
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of novel spiroheterocycles via diastereoselective aziridination of cyclic enones†
Kai-Chuan Yangabc,
Jun-Long Li *ab,
Xu-Dong Shenb,
Qiang Lib,
Hai-Jun Lengb,
Qian Huangb,
Peng-Kun Zhengb,
Xiao-Jun Gou*b and
Yong-Gang Zhi*ac
*ab,
Xu-Dong Shenb,
Qiang Lib,
Hai-Jun Lengb,
Qian Huangb,
Peng-Kun Zhengb,
Xiao-Jun Gou*b and
Yong-Gang Zhi*ac
aChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, PR China. E-mail: zyggroup@hotmail.com
bAntibiotics Research and Re-evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, Chengdu University, Chengdu 610052, PR China. E-mail: lijunlong709@hotmail.com; gouxj@163.com
cUniversity of Chinese Academy of Science, Beijing 100049, P. R. China
First published on 13th April 2017
Abstract
A collection of novel spiroheterocycles with strained aziridine moieties have been facilely synthesized by using easily accessible starting materials under mild reaction conditions. This procedure is robust, scalable and highly diastereoselective, and also features broad substrate scope. In addition, some of the products show promising in vitro biological activity against a spectrum of pathogens, which might be considered as a clue for the discovery of new antimicrobial agents.
Multifunctionalized aziridines, due to their unique and strained core structure, have provoked great interest in both pharmaceutical science and synthetic chemistry.1 In particular, polycyclic heterocycles containing an aziridine moiety were frequently seen in many bioactive natural products and drug candidates.2 For example, albomitomycin C, a representative aziridine alkaloid, was isolated from Streptomyces caespitosus culture broth, and can be used as a neoplasm inhibitor (Fig. 1, I). Antibiotic AX-2 which was derived from mitomycin C3 shows significant anticancer activity (Fig. 1, II). On the other hand, spiropyrrolines have also been demonstrated to be a privileged pharmacophore among various therapeutic chemicals.4 Merging these two interesting chemical building blocks can lead to a fascinating structural motif named spiro[aziridine-pyrrolines] (SAP), which has been successfully applied in the design of new antimicrobial agents in recent years (e.g. Fig. 1, III and IV).5 However, efficient synthesis of such SAP derivatives has not received as much attention as other spirocycles,6 despite their synthetic usefulness and great potential in medicinal chemistry.
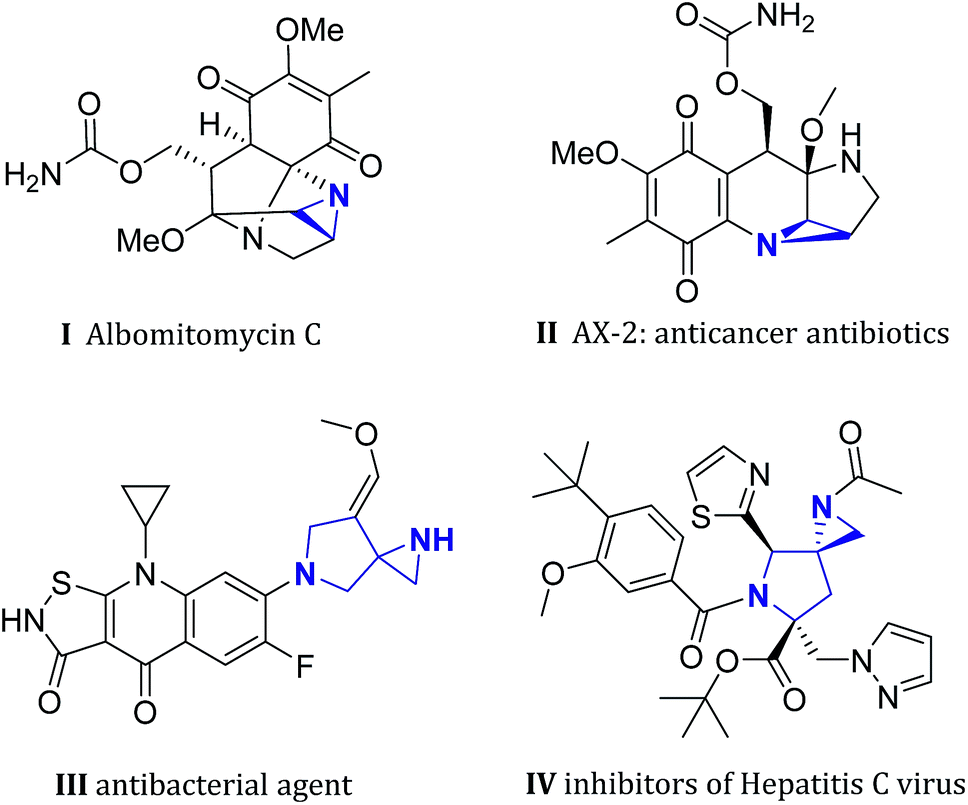 | ||
| Fig. 1 Selected natural products and bioactive molecules containing (spiro-)aziridine core structures.3,5 | ||
