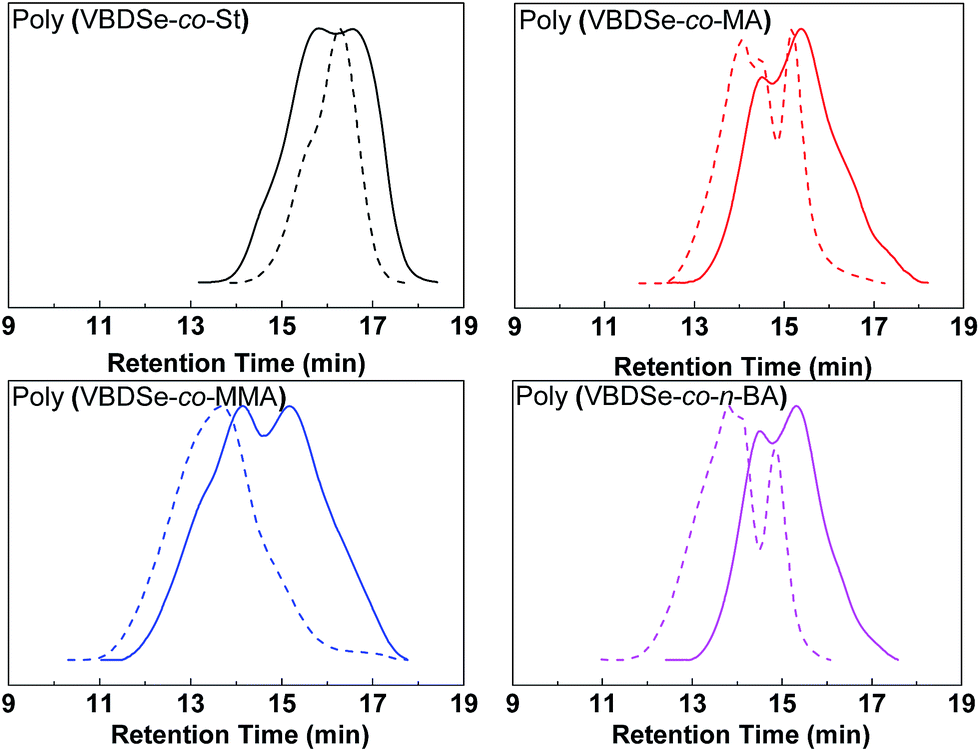 | ||
| Fig. 3 SEC curves of hyperbranched copolymers synthesized by Se-RAFT-SCVP corresponding to Table 2. [M]0:[VBDSe]0 = 10:1 (solid line) or 100:1 (dashed line). | ||
Structure of the polymers obtained by copolymerization was also investigated by UV-vis analyses. Fig. S4† shows typical UV-vis absorption spectra of copolymers. Strong peaks between 310 to 380 nm, corresponding to the absorption of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se bond, were found indicating the copolymers are ended with diselenocarbonate. Furthermore, the molecular weight increased from 5800 g mol−1 to 105000 g mol−1 after chain extension reaction, demonstrating the activity of diselenocarbonates end group of the hyperbranched copolymer (Fig. S5 and S6†).
Se bond, were found indicating the copolymers are ended with diselenocarbonate. Furthermore, the molecular weight increased from 5800 g mol−1 to 105000 g mol−1 after chain extension reaction, demonstrating the activity of diselenocarbonates end group of the hyperbranched copolymer (Fig. S5 and S6†).
