
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphinito palladium(II) complexes as catalysts for the synthesis of 1,3-enynes, aromatic alkynes and ynones
R. E. Islas a,
J. Cárdenas*a,
R. Gaviñoa,
E. García-Ríosa,
L. Lomas-Romerob and
J. A. Morales-Serna*b
a,
J. Cárdenas*a,
R. Gaviñoa,
E. García-Ríosa,
L. Lomas-Romerob and
J. A. Morales-Serna*b
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Ciudad de México, 04510, Mexico. E-mail: rjcp@unam.mx; Fax: +52 55 5616 2217; Tel: +52 55 56224413
bDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco No. 186, Ciudad de México, C. P. 09340, Mexico. E-mail: joseantonio.moralesserna@xanum.uam.mx; Fax: +52 55 53189000; Tel: +52 55 5318 9593
First published on 2nd February 2017
Abstract
An air-stable phosphinito palladium(II) complex (Ph1-Phoxide) has been found to be an efficient catalyst in the formation of C–C bonds. The coupling of terminal alkynes formed gem-1,3-enynes as the only reaction products. Aromatic alkynes can be synthesized from the coupling of terminal alkynes and haloaromatic compounds (Sonogashira coupling). The phosphinito palladium(II) complex also catalyses the coupling between acyl chlorides and terminal alkynes (Sonogashira coupling), furnishing ynones in excellent yields.
1. Introduction
Secondary phosphine oxides (SPOs)1 are a class of pre-ligands2 that are valued for their stability in air and in moisture.3 Because of the tautomeric equilibrium between the pentavalent phosphorus oxide 1 and the trivalent phosphinous acid 2 (Scheme 1),4 SPOs form hydrogen bond-stabilized bidentate ligands.2,5 The shift of this equilibrium in favour of either species, and therefore the formation of transition metal complex 4, depends on metal coordination,6 the solvents used in the process7 and the phosphine substituents.8 The formation of 4 can be rationalized as the coordination of phosphinous acid 2 and phosphinito ligand 3, which in turn, have their origin in two SPOs. A strong symmetric hydrogen bridge is formed in the presence of a metal, leading to the formation of mononuclear metal complex 4 (Scheme 1).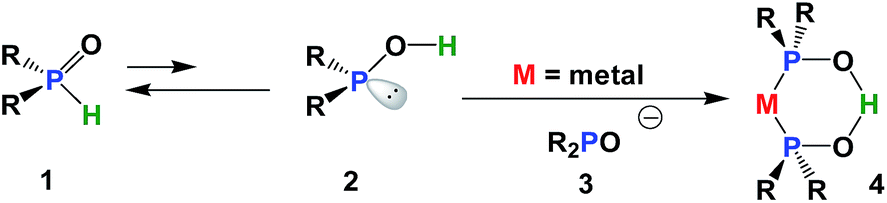 | ||
| Scheme 1 Tautomerization of SPOs and chelate formation. | ||
Many phosphinito transition metal complexes have been synthesized,2 including complexes of palladium,9 platinum,10 molybdenum,5e nickel,5b gold,3c rhodium5d,6e and ruthenium.11 Applications of SPO Pd complexes in catalysis include Negishi cross-coupling (POPd1, POPd5 and POPd7),12 conjugate addition of arylsiloxanes13 to α,β-unsaturated carbonyl compounds (POPd1),14 Suzuki–Miyaura (POPd1, POP7 and Ph1-Phoxide)15 and Stille and Hiyama-type coupling (POPd1)16 (Fig. 1).
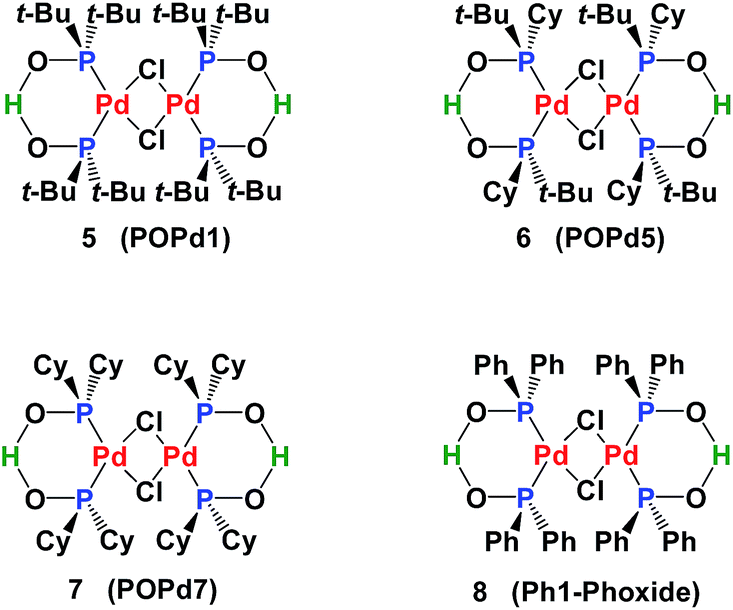 | ||
| Fig. 1 Structures of SPO Pd complexes. | ||
SPO Pd complex 8 exhibits high catalytic activity in Heck-type reactions as well as high stability towards moisture and air.17 The SPOs Pd complex 8 was first synthesized by Dixon18 in 1971 and then later synthesized and crystallized by many groups.19 From a structural view point, 8 can be considered as a dimer of complex 4. However, our synthesis of 8 was achieved by hydrolysis of the palladium chlorodiphenylphosphine complex 10 (Scheme 2). Fig. 2 shows the NMR spectra (1H, 13C and 31P) of complex 8, which is a homogeneous yellow solid.
