
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of Co3O4 nanoparticles in thin porous carbon shells from metal–organic frameworks for enhanced electrochemical performance†
Bin Qiu,
Wenhan Guo,
Zibin Liang,
Wei Xia,
Song Gao,
Qingfei Wang,
Xiaofeng Yu,
Ruo Zhao and
Ruqiang Zou *
*
Beijing Key Laboratory for Theory and Technology of Advanced Battery Materials, Department of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, P. R. China. E-mail: rzou@pku.edu.cn
First published on 27th February 2017
Abstract
Cobalt oxides, typically Co3O4, have received considerable attention due to their high theoretical capacity as anode materials for Li-ion batteries. However, their poor electron conductivity and large volume change upon the insertion/removal of Li+ ions limit their practical application. Carbon coating is widely used to improve the electrochemical performance of materials and release the strain during the lithiation/delithiation processes, in which the thickness of the coating carbon shell has a vital role in determining the performance of the material. In this study, Co3O4 nanoparticles coated with a thin carbon shell are obtained from the metal–organic framework (MOF) precursor Co-MOF-74 via a sequential two-step carbonization process, where carbon oxides, e.g., CO2, are used as the oxidation atmosphere in the second step. The carbon content and shell thickness are controlled by changing the calcination time. The electrode containing a certain carbon content (3.17 wt%) exhibits a capacity of 1137 mA h g−1 after 100 cycles tested at 100 mA g−1 between 0.005 and 3.0 V. This enhanced electrochemical performance is attributed to the well-dispersed nanosized Co3O4 particles and thin carbon shell coating on the electrode surface, which shorten the Li+ ion diffusion length and enhance the electron conductivity of the hybrid.
Introduction
Lithium ion batteries (LIBs) have attracted considerable attention with the development of mobile electronics and electric vehicles.1 Numerous studies have focused on electrode materials, separators, and electrolytes.2,3 Specifically, the anode material is regarded as one of the key elements in the construction of lithium ion batteries. Since Poizot et al. first reported the mechanism of transition oxides (CoO, FeO, NiO and CuO) as lithium storage materials,4 metal oxides have attracted great attention due to their high theoretical capacities (much higher than that of graphite ∼372 mA h g−1) and low cost.5,6 In particular, oxides of cobalt (CoO, Co3O4, etc.) have been widely investigated, and much progress has been made in Li-ion storage capacities.7–10 However, there are still some disadvantages, such as fast capacity fading and large volume change during the lithiation and delithiation process, that severely limit their further application. To address these issues, two main types of strategies have been adopted. The first method focuses on the design of special structures (hollow spheres and polyhedral structures).11,12 The special structures of electrode materials are believed to facilitate the diffusion of lithium ions and release the mechanical strain.13 The second approach is carbon coating,14 where carbon acts as a layer to buffer large volume changes, mitigate the aggregation of particles and increase the electronic conductivity of electrodes.15Metal–organic frameworks (MOFs) are a family of microporous crystalline polymers with a high surface area and porosity, and are constructed from metal-containing clusters and organic ligands.16 They have been widely used in gas storage/separation, chemical sensing, heterogeneous catalysis, bioimaging, drug delivery and luminescence.17,18 Inheriting these properties, MOF-derived materials, including porous carbon, metals, metal oxides and metal sulphides, also show high surface areas, high pore volumes and tunable pore size distributions.19 Furthermore, the obtained metals/metal oxides are well-dispersed in the carbon matrix, which contributes to a good electrochemical performance. Recently, transition metal oxides (Fe2O3, Co3O4, NiO and MnO) made from MOFs have been widely used as anodes for lithium-ion batteries.20–25 In the preparation process for these methods, metal oxides/carbon hybrids are derived from MOFs mainly through two ways. The first way is through a sequential two-step heat treatment in which metal/carbon hybrids are first obtained under inert atmosphere, and then air is used as the oxidation gas to obtain metal oxides/carbon hybrids.26,27 The second is the direct carbonization of MOFs under air atmosphere.28–30 In both methods, control over the content and thickness of the carbon coating is essential for the battery performance of the final metal oxides/carbon hybrid electrodes. A thin porous carbon shell layer may enhance the electrochemical properties, but a thick amorphous carbon coating on the electrode may hinder the transportation of Li+ through the interface between the electrolyte and electrode solid phase, leading to the depletion of battery performance.31a Metal oxides can be obtained through carbonization under a CO2 atmosphere. Using CO2 as the oxidation atmosphere, carbon can be kept and at the same time the metal can be oxidized to metal oxide with a carbon coating on its surface.31b,c This method is very convenient and could open a new way for the synthesis of metal oxides/carbon composites derived from metal organic frameworks. To date, no reports have focused on controlling the surface carbon coating of electrodes. Herein, we develop a simple method to prepare metal oxides/carbon hybrids derived from a cobalt MOF via two heat-treatment steps, using carbon oxides (CO2) as the oxidation gas after pyrolysis under an inert atmosphere. Annealing in CO2 converts the Co species into Co3O4, while the excess carbon matrix is consumed. By controlling the carbonization time and temperature, Co3O4 particles with a certain carbon coating are obtained through calcination of the rod-like Co-MOF-74 crystals, whose structure is composed of infinite Co3O3(CO2)3 rods and 2,5-dioxidoterephthalate linkers, where each metal ion is coordinated to three carboxyl and two hydroxyl groups and a coordinated ligand [N,N-dimethylformamide (DMF) or water].32 The particle size of Co3O4 is around 12 nm and this oxide is homogeneously dispersed in the carbon matrix. As expected, when evaluated as an anode for lithium ion batteries, this hybrid exhibits a higher specific capacity, better cyclability and excellent rate performance than the thick carbon-coated electrodes and pure Co3O4 electrodes.
Experimental
Synthesis of Co3O4@C composites
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, 100 mL) in a 125 mL Teflon-lined steel autoclave. The autoclave was tightly capped and heated in an oven at 100 °C for 24 h. The as-prepared red-orange crystals were filtered and washed with N,N-dimethylformamide (DMF) and methanol, sequentially, and finally dried at 150 °C under vacuum for 12 h to obtain the rod-shaped Co-MOF-74 crystals.