Over the past decades, great efforts have been made to construct aziridines in a rapid and straightforward manner.7 Significant advancement has been achieved in this adventure mostly based on three synthetic strategies: intramolecular substitution, direct aziridination of imines and aziridination of alkenes. Notably, it is necessary to pre-install two vicinal functional groups (an amine and a leaving group) in one molecule before using the intramolecular substitution strategy, which requires at least two chemical steps (Scheme 1a).7a The direct aziridination of imines, on the other hand, provides a variety of generally accepted and well-established methods for manufacturing the desired products; for example, the aza-Corey–Chaykovsky reaction, which involves the addition of sulfur ylides to imines, has been regarded as a robust and powerful protocol in the synthesis of aziridines since the middle of last century (Scheme 1b).8 Alternatively, the aziridination of alkenes is also a particularly appealing synthetic strategy because of the ready availability of various olefinic starting materials; however, such a process is relatively less studied, and the organo-promoted or -catalyzed reactions with this strategy has not been systematically explored until the pioneering work of Loreto,9a–f Prabhakar9g,h and Córdova,9i respectively. Among these studies, there are still a lot of remaining problems7 in terms of the reaction efficiency, substrate compatibility and the stereoselectivity (Scheme 1c). Thus, the development of further efficient routes to novel structures of aziridines from easily accessible alkenes is highly desirable.
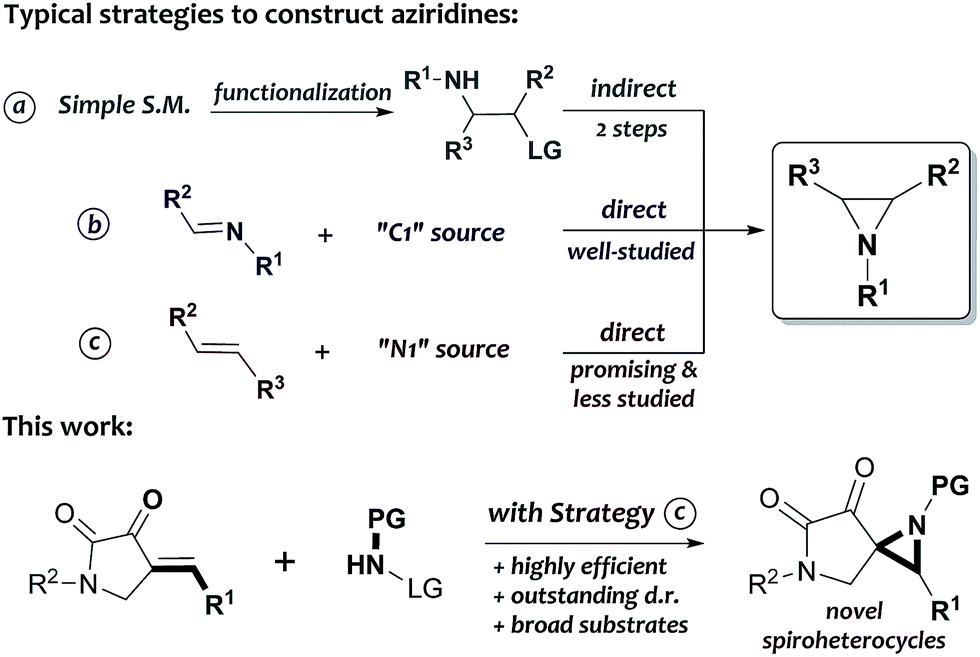 | ||
| Scheme 1 Assembly of spiro[aziridine-pyrrolines] derivatives via aziridination of alkenes. | ||
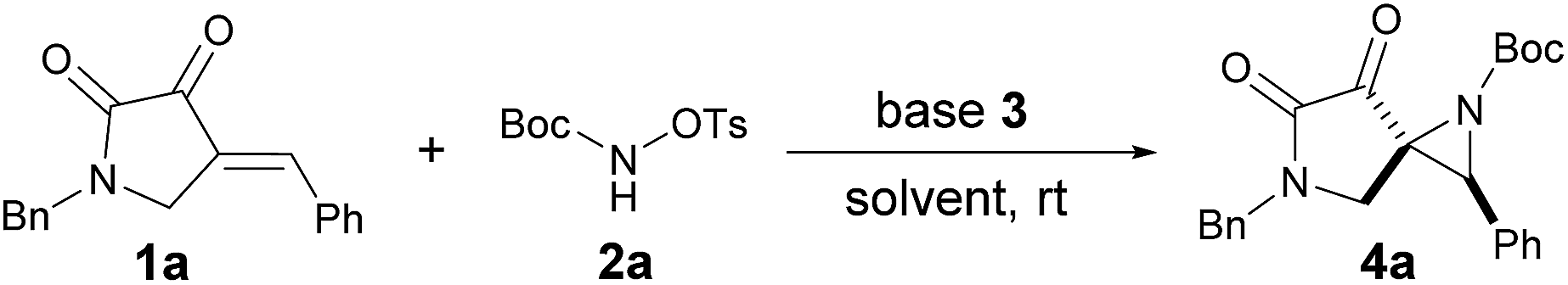
As part of our continuing interest in constructing medicinal relevant frameworks by using small nonmetal organic molecules,10 we describe herein a base-promoted highly diastereoselective aziridination of cyclic electron-deficient alkenes, which leads to facile synthesis of novel SAP derivatives. Initially, we investigated the feasibility of this approach by evaluating the reaction between readily available enone 1a10a,b and the modified carbamate 2a. To our delight, the reaction proceeded smoothly in the presence of DABCO in DCM at room temperature, affording the desired product 4a in 52% yield as a single diastereoisomer (Table 1, entry 1). This result encouraged us to further investigate the solvent effect (Table 1, entries 2–6), and chloroform was found to be the most appropriate (Table 1, entry 5). Changing other organic base (Table 1, entries 7–10) or using inorganic base (Table 1, entries 11 and 12) led to inferior results. Furthermore, the yield would be slightly dropped if less amount of base was used (Table 1, entry 13).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base 3 | d.r.b | Yieldc (%) |