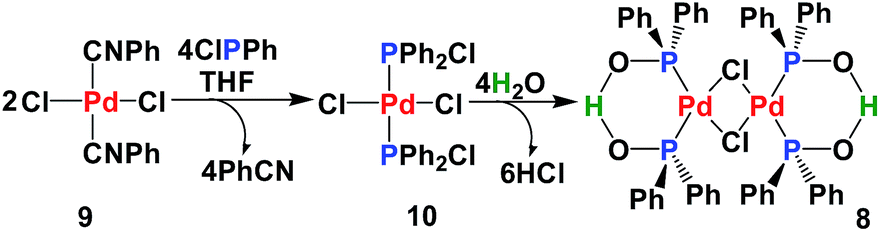 | ||
| Scheme 2 Synthesis of the SPO Pd complex Ph1-Phoxide. | ||
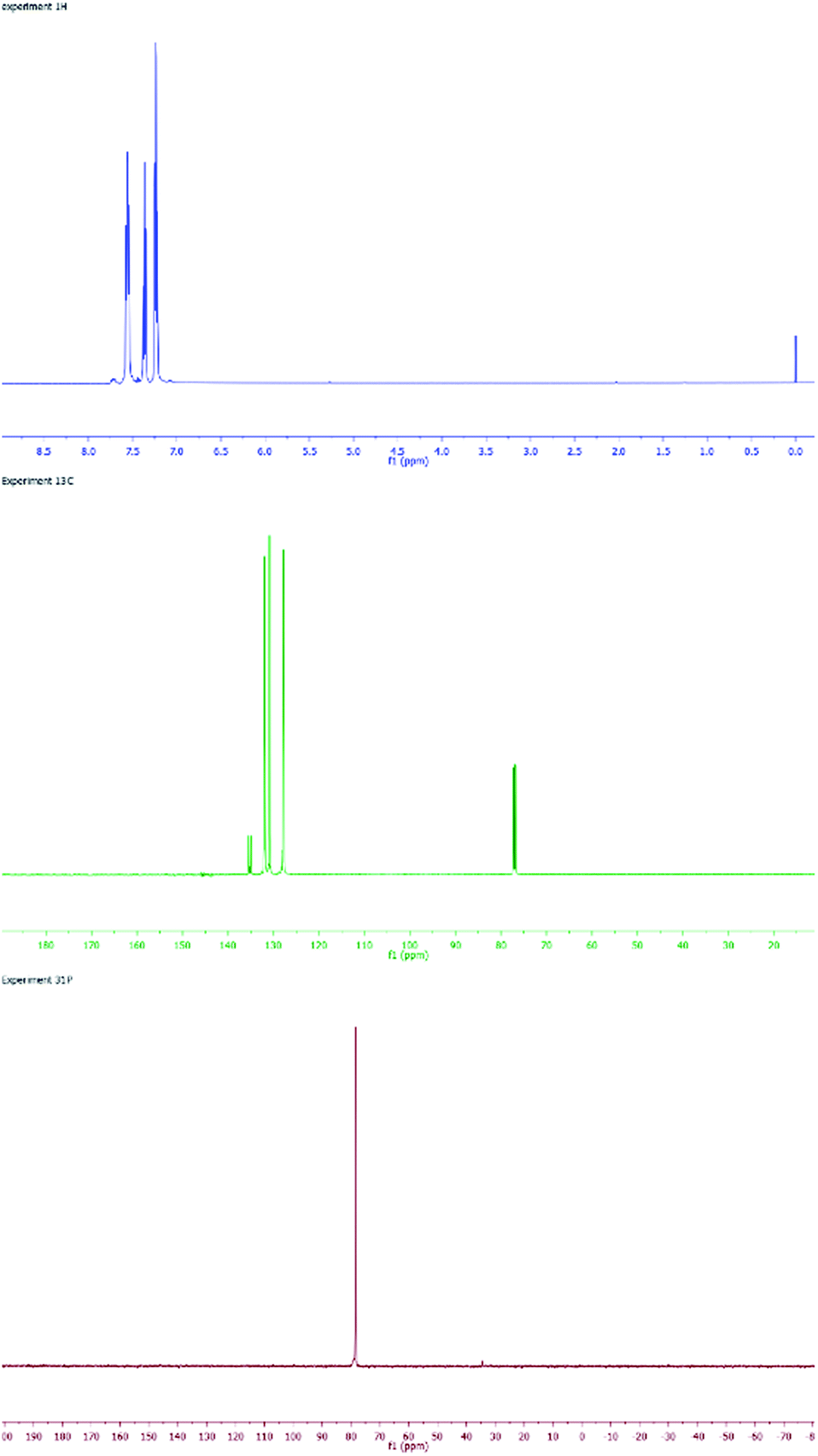 | ||
| Fig. 2 NMR spectra of complex 8: 1H (blue), 13C (green) and 31P (red). | ||
Driven by our interest in the catalytic properties of SPO Pd complex 8 in organic reactions and its application in the synthesis of natural products,17 we report herein the results obtained in the application of complex 8 as a catalyst for the synthesis of 1,3-enynes, aromatic-alkynes and ynones (Sonogashira coupling). Additionally, we report the utility of complex 8 in the synthesis of anemarchalconyn, a natural product isolated from the rhizomes of Anemarrhena asphodeloides. To the best of our knowledge, the use of the Pd SPO complex 8 in these reactions has not been reported previously.
2. Results and discussion
2.1. Synthesis of 1,3-enynes
Initially, we were interested in evaluating the ligation properties of the secondary phosphine oxides in complex 8, with the thought that the sterically demanding groups in that complex influence the selectivity of the reaction. The first reaction examined was the coupling of two alkynes to obtain 1,3-enynes.20,21 As shown in Table 1, the dimerization reaction of alkynes in the presence of 5 mol% complex 8, NaOAc and toluene as the solvent at 100 °C affords 1,3-enynes 12a–12j in good to excellent yield. In all cases, the use of a microwave reactor allowed for a significant reduction in reaction time, from 18 h to 30 minutes, without affecting yield.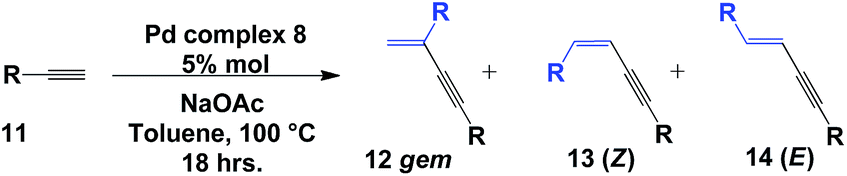
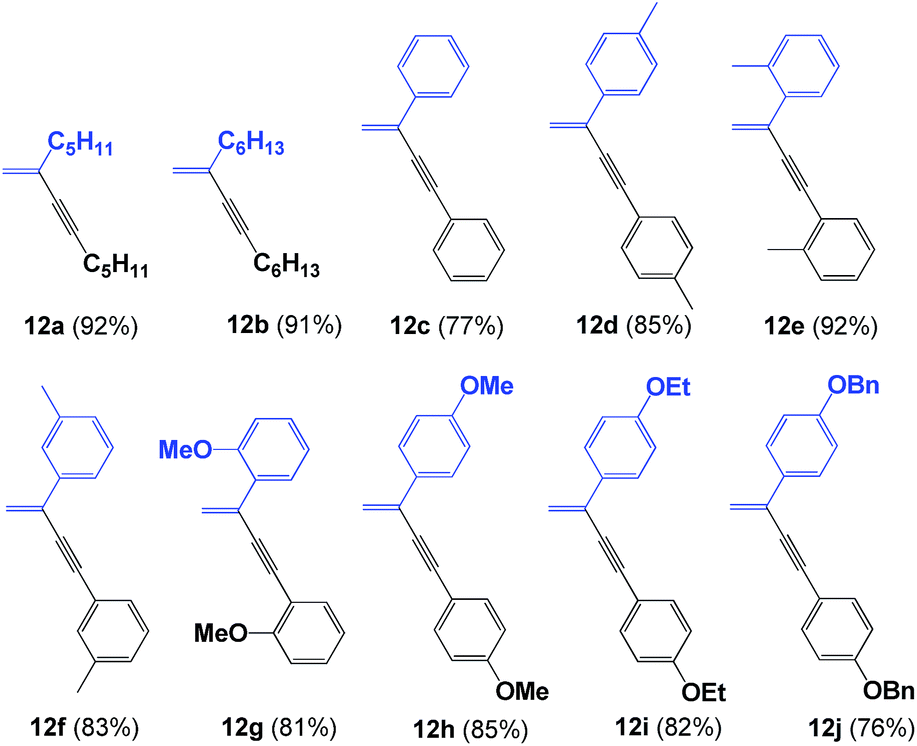
This alkyne dimerization reaction can result in three different isomers, gem, (Z) and (E), depending on the reaction conditions and/or the catalyst. In this case, only the gem isomer was formed, which suggests that the presence of the sterically bulky group on the SPO has an effect on the regioselectivity of the reaction. The same reaction in the presence of Pd(OAc)2 and PPh3 gives a mixture of the three isomers.