:1:1, 100 mL) in a 125 mL Teflon-lined steel autoclave. The autoclave was tightly capped and heated in an oven at 100 °C for 24 h. The as-prepared red-orange crystals were filtered and washed with N,N-dimethylformamide (DMF) and methanol, sequentially, and finally dried at 150 °C under vacuum for 12 h to obtain the rod-shaped Co-MOF-74 crystals.Materials characterization
The obtained products were characterized via XRD (BRUKER D8 ADVANCED, Cu Kα = 1.54 Å) at a scan rate of 0.02° s−1 from 10° to 80°. The operating voltage and current were kept at 40 kV and 40 mA, respectively. The BET surface area and pore volume of the materials were calculated from isothermal nitrogen sorption measurements at 77 K using a Quantachrome Autosorb-IQ gas adsorption analyzer. The general morphologies of the synthesized composites were examined via SEM using an SEM-HITACHI S-4800 and TEM using an FEI Tecnai T20. HRTEM images were recorded using an FEI Tecnai F20. Raman spectroscopic analysis was performed with a LabRAM HR800 with the laser excitation energy of 632.8 nm. Elemental analysis was performed on a Vario EL elemental analyzer. XPS measurements were performed on a Kratos Axis Ultra Imaging photoelectron spectrometer using a monochromatic Al Kα line (1486.7 eV). TGA was carried out on an SDT Q600 analyzer at a heating rate of 5 °C min−1 in a flowing air atmosphere.Cell assembly and testing
The working electrode was composed of active material (80 wt%), Super P (10 wt%) and poly(vinylidene difluoride) (PVDF, 10 wt%). These materials were dissolved in N-methyl pyrrolidone (NMP) and sonicated until a homogenous slurry was formed. The slurry was then spread onto nickel foam, and pressed and dried under vacuum at 120 °C for 12 h. After the drying process, the foam was further pressed under a pressure of 20 MPa. Coin cells were finally assembled in an argon-filled glovebox with the as-prepared materials as the test electrode, metallic lithium as the counter and reference electrode, 1 M LiPF6 in EC:DMC (1:1 in volume) as the electrolyte, and Whatman GF/D borosilicate glass-fiber sheets as the separator. The coin cells were cycled galvanostatically in the voltage range of 0.005–3.00 V at a current density of 100 mA g−1 with a multichannel battery test system (NEWARE). Cyclic voltammetry measurements were conducted with an RST electrochemical workstation at the scanning rate of 0.5 mV s−1 between 0.005 V and 3.00 V (vs. Li/Li+). Electrochemical impedance spectroscopy (EIS) was conducted on a Zahner Zennium electrochemical workstation in the frequency range from 0.01 Hz to 100000 Hz using a sine wave with an amplitude of 5.0 mV.
Results and discussion
The formation process for the Co3O4@C composite is illustrated in Scheme 1. Typically, Co-MOF-74 was first synthesized under hydrothermal conditions according to the reported method. The obtained Co-MOF-74 rods were first carbonized under an inert atmosphere for 2 h to be derived into the Co@C hybrids.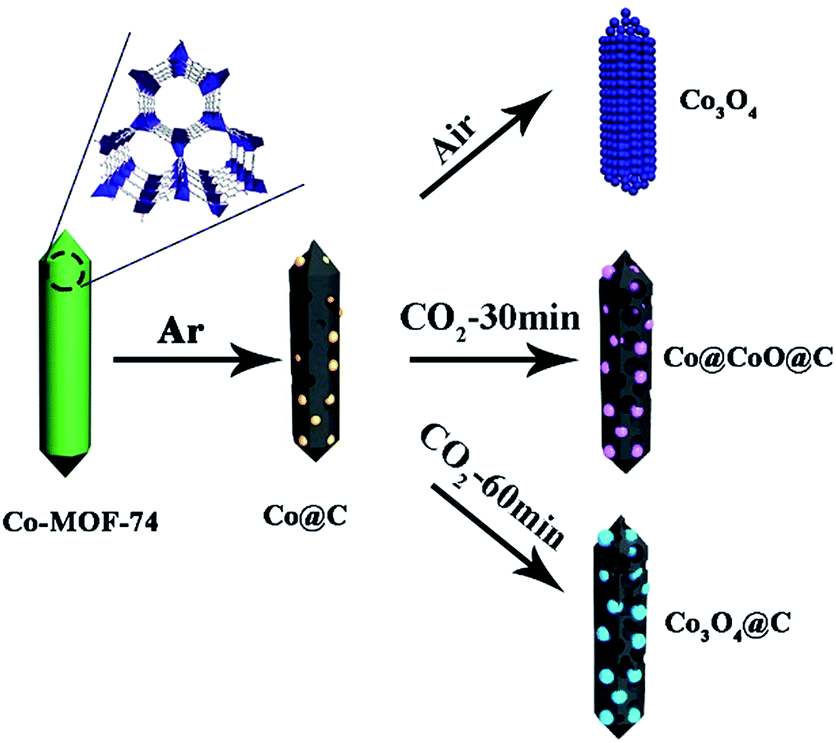 | ||
| Scheme 1 Schematic of the synthetic process of the Co3O4@C composite. | ||
The Co@C samples were further calcined under a carbon dioxide flow for a certain time, where cobalt particles were oxidized into cobalt oxides, and at the same time the thick carbon shells around the metal species were also consumed via oxidation. Finally, a thin carbon shell coated Co3O4 hybrid was formed (denoted as Co3O4@C). It is interesting to find that the final Co3O4@C composites still exhibited rod-like morphology, and the as-formed Co3O4 nanoparticles were homogeneously dispersed in the carbon matrix. The as-prepared materials showed mesoporous structures, which could facilitate the diffusion of Li+ and electrolyte to enhance the kinetic properties of electrodes. For comparison, samples with a thick carbon shell (denoted as Co@CoO@C) and without the carbon coating (denoted as Co3O4) were prepared by shortening the CO2 annealing time and direct oxidation in air, respectively. The