| a Unless otherwise noted, reactions were performed with 0.1 mmol of 1a, 0.15 mmol of 2a, and 0.15 mmol of base 3 in 1 mL solvent at room temperature overnight.b The diastereomeric ratio was determined by 1H NMR spectroscopy of the crude reaction mixture.c Isolated yield.d 10 mol% of TBAB was added as phase transfer catalyst.e 0.1 mmol of DABCO was used as the base. | ||||
| 1 | DCM | DABCO | >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 :5 |
52 |
| 2 | DCE | DABCO | >95:5 |
57 |
| 3 | CH3CN | DABCO | >95:5 |
57 |
| 4 | THF | DABCO | >95:5 |
82 |
| 5 | CHCl3 | DABCO | >95:5 |
93 |
| 6 | Toluene | DABCO | >95:5 |
89 |
| 7 | CHCl3 | TMG | >95:5 |
91 |
| 8 | CHCl3 | DBU | >95:5 |
60 |
| 9 | CHCl3 | TEA | >95:5 |
83 |
| 10 | CHCl3 | DIPEA | >95:5 |
85 |
| 11d | CHCl3 | K2CO3 | >95:5 |
35 |
| 12d | CHCl3 | KOH | >95:5 |
50 |
| 13e | CHCl3 | DABCO | >95:5 |
85 |
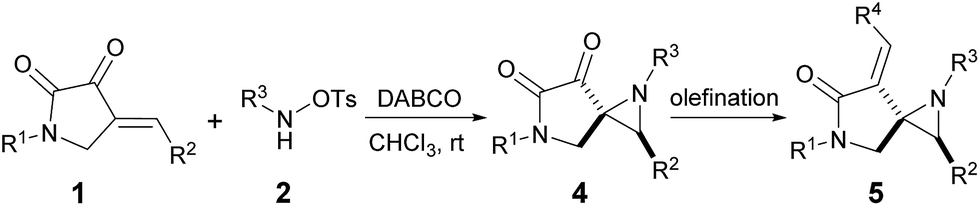
Having established the optimal reaction conditions (Table 1, entry 5), we set out to explore the generality of the [2 + 1] cycloadditions. Due to the ketone group that was adjacent to the aziridine moiety, some of the spiro-products were found to be somewhat unstable unless stored under low temperature, which limits the relevant biological study of such frameworks. Thus, a reliable Horner–Wadsworth–Emmons (HWE) reaction was utilized to transform this ketone group to the corresponding α,β-unsaturated ester as the final bench-stable product 5. As summarized in Table 2, a variety of cyclic enones 1 bearing either electron-withdrawing (Table 2, entries 2–9) or electron-donating (Table 2, entries 10–12) groups at different positions of the phenyl ring reacted efficiently to afford the desired products 5b–5l in excellent diastereoselectivities and nice isolated yields. The reaction was also suitable for enone substrates with polycyclic aromatics, such as 1-naphthyl and 2-naphthyl rings (Table 2, entries 13 and 14). It was revealed that the N-protecting groups of the enones have limited effect on the outcome of this reaction (Table 2, entries 15 and 16). On the other hand, in terms of the nucleophile, the Cbz group on the nitrogen atom could also be well tolerated (Table 2, entry 17). Furthermore, by using Wittig reaction, product 4a could be easily transformed to the corresponding α,β-unsaturated lactam 5r which contains an interesting exocyclic terminal alkenes (Table 2, entry 18).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | R3 | R4 | Yieldb (%) |