The proposed mechanism for the enyne formation is shown in Scheme 3. The first step involves the rupture of the Ph1-Phoxide 8 by sodium acetate to form the catalytic precursor A. This precursor promotes ligand exchange with the alkyne substrates B and C–C coupling between them, generating species C. The enyne is obtained by insertion of another alkyne substrate molecule, regenerating the proposed catalytic species B (Scheme 3).20
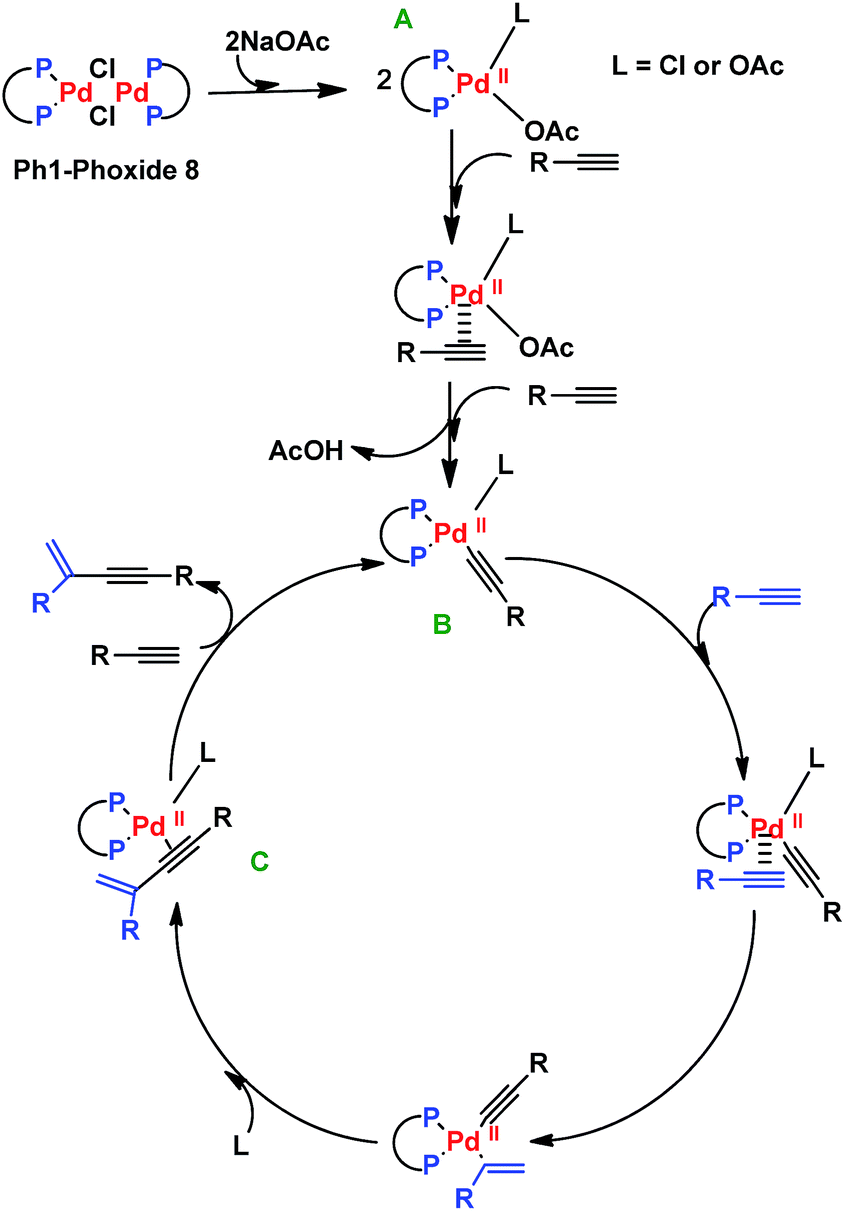 | ||
| Scheme 3 Proposed mechanism for 1,3-enynes formation. | ||
2.2. Synthesis of aromatic-alkynes
Following these results, we extended the use of SPO Pd complex 8 towards the coupling of terminal alkynes to aromatic rings (Sonogashira coupling).22,23 Iodobenzene derivatives were tested first, as they are often used as model substrates for a class of reactions. The phenyl-alkynes 14a–14f were isolated in excellent yields (90%, Table 2). The yield decreased to 80% when substituted bromobenzenes were used. No product was observed with chlorobenzene derivatives. When the reactions were carried out in the absence of CuI, only alkyne dimers were isolated. Again, the use of a microwave reactor allowed for a significant reduction in reaction time, from 18 h to 20 minutes, without affecting yield.
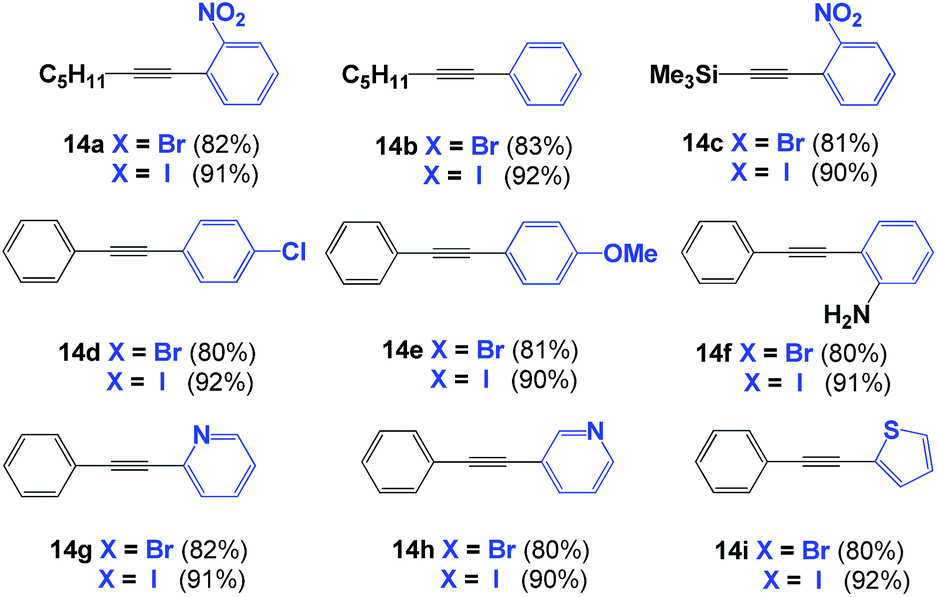
The reaction also works with halogenated heterocycles. For example, 2- and 3-halopyridines were alkynylated under the same reaction conditions; both products were obtained in excellent yield. Notably, alkynylation of the 3-position of pyridine is known to be a difficult reaction, but this reaction proceeds smoothly in the presence of complex 8 (90% for iodo- and 80% for bromo-substituted aryls). The reaction also tolerates halothiophenes, providing the alkyne-aryl adduct in excellent yield (Table 2).