powder X-ray diffraction (XRD) patterns of the as-synthesized Co-MOF-74 crystals were well-consistent with the simulated patterns, as shown in Fig. S1 in the ESI† which confirms the high quality of the obtained samples. The SEM images (Fig. S2, ESI†) show that all the Co-MOF-74 crystals possess a rod-like morphology with smooth surfaces, clear facets and high length–diameter ratio, which suggest the high crystallinity of the MOFs. A typical type-I isothermal curve is observed from the nitrogen adsorption/desorption measurement of the Co-MOF-74 rods, with a calculated Brunauer–Emmett–Teller (BET) surface area of 1025 m2 g−1 and pore volume of 0.48 cm3 g−1, which confirm the microporosity of the MOFs (Fig. S3a, ESI†). Fig. S3b† shows a narrow pore size distribution (fitted using the Nonlinear Density Functional Theory (NLDFT) model) with only one peak at ∼0.5 nm, which corresponds to the one dimensional (1D) channels in the hexagonal MOF-74 matrix. The thermal treatment procedure was established according to the thermogravimetric analysis (TGA) results. From the TGA curve of pure Co-MOF-74 under an inert atmosphere, the first weight loss before 100 °C came from the evaporation of remnant solvents, such as DMF and ethanol, whereas the second weight loss at around 500 °C was due to the destruction of the MOF structures (Fig. S4, ESI†). The MOFs worked as a sacrificial self-template and underwent complicated reactions during the pyrolysis process, where the carbonization of organic components and reduction of metal species happened simultaneously. To ensure the complete reduction of Co and remove unstable carbon species, a high temperature of 700 °C was chosen for the first thermal treatment step. The diffraction peaks of the products match well with the pure Co phase with a cubic structure, which confirms the formation of metallic Co (Fig. S5, ESI†) and the rod shape of the mother crystals was also well-reserved during this process (Fig. S6, ESI†). The obtained Co@C samples were further treated in different atmospheres of air and CO2, separately (Fig. S7, ESI†). It is clear that under air atmosphere, the Co nanoparticles were easily oxidized at a low temperature of 150 °C, accompanied by the complete removal of the carbon matrix. This process was completed at 250 °C, leaving only a trace amount of carbon in the final products (Table S1, ESI†). On the other hand, under the much milder oxidation atmosphere of flowing CO2, the gradual oxidation of Co nanoparticles and carbon evaporation happened simultaneously at a much slower rate until 600 °C, where an abrupt weight loss took place due to the vigorous reaction between the carbon matrix and CO2. Accordingly, the temperatures of 250 °C and 500 °C were chosen for annealing in air and CO2 atmosphere, respectively, in the second thermal treatment step.
The crystal and phase information of the as-synthesized Co@C, Co3O4, Co@CoO@C and Co3O4@C composites were confirmed by XRD, as shown in Fig. 1. After the first heat treatment step, the Co@C composite was obtained, where nanosized cobalt nanoparticles formed in situ and dispersed uniformly in the porous carbon matrix. After the oxidation process, the XRD pattern of the Co3O4 composites is consistent with the theoretical patterns of Co3O4 with a face-centered cubic lattice (JCPDF No. 42-1467), whereas the Co3O4 and Co3O4@C composites show similar diffraction peaks, which indicates that the main phase of both nanocomposites is identical. It is noteworthy that no diffraction peaks of CoO or Co were present for these two samples, which proves the complete oxidation of the Co species. On the other hand, for the Co@CoO@C nanocomposites, it is evident that there are strong peaks belonging to metallic Co, whereas the other weak peaks are attributed to CoO species. The diffraction of Co3O4 was not observed, which implies that the Co nanoparticles were only partially oxidized. These results indicate that the oxidation of Co species in CO2 proceeds stepwise: first in the metastable CoO phase, and then after the complete consumption of metallic Co, the oxidation of CoO in the thermodynamically-stable Co3O4 phase could take place. It is proven that control over the annealing time in CO2 is crucial for the phase composition of the final products. A shortened annealing time could lead to the incomplete consumption of metallic Co and halfway oxidation into a metastable CoO phase due to more sluggish reaction kinetics in CO2 compared to that in air, which is consistent with TGA results.
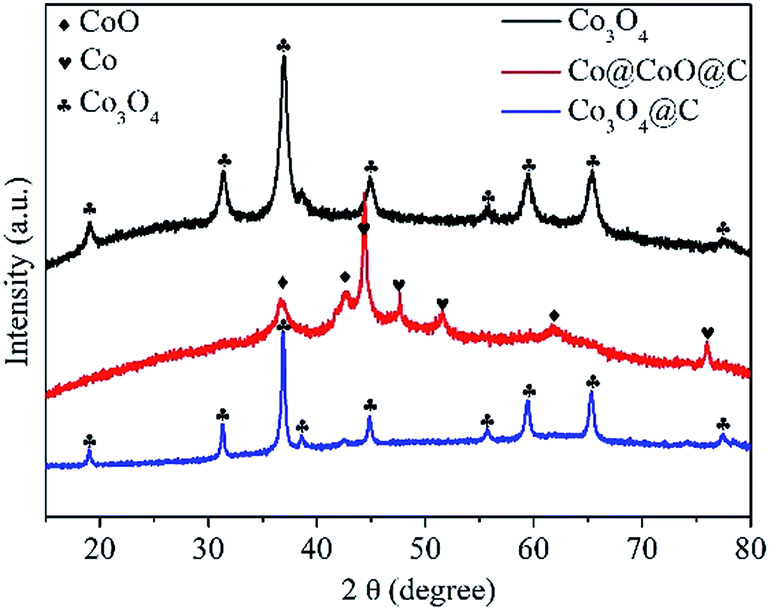 | ||
| Fig. 1 XRD patterns of the Co3O4, Co@CoO@C and Co3O4@C composites derived from the pyrolysis of Co-MOF-74 under various gas atmospheres and temperatures. | ||
The typical scanning electron microscopy (SEM), transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) images of the Co3O4, Co@CoO@C and Co3O4@C composites are shown in Fig. 2. The inserted pictures show an individual particle of each composite. In Fig. 2a–c, it is shown that the original rod shape of the mother MOFs is well-reserved for all three composites, although some cracks could be seen in the Co3O4 sample, which are caused by the severe structural change during the direct heat treatment in air. No such cracks were found in the Co@CoO@C and Co3O4@C composites, and some irregular edges indicate the residue of amorphous carbon, which is also evidenced by the TEM images. Fig. 2d–f show the TEM images of the Co3O4, Co@CoO@C and Co3O4@C composites, respectively. From Fig. 2d it can be seen that the rods of Co3O4 are actually composed of aggregated Co3O4 nanoparticles with an average size of ca. 12 nm. In the HRTEM image in Fig. 2g, lattice fringes are observed with the measured lattice spacing of 0.233 nm, which correspond to the (222) crystal planes of Co3O4, and it is clear that the Co3O4 particles agglomerated without any coating on their surface. On the contrary, for the CO2-treated samples, spherical cobalt oxide nanoparticles are dispersed in the carbon matrix, as shown in Fig. 2e and f. The carbon layer coating on the surface of the cobalt oxide particles could be easily distinguished for both Co@CoO@C and Co3O4@C from the HRTEM images in Fig. 2h and i, respectively. For the Co@CoO@C sample, the thickness of the carbon shells is ∼14 nm, whereas for Co3O4@C much thinner shells ranging from 2–6 nm are observed. These results imply that by controlling the calcination time in the CO2 flow at moderate temperature, a certain amount of carbon could be controlled using our method. Just by changing the carbonization time, the content and thickness of the coated carbon could be controlled to an optimal level to facilitate the diffusion of Li+ through the shell and conductivity of the hybrids for enhanced electrochemical performances. The Raman spectra of the as-synthesized composites were investigated to illustrate the graphitization degree of carbon (Fig. S8, ESI†). As shown by the Raman spectra, the Co@C, Co3O4@C and Co@Co@C samples display two distinguishable peaks in the range of 1000–2000 cm−1. The peak located at around 1350 cm−1 is attributed to the D band, whereas the peak at around 1590 cm−1 is from the G band. An increased intensity ratio in the ID/IG ratios indicates the transitional stage from amorphous carbon to crystalline graphite.34 The ID/IG ratio is close to 0.86 for Co@C and about 0.91 for Co@CoO@C, whereas for Co3O4@C the ratio is 1.34 (Fig. S8, ESI†). This indicates that the Co3O4@C samples possess the highest graphitization degree, and more defects and edges exposed for carbon, which could enhance the electron conductivity of these hybrids and give more sites to store Li+. Element analysis was conducted to determine the carbon content in the hybrids, and the results are shown in Table S1 (ESI†). The carbon content for the Co@C samples is 12.38 wt%, whereas after CO2 treatment for 30 min, the content decreases to 10.7 wt%. When the treatment time increased to 60 min, the percentage was lowered to only 3.17 wt%. For pure Co3O4 samples, only a trace amount of carbon was detected. These results are consistent with the HRTEM image analysis that the content and thickness of carbon shell are controlled by changing the calcination atmosphere and time.
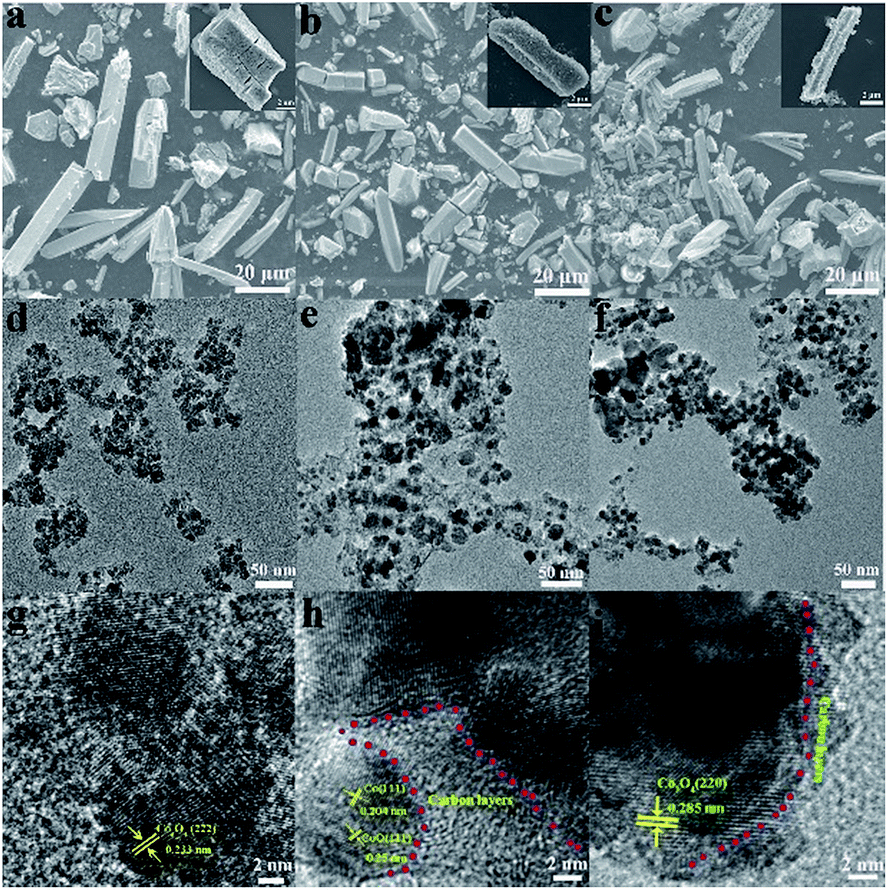 | ||
| Fig. 2 SEM images of the (a) Co3O4, (b) Co@CoO@C and (c) Co3O4@C composites. TEM images of the (d) Co3O4, (e) Co@CoO@C and (f) Co3O4@C composites. HRTEM images of the (g) Co3O4, (h) Co@CoO@C and (i) Co3O4@C composites. | ||
X-ray photoelectron spectroscopy (XPS) was employed to further identify the surface composition and the oxidation state of Co in the composites (Fig. 3). The full XPS spectra show the existence of Co, C and O, which is in accordance with the element analysis, as shown in Fig. 3a. The peak intensity of carbon for Co3O4@C is much weaker than that of the Co@CoO@C sample, which indicates a lower carbon content in the former hybrid, whereas the pure Co3O4 sample shows almost no carbon signal, which confirms its ultra-low carbon content. In the high-resolution XPS spectrum of Co 2p for the Co3O4 composites in Fig. 3b, there are two peaks located at 795.4 eV (Co 2p1/2) and a low energy band at 780.4 eV (Co 2p3/2). The energy difference between the Co 2p1/2 and Co 2p3/2 splitting is 15 mV, which indicates the existence of Co2+ and Co3+ and corresponds to the existence of Co3O4. Two satellite peaks near the Co 2p peaks are also observed.35 The Co 2p spectrum of the Co3O4@C composite is similar to that of Co3O4, which is consistent with the XRD patterns that both samples contain Co3O4, as shown in Fig. 3c. However, in the Co 2p spectrum of the Co@CoO@C composite in Fig. 3d, there are two peaks corresponding to the high energy band (796.1 eV) and low energy band (780.2 eV), which are attributed to the Co 2p1/2 and Co 2p3/2 binding energies of CoO. These results confirm the existence of CoO in the composites.36
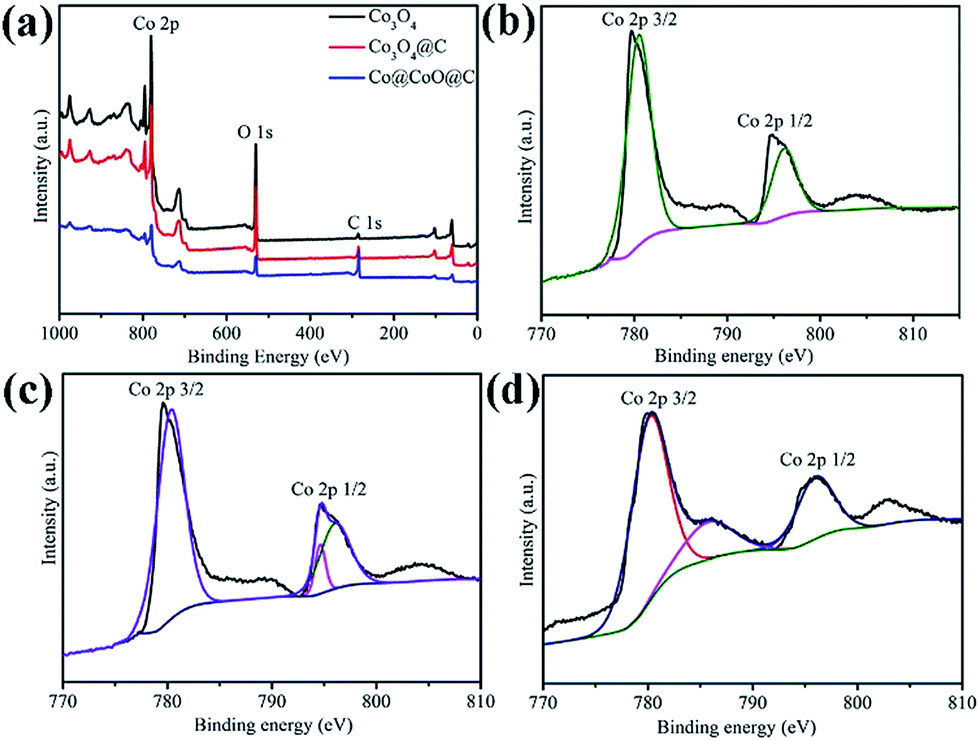 | ||
| Fig. 3 View of (a) full XPS spectra of the Co3O4, Co3O4@C and Co@CoO@C composites. High-resolution XPS spectra of Co 2p for the (b) Co3O4, (c) Co3O4@C and (d) Co@CoO@C composites. | ||
N2-sorption measurements were conducted to investigate the specific surface areas and porous features of all the composites, as shown in Fig. 4 and S9 (ESI†). All the composites exhibit type IV isotherms (IUPAC definition) with evident hysteresis loops, which indicate their mesoporous nature. The specific surface areas were calculated based on the multi-point BET method, and the NLDFT method was used to analyze the pore size distribution for all the samples. The Co@C composite shows a high BET surface area of 133 m2 g−1 (Fig. S9a, ESI†) and pore volume of 0.23 cm3 g−1, with a single pore size distribution peak from 3.0 nm to 15.0 nm (Fig. S9b, ESI†). After CO2 calcination, the surface areas decrease from 121 m2 g−1 for Co@CoO@C to 76 m2 g−1 for Co3O4@C as the heat treatment time is extended from 30 min to 60 min, which is due to the consumption of the amorphous porous carbon matrix. The Co3O4 sample, on the other hand, presents a high surface area of 101 m2 g−1. The pore size distribution of these three samples shows generally two types of pores: a micropore of ca. 2.0 nm and another larger pore from 2.5 nm to the mesoporous range. The pore size ranges from 1.1 to 4.2 nm for the Co@CoO@C composite, whereas for the Co3O4@C composite the pore size is between 1.5 and 2.7 nm. The Co3O4@C composite shows less mesopores than Co@CoO@C due to the collapse of the unstable hole walls during the heat treatment. The Co3O4 sample, however, shows a larger mesopore component ranging from 2.5 to 11.0 nm, which originates from the vigorous gas evolution in direct air calcination. The mesoporous structure of these composites may be beneficial for improving electrochemical performances by facilitating the diffusion of electrolyte into the matrix and exposing more active sites reacting with Li+.
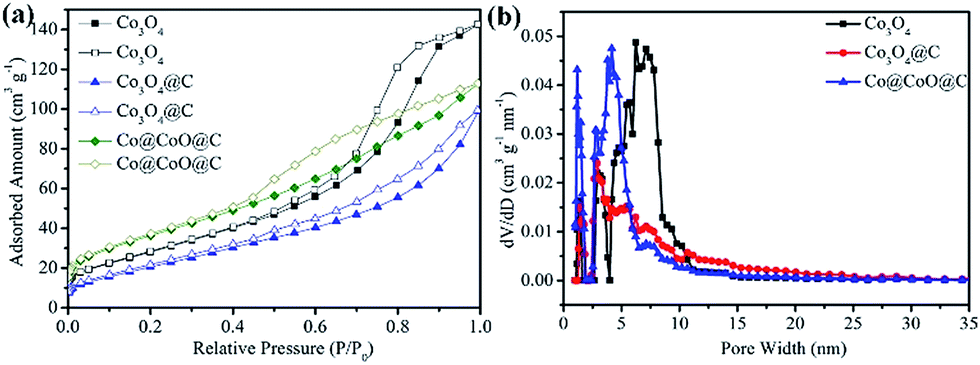 | ||
| Fig. 4 (a) N2 adsorption/desorption isotherm curves and (b) pore size distributions of the Co3O4, Co3O4@C and Co@CoO@C composites. | ||
The electrochemical performance of the Co3O4, Co@CoO@C and Co3O4@C composites were investigated, as shown in Fig. 5. The first and second charge–discharge curves of the Co3O4@C composite were tested at a current density of 100 mA g−1 between 0.005 and 3.0 V (Fig. 5a). During the first cycle, the plateau at 1.0 V is ascribed to the lithiation of Co3O4 forming LixCo3O4.37 Although the voltage was further decreased, the plateau below 1.0 V is followed by a long slope corresponding to the conversion reaction and the formation of an SEI film on the surface of the electrode, particularly for metal oxides materials.38 The initial charge and discharge capacities were 1165 mA h g−1 and 1857 mA h g−1, respectively, showing an irreversible capacity loss of 37%. Fig. 5b shows that the first charge and discharge capacities were 448 and 822 mA h g−1 for the Co@CoO@C composite, respectively, and the irreversible capacity loss was about 45%. The first charge and discharge capacities were 594 and 961 mA h g−1, respectively, with the irreversible capacity loss of 38% for Co3O4 electrode in Fig. 5c. All these electrodes show large irreversible capacities, which may be attributed to the formation of a solid electrolyte interphase (SEI) film during the discharge process and to the irreversible decomposition of the electrolyte.39 However, the Co@CoO@C electrodes show a larger capacity loss than the Co3O4 and Co3O4@C electrodes. The high content of amorphous carbon in this composite that could react with lithium consumed more lithium-ions, which led to the large capacity loss during the first cycle.40 After the first cycle, the coulombic efficiencies above 98% of the electrodes indicate good reversibility of these electrodes. To further investigate the electrochemical process, CV profiles of the electrodes are shown in Fig. 5d–f between 0.005 and 3.0 V at a scan rate of 0.5 mV s−1. In the first cycle, the peak located at 0.6 V for the Co3O4@C electrode is attributed to the formation of an SEI film on the electrode surface in Fig. 5d. During the second cycle, there is a peak located at 0.75 V, which is attributed to the lithiation of Co3O4, and an oxidation peak located at 2.1 V corresponding to the delithiation process and the recovery of Co3O4.41 Both the reduction and oxidation peaks shift in the subsequent cycle, which is common for metal oxide electrodes. These results are consistent with the charge–discharge plateaus. There are several CV profiles for the Co@CoO@C electrode, as shown in Fig. 5e, and it is evident that during the first cycle, there is a peak located at 0.5 V corresponding to the formation of an SEI film after the first cycle. The reduction peak-shift to 0.7 V is attributed to the lithiation of CoO to form LixCoO.42 After the first cycle, there are two cathodic peaks located at 0.7 V and 1.3 V, which correspond to the electrochemical reaction between CoO and Li+.43 For the pure Co3O4 electrode, the 0.7 V peak is also attributed to the formation of an SEI film on the electrode surface. After the first cycle, the weak cathodic peak shifts to 1.0 V during the following cycles, which is also consistent with the Co3O4 electrode. However, the anodic peaks for the Co3O4 electrode located at 2.2 V are from the oxidation of Co to Co3O4 in Fig. 5f. The long-term stability cycling performance of the Co3O4, Co3O4@C and Co@CoO@C electrodes is presented in Fig. 5g. The second charge and discharge capacities are 1167 mA h g−1 and 1255 mA h g−1 for the Co3O4@C electrode, and even after 100 cycles the charge capacity remains at 1095 mA h g−1 and the discharge capacity is 1137 mA h g−1, which correspond to a high capacity retention of 96% compared with the second cycle charge capacity. This superior performance is comparable with previous reports (Table S2, ESI†). However, the pure Co3O4 and Co@CoO@C electrodes only exhibit a discharge capacity of 593 and 355 mA h g−1, respectively, after the 100 cycles test. From the HRTEM test, it is clear that the carbon shell thickness of the Co3O4@C composite is 2–6 nm, whereas the thickness of the Co@CoO@C composite is about 14 nm, in which a thin carbon shell could facilitate the diffusion of mass and electrons. These results confirm that the thin carbon shell coating benefits the electrochemical performance by enhancing electron conductivity and making full use of the active materials. However, for the Co@CoO@C electrode, pure Co particles dominate the main part of the entire hybrid, which do not contribute to the electrochemical performance of the electrode. The rate performances of the electrodes are shown in Fig. 5h. The current range is from 100 mA g−1 to 1 A g−1. The reversible capacity reached 1090, 1020, 867, 682, and 447 mA h g−1 when tested at 100, 200, 300, 500, and 1000 mA g−1, respectively. When the current density decreased to 100 mA g−1, the revisable capacity returned to 1084 mA h g−1, which indicates the stable structure of the electrodes.44 Notably, the capacities of the pure Co3O4 electrode were far less than those of the Co3O4@C electrode. The reversible capacity reached 730, 640, 510, 383, and 240 mA h g−1 when tested at the same current density as that of Co3O4@C. After the current decreases to 100 mA g−1, the revisable capacity goes back to 704 mA h g−1. These results confirm that the rate performance of the Co3O4@C electrode is better than that of the Co3O4 electrode, which implies good structure stability of the Co3O4@C electrode. It is inferred that the thin carbon shell coating on the electrode inhibits capacity decay by buffering the volume changes during the lithium insertion and extraction processes.45 Electrochemical impedance spectroscopy (EIS) was employed to investigate the good electrochemical performance of these three electrodes, as shown in Fig. 5i. All three plots in this figure are composed of a semicircle in the high to medium frequency region, followed by a sloped line in the low frequency region. The semicircle is associated with the charge transfer resistance at high frequency. The impedance is associated with the charge transfer resistance between the electrode and electrolyte and the slope line at the low frequency is associated with the resistance in the electrode.46,47 The resistance of Co3O4@C was significantly lower than that of the Co3O4 electrode, whereas the resistance of the Co@CoO@C electrode was almost the same as that of the Co3O4@C electrode. We used the equivalent circuit to fit the data. Rs and Rct are the ionic resistance of the electrode and the charge transfer resistance, respectively.48a ZW is the Warburg impedance, and CPE is the constant phase-angle element involving the double layer capacitance.48 From the calculated data, Rct for Co3O4, Co@CoO@C, and Co3O4@C is 14.73 Ω, 4.26 Ω, 3.70 Ω, respectively. It is clear that the Rct of the Co3O4@C composite is smaller than that of the Co3O4 and Co@CoO@C composites, which indicates good electrolyte infiltration and charge-transport capability (Fig. S10, ESI†). The electronic conductivity was enhanced and it thus enabled easier charge transfer between the electrode and electrolyte interface in the Co3O4@C electrode. Thus, carbon coating is important to enhance the electron conductivity to improve the electrochemical performance.
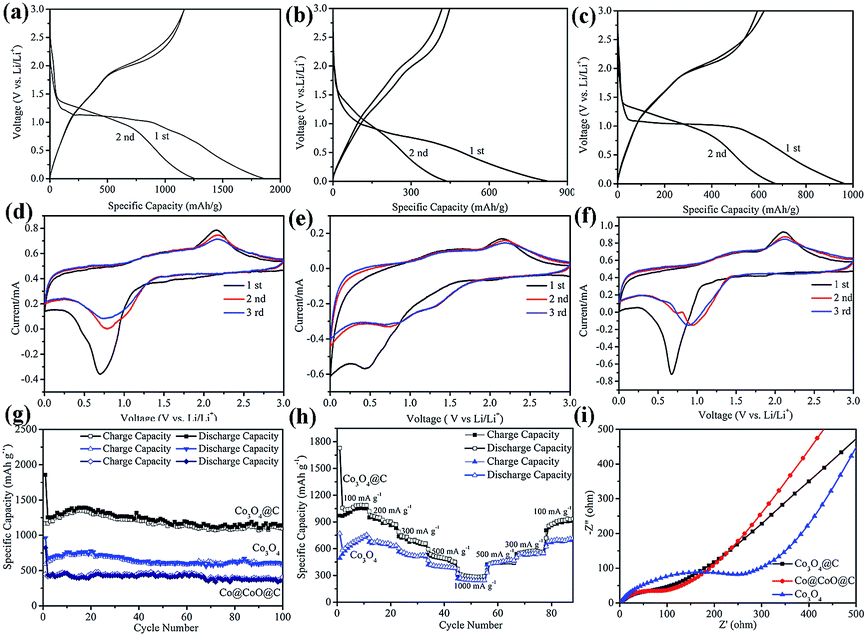 | ||
| Fig. 5 Electrochemical properties of the (a) first and second charge–discharge curves of Co3O4@C, (b) Co@CoO@C and (c) Co3O4 electrodes. Cyclic voltammogram (CV) profiles of the (d) Co3O4@C, (e) Co@CoO@C and (f) Co3O4 electrodes. Cycling performances at (g) a current density of 100 mA g−1 and (h) varied current densities from 0.1 to 1.0 A g−1. (i) Electrochemical impedance spectra of the three electrodes. | ||
Conclusions
In summary, thin carbon shell coated Co3O4 particles were prepared via the CO2 oxidization of porous MOF materials. The as-synthesized Co3O4@C electrode exhibits a higher specific capacity and excellent rate performance compared to the bare Co3O4 electrode, which is attributed to the small particle size and thin carbon shell buffering the volume change the during lithiation and delithiation processes as well as enhancing the electron conductivity. This study provides a new method to fabricate composites derived from porous MOF nanostructures and it may provide a new approach for the synthesis of metal oxide composites with enhanced properties for energy storage in lithium-ion batteries.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 51322205 and 21371014) and the National Program for Support of Top-notch Young Professionals, and the Beijing Municipal Science & Technology Commission Program (Z151100000915074).References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, R. Q. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- M. Barghamadi, A. S. Best, A. I. Bhatt, A. F. Hollenkamp, M. Musameh, R. J. Reesc and T. Ruthera, Energy Environ. Sci., 2014, 7, 3902–3920 CAS.
- P. Poizot, S. Laruelle, S. Grugeon and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A1212–A1217 CrossRef CAS.
- J. Jiang, Y. Y. Li, J. P. Liu, X. T. Huang, C. Z. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- J. F. Zhang, W. Y. Ren, Y. Y. Zhou, P. Li, L. Xu, D. M. Sun, P. Wu, Y. M. Zhou and Y. W. Tang, Chem.–Eur. J., 2016, 22, 9599–9606 CrossRef CAS PubMed.
- K. M. Nam, J. H. Shim, D. W. Han, H. S. Kwon, Y. M. Kang, Y. Li, H. Song, W. S. Seo and J. T. Park, Chem. Mater., 2010, 22, 4446–4454 CrossRef CAS.
- A. K. Rai, J. Gim, L. T. Anh and J. Kim, Electrochim. Acta, 2013, 100, 63–71 CrossRef CAS.
- J. Y. Wang, N. L. Yang, H. J. Tang, Z. H. Dong, Q. Jin, M. Yang, D. Kisailus, H. J. Zhao, Z. Y. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef CAS PubMed.
- H. H. Li, Z. Y. Li, X. L. Wu, L. L. Zhang, C. Y. Fan, H. F. Wang, X. Y. Li, K. Wang, H. Z. Sun and J. P. Zhang, J. Mater. Chem. A, 2016, 4, 8242–8248 CAS.
- J. Y. Wang, N. L. Yang, H. J. Tang, Z. H. Dong, Q. Jin, M. Yang, D. Kisailus, H. J. Zhao, Z. Y. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 125, 6545–6548 CrossRef.
- H. Guo, T. T. Li, W. W. Chen, L. X. Liu, X. J. Yang, Y. P. Wang and Y. C. Guo, Nanoscale, 2014, 6, 15168–15174 RSC.
- F. Zhang, C. Z. Yuan, J. J. Zhu, J. Wang, X. G. Zhang and X. W. Lou, Adv. Funct. Mater., 2013, 23, 3909–3915 CrossRef CAS.
- F. B. Hao, Z. W. Zhang and L. W. Yin, ACS Appl. Mater. Interfaces, 2013, 5, 8337–8344 CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- Y. Zhao, Z. Song, X. Li, Q. Sun, N. Cheng, S. Lawes and X. Sun, Energy Storage Materials, 2016, 2, 35–62 CrossRef.
- X. D. Xu, R. G. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- F. C. Zheng, G. L. Xia, Y. Yang and Q. W. Chen, Nanoscale, 2015, 7, 9637–9645 RSC.
- F. Zou, Y. M. Chen, K. W. Liu, Z. T. Yu, W. F. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377–386 CrossRef CAS PubMed.
- W. X. Guo, W. W. Sun and Y. Wang, ACS Nano, 2015, 9, 11462–11471 CrossRef CAS PubMed.
- H. Li, M. Liang, W. W. Sun and Y. Wang, Adv. Funct. Mater., 2016, 26, 1098–1103 CrossRef CAS.
- G. Huang, F. F. Zhang, X. C. Du, Y. L. Qin, D. M. Yin and L. M. Wang, ACS Nano, 2015, 9, 1592–1599 CrossRef CAS PubMed.
- W. Xia, R. Q. Zou, L. An, D. G. Xia and S. J. Guo, Energy Environ. Sci., 2015, 8, 568–576 CAS.
- A. Aijaz, J. Masa, C. Rosler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087–4091 CrossRef CAS PubMed.
- R. B. Wu, X. K. Qian, K. Zhou, J. Wei, J. Lou and P. M. Ajayan, ACS Nano, 2014, 8, 6297–6303 CrossRef CAS PubMed.
- F. Zou, X. L. Hu, Z. Li, L. Qie, C. C. Hu, R. Zeng, Y. Jiang and Y. H. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed.
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang, S. G. Wi, B. Park and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394–7397 CrossRef CAS PubMed.
- (a) Q. F. Wang, R. Q. Zou, W. Xia, J. Ma, B. Qiu, A. Mahmood, R. Zhao, Y. Y. C. Yang, D. G. Xia and Q. Xu, Small, 2015, 11, 2511–2517 CrossRef CAS PubMed; (b) S. H. Ren, R. Prakash, D. Wang, V. S. K. Chakravadhanula and M. Fichitner, ChemSusChem, 2012, 5, 1397–1400 CrossRef CAS PubMed; (c) N. Yan, X. Zhou, Y. Li, F. Wang, H. Zhong, H. Wang and Q. W. Chen, Sci. Rep., 2013, 3, 3392 Search PubMed.
- L. J. Wang, H. X. Deng, H. Furukawa, F. Gándara, K. E. Cordova, D. Peri and O. M. Yaghi, Inorg. Chem., 2014, 53, 5881–5883 CrossRef CAS PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- R. Gao, Z. Y. Li, X. L. Zhang, J. C. Zhang, Z. B. Hu and X. F. Liu, ACS Catal., 2016, 6, 400–406 CrossRef CAS.
- X. Yao, G. L. Guo, Y. Zhao, Y. Zhang, S. Y. Tan, Y. F. Zeng, R. Q. Zou, Q. Y. Yan and Y. L. Zhao, Small, 2016, 12, 3849–3860 CrossRef CAS PubMed.
- S. L. Xiong, J. S. Chen, X. W. Lou and H. C. Zeng, Adv. Funct. Mater., 2012, 22, 861–871 CrossRef CAS.
- Y. H. Dou, J. T. Xu, B. Y. Ruan, Q. N. Liu, Y. D. Pan, Z. Q. Sun and S. X. Dou, Adv. Energy Mater., 2016, 6, 1501835 CrossRef.
- J. L. Tang, V. Etacheri and V. G. Pol, ACS Sustainable Chem. Eng., 2016, 4, 2624–2631 CrossRef CAS.
- I. Sultana, M. M. Rahman, T. Ramireddy, N. Sharma, D. Poddar, A. Khalid, H. Z. Zhang, Y. Chen and A. M. Glushenkov, ACS Appl. Mater. Interfaces, 2015, 7, 20736–20744 CAS.
- S. H. Wang, M. Q. Chen, Y. Y. Xie, Y. N. Fan, D. W. Wang, J. J. Jiang, Y. G. Li, H. Grutzmacher and C. Y. Su, Small, 2016, 12, 2365–2375 CrossRef CAS PubMed.
- Y. F. Dong, S. H. Liu, Z. Y. Wang, Y. Liu, Z. B. Zhao and J. S. Qiu, RSC Adv., 2015, 5, 8929–8932 RSC.
- G. Y Huang, S. M. Xu, S. S. Lu, L. Y. Li and H. Y. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 7236–7243 Search PubMed.
- Y. Zhao, X. F. Li, B. Yan, D. B. Xiong, D. J. Li, S. Lawes and X. L. Sun, Adv. Energy Mater., 2016, 1502175 CrossRef.
- Y. M. Liu, X. Y. Zhao, F. Li and D. G. Xia, Electrochim. Acta, 2011, 56, 6448–6452 CrossRef CAS.
- G. Chen, Z. Y. Wang and D. G. Xia, Chem. Mater., 2008, 20, 6951–6956 CrossRef CAS.
- (a) D. Wang, Q. Wang and T. Wang, Inorg. Chem., 2011, 50, 6482–6492 CrossRef CAS PubMed; (b) Y. Tan, Q. Gao, Z. Li, Q. Tian, W. Qian, C. Yang and H. Zhang, Sci. Rep., 2016, 6, 26460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28296b |
| This journal is © The Royal Society of Chemistry 2017 |