| a Unless otherwise noted, reactions were performed with 0.2 mmol of 1, 0.3 mmol of 2 and 0.3 mmol of DABCO in 2 mL CHCl3 at rt overnight. The diastereomeric ratio of 4 was determined to be >95:5 by 1H NMR spectroscopy of the crude reaction mixture. The HWE reaction was used for the olefination process, for details, see ESI.b Isolated yield of the aziridination product 4, and the data in parentheses refers to the isolated yield of the olefination product 5.c The structure of 5d was determined by X-ray diffraction analysis, and others were determined by analogy.d Using methyltriphenylphosphonium bromide as the Wittig reagent for the olefination process, see ESI. Bn: benzyl; PMB: p-methoxybenzyl; Boc: t-butyloxy-carboryl; Cbz: carboxybenzyl. |
||||||
| 1 | 4a/5a | Bn | C6H5 | Boc | CO2Et | 93 (91) |
| 2 | 4b/5b | Bn | 4-FC6H4 | Boc | CO2Et | 87 (88) |
| 3 | 4c/5c | Bn | 4-ClC6H4 | Boc | CO2Et | 89 (91) |
| 4c | 4d/5d | Bn | 4-BrC6H4 | Boc | CO2Et | 88 (89) |
| 5 | 4e/5e | Bn | 4-NO2C6H4 | Boc | CO2Et | 89 (90) |
| 6 | 4f/5f | Bn | 3-ClC6H4 | Boc | CO2Et | 85 (89) |
| 7 | 4g/5g | Bn | 3-BrC6H4 | Boc | CO2Et | 87 (91) |
| 8 | 4h/5h | Bn | 2-ClC6H4 | Boc | CO2Et | 81 (84) |
| 9 | 4i/5i | Bn | 2,4-Cl2C6H3 | Boc | CO2Et | 86 (89) |
| 10 | 4j/5j | Bn | 4-MeC6H4 | Boc | CO2Et | 90 (89) |
| 11 | 4k/5k | Bn | 3-MeC6H4 | Boc | CO2Et | 88 (90) |
| 12 | 4l/5l | Bn | 3-MeOC6H4 | Boc | CO2Et | 84 (89) |
| 13 | 4m/5m | Bn | 1-Naphthyl | Boc | CO2Et | 80 (87) |
| 14 | 4n/5n | Bn | 2-Naphthyl | Boc | CO2Et | 86 (90) |
| 15 | 4o/5o | PMB | C6H5 | Boc | CO2Et | 89 (91) |
| 16 | 4p/5p | Allyl | C6H5 | Boc | CO2Et | 80 (81) |
| 17 | 4q/5q | Bn | C6H5 | Cbz | CO2Et | 90 (91) |
| 18d | 4a/5r | Bn | C6H5 | Boc | H | 93 (81) |
To further illustrate the robustness and practicality of this methodology, the aziridination reaction with 1a was scaled up to 1.0 gram under optimal conditions. Gratifyingly, the desired spiroaziridine 4a and its derivative 5a were smoothly obtained with excellent diastereoselectivity in 88% and 87% yield, respectively (Scheme 2a). It should be worth highlighting that such spiroaziridine skeleton could also be easily transformed to the spiro[lactam-oxazolidinone] core structure; as illustrated in Scheme 2b, in the presence of Cu(OTf)2 as Lewis acid catalyst, the corresponding product 6 was synthesized in high yield, albeit with moderate diastereoselectivity. Moreover, structural correctness of the spiroaziridines and the relative configuration of the adjacent stereocenters were confirmed by X-ray diffraction analysis of the representative product 5d (Fig. 2, for details, see ESI†).