Since the thermal properties of hyperbranched copolymers usually differ from their linear analogue, the thermal properties of the hyperbranched copolymers were characterized by DSC and TGA. These copolymers have glass transition temperatures in a temperature range varying from −44 to 124 °C by changing the ratio of different monomers (Fig. S7†). In DSC curves of these copolymers, only one obvious glass transition temperature (Tg) was observed, suggesting good compatibility of VBDSe and other monomer units in these hyperbranched copolymers. With scaling-up the monomer ratio from 10 to 100 or for decreasing DB, a slight increase of the Tg or a more significant increased Tg were noted in entry 2 and entry 4, respectively. On the contrary, decreased Tg was noted in entries 6 and 8. Normally, with decreasing DB, the free volume of branched polymers decreased due to the reduced number of chain ends, and this result in an increase of the Tg value.10d In this study, both the composition and DB were found affecting the glass transition of the hyperbranched copolymers. With the decreasing DB and increasing molecular weights, the Tg of copolymers were found to be more close to the linear polymers. Furthermore, after chain extension of poly(VBDSe-co-St) with n-BA, the Tg was shifting from 88.2 °C to −44.0 °C, which is much closer to the value for a linear PBA (Fig. 4). In the TGA curves of these branched copolymers, two significant stages of thermal decomposition were observed. The first stage in which the maximum decomposition temperatures (Tmax) appeared between 200 and 340 °C could be ascribed to the decomposition of the diselenocarbonate functionality present in terminal groups. At the second stage, at about 350 °C, the residual polymers began to decompose, and this decomposition is completed at around 450 °C. The thermal stability of the various branched copolymers was different, suggesting its dependence on topology and chemical composition (Fig. 5).