2.3. Synthesis of ynones
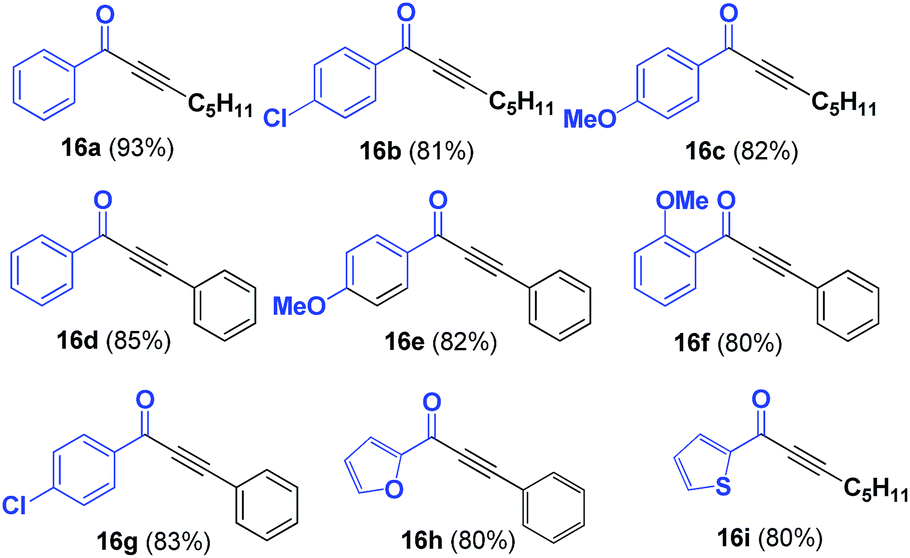
SPO complex 8 was also applied in the synthesis of α,β-acetylenic carbonyl compounds (ynones), using highly reactive acyl chlorides as substrates. Ynones are often employed as key intermediates in natural product synthesis.24,25 In this case, the ynones were obtained in excellent yield when the reaction was performed in the presence of triethylamine in toluene at 100 °C; the scope of the reaction was then examined using these optimum reaction conditions. Ynones 16a–16i were obtained in high yield (Table 3). When the reactions were carried out with microwave heating, similar yields were obtained in 1 h.
It is noteworthy that when the reaction was carried out with an alkyl acyl chloride (butyryl chloride 17), the only product observed was the β-lactone 19. This product could be explained as a result of [2 + 2] cycloaddition of ketene 18, which was formed from alkyl acyl chloride 17 in presence of triethtylamine (Scheme 4).26
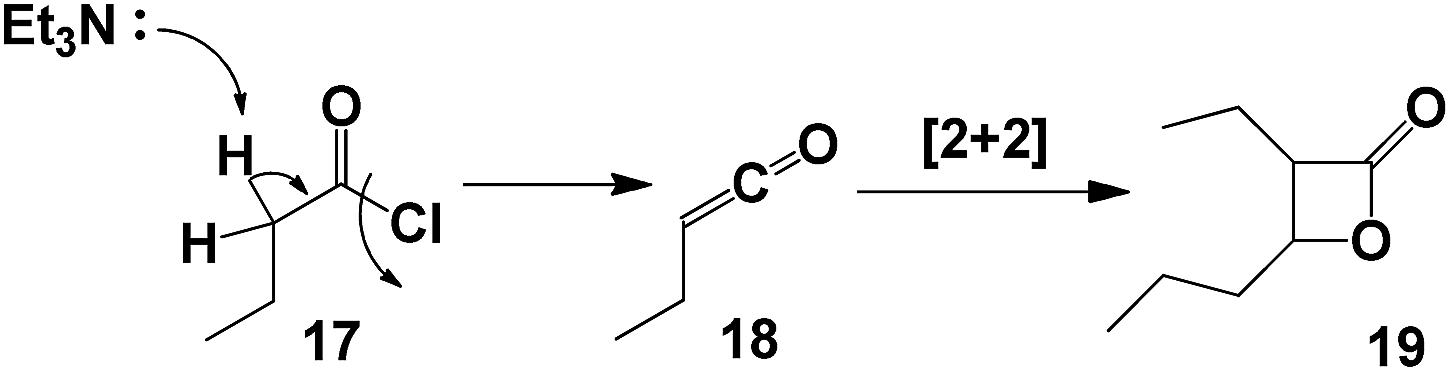 | ||
| Scheme 4 Reaction of alkyl acyl chloride in presence of TEA. | ||
The mechanism involved in both ynones and phenyl-alkynes synthesis is proposed as follows: the formation of the catalytic species D is performed in situ by rupture of the dimeric complex 8. Then species E is formed by oxidative addition of an acyl chloride or an aryl halide. A trasmetallation reaction between the complex E and the copper acetylide generates species F. Finally, the products are formed by reductive elimination, which regenerates the palladium catalyst (Scheme 5).22
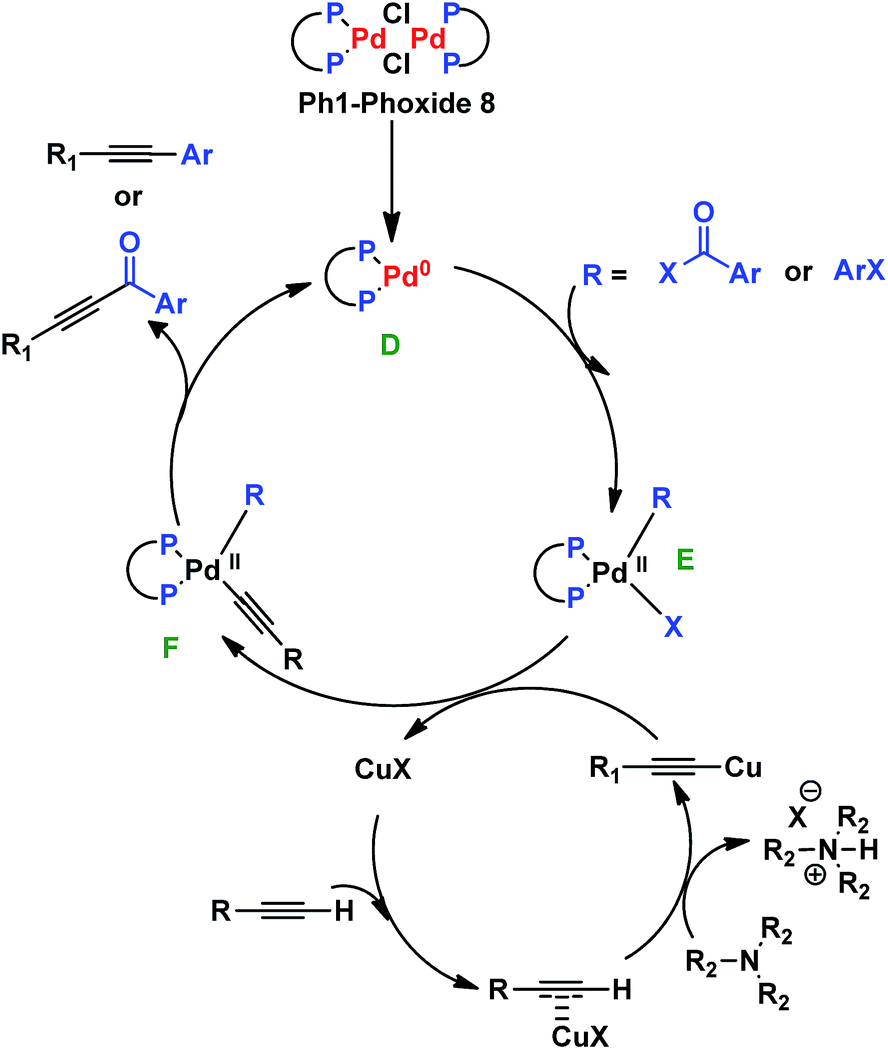 | ||
| Scheme 5 Proposed mechanism for aromatic-alkynes and ynones formation. | ||
2.4. Synthesis of anemarchalconyn
With optimized conditions for ynone formation and aryl alkynylation, we decided to apply our methodology to synthesis of a natural product that would utilize SPO complex 8 in two separate steps. Anemarchalconyn (16k, Scheme 6) was isolated from the rhizomes of Anemarrhena asphodeloides and has inhibitory effects on the adipogenic differentiation of preadipocyte 3T3-L1 cells, with an IC50 value of 5.3 μM.27 The synthesis began with the methylation of 4-iodophenol 20, followed by the addition of ethynyltrimethylsilane to 4-iodoanisole 21 in the presence of palladium complex 8, generating intermediate 22 in 80% yield. The silyl group was removed using basic conditions to give alkyne 14j in 90% yield. Oxidation of p-anisyl alcohol 23, followed by activation of carbonyl group, afforded acyl chloride 25. The key step in the synthesis was the coupling between alkyne 14j and acyl chloride 25 catalysed by the palladium complex 8, giving ynone 16j in 80% yield. Finally, removal of the methyl ether protecting group furnished anemarchalconyn (16k) in 35% yield (Scheme 1). The 1H and 13C NMR spectroscopic data matched those reported for the natural product.27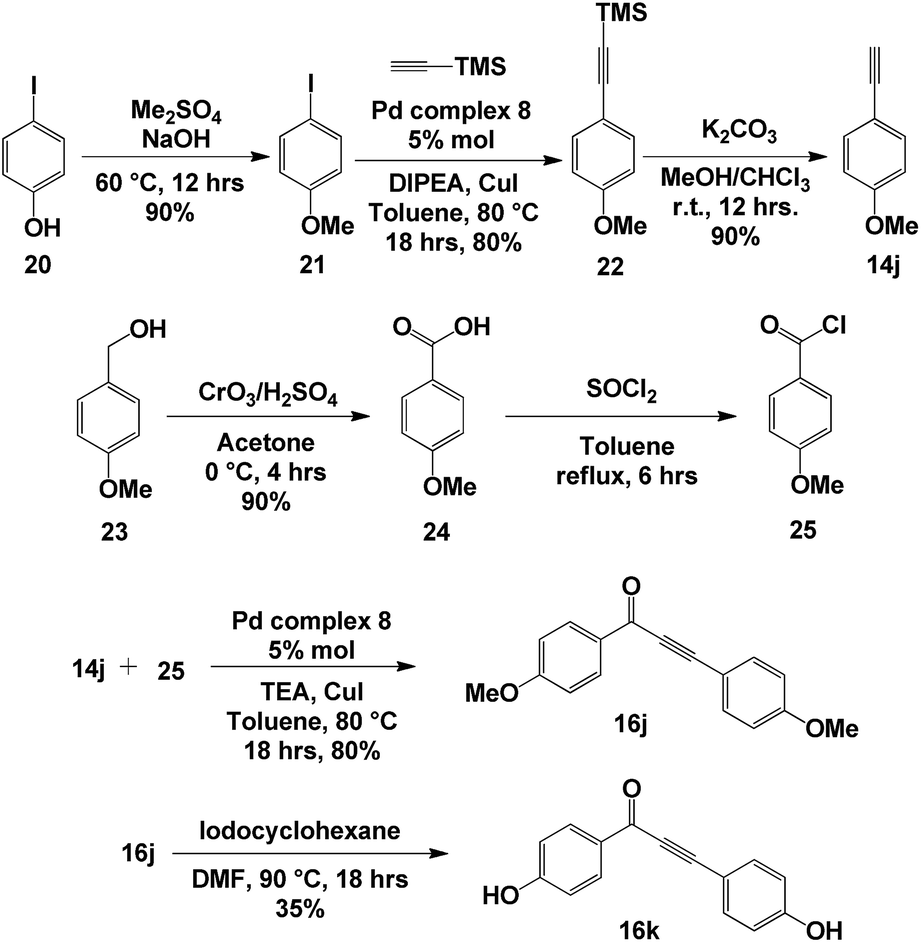 | ||