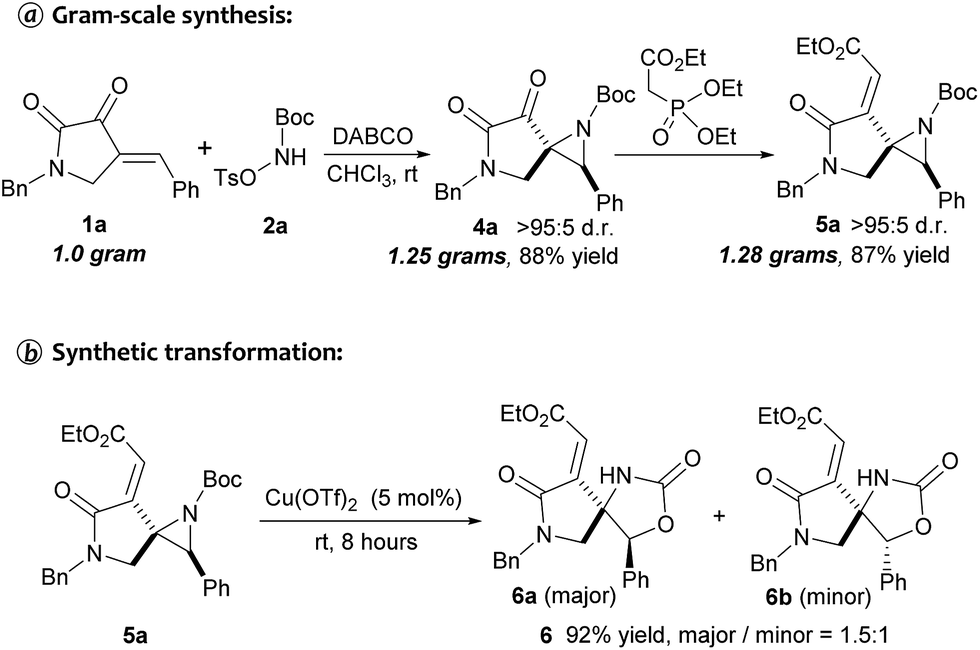 | ||
| Scheme 2 Gram-scale synthesis of spiroaziridines 4a and 5a, and the synthetic transformation of 5a. | ||
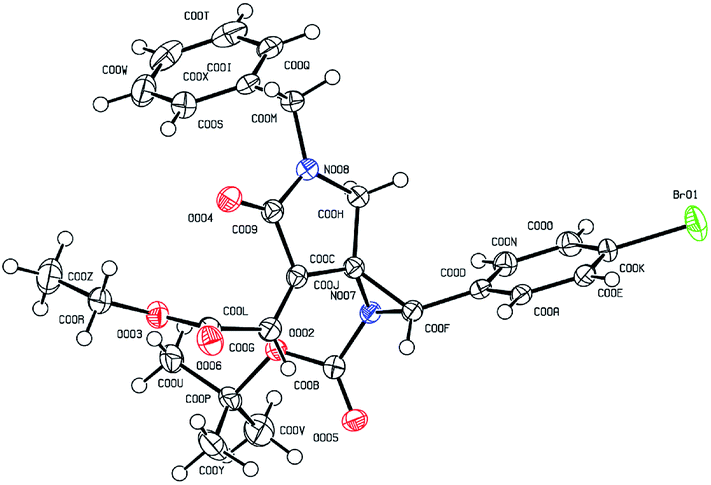 | ||
| Fig. 2 Single crystal X-ray diffraction analysis of product 5d.11 | ||
In addition, we have demonstrated that the enantioenriched spiroaziridine 5a could be synthesized by using the readily available and relatively inexpensive quinidine as the chiral Brønsted base (Scheme 3). However, considering its moderate enantioselectivity and high loading of the chiral base, further optimization is still in urgent demand and currently underway in our lab.