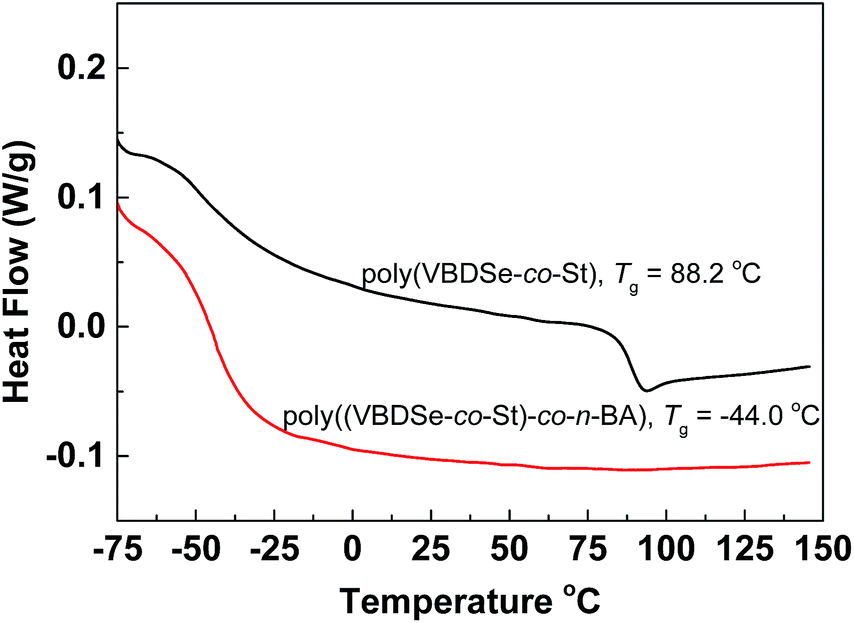 | ||
| Fig. 4 DSC curves of hyperbranched poly(VBDSe-co-St) and poly(VBDSe-co-St)-co-n-BA after chain extension with n-BA using a feed ratio: [n-BA]0:[macro-CTA]0:[AIBN]0 = 500:1:0.5. | ||
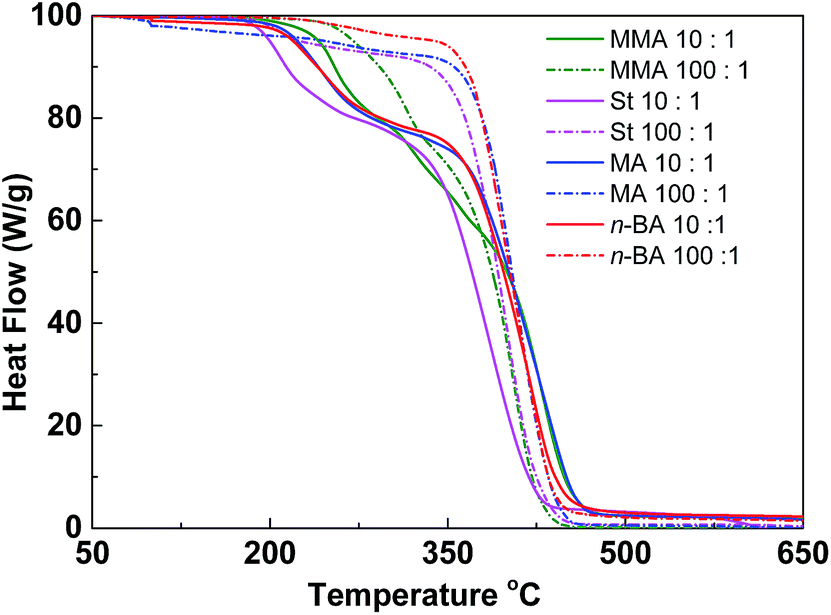 | ||
| Fig. 5 TGA curves of hyperbranched copolymers synthesized by Se-RAFT-SCVP. [M]0:[VBDSe]0 = 10:1 (solid line) or 100:1 (dashed line). | ||
Poly(N-isopropylacrylamide) (PNIPAM) is a well-known water soluble polymer with low critical solution temperature (LCST) around 32 °C. Highly branched PNIPAM have been synthesized and used for wide applications such as drug delivery or gene transfection.10b,14 Herein, we prepared branched PNIPAM by Se-RAFT and described the dramatic influence of chain architectures on the resulting responsive solution behavior. The structures of copolymers were determined by SEC, 1H NMR and UV spectra (Fig. S8–S10†). The end group and the phase transitions of the branched PNIPAM in water were investigated by UV-vis and the results are shown in Fig. 6 and S10.† The LCST of the hyperbranched copolymers increased with increasing molecular weight. Indeed, for the polymer prepared with [VBDSe]0/[NIPAM]0 = 1/40, the LCST was approximately 24.4 °C, and the one prepared with [VBDSe]0/[NIPAM]0 = 1/100 showed a transition around 29.1 °C. The phase transition temperature (Tp) of the hyperbranched copolymers were significantly lower than those typically associated with linear polymers.15 Indeed, solubility of the polymer, with an increased degree of branching prepared with [VBDSe]0/[NIPAM]0 = 1/10, is too low to measure its Tp.14a,c On the basis of previous reports of the effect of chain transfer agent derived end groups on the solubility behavior of polymers prepared by RAFT, the phase transitions of the hyperbranched PNIPAM end-capped by selenol were investigated after aminolysis. Interestingly, the LSCT observed for the selenol end-capped polymer after aminolysis ([VBDSe]0/[NIPAM]0 = 1/40) is 