| Scheme 6 Synthesis of anemarchalconyn. | ||
3. Conclusions
In conclusion, we have demonstrated that an air-stable palladium(II) phosphinito complex 8 (Ph1-Phoxide), was able to catalyse the regioselective dimerization of alkynes to obtain gem-1,3-enynes, the coupling of haloaromatics and acyl chlorides and the formation of ynones from alkynes and acyl chlorides. Furthermore, the methodology was applied to the synthesis of anemarchalconyn, an aromatic ynone isolated from the rhizomes of Anemarrhena asphodeloides.4. Experimental section
4.1. General methods
All reactions were conducted under a dried argon stream. The reagents were purchased from Aldrich Chemical Co, St. Louis Missouri, United States, and used without further purification unless stated otherwise. Yields refer to the chromatographically and spectroscopically (1H and 13C) homogeneous materials, unless otherwise stated. All glassware was flame-dried before use. The reactions were monitored by TLC carried out on 0.25 mm E. Merck silica gel plates. The developed TLC plates were visualised under a short-wave UV lamp or by heating after they were dipped in Ce(SO4)2. Flash column chromatography (FCC) was performed using silica gel (230–400) and employed a solvent polarity correlated with the TLC mobility. NMR experiments were conducted on a Varian 300 MHz instrument in CDCl3 (99.9% D) as the solvent; the chemical shifts (δ) were referenced to CDCl3 (7.26 ppm 1H, 77.00 ppm 13C) or Me4Si (0.00 ppm). The chemical shifts are reported with respect to the solvent peak in parts per million (ppm). Microwave irradiation experiments were performed on a Monowave 450-Anton Paar, using standard sealed microwave glass vials.4.2. General procedures
4.3. Characterization data
4.4. Synthesis of anemarchalconyn
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were added to ((4-methoxyphenyl)ethynyl)trimethylsilane 22 (200 mg, 0.98 mmol) in a 50 mL recovery flask. The suspension was stirred at room temperature for 18 h. After that time, the reaction mixture was diluted with EtOAc (25 mL), washed successively with 10% citric acid solution (2 × 20 mL) and brine (3 × 20 mL), dried over magnesium sulphate, filtered and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel using hexane–EtOAc, to give 116 mg of 14j (90% yield). 1H NMR (300 MHz, CDCl3): δ 7.46 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 3.84 (s, 3H), 3.02 (s, 1H); 13C NMR (75.5 MHz, CDCl3): δ 159.9, 133.5, 114.2, 113.9, 93.9, 83.6, 55.3. Anal. calcd for C9H8O: C, 81.79; H, 6.10. Found: C, 81.76; H, 6.08.