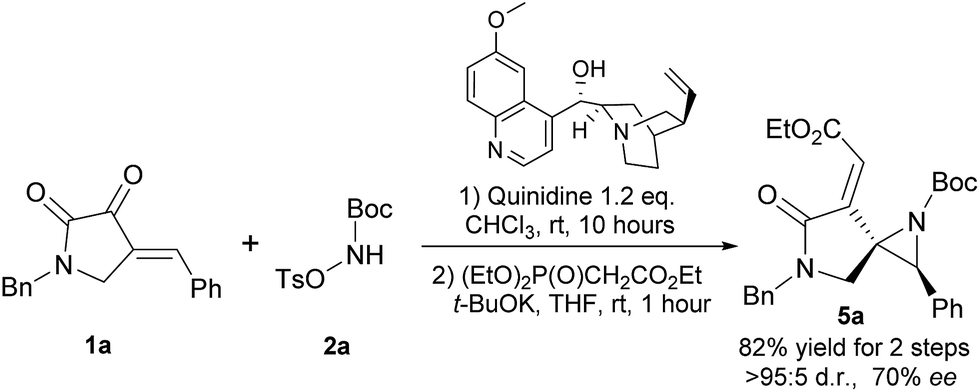 | ||
| Scheme 3 Asymmetric synthesis of chiral spiroaziridine 5a by using commercially available quinidine. | ||
The collection of new compounds 5a–5r was screened for in vitro antibacterial activity against Staphylococcus aureus (ATCC 25923), methicillin-resistant Staphylococcus aureus (MRSA, clinic isolates) and Proteus mirabilis (clinic isolates).12 It was revealed that some of these spiroaziridines exhibited promising bioactivity, with the minimum inhibitory concentrations (MICs) value ranging from 8 to 128 μg mL−1. Particularly, compound 5c shows obvious in vitro antibacterial activity against MRSA which is regarded as a clinically important pathogen (Table 3).
| Entry | Compound | MICb (μg mL−1) | ||
|---|---|---|---|---|
| S. aureusc | MRSAd | P. mirabilise | ||
| a Broth dilution method was used, for details, see ESI.b MIC: minimum inhibitory concentration.c S. aureus: Gram-positive, MIC of amoxicillin: 0.5 μg mL−1 (positive control).d MRSA: Gram-positive, MIC of amoxicillin: 16 μg mL−1 (positive control).e P. mirabilis: Gram-negative, MIC of amoxicillin: 4 μg mL−1 (positive control). | ||||
| 1 | 5a | 16 | 128 | >128 |
| 2 | 5b | 16 | >128 | >128 |
| 3 | 5c | 8 | 16 | 64 |
| 4 | 5d | 8 | 128 | 128 |
| 5 | 5e | 8 | >128 | >128 |
| 6 | 5f | 64 | 128 | >128 |
| 7 | 5g | 32 | 128 | >128 |
| 8 | 5h | 16 | 64 | 32 |
| 9 | 5i | 8 | 64 | 128 |
| 10 | 5j | 8 | >128 | >128 |
| 11 | 5k | 16 | >128 | >128 |
| 12 | 5l | 16 | >128 | >128 |
| 13 | 5m | 8 | 128 | 64 |
| 14 | 5n | 8 | 64 | 16 |
| 15 | 5o | 16 | 128 | >128 |
| 16 | 5p | 128 | >128 | >128 |
| 17 | 5q | 64 | 128 | 128 |
| 18 | 5r | 32 | 128 | >128 |
Conclusions
In summary, we have successfully developed a highly diastereoselective aziridination reaction of cyclic enones under mild condition, giving an efficient access to a class of novel spiroaziridines. This approach is highly practical and functional group tolerant, which allows for large-scale synthesis and rapid assembly of substituent divergent products from simple, readily available starting materials. Preliminary biological studies of the synthesized compounds showed promising in vitro antibacterial activity against S. aureus, MRSA and P. mirabilis. Asymmetric organocatalysis of this aziridination process and further medicinal chemistry studies upon these novel structures are our current research interests, and the result will be reported in due course.Acknowledgements