0.3 degree higher than the polymer before aminolysis (Fig. 6). A similar result was obtained for the polymer prepared with [VBDSe]0/[NIPAM]0 = 1/100. It was reported that the end group in these polymers is having a weak effect on the LCST, and that the molecular weight decrease and presence of hydrophobic group in the main chain result in a diminution of the LCST.16
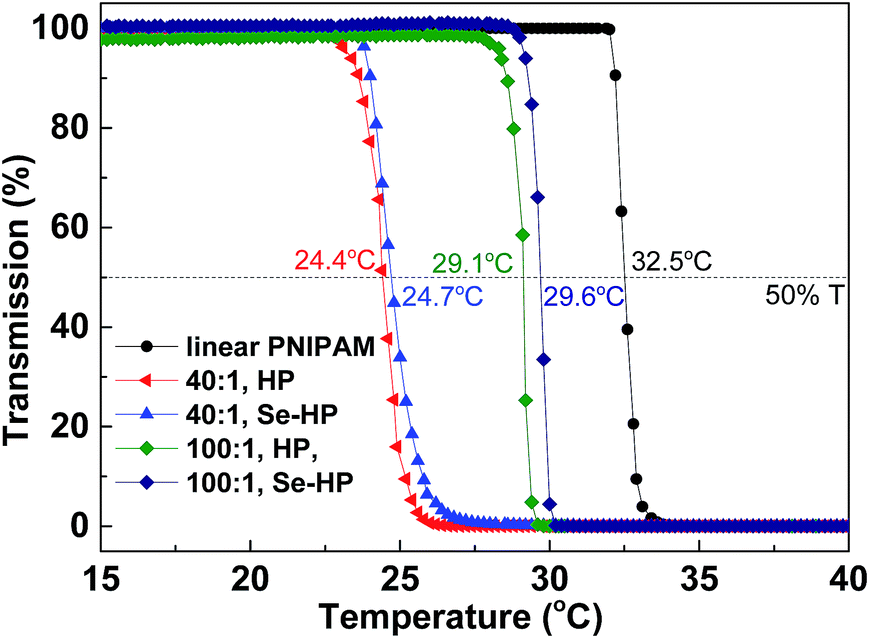 | ||
| Fig. 6 LCST of the hyperbranched poly(VBDSe-co-NIPAM) before (HP) and after (Se-HP) aminolysis characterized by UV-vis. | ||
The products after aminolysis were selenol-terminated hyperbranched copolymers that were susceptible to diselenide formation under air.12a Therefore, similar to thiol group (–SH), coupling of the selenol-terminated chains could lead to reconstruction of the hyperbranched architecture and a transition to the gel state.14a To confirm its redox responsiveness, the diselenide linked hydrogel was subjected to reduction with dithiothreitol (DTT) and oxidation with air. The redox process is shown in Fig. 7. Recovery of the gelling behavior was consistent with at least the partial formation of diselenide linked hyperbranched copoly(VBDSe-co-NIPAM). The LCST of the copolymers, coupled with the reversibility of diselenide formation, make these gels both thermo- and redox-responsive. Indeed, many biological processes are governed by interconversion of selenols and diselenides, suggesting this hydrogel can potentially be utilized as new platform for targeted drug delivery and combined therapy.4,17
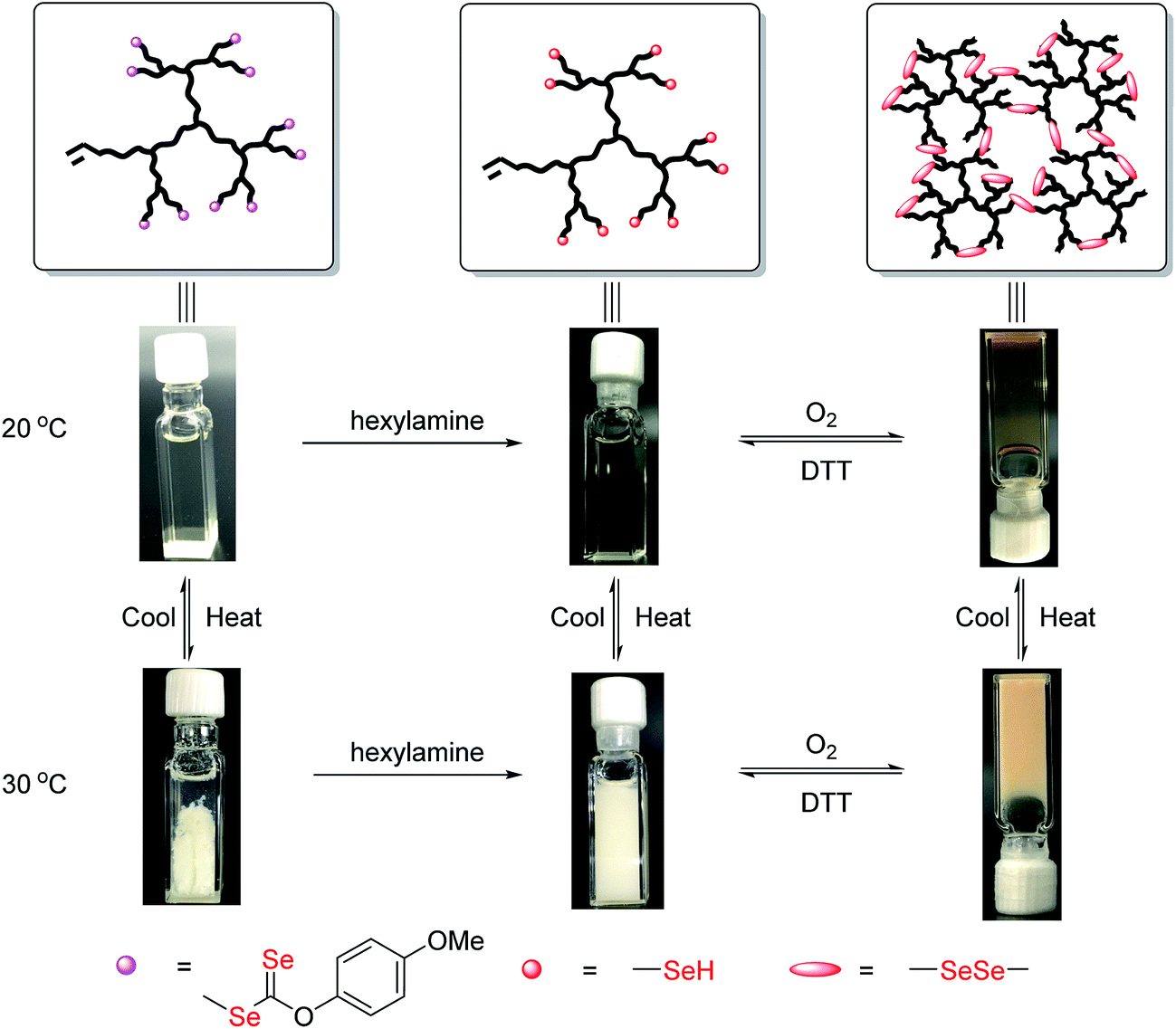 | ||
| Fig. 7 Sol to gel transition of hyperbranched poly(VBDSe-co-NIPAM) in response to thermal and redox stimuli ([NIPAM]0:[VBDSe]0 = 40:1). | ||
Conclusion