:1) were added to ((4-methoxyphenyl)ethynyl)trimethylsilane 22 (200 mg, 0.98 mmol) in a 50 mL recovery flask. The suspension was stirred at room temperature for 18 h. After that time, the reaction mixture was diluted with EtOAc (25 mL), washed successively with 10% citric acid solution (2 × 20 mL) and brine (3 × 20 mL), dried over magnesium sulphate, filtered and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel using hexane–EtOAc, to give 116 mg of 14j (90% yield). 1H NMR (300 MHz, CDCl3): δ 7.46 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 3.84 (s, 3H), 3.02 (s, 1H); 13C NMR (75.5 MHz, CDCl3): δ 159.9, 133.5, 114.2, 113.9, 93.9, 83.6, 55.3. Anal. calcd for C9H8O: C, 81.79; H, 6.10. Found: C, 81.76; H, 6.08.Acknowledgements
The authors are very grateful for economic support acquired from CONACyT (Project-255819) and PRODEP-SEP (Project-DSA/103.5/16/10288). JAMS is grateful to the Royal Society (UK) for the International Alumni Grant.Notes and references
- (a) T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771 CrossRef CAS; (b) V. K. Jain and L. Jain, Coord. Chem. Rev., 2005, 249, 3075 CrossRef CAS.
- T. Achard, Chimia, 2016, 70, 8 CrossRef CAS PubMed.
- (a) C.-Y. Hu, Y.-Q. Chen, G.-Y. Lin, M.-K. Huang, Y.-C. Chang and F.-E. Hong, Eur. J. Inorg. Chem., 2016, 3131 CrossRef CAS; (b) Y.-C. Chang, C.-Y. Hu, Y.-H. Liang and F.-E. Hong, Polyhedron, 2016, 105, 123 CrossRef CAS; (c) I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2015, 137, 7718 CrossRef CAS PubMed; (d) E. Rafter, T. Gutmann, F. Löw, G. Buntkowsky, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2013, 3, 595 RSC; (e) B. Kurscheid, B. Neumann, H.-G. Stammler and B. Hoge, Chem.–Eur. J., 2011, 17, 14935 CrossRef CAS PubMed; (f) L. Ackermann, Synthesis, 2006, 1557 CrossRef CAS.
- L. Copey, L. Jean-Gérard, B. Andrioletti and E. Framery, Tetrahedron Lett., 2016, 57, 543 CrossRef CAS.
- (a) L. V. Graux, M. Giorgi, G. Buono and H. Clavier, Organometallics, 2015, 34, 1864 CrossRef CAS; (b) N. P. N. Wellala and H. Guan, Org. Biomol. Chem., 2015, 13, 10802 RSC; (c) Y.-C. Chang, W.-C. Chang, C.-Y. Hu and F.-E. Hong, Organometallics, 2014, 33, 3523 CrossRef CAS; (d) P. M. Castro, H. Gulyás, J. Benet-Buchholz, C. Bo, Z. Freixa and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2011, 1, 401 RSC; (e) D. Martin, D. Moraleda, T. Achard, L. Giordano and G. Buono, Chem.–Eur. J., 2011, 17, 12729 CrossRef CAS PubMed.
- (a) J. Weismann, L. T. Scharf and V. H. Gessner, Organometallics, 2016, 35, 2507 CrossRef CAS; (b) L. V. Graux, M. Giorgi, G. Buono and H. Clavier, Dalton Trans., 2016, 45, 6491 RSC; (c) D. Zell, S. Warratz, D. Gelman, S. J. Garden and L. Ackermann, Chem.–Eur. J., 2016, 22, 1248 CrossRef CAS PubMed; (d) N. Allefeld, J. Bader, B. Neumann, H.-G. Stammler, N. Ignat’ev and B. Hoge, Inorg. Chem., 2015, 54, 7945 CrossRef CAS PubMed; (e) A. Christiansen, D. Selent, A. Spannenberg, M. Köckerling, H. Reinke, W. Baumann, H. Jiao, R. Franke and A. Börner, Chem.–Eur. J., 2011, 17, 2120 CrossRef CAS PubMed.
- (a) J. Meeuwissen, R. J. Detz, A. J. Sandee, B. de Bruin and J. N. H. Reek, Dalton Trans., 2010, 39, 1929 RSC; (b) C. Waloch, J. Wieland, M. Keller and B. Breit, Angew. Chem., Int. Ed., 2007, 46, 3037 CrossRef CAS PubMed; (c) B. Patel, S. J. A. Pope and G. Reid, Polyhedron, 1998, 17, 2345 CrossRef CAS; (d) J. A. S. Duncan, T. A. Stephenson, M. D. Walkinshaw, D. Heden and D. M. Roundhill, J. Chem. Soc., Dalton Trans., 1984, 801 RSC; (e) J. A. S. Duncan, T. A. Stephenson, W. B. Beaulieu and D. M. Roundhill, J. Chem. Soc., Dalton Trans., 1983, 1755 RSC; (f) J. A. S. Duncan, D. Hedden, D. M. Roundhill, T. A. Stephenson and M. D. Walkinshaw, Angew. Chem., Int. Ed. Engl., 1982, 21, 452 CrossRef; (g) D. M. Roundhill, R. P. Sperline and W. B. Beaulieu, Coord. Chem. Rev., 1978, 26, 263 CrossRef CAS.