Financial support from National Natural Science Foundation of China (no. 21502009), the Science & Technology Department of Sichuan Province (no. 2017JQ0032), Scientific Research Fund of Sichuan Provincial Education Department (no. 16ZB0430), “Thousand Talents Program” of Sichuan Province, and the Start-up Fund of Chengdu University (no. 2081915026, 2080515047) is gratefully acknowledged.Notes and references
- For selected reviews, see: (a) G. Callebaut, T. Meiresonne, N. De Kimpe and S. Mangelinckx, Chem. Rev., 2014, 114, 7954 CrossRef CAS PubMed; (b) S. Stanković, M. D'hooghe, S. Catak, H. Eum, M. Waroquier, V. Van Speybroeck, N. De Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643 RSC; (c) P. Lu, Tetrahedron, 2010, 66, 2549 CrossRef CAS; (d) C. Schneider, Angew. Chem., Int. Ed., 2009, 48, 2082 CrossRef CAS PubMed; (e) A. Padwa, in Comprehensive Heterocyclic Chemistry III, Elsevier, Oxford, 2008 Search PubMed; (f) G. S. Singh, M. D'hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS PubMed.
- For leading reviews on bioactive molecules containing an aziridine moiety, see: (a) F. M. D. Ismail, D. O. Levitsky and V. M. Dembitsky, Eur. J. Med. Chem., 2009, 44, 3373 CrossRef CAS PubMed; (b) P. A. S. Lowden, Aziridine Nature Products-Discovery, Biological Activity and Biosynthesis, in Aziridine and Epoxides in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) S. K. Carter and S. T. Crooke, Mitomycin C: Current Status and New Developments, Academic Press, New York, 1979 Search PubMed; (b) M. Kono, Y. Saitoh and K. Shirahata, J. Am. Chem. Soc., 1987, 109, 7224 CrossRef CAS , and the references therein.
- For recent reviews, see: (a) S. A. A. El Bialy, H. Braun and L. F. Tietze, Synthesis, 2004, 14, 2249 Search PubMed; (b) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed, and the references therein; for selected examples, see: (c) J.-M. Adam, C. Bissantz, U. Grether, A. Kimbara, M. Nettekoven, S. Roever and M. Rogers-Evans, U.S. Pat. Appl., 0116236, 2013; (d) L. A. M. Johansson, PCT Int. Appl., 011285, 2013; (e) R. Xu, M. Czarniecki, J. de Man, J. Pan, L. Qiang, Y. Root, S. Ying, J. Su, X. Sun, Y. Zhang, T. Yu, Y. Zhang, T. Hu and S.-H. Chen, Tetrahedron Lett., 2011, 52, 3266 CrossRef CAS; (f) L. Chen, L. Feng, S. Feng, L. Gao, T. Guo, M. Huang, C. Liang, Y. Liu, L. Wang, J. C. Wong, J. Z. Wu, X. Wu, H. Yun and X. Zheng, PCT Int. Appl., 020993, 2013; (g) L. F. Tietze and H. Schirok, J. Am. Chem. Soc., 1999, 121, 10264 CrossRef CAS; (h) M. L. R. Heffernan, L. W. Hardy, F. X. Wu, L. D. Saraswat and K. L. Spear, PCT Int. Appl., 170845, 2012.
- (a) Y. S. Or, L. Ying, C. Wang, J. Long and Y.-L. Qiu, PCT Int. Appl., 003009, 2009; (b) B. J. Bradbury, M. Deshpande, M. J. Pucci, Q. Wang, J. A. Wiles, Y. Song, A. Hashimoto and E. Lucien, U.S. Pat., Appl., 0100215, 2006.