In summary, a vinyl contained diselenocarbonate Se-RAFT agent was synthesized and used as a chain-transfer monomer to initiate the polymerization of vinyl monomers to obtain new selenium-containing branched polymers. 1H NMR and SEC measurements confirmed the branched structure, and the results showed that the degrees of branching depended on the ratio of monomer to the Se-RAFT agent. Se-RAFT-SCVP of NIPAM afforded thermo-responsive hyperbranched copolymer. The LCST of the copolymers could be adjusted by the control over the feed ratio. After aminolysis, a selenol capped branched PNIPAM was obtained and the polymer chains can be crosslinked with each other by the formation of dynamic diselenide bonds. The formed gel has potential application in biomaterials for its thermo- and redox-responsive properties.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21302132, 21374067, 21474068), Natural Science Foundation of Jiangsu Province (BK20130296), the China Scholarship Council (No. 201506920009), the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Suzhou Key Lab of Macromolecular Design and Precision Synthesis.Notes and references
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647 CrossRef CAS PubMed.
- X. Huang, X. Liu, Q. Luo, J. Liu and J. Shen, Chem. Soc. Rev., 2011, 40, 1171 RSC.
- S. B. Ji, W. Cao, Y. Yu and H. P. Xu, Adv. Mater., 2015, 27, 7740 CrossRef CAS PubMed.
- (a) J. Y. Liu, Y. Pang, J. Chen, P. Huang, W. Huang, X. Y. Zhu and D. Y. Yan, Biomaterials, 2012, 33, 7765 CrossRef CAS PubMed; (b) J. Y. Liu, Y. Pang, Z. Y. Zhu, D. L. Wang, C. T. Li, W. Huang, X. Y. Zhu and D. Y. Yan, Biomacromolecules, 2013, 14, 1627 CrossRef CAS PubMed.
- X. Zhang, H. P. Xu, Z. Y. Dong, Y. P. Wang, J. Q. Liu and J. C. Shen, J. Am. Chem. Soc., 2004, 126, 10556 CrossRef CAS PubMed.
- R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533 RSC.
- (a) H. P. Xu, J. Gao, Y. P. Wang, Z. Q. Wang, M. Smet, W. Dehaen and X. Zhang, Chem. Commun., 2006, 796 RSC; (b) J. Thomas, Z. Y. Dong, W. Dehaen and M. Smet, Macromol. Rapid Commun., 2012, 33, 2127 CrossRef CAS PubMed; (c) Y. Fu, J. Y. Chen, H. P. Xu, C. Van Oosterwijck, X. Zhang, W. Dehaen and M. Smet, Macromol. Rapid Commun., 2012, 33, 798 CrossRef CAS PubMed.