- (a) N. Allefeld, M. Grasse, N. Ignat’ev and B. Hoge, Chem.–Eur. J., 2014, 20, 8615 CrossRef CAS PubMed; (b) W.-J. Yoo and S. Kobayashi, Green Chem., 2013, 15, 1844 RSC; (c) B. Hoge, J. Bader, H. Beckers, Y. S. Kim, R. Eujen, H. Willner and N. Ignat’ev, Chem.–Eur. J., 2009, 15, 3567 CrossRef CAS PubMed; (d) B. Hoge, P. Garcia, H. Willner and H. Oberhammer, Chem.–Eur. J., 2006, 12, 3567 CrossRef CAS PubMed; (e) B. Hoge, S. Neufeind, S. Hettel, W. Wiebe and C. Thosen, J. Organomet. Chem., 2005, 690, 2382 CrossRef CAS.
- Y.-Y. Chang and F.-E. Hong, Tetrahedron, 2013, 69, 2327 CrossRef CAS.
- (a) H. Gulyás, I. Rivilla, S. Curreli, Z. Freixa and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2015, 5, 3822 RSC; (b) M. Karanik, D. Lesage, Y. Gimbert, P. Nava, S. Humbel, L. Giordano, G. Buono and J.-C. Tabet, Organometallics, 2011, 30, 4814 CrossRef CAS; (c) T. J. Ahmed, B. R. Fox, S. M. M. Knapp, R. B. Yelle, J. J. Juliette and D. R. Tyler, Inorg. Chem., 2009, 48, 7828 CrossRef CAS PubMed; (d) M. North, A. W. Parkins and A. N. Shariff, Tetrahedron Lett., 2004, 45, 7625 CrossRef CAS; (e) X. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327 CrossRef CAS PubMed; (f) L.-B. Hand, N. Choi and M. Tanaka, Organometallics, 1996, 15, 3259 CrossRef.
- J. Coetzee, G. R. Eastham, A. M. Z. Slawin and D. J. Cole-Hamilton, Dalton Trans., 2014, 43, 3479 RSC.
- H. Xu, K. Ekoue-Kovi and C. Wolf, J. Org. Chem., 2008, 73, 7638 CrossRef CAS PubMed.
- C. Wolf and R. Lerebours, Org. Lett., 2004, 6, 1147 CrossRef CAS PubMed.
- R. Lerebours and C. Wolf, Org. Lett., 2007, 9, 2737 CrossRef CAS PubMed.
- C. Wolf and K. Ekoue-Kovi, Eur. J. Org. Chem., 2006, 1917 CrossRef CAS.
- C. Wolf and R. Lerebours, J. Org. Chem., 2003, 68, 7077 CrossRef CAS PubMed.
- (a) J. A. Morales-Serna, A. Zúñiga-Martínez, M. Salmón, R. Gaviño and J. Cárdenas, Synthesis, 2012, 44, 446 CrossRef CAS; (b) A. Sauza, J. A. Morales-Serna, M. García-Molina, R. Gaviño and J. Cárdenas, Synthesis, 2012, 44, 272 CAS.
- K. R. Dixon and A. D. Rattray, Can. J. Chem., 1971, 49, 3997 CrossRef CAS.
- (a) P. Bergamini, V. Bertolasi, M. Cattabriga, V. Ferretti, U. Loprieno, N. Mantovani and L. Marvelli, Eur. J. Inorg. Chem., 2003, 5, 918 CrossRef; (b) T. G. Gebauer, K. Frenzen and Z. Dehnicke, Journal of Crystallography–Crystalline Materials, 1995, 210, 539 CAS; (c) T. Ghaffar, A. Kieszkiewicz, S. C. Nyburg and A. W. Parkins, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 697 CrossRef.
- B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212 RSC.
- For recent synthesis of 1,3-enynes, see: (a) N.-N. Wang, L.-R. Huang, W.-J. Hao, T.-S. Zhang, G. Li, S.-J. Tu and B. Jiang, Org. Lett., 2016, 18, 1298 CrossRef CAS PubMed; (b) M. L. N. Rao, P. Dasgupta and V. N. Murty, RSC Adv., 2015, 5, 24834 RSC; (c) J.-Y. Jiao, X.-G. Zhang and X.-H. Zhang, Tetrahedron, 2015, 71, 9245 CrossRef CAS; (d) A. Ikeda, M. Omote, K. Kusumoto, A. Tarui, K. Sato and A. Ando, Org. Biomol. Chem., 2015, 13, 8886 RSC; (e) J. H. Kim, Y. R. Jeong, S. L. Jeon and I. H. Jeong, J. Fluorine Chem., 2014, 167, 166 CrossRef CAS; (f) O. S. Morozov, A. F. Asachenko, D. V. Antonov, V. S. Kochurov, D. Y. Paraschuk and M. S. Nechaev, Adv. Synth. Catal., 2014, 356, 2671 CrossRef CAS; (g) P. Sun, H. Yan, L. Lu, D. Liu, G. Rong and J. Mao, Tetrahedron, 2013, 69, 6969 CrossRef CAS; (h) Y. Zhu, T. Li, X. Qu, P. Sun, H. Yang and J. Mao, Org. Biomol. Chem., 2011, 9, 7309 RSC; (i) Y. Wen, A. Wang, H. Jiang, S. Zhu and L. Huang, Tetrahedron Lett., 2011, 52, 5736 CrossRef CAS.
- For reviews about Sonogashira reaction, see: (a) L. Boulton, in Synthetic Methods in Drug Discovery, ed. D. C. Blakemore, P. M. Doyle and Y. M. Fobian, RSC, 2016, vol. 1, ch. 4, pp. 122–142 Search PubMed; (b) M. B. Nielsen, Synthesis, 2016, 48, 2732 CrossRef CAS; (c) M. Karak, L. C. A. Barbosa and G. C. Hargaden, RSC Adv., 2014, 4, 53442 RSC; (d) A. M. Thomas, A. Sujatha and G. Anilkumar, RSC Adv., 2014, 4, 21688 RSC; (e) D. Wang and S. Gao, Org. Chem. Front., 2014, 1, 556 RSC; (f) M. Bakherad, Appl. Organomet. Chem., 2013, 27, 125 CrossRef CAS; (g) R. Chinchilla and C. Naájera, Chem. Soc. Rev., 2011, 40, 5084 RSC; (h) B. K. Singh, N. Kaval, S. Tomar, E. Van der Eycken and V. S. Parmar, Org. Process Res. Dev., 2008, 12, 468 CrossRef CAS; (i) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834 CrossRef CAS PubMed.