- For selected reviews, see: (a) V. I. Minkin, Chem. Rev., 2004, 104, 2751 CrossRef CAS PubMed; (b) R. Rios, Chem. Soc. Rev., 2012, 41, 1060 RSC; (c) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC; (d) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (e) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC; (f) A. K. Franz, N. V. Hanhan and N. R. Ball-Jones, ACS Catal., 2013, 3, 540 CrossRef CAS; (g) D. Cheng, Y. Ishihara, B. Tan and C. F. Barbas III, ACS Catal., 2014, 4, 743 CrossRef CAS; (h) V. A. D'yakonov, O. A. Trapeznikova, A. de Meijere and U. M. Dzhemilev, Chem. Rev., 2014, 114, 5775 CrossRef PubMed.
- For selected reviews, see: (a) H. Pellissier, Adv. Synth. Catal., 2014, 356, 1899 CrossRef CAS; (b) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed; (c) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881 CrossRef CAS PubMed; (d) X. L. Hou, J. Wu, R. H. Fan, C. H. Ding, Z. B. Luo and L. X. Dai, Synlett, 2006, 37, 181 CrossRef; (e) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353 CrossRef CAS.
- (a) M. Carducci, S. Fioravanti, M. A. Loreto, L. Pellacani and P. A. Tardella, Tetrahedron Lett., 1996, 37, 3777 CrossRef CAS; (b) A. Fazio, M. A. Loreto and P. A. Tardella, Tetrahedron Lett., 2001, 42, 2185 CrossRef CAS; (c) T. Gasperi, M. A. Loreto, P. A. Tardella and E. Veri, Tetrahedron Lett., 2003, 44, 4953 CrossRef CAS; (d) M. A. Loreto, A. Migliorini, P. A. Tardella and A. Gambacorta, Eur. J. Org. Chem., 2007, 2365 CrossRef CAS; (e) I. Ammetto, T. Gasperi, M. A. Loreto, A. Migliorini, F. Palmarelli and P. A. Tardella, Eur. J. Org. Chem., 2009, 6189 CrossRef CAS; (f) T. Gasperi, M. A. Loreto, A. Migliorini and C. Ventura, Eur. J. Org. Chem., 2011, 385 CrossRef CAS; (g) J. Aires-de-Sousa, A. M. Lobo and S. Prabhakar, Tetrahedron Lett., 1996, 37, 3183 CrossRef CAS; (h) J. Aires-de-Sousa, S. Prabhakar, A. M. Lobo, A. M. Rosa, M. J. S. Gomes, M. C. Corvo, D. J. Williams and A. J. P. White, Tetrahedron: Asymmetry, 2001, 12, 3349 CrossRef; for pioneering work of asymmetric aziridination via iminium/enamine catalysis, see: (i) J. Vesely, I. Ibrahem, G.-L. Zhao, R. Rios and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 778 CrossRef CAS PubMed.
- For our recent success in developing metal-free reactions to assemble medicinally relevant structures, see: (a) J.-L. Li, K.-C. Yang, Y. Li, Q. Li, H.-P. Zhu, B. Han, C. Peng, Y.-G. Zhi and X.-J. Gou, Chem. Commun., 2016, 52, 10617 RSC; (b) J.-L. Li, Q. Li, K.-C. Yang, Y. Li, L. Liang, B. Han, C. Peng and X.-J. Gou, RSC Adv., 2016, 6, 38875 RSC; (c) J.-L. Li, B. Sahoo, C.-G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10515 CrossRef CAS PubMed; (d) J.-L. Li, S.-L. Zhou, P.-Q. Chen, L. Dong, T.-Y. Liu and Y.-C. Chen, Chem. Sci., 2012, 3, 1879 RSC.
- CCDC 1479201 (5d) contains the supplementary crystallographic data for this paper.†.
- For motivation of this antibacterial activity study of the spiroaziridines, see ESI.† The bioactivity was assessed by using broth dilution method, for relative references, see: (a) L. Ouyang, Y. Huang, Y. Zhao, G. He, Y. Xie, J. Liu, J. He, B. Liu and Y. Wei, Bioorg. Med. Chem. Lett., 2012, 22, 3044 CrossRef CAS PubMed; (b) Farhanullah, T. Kang, E.-J. Yoon, E.-C. Choi, S. Kim and J. Lee, Eur. J. Med. Chem., 2009, 44, 239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for new compounds and crystallographic data in CIF or other electronic formats. CCDC 1479201. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28508b |
| This journal is © The Royal Society of Chemistry 2017 |