- (a) J. M. Frechet, M. Henmi, I. Gitsov and S. Aoshima, Science, 1995, 269, 1080 CrossRef CAS PubMed; (b) T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873 CrossRef PubMed.
- (a) C. J. Hawker, J. M. J. Frechet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763 CrossRef CAS; (b) S. G. Gaynor, S. Edelman and K. Matyjaszewski, Macromolecules, 1996, 29, 1079 CrossRef CAS; (c) Z. M. Wang, J. P. He, Y. F. Tao, L. Yang, H. J. Jiang and Y. L. Yang, Macromolecules, 2003, 36, 7446 CrossRef CAS.
- (a) B. L. Liu, A. Kazlauciunas, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 2131 CrossRef CAS; (b) S. Carter, B. Hunt and S. Rimmer, Macromolecules, 2005, 38, 4595 CrossRef CAS; (c) L. Tao, J. Q. Liu, B. H. Tan and T. P. Davis, Macromolecules, 2009, 42, 4960 CrossRef CAS; (d) C. B. Zhang, Y. A. Zhou, Q. A. Liu, S. X. Li, S. Perrier and Y. L. Zhao, Macromolecules, 2011, 44, 2034 CrossRef CAS; (e) J. Schmitt, N. Blanchard and J. Poly, Polym. Chem., 2011, 2, 2231 RSC; (f) J. Han, S. P. Li, A. J. Tang and C. Gao, Macromolecules, 2012, 45, 4966 CrossRef CAS; (g) S. G. Roy and P. De, Polym. Chem., 2014, 5, 6365 RSC; (h) B. Mo, H. H. Liu, X. D. Zhou and Y. L. Zhao, Polym. Chem., 2015, 6, 3489 RSC; (i) J. B. Huang, L. H. Lin, H. Liang and J. Lu, Polym. Chem., 2015, 6, 4020 RSC; (j) J. A. Alfurhood, H. Sun, P. R. Bachler and B. S. Sumerlin, Polym. Chem., 2016, 7, 2099 RSC.
- J. D. Zeng, J. Zhu, X. Q. Pan, Z. B. Zhang, N. C. Zhou, Z. P. Cheng, W. Zhang and X. L. Zhu, Polym. Chem., 2013, 4, 3453 RSC.
- (a) F. Gao, X. Q. Pan, J. Zhu, Z. B. Zhang, W. Zhang and X. L. Zhu, Polym. Chem., 2015, 6, 1367 RSC; (b) Z. Cai, W. Lu, F. Gao, X. Pan, J. Zhu, Z. Zhang and X. Zhu, Macromol. Rapid Commun., 2016, 37, 865 CrossRef CAS PubMed.
- (a) J. J. Lu, N. C. Zhou, X. Q. Pan, J. Zhu and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 504 CrossRef CAS; (b) K. Takagi, Y. Nishikawa, T. S. Kwon, H. Kunisada and Y. Yuki, Polym. J., 2000, 32, 970 CrossRef CAS.
- (a) W. Tao and L. F. Yan, J. Appl. Polym. Sci., 2010, 118, 3391 CrossRef CAS; (b) S. Carter, S. Rimmer, A. Sturdy and M. Webb, Macromol. Biosci., 2005, 5, 373 CrossRef CAS PubMed; (c) A. P. Vogt and B. S. Sumerlin, Macromolecules, 2008, 41, 7368 CrossRef CAS.
- (a) H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS; (b) Y. Xia, N. A. Burke and H. D. Stöver, Macromolecules, 2006, 39, 2275 CrossRef CAS.
- R. Plummer, D. J. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379 CrossRef CAS.
- (a) T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS; (b) J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437 CrossRef CAS PubMed; (c) X. Cheng, Y. Jin, T. Sun, R. Qi, B. Fan and H. Li, RSC Adv., 2015, 5, 4162 RSC; (d) J. Luan, W. Shen, C. Chen, K. Lei, L. Yu and J. Ding, RSC Adv., 2015, 5, 97975 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28565a |
| This journal is © The Royal Society of Chemistry 2017 |