- For some recent and selected examples about Sonogashira reaction, see: (a) H. Deol, S. Pramanik, M. Kumar, I. A. Khan and V. Bhalla, ACS Catal., 2016, 6, 3771 CrossRef CAS; (b) V. N. Mikhaylov, V. N. Sorokoumov, K. A. Korvinson, A. S. Novikov and I. A. Balova, Organometallics, 2016, 35, 1684 CrossRef CAS; (c) M. Gazvoda, M. Virant, A. Pevec, D. Urankar, A. Bolje, M. Kočevar and J. Košmrlj, Chem. Commun., 2016, 52, 157 RSC; (d) S. Pradhan, S. Dutta and R. P. John, New J. Chem., 2016, 40, 7140 RSC; (e) H. Salemi, B. Kaboudin, F. Kazemi and T. Yokomatsu, RSC Adv., 2016, 6, 52656 RSC; (f) C. I. Ezugwu, B. Mousavi, M. A. Asrafa, A. Mehta, H. Vardhan and F. Verpoort, Catal. Sci. Technol., 2016, 6, 2050 RSC; (g) A. M. Mak, Y. H. Lim, H. Jong, Y. Yang, C. W. Johannes, E. G. Robins and M. B. Sullivan, Organometallics, 2016, 35, 1036 CrossRef CAS; (h) M. Trunk, A. Herrmann, H. Bildirir, A. Yassin, J. Schmidt and A. Thomas, Chem.–Eur. J., 2016, 22, 7179 CrossRef CAS PubMed; (i) D.-C. Wang, H.-X. Wang, E.-J. Hao, X.-H. Jiang, M.-S. Xie, G.-R. Qu and H.-M. Guo, Adv. Synth. Catal., 2016, 358, 494 CrossRef CAS; (j) M. Bagherzadeh, N. Mousavi, M. Zare, S. Jamali, A. Ellern and L. K. Woo, Inorg. Chim. Acta, 2016, 451, 227 CrossRef CAS; (k) Y. Kelgokmen and M. n Zora, RSC Adv., 2016, 6, 4608 RSC.
- For reviews about synthesis of ynones, see: (a) R. E. Whittaker, A. Dermenci and G. Dong, Synthesis, 2016, 48, 161 CAS; (b) C. F. Gers-Panther and T. J. J. Müller, Adv. Heterocycl. Chem., 2016, 120, 67 CrossRef.
- For recent synthesis of ynones, see: (a) M. N. Soltani Rad, J. Saudi Chem. Soc., 2016 DOI:10.1016/j.jscs.2016.06.001; (b) S. Tang, L. Zeng, Y. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 15850 CrossRef CAS PubMed; (c) H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374 CrossRef CAS PubMed; (d) X.-H. Ouyang, R.-J. Song, C.-Y. Wang, Y. Yang and J.-H. Li, Chem. Commun., 2015, 51, 14497 RSC; (e) R.-Y. Zhang, L.-Y. Xi, L. Zhang, S.-Y. Chen and X.-Q. Yu, Tetrahedron, 2015, 71, 6176 CrossRef CAS; (f) P.-F. Wang, Y.-S. Feng, Z.-F. Cheng, Q.-M. Wu, G.-Y. Wang, L.-L. Liu, J.-J. Dai, J. Xu and H.-J. Xu, J. Org. Chem., 2015, 80, 9314 CrossRef CAS PubMed; (g) R.-Y. Zhang, L.-Y. Xi, L. Zhang, S.-Y. Chen and X.-Q. Yu, Tetrahedron, 2015, 71, 6176 CrossRef CAS; (h) C. L. Taylor and Y. Bolshan, J. Visualized Exp., 2015, 96, e52401, DOI:10.3791/52401; (i) Z. Wang, L. Li and Y. Huang, J. Am. Chem. Soc., 2014, 136, 12233 CrossRef CAS PubMed; (j) E. Mohammadi, B. Movassagh and M. Navidi, Helv. Chim. Acta, 2014, 97, 70 CrossRef CAS; (k) C. Taylor and Y. Bolshan, Org. Lett., 2014, 16, 488 CrossRef CAS PubMed; (l) J. A. Morales-Serna, A. Sauza, G. Padrón de Jesús, R. Gaviño, G. García de la Mora and J. Cárdenas, Tetrahedron Lett., 2013, 54, 7111 CrossRef CAS.
- R. J. Cox, D. J. Ritson, T. A. Dane, J. Berge, J. P. H. Charmantc and A. Kantacha, Chem. Commun., 2005, 1037 RSC.
- U. J. Youn, Y. S. Lee, H. Jeong, J. Lee, J.-W. Nam, Y. J. Lee, E. S. Hwang, J.-H. Lee, D. Lee, S. S. Kang and E.-K. Seo, J. Nat. Prod., 2009, 72, 1895 CrossRef CAS PubMed.
- S. Karabulut, B. Özgün Öztürk and Y. Imamoglu, J. Organomet. Chem., 2010, 695, 2161 CrossRef CAS.
- O. V. Zatolochnaya, E. G. Gordeev, C. Jahier, V. P. Ananikov and V. Gevorgyan, Chem.–Eur. J., 2014, 20, 9578 CrossRef CAS PubMed.
- C. Xu, W. Du, Y. Zeng, B. Dai and H. Guo, Org. Lett., 2014, 16, 948 CrossRef CAS PubMed.
- C. V. Ramana, P. Patel, K. Vanka, B. Miao and A. Degterev, Eur. J. Org. Chem., 2010, 5955 CrossRef CAS.
- X. Qu, T. Li, P. Sun, Y. Zhu, H. Yang and J. Mao, Org. Biomol. Chem., 2011, 9, 6938 CAS.
- Y. Yamane, X. Liu, A. Hamasaki, T. Ishida, M. Haruta, T. Yokoyama and M. Tokunaga, Org. Lett., 2009, 11, 5162 CrossRef CAS PubMed.
- R. Bernini, S. Cacchi, G. Fabrizi, G. Forte, F. Petrucci, A. Prastaro, S. Niembro, A. Shafir and A. Vallribera, Green Chem., 2010, 12, 150 RSC.
- F. Roschangar, J. Liu, E. Estanove, M. Dufour, S. Rodríguez, V. Farina, E. Hichey, A. Hossain, P.-J. Jones, H. Lee, B. Z. Lu, R. Varsolona, J. Schöder, P. Beaulieu, J. Gillard and C. H. Senayake, Tetrahedron Lett., 2008, 49, 363 CrossRef CAS.
- H.-J. Chen, Z.-Y. Lin, M.-Y. Li, R.-J. Lian, Q. W. Xue and J.-L. Chung, Tetrahedron, 2010, 66, 7755 CrossRef CAS.
- H. Kim and P. H. Lee, Adv. Synth. Catal., 2009, 351, 2827 CrossRef CAS.
- K. Park, G. Bae, J. Moon, J. Choe, K. Song and S. Lee, J. Org. Chem., 2010, 75, 6244 CrossRef CAS PubMed.
- B. Wang, M. Bonin and L. Micouin, J. Org. Chem., 2005, 70, 6126 CrossRef CAS PubMed.
- J.-Y. Chen, T.-C. Lin, S.-C. Chen, A.-J. Chen, C.-Y. Moub and F.-Y. Tsai, Tetrahedron, 2009, 65, 10134 CrossRef CAS.
- L. Zuo, S. Yao, W. Wang and W. Duan, Tetrahedron Lett., 2008, 49, 4054 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |