DOI:
10.1039/C6RA28142G
(Paper)
RSC Adv., 2017,
7, 13458-13466
Efficient cocktail chemotherapy by co-delivery of a hydrogen sulfide-releasing aspirin prodrug and paclitaxel via single nanoparticles
Received
13th December 2016
, Accepted 12th February 2017
First published on 28th February 2017
Abstract
Cocktail treatment has become an effective multidrug medication therapy for many diseases, because of the additive or synergistic effect of each medicine and relief from adverse effects of drugs. Nanotechnology offers an unparalleled opportunity for co-delivery strategies with the ability to simultaneously release various drugs and controllably unify the pharmacokinetics. In this work, we formulated a cocktail composed of hydrogen sulfide-releasing aspirin (HS-ASP) and paclitaxel (PTX) in a single delivery system, which is fabricated by a biodegradable block copolymer (i.e., methoxy poly(ethylene glycol)–polycaprolactone). The resulting drug cocktail-loaded nanoparticles possess small and narrow size distribution (∼44 nm), high dual-drug encapsulation efficiency, pH-sensitivity and simultaneous drug release capability. The co-delivery of HS-ASP and PTX has significantly decreased the IC50 value of PTX on LL/2 cells by 17.7-fold compared with single free PTX and by 5.4-fold compared with PTX NPs. Meanwhile, its cellular uptake is improved by 2.67-fold compared with free HS-ASP/FITC-PTX, and it enhanced the cellular apoptosis (53.62%) as well as the cell cycle blocking at G2/M phase (47.9%) relative to other formulations. Confocal laser scanning results show that HS-ASP inhibited the mitosis of LL/2 cells, thereby sensitizing the cells to the tubulin disruption effect, which is a major apoptosis mechanism of PTX. Overall, such a combinational and controlled release strategy is expected to hold great potential for clinical use in cancer therapy.
Introduction
Single chemotherapeutic modality has been widely used for anti-cancer therapy. However, due to the physiological complexity of tumours, single chemotherapy may not be sufficient for effective treatment, which leads to many drawbacks, such as high toxicity, drug resistance, and limited clinical use.1 To overcome these issues, the combination of two or more therapeutic drugs has been developed with different pharmacological mechanism to reduce the dosage of each agent, to realize synergistic therapeutic efficacy, to decrease unwanted side effects, and to achieve long-term prognosis.2–4
Hydrogen sulfide-releasing aspirin prodrug (HS-ASP), a recently developed aspirin derivative, is promising as an anti-cancer agent. It consists of a traditional aspirin molecule covalently bound to an H2S-releasing moiety (5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione). HS-ASP demonstrated anti-proliferation effects through the induction of G0/G1 arrest and apoptosis, down-regulation of NF-κB, reduction of thioredoxin reductase activity, and increase of reactive oxygen species level.5 This compound is also able to up-regulate Nrf2, NQO1, UGT, and GST levels, and co-induce multiple phase-II metabolizing enzymes.6 Furthermore, HS-ASP has the capability of inhibiting the tumour growth in breast cancer cell (i.e., MDA-MB 231) and other mammary cell-derived tumour xenograft mouse models.5 HS-ASP has particular advantages over other conventional chemotherapeutic drugs, in terms of its NSAID based structure and alleviative gastrointestinal side effects of NSAIDs. HS-ASP also allows minimal side-effects even at high doses,5 which is promising for in vivo applications All of those findings suggest that HS-ASP can serve as a potential safe additive for cancer prevention and treatment.
Paclitaxel (PTX) is one of the most effective chemotherapeutic agents against many forms of cancer.1 Significant progress has been achieved in the combination therapy using PTX with other agents, including doxorubicin, Herceptin, siRNA, and other cytokine.7–9 We therefore hypothesize that the cocktail of PTX with HS-ASP can generate synergistic effect in antitumor therapy. However, the limited water-solubility and tumour selectivity of PTX and HS-ASP may result in unsatisfactory efficiency,10 and the application of combination therapy is often encumbered by the varying pharmacokinetics among different drugs.
Over the past decades, nanocarrier-based drug delivery has gained a great attention because of increased water solubility for encapsulated drugs, enhanced drug accumulation at tumour site, minimized side effects and optimized therapeutic efficacy.11–14 These nanocarriers can also exploit passive accumulation route to accomplish the tumour-specific delivery through the enhanced permeability and retention (EPR) effect.15 Moreover, when multiple drugs are delivered in single vehicles, the synergistic effects can be envisaged for cancer therapy. In this context, biodegradable polymersomes loaded with both PTX and DOX have been reported to deeply penetrate tumours, leading to the shrinkage of tumors.16,17 In addition, nanoparticles (NPs) are composed of biocompatible and biodegradable copolymers (e.g., poly(ethylene glycol)-block-polycaprolactone (PEG–PCL)). Biodegradable polymersomes have been widely studied as drug delivery carriers to alter the pharmacokinetics, increase the concentration of drugs in tumours, suppress tumour growth and metastasis, and prolong survival in tumour-bearing mouse model.15,18 The cloaking of hydrophilic PEG chains on particle surface endows the “stealth” property, while the hydrophobic PCL segments can host diverse and multiple drugs. These findings suggest the promising application of PEG–PCL NPs for anti-tumour drug delivery. However, so far, there is no report about PEG–PCL NPs co-delivering HS-ASP and PTX for cancer therapy.
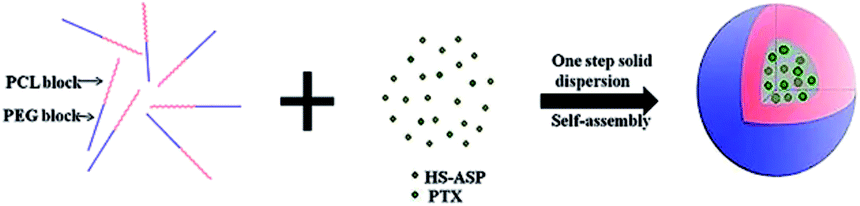
In this study, we aim at presenting a novel cocktail strategy using the mPEG–PCL (Mw 2000–6600, copolymer ratio 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) NPs co-delivering HS-ASP and PTX for cancer therapy. To achieve simultaneous delivery, as shown in Fig. 1, nanoparticles with a core–shell structure are designed with two functional components: (1) an mPEG shell water-solubility improvement and cloaking; (2) a PCL core for hosting adequate PTX and HS-ASP. We hypothesize that co-delivery of HS-ASP and PTX by mPEG–PCL NPs promotes simultaneous drug release, enhances endocytosis, modulates different signalling pathways, and induces synergetic responses, which thus maximizes the therapeutic effect.
:50) NPs co-delivering HS-ASP and PTX for cancer therapy. To achieve simultaneous delivery, as shown in Fig. 1, nanoparticles with a core–shell structure are designed with two functional components: (1) an mPEG shell water-solubility improvement and cloaking; (2) a PCL core for hosting adequate PTX and HS-ASP. We hypothesize that co-delivery of HS-ASP and PTX by mPEG–PCL NPs promotes simultaneous drug release, enhances endocytosis, modulates different signalling pathways, and induces synergetic responses, which thus maximizes the therapeutic effect.
 |
| | Fig. 1 Scheme of HS-ASP/PTX mPEG–PCL nanoparticle preparation. | |
Experimental methods
Materials
Paclitaxel, polyvinyl alcohol (PVA, Mw 30000–70000), fluorescein isothiocyanate (FITC) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (methyl thiazolyl tetrazolium, MTT), were purchased from Sigma (USA). mPEG–PCL (Mw 2000–6600, copolymer ratio 50:50) was purchased from Jinan Daigang biomaterials (China). Tubulin-Tracker Red and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Beyotime (China). Roswell Park Memorial Institute 1640 medium (RPMI 1640, Gibco, USA), acetonitrile (HPLC grade, Fisher Scientific, UK) and propidium iodide (PI, J&K Scientific, China) were used without further purification. 4-(5-Thioxo-5H-1,2-dithiol-3-yl)-phenyl-2-acetoxy-benzoate (HS-ASP) was synthesized and purified according to literature,19 FITC labelled PTX (FITC-PTX) was synthesized and purified according to literature.15 All the materials used in this study were analytic reagent (AR) grade and used as received. LL/2 cells (murine Lewis lung cancer cells) were purchased from the American Type Culture Collection (ATCC; Rockville, MD), which grew in RPMI 1640 supplement with 10% fetal bovine serum (FBS). The cell cultures were maintained in a 37 °C incubator with a humidified 5% CO2 atmosphere.
Methods
Preparations of HS-ASP/PTX mPEG–PCL NPs. mPEG–PCL NPs loaded with both HS-ASP/PTX or respective drug alone were prepared using a one-step solid dispersion method. Briefly, the predetermined amounts of HS-ASP and/or PTX and mPEG–PCL (Mw 2000–6600, copolymer ratio 50:50) were dissolved in dehydrated alcohol under mild stirring. Then, the organic solvent was evaporated in a rotary evaporator at 60 °C. During this process, HS-ASP and/or PTX were distributed in mPEG–PCL copolymer as amorphous substances.Subsequently, the mixture obtained after co-evaporation was dissolved in 1% PVA aqueous solution at 45 °C to self-assemble into NPs. After ultrasonication for 10 min, the NPs were collected by filtration through filters (pore diameter 0.22 μm) and dialysis for 1 h at room temperature to remove the unloaded drugs. The collected samples were lyophilized and stored at 4 °C before characterization or use for in vitro experiments.
Characterization of HS-ASP/PTX NPs
Encapsulation efficiency. After the drugs were extracted from the micelles by methanol, the final concentration of HS-ASP/PTX was measured by high-performance liquid chromatography (HPLC) analysis. Chromatographic separations were carried out on a C18 column (250 × 4.6 mm, 5 μm) and samples were eluted using acetonitrile and water (70:30, v/v) at a flow rate of 1.0 mL min−1 with the UV detector at 227 nm (PTX) and 435 nm (HS-ASP). The encapsulation efficiency of HS-ASP/PTX in NPs was calculated according to the following equation:
| EE (%) = Wencapsulated /Wtotal × 100% |
where, Wtotal is the total amount of HS-ASP/PTX in NPs before purification and Wencapsulated is the amount of HS-ASP/PTX encapsulated in the NPs after purification.
Mean particle sizes and zeta potential. The particle size distribution and zeta potential were measured by dynamic light scattering detector (Nano-ZS, Malvern, UK). At least three different batches were analysed to obtain an average value and the related standard deviation. The morphology of prepared NPs was observed by transmission electron microscope (TEM). NPs were diluted with distilled water and dropped on a copper grid covered with nitrocellulose. The samples were negatively stained with phosphotungstic acid and dried at room temperature, after which images of prepared NPs were taken by TEM (H-6009IV, Hitachi, Japan).
Differential scanning calorimetry (DSC) tests. To investigate whether the drugs were successfully entrapped into mPEG–PCL NPs, differential scanning calorimetry was carried out using thermogravimetric analyser (Q2000, TA, USA) in nitrogen at a heating rate of 10 °C min−1 from 20 °C to 260 °C. For each test, 2–4 mg of freeze-dried blank NPs, PTX NPs, HS-ASP NPs, HS-ASP/PTX co-loaded NPs, physical mixture of HS-ASP NPs and PTX powder, physical mixture of PTX NPs and HS-ASP powder, or physically mixture of blank NPs, HS-ASP powder and PTX powder were loaded in the aluminium pan and respectively analysed. The relationship was obtained in terms of heat flow and heating temperature. An empty sealed pan was used as the reference.
In vitro release kinetics study. The in vitro release of the HS-ASP/PTX loaded NPs was monitored by a dialysis method. 1 mL drug-loaded NPs were transferred to dialysis bags (MWCO: 3500 Da) placed in 400 mL of PBS (pH 7.4 or 5.0) with 5% Tween 80, and stirred at 110 rpm at 37 °C. The initial concentrations of HS-ASP and PTX in the formulations were determined to be 2 and 0.4 mg mL−1, respectively. At scheduled intervals, 1 mL of the dialysis medium was collected and the same volume of fresh PBS was added immediately. The concentration of HS-ASP or PTX in dialysis medium was monitored by HPLC assay. The release percentage was calculated according to the following equation:
| Release percentage (%) = (Wrelease/Wtotal) × 100% |
where, Wtotal is the total amount of HS-ASP or PTX in NPs, and Wrelease is the amount of HS-ASP or PTX released from NPs into dialysis medium at the scheduled time.
Synergistic cytotoxicity assay. LL/2 cells were maintained onto 96-well plates at 37 °C in humanized atmosphere containing 5% CO2 for 24 h in RMPI 1640 medium, which was supplemented with 10% (v/v) fetal bovine serum. The culture media were replaced with media containing different concentration of blank NPs, PTX in solution, PTX NPs, HS-ASP/PTX in solution, and HS-ASP/PTX NPs, respectively. Cells cultured with only RMPI 1640 media were used as control. After 48 h of incubation, the amount of viable cells was evaluated via MTT assay. Each experiment was repeated three times in triplicate (n = 9). To test synergism, PTX and HS-ASP combination index (CI) was determined with the classic isobologram equation of Chou and Talalay.20
Induction of apoptosis in vitro and cell cycle analysis. The apoptosis of the cells and the distribution of DNA in the cell cycle were studied by flow cytometry. 3 × 105 LL/2 cells per well were seeded in 6-well plates 24 h before the experiment. The cells were exposed to blank NPs, HS-ASP in solution (30 μg mL−1), PTX in solution (3 μg mL−1), or a combination of both drugs either in solution (free HS-ASP/PTX) or in formulation (HS-ASP/PTX NPs) with drug concentration in the proportion of (30 + 3) μg mL−1, and incubated for additional 24 h at 37 °C. Cells treated with only medium were used as control. After incubation period, the cells were washed with DPBS twice, and suspended in 500 μL DPBS, which consists of 1 μL of propidium iodide (PI, 10 μg μL−1), 1 μL of RNase A (10 μg μL−1), and 0.5% Tween-20. Then, the cells were incubated for 1 h at room temperature in the dark before analysis. Cell apoptosis and cycle distribution were determined by analyzing 10000 ungated cells using a Flow Cytometer (C6, Accuri, USA) and CFlow software (Accuri, USA). All experiments were performed in triplicate.
Cellular uptake of mPEG–PCL NPs. We chose PTX-FITC as a probe to study the cellular uptake of drugs in mPEG–PCL NPs by fluorescence determine. LL/2 cells at log phase were seeded in 6-well plates at a density of 3 × 105 cells per well and cultured for 24 h before experiment. Then, the growth medium was removed, and the cells were treated with 2 mL of serum-free medium containing blank micelles, equivalent concentration of HS-ASP and PTX-FITC (HS-ASP = 30 μg mL−1, PTX = 3 μg mL−1) either in the form of free HS-ASP/PTX-FITC solution or HS-ASP/PTX-FITC NPs, respectively, and incubated for 4 h at 37 °C in CO2 incubator. Subsequently, the cells were washed three times with cold DPBS (Beyotime, China) to remove the excess of unbound drugs. After that, cells were fixed with cold acetone, washed again with cold DPBS, stained with DAPI (Beyotime, China). The quantitative determination of intracellular FITC-PTX fluorescence and images were analyzed using an ArrayScan® VTI HCS reader (Thermo Scientific, USA).
Confocal laser scanning of cellular tubulin pattern. To further explore the possible mechanism for the synergistic therapeutic effect, we evaluated the effect of HS-ASP with or without PTX on cellular tubulin pattern. Briefly, LL/2 cells were seeded onto a borosilicate chambered cover glass at a density of 5 × 105 cells per well at 37 °C for 24 h, then cultured with HS-ASP NPs (HS-ASP = 30 μg mL−1), PTX NPs (PTX = 3 μg mL−1) and HS-ASP/PTX NPs (HS-ASP = 30 μg mL−1, PTX = 3 μg mL−1) for 4 h. Cells were washed three times with cold DPBS, fixed with cold acetone, and then washed again. After that, cells were stained with Tubulin-Tracker Red (Beyotime, China) for 1 h and DAPI for 5 min according to the standard process provided by the company. The cells were washed three times by DPBS before image acquisition. Fluorescence images were acquired with a confocal microscope (DM6000 CS, Leica, Germany).
Statistics analysis. Statistical analyses were performed using a Student's t-test. The differences were considered significant for p values < 0.05 and very significant for p values < 0.005.
Results and discussion
Physicochemical characterization of drug-loaded NPs
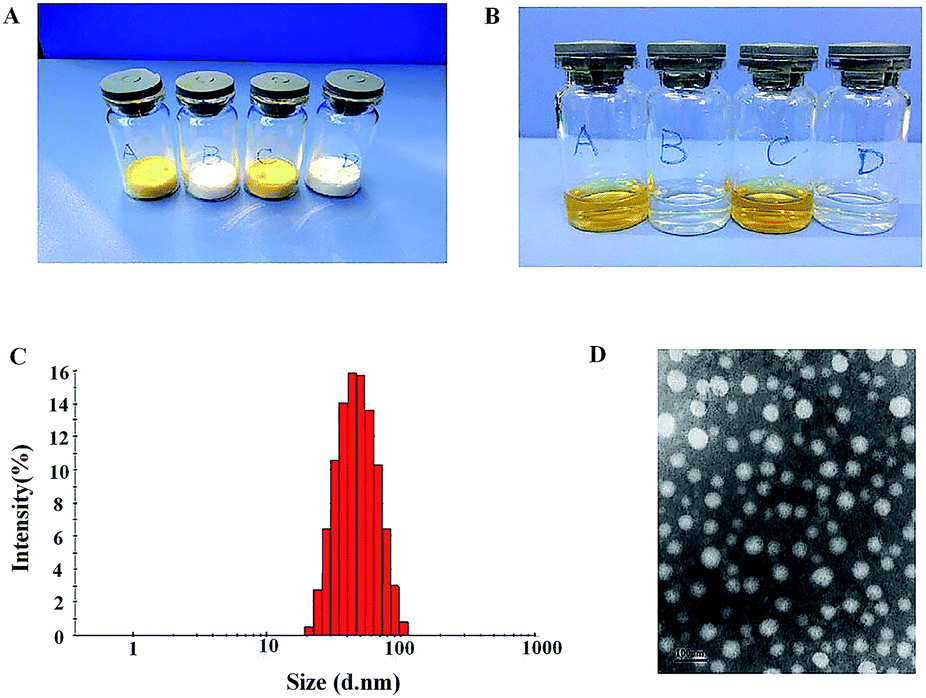
Synergistic combination of two or more drugs attracts great attentions because this strategy can potentiate individual drugs or overcome the undesirable toxicity and other side effects that limit the utility of many potential drug.21 Therefore, it is essential to develop a co-delivery system which allows to formulate multiple drugs in single carriers and to simultaneously transport them to cancer cells. Nanosized carriers have been widely explored for the co-delivery of chemically disparate drugs. Like other amphiphilic copolymers, mPEG–PCL can be assembled in aqueous solutions to form core–shell structure. The stealth property of particle surface makes the produced NPs to evade detection and uptake by the reticuloendothelial system (RES) and to achieve longevity during systemic circulation.22 As shown in Fig. 1, HS-ASP/PTX NPs are prepared by a one-step solid dispersion method, which is highly reproducible. The appearance of the prepared NPs is presented in Fig. 2A and B. These NPs could be lyophilized into powder forms without any adjuvants (Fig. 2A), and the re-suspended NPs are stable and homogeneous (Fig. 2B). In Fig. 2B, the clear solutions of re-dissolved HS-ASP NPs (A), PTX NPs (B), HS-ASP/PTX NPs (C) and blank NPs (D) are observed. According to the particle size distribution measured by DLS (Fig. 2C), the HS-ASP/PTX NPs exhibit a very narrow particle size distribution. The TEM image of the HS-ASP/PTX NPs is shown in Fig. 2D, which reveals that the NPs are spherical and the diameter is in good agreement with the results of the DLS. Table 1 discloses that loading of double drugs resulted in slight increase of the mean particle size when compared with HS-ASP NPs and PTX NPs, while a narrow distribution is maintained. When the ratio of PTX/HS-ASP/mPEG–PCL copolymer is fixed at 1/10/200 (w/w/w), the average particle size of the prepared NPs is 43.99 ± 0.36 nm with a polydispersity index (PDI) of 0.119 ± 0.012 (Table 1). Moreover, the encapsulation efficiency (EE, %) of the obtained HS-ASP/PTX NPs are 93.16 ± 3.64% for PTX, and 52.47 ± 1.83% for HS-ASP, respectively. This co-loading method does not significantly reduce the EE of PTX relative to PTX NPs (95.25 ± 2.46%). In this study, NPs composed of mPEG–PCL has high solubility and EE values toward HS-ASP and PTX, and it is expected that NPs with small sizes might use the EPR effect to preferentially accumulate at tumour sites. The zeta-potentials of different drug-loaded NPs are also summarized in Table 1. The negative surface charge of HS-ASP/PTX NPs (−13.4 ± 0.38 mV) might contribute to a better blood compatibility and prolonged circulation time of NPs due to the reduced clearance by RES.23
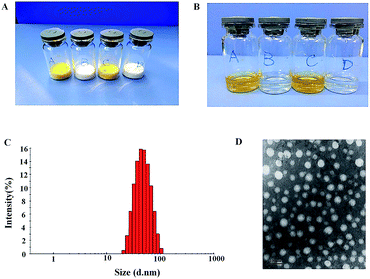 |
| | Fig. 2 Characteristics of mPEG–PCL NPs. (A) The appearance of freeze-dried powders in various dosage forms ((A) HS-ASP NPs; (B) PTX NPs; (C) HS-ASP/PTX NPs; (D) blank NPs); (B) the appearance of re-dispersed NPs in various dosage forms ((A) HS-ASP NPs; (B) PTX NPs; (C) HS-ASP/PTX NPs; (D) blank NPs); (C) size distribution of HS-ASP/PTX NPs; (D) typical TEM images of HS-ASP/PTX NPs. | |
Table 1 Physicochemical characteristics of NPs formulation
| Formulation |
Mean sizea (nm) |
Polydispersity index |
Zeta potentialb (mV) |
Encapsulation efficiencyc (%) |
| Mean size in nm as measured by dynamic laser spectroscopy. Zeta potential in mV as measured by zetasizer. Encapsulation efficiency of drugs (expressed as %) was estimated by HPLC. For HS-ASP. For PTX. |
| HS-ASP NPs |
26.61 ± 0.43 |
0.099 ± 0.009 |
−12.9 ± 0.36 |
59.22 ± 3.81d |
| PTX NPs |
36.72 ± 4.86 |
0.176 ± 0.089 |
−1.35 ± 0.34 |
95.25 ± 2.46e |
| HS-ASP/PTX NPs |
43.99 ± 0.36 |
0.119 ± 0.012 |
−13.4 ± 0.38 |
52.47 ± 1.83d, 93.16 ± 3.64e |
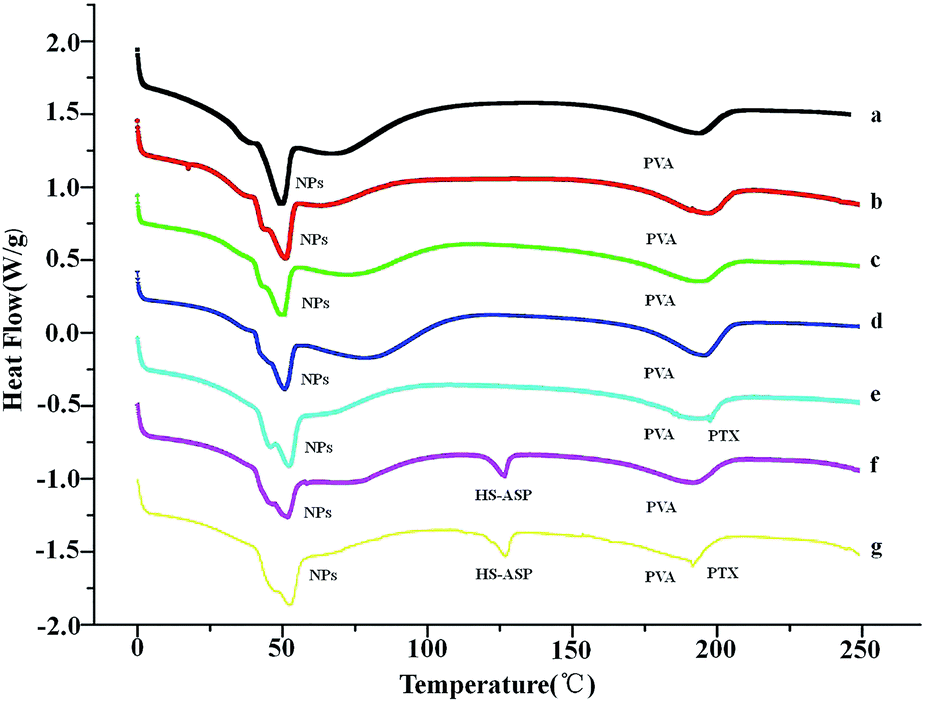
DSC analysis is performed to confirm the successful entrapment of both drugs within mPEG–PCL NPs. Fig. 3 shows the endothermic curves of lyophilized HS-ASP NPs (a), PTX NPs (b), HS-ASP/PTX NPs (c), blank NPs (d), HS-ASP NPs mixed with PTX (e), PTX NPs mixed with HS-ASP (f) and blank NPs mixed with PTX/HS-ASP (g). As the temperature increases, blank mPEG–PCL polymer (d) reaches its melting temperature (Tm) which can be seen as endothermic peaks at 50.67 °C, and the emulgator PVA reaches its Tm at 195.62 °C. The dual drug loaded NPs and single drug loaded NPs show similar endothermic peaks when compare with blank NPs, and no other endothermic peak is observed. In contrast, when NPs are physically mixed with HS-ASP or PTX, endothermic peaks of free PTX appear at 197.44 °C and that of HS-ASP appear at 126.48 °C. These results indicate that both drugs are successfully entrapped and appeared in their native forms without chemical conjugation with the polymer backbones.
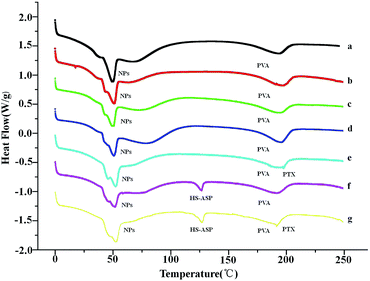 |
| | Fig. 3 DSC curves of HS-ASP NPs (a), PTX NPs (b), HS-ASP/PTX NPs (c), blank NPs (d), HS-ASP NPs mixed with PTX (e), PTX NPs mixed with HS-ASP (f), and blank NPs mixed with HS-ASP/PTX (g). | |
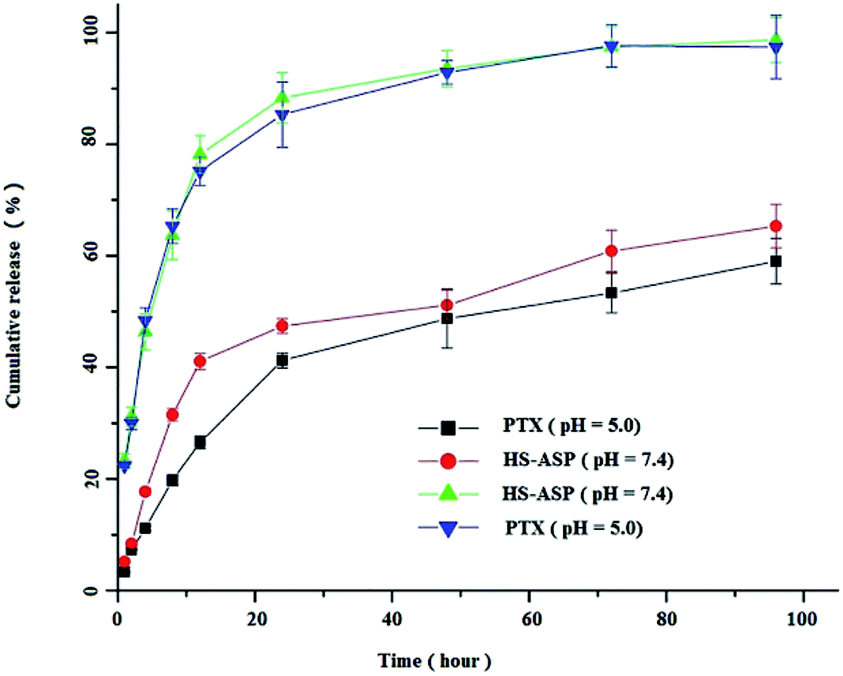
To the best of our knowledge, the cellular uptake of NPs most likely occurs via endocytosis or potentially pinocytosis, in which lysosomal (pH 4–5) processing and the subsequent release of drug may occur at low pH. Meanwhile, the pH of tumour is always lower than that in the normal physiological environment. Therefore, we selected the pH 5.0 and 7.4 media to examine the release profiles of HS-ASP/PTX. As shown in Fig. 4, the in vitro release profiles of both HS-ASP and PTX from mPEG–PCL NPs are obtained within pH 7.4–5.0. Both HS-ASP and PTX release processes are greatly affected by the environmental acidity. At pH 5.0, 88.2 ± 5.5% of HS-ASP and 84.6 ± 6.4% of PTX are respectively released from HS-ASP/PTX NPs within 24 h, which are close to 95% of the total drug content being released within the first 48 h. However, the releasing of HS-ASP and PTX at pH 7.4 are slower and sustainable. Approximately 47.8 ± 2.1% of HS-ASP and 41.5 ± 2.3% of PTX are respectively released from NPs within 24 h. The initial burst release is followed by a slower sustained release of drug presented inside the core of NPs.
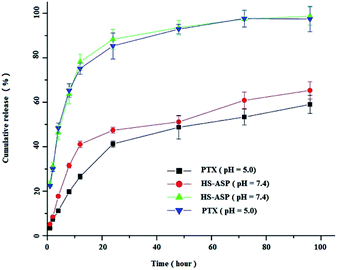 |
| | Fig. 4 In vitro drug release of HS-ASP and PTX from mPEG–PCL NPs at 37 °C and pH 7.4 or pH 5.0. | |
The change in the structure of drugs usually leads to the morphological change of nanocarrier, which reduces the stability of the drug–nanocarrier system. From Fig. 4, it is inferred that HS-ASP could accept the protons under acidic condition, and consequently hydrophobic HS-ASP tends to form a hydrophilic structure. Accordingly, the structure and corresponding stability of the drug–nanocarrier complexes could be affected, which might explain the higher drug release behaviour at pH 5. This phenomenon suggests that the drug release might be slower in blood circulation (pH 7.4) but faster in lysosome and tumour (pH 4–5). As a result, the co-delivery system could reduce the clearance of drugs by circulation system but increase the accumulation of drugs at tumour sites. In addition, the similar release profiles is obtained from both HS-ASP and PTX indicates that the co-delivery system provides a possibility of their synergistic effect.
Synergistic cytotoxicity by combination of PTX and HS-ASP
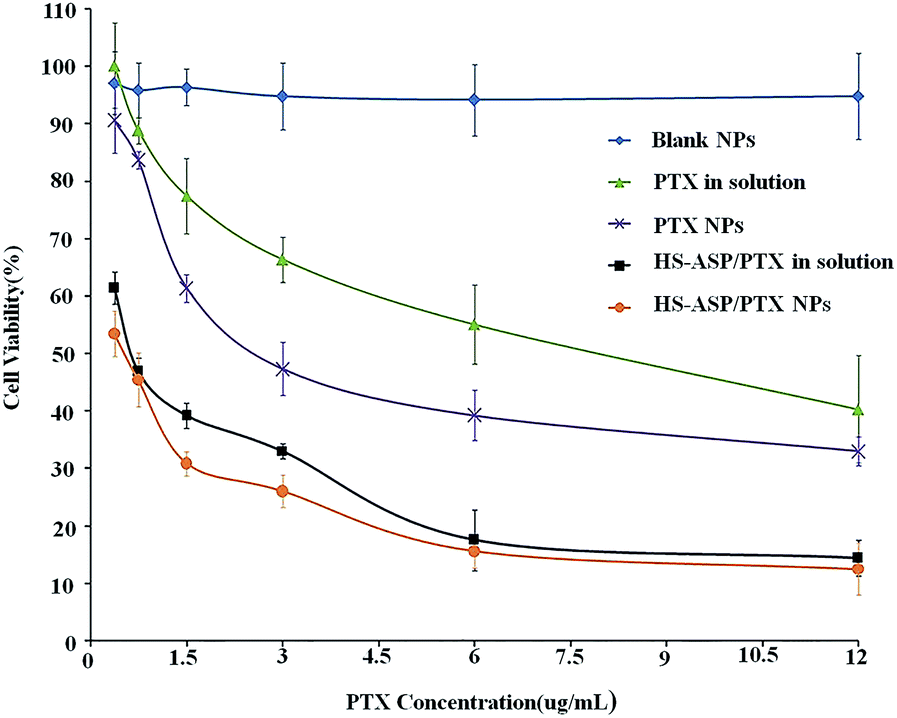
The in vitro cytotoxicity of the different nanoparticle formulations is investigated against LL/2 cell line. The survival rate histograms of LL/2 cells after incubation with PTX as a single agent or combination with HS-ASP in solution or nanoparticle formulations at 48 h are shown in Fig. 5. Blank mPEG–PCL NPs (♦) have no obvious cytotoxic effects on the cells at the corresponding concentrations used. All the other formulations show concentration dependent tumour cell proliferation inhibition effect. Dual-drug-loaded NPs (●) show better proliferation inhibition effect over HS-ASP/PTX in the solution (■). As shown in Table 2, the IC50 on LL/2 cells of PTX in solution (free PTX) and PTX NPs are 8.16 ± 0.63 and 2.57 ± 0.42 μg mL−1, respectively. The DMSO solution of PTX requires more than 3-fold dose of drug to achieve the same cytotoxicity as the nanoparticle formulation. The IC50 of HS-ASP in solution (free HS-ASP) and HS-ASP NPs are determined to be 25.62 ± 1.88 and 15.33 ± 1.06 μg mL−1, respectively. The DMSO solution of HS-ASP requires more than 1.5-fold dose of drug to achieve the same cytotoxicity as the nanoparticle formulation. The IC50 of PTX equivalent in HS-ASP/PTX in solution and HS-ASP/PTX NPs reach 0.53 ± 0.07 μg mL−1 and 0.46 ± 0.05 μg mL−1, respectively. These are significantly decreased compared to single PTX in solution (15.4-fold) or PTX NPs (5.4-fold), respectively. Combination of two agents decrease the amount of each drug but achieve a better cytotoxic efficiency. Furthermore, the cytotoxic on LL/2 cells of HS-ASP/PTX in NP formulation is enhanced by 13% compared with HS-ASP/PTX in solution in vitro.
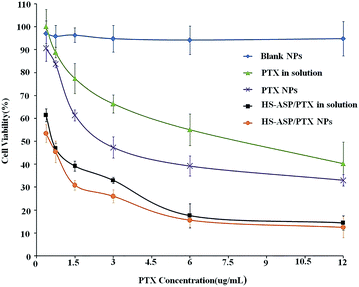 |
| | Fig. 5 Dose dependent cytotoxicity of different concentration of blank NPs (♦), PTX in solution (▲), PTX NPs (✗), HS-ASP/PTX in solution (■) and HS-ASP/PTX NPs (●) on LL/2 cells. The extent of growth inhibition was measured after 48 h using MTT assay. The percentage of survival was determined by normalizing the absorbance of controls to 100%. | |
Table 2 IC50 doses of HS-ASP/PTX in solution or in nano-formulation, either single or in combination on LL/2 cells
| Sample |
IC50a (μg mL−1) |
| Data as mean ± SD, n = 6. *p < 0.005, PTX NPs vs. PTX in solution, and HS-ASP NPs vs. HS-ASP NPs; **p < 0.005, PTX equivalent in HS-ASP/PTX in solution or in HS-ASP/PTX NPs vs. PTX in solution or PTX NPs. |
| PTX in solution |
8.16 ± 0.63 |
| PTX NPs |
2.57 ± 0.42* |
| HS-ASP in solution |
25.62 ± 1.88 |
| HS-ASP NPs |
15.33 ± 1.06* |
| PTX equivalent in HS-ASP/PTX in solution |
0.53 ± 0.07** |
| PTX equivalent in HS-ASP/PTX NPs |
0.46 ± 0.05** |
To test synergism, PTX and HS-ASP combination index (CI) is determined with the classic isobologram equation of Chou and Talalay.20
where, “
a” is the PTX IC
50 in combination with HS-ASP at concentration “
b”;
A is the PTX IC
50 and
B is the HS-ASP IC
50. When the CI is lower than 1, a synergistic effect is observed. CI = 1 corresponds to an additive effect, and when the CI is greater than 1 an antagonistic effect was observed. With respect to the LL/2 cell line, at a ratio of HS-ASP
:
PTX = 10
:
1, the CI is 0.26 at variable concentrations of PTX, suggesting a effective synergistic combination treatment. These results demonstrate a dramatic improvement in the efficacy of PTX when combined with HS-ASP.
Enhanced cell cycle arrest and apoptosis by HS-ASP/PTX NPs
To further confirm the therapeutic potential of combination therapy, cell cycle inhibition study was performed using flow cytometry. Table 2 shows the relative cell cycle blocking activities upon treatment with single drug and dual drugs in solution and in NP formulations. Both the formulations of HS-ASP shows more cell cycle arrest at G0/G1 phase (58.8% and 56.8%), whereas both the formulations of PTX shows more cell cycle arrest at G2/M phase (35.9% and 36.5%) when compared with the control and blank NP groups, which are consistent with the previous reports.5,24 One interesting phenomenon is that the extent of cell cycle blocking at G2/M phase is the highest (44.5% for solution and 47.9% for NPs) when the cells are treated with dual drug formulations as compared to single drug formulations. These results are in agreement with the cell viability, suggesting that simultaneous formulation with HS-ASP and PTX in single NPs produce more potential therapeutic effects. Moreover, in the case of dual drug treatment, the blocking at G2/M phase play a major role in cell cycle arrest, indicating that HS-ASP sensitize the cells to PTX and, thus, to augment the activity of PTX on cell cycle (Table 3).
Table 3 Effect of HS-ASP, PTX, and drug loaded mPEG–PCL NPs on cell cyclea
| Formulations |
Cell cycle/% |
| G0/G1 |
G2/M |
| Data as mean ± SD, n = 3. *p < 0.05, free HS-ASP or HS-ASP NPs vs. control or blank NPs; **p < 0.005, free PTX, PTX NPs, free HS-ASP/PTX or HS-ASP/PTX NPs PTX vs. control or blank NPs. |
| Control |
50.9 ± 2.5 |
14.5 ± 1.7 |
| Blank NPs |
52.4 ± 3.6 |
16.0 ± 2.3 |
| Free HS-ASP (30 μg mL−1) |
58.8 ± 3.1* |
15.8 ± 2.0 |
| HS-ASP NPs (30 μg mL−1) |
56.8 ± 4.7* |
16.8 ± 1.2 |
| Free PTX (3 μg mL−1) |
9.8 ± 0.8 |
35.9 ± 2.6** |
| PTX NPs (3 μg mL−1) |
14.2 ± 1.1 |
36.5 ± 3.4** |
| Free HS-ASP (30 μg mL−1)/PTX (3 μg mL−1) |
12.1 ± 0.7 |
44.5 ± 3.8** |
| HS-ASP (30 μg mL−1)/PTX (3 μg mL−1) NPs |
18.2 ± 1.3 |
47.9 ± 2.6** |
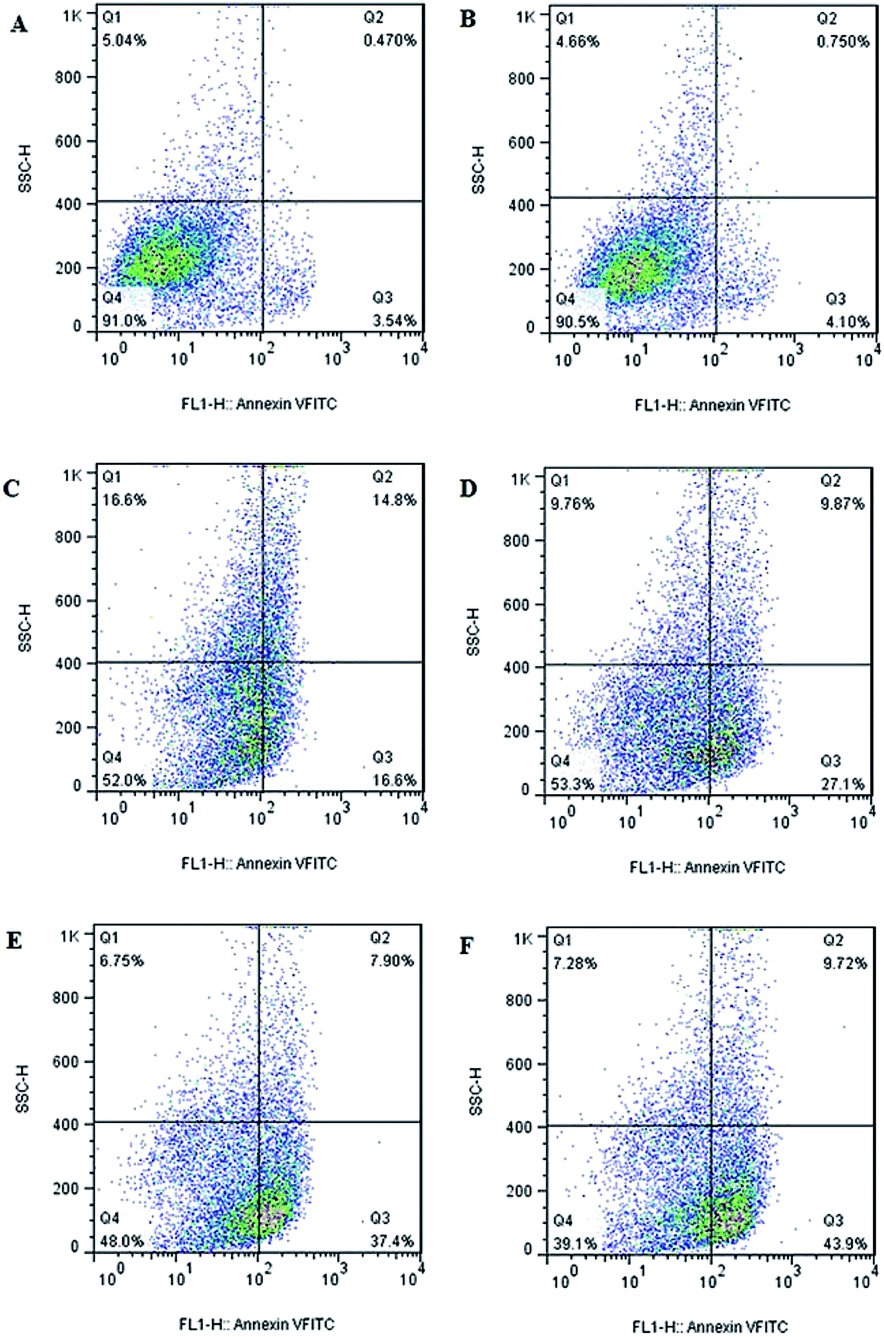
Since cell apoptosis may be one of the consequences of cell-cycle arrest, the cellular apoptosis event is detected using PI staining and flow cytometry. Flow cytometry plots of LL/2 cells treats with different formulations (Fig. 6) reveals that there is no obvious change in cellular apoptosis in blank NPs group (4.85%) when compared to the control (4.01%). However, obvious enhancement is observed in cellular apoptosis when HS-ASP and PTX are co-administered in solution (45.3%) relative to the respective single drug in solution (31.4% and 36.97%). In addition, when HS-ASP and PTX are co-loaded in NPs, the cellular apoptosis further enhances (53.62%) in contrast to all the other formulations, which agrees well with the result of cell viability assay and cell cycle arrest study. Therefore, HS-ASP and PTX co-loaded NPs inhibit proliferation of LL/2 cells by a combined induction of G2/M arrest and apoptosis. Combining HS-ASP with PTX is successful in enhancing the antitumor effect in vitro as shown by the dramatic improvement of cell viability inhibition, cell cycle arrest, and cell apoptosis efficacy. With enhanced antitumor effect, the lower dosage of chemotherapeutics may induce a reduction of side effects on normal tissues.2–4
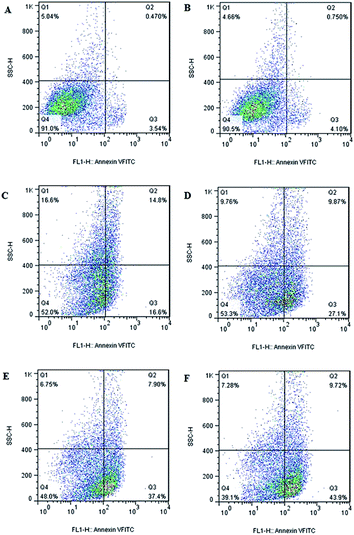 |
| | Fig. 6 Detection of apoptosis by flow cytometry. (A) Control; (B) blank NPs; (C) HS-ASP 30 μg mL−1 in solution; (D) PTX 3 μg mL−1 in solution; (E) HS-ASP 30 μg mL−1 and PTX 3 μg mL−1 in solution; (F) HS-ASP 30 μg mL−1 and PTX 3 μg mL−1 NPs. | |
Enhanced endocytosis of HS-ASP/FITC-PTX NPs
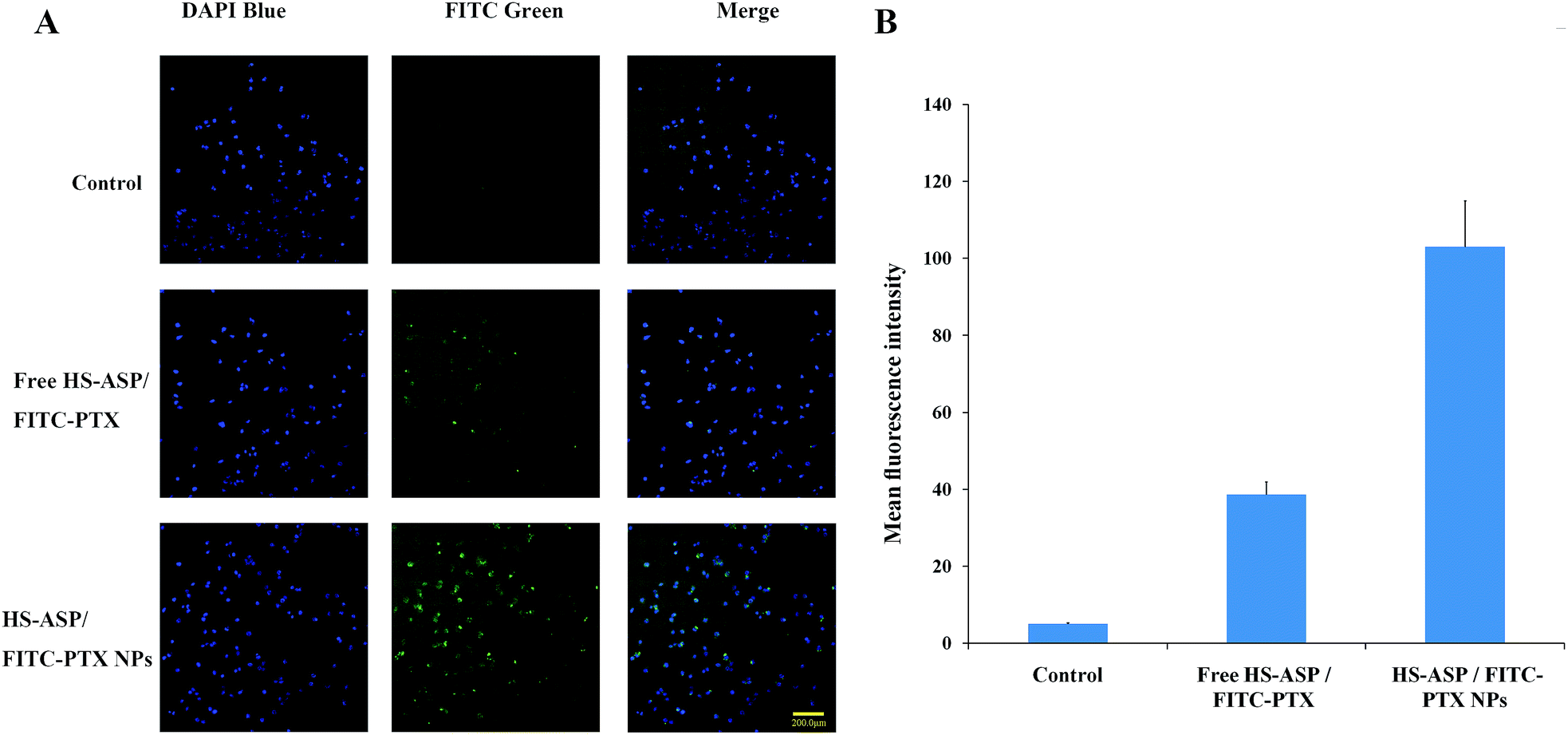
To investigate the mechanism of the enhanced cytotoxic activity of the NPs, cellular uptake study of the dual agents in solution (free HS-ASP/FITC-PTX) or in NPs (HS-ASP/FITC-PTX NPs) were conducted by visualizing and detecting the intrinsic green fluorescence of FITC-PTX using HCS reader. Fig. 7A presents the images of cells treated with blank NPs as control, HS-ASP/FITC-PTX in solution and HS-ASP/FITC-PTX NPs after 4 h treatment. The cells in control group do not show any green fluorescence. Only a negligible fluorescence signal is observed in cells treating with the solution containing HS-ASP/FITC-PTX. By sharp contrast, the HS-ASP/FITC-PTX NP-treated cells shows a strong green fluorescence signal. Furthermore, quantitative analyses (Fig. 7B) also confirm that the cellular uptake of drugs is enhanced by 2.67-fold upon treating with mPEG–PCL NPs in comparision to that treated with HS-ASP/FITC-PTX in solution (103.0 ± 11.9 vs. 38.6 ± 3.3, the mean fluorescence intensity). These results imply that encapsulation of drugs into mPEG–PCL NPs could enhance the delivery of drugs into cells, thereby augmenting the anticancer cytotoxicity. Diao et al.25 reported that doxorubicin-loaded PEG–PCL micelles elevated the intracellular concentration of doxorubicin through different transport process from doxorubicin solution, and enhanced drug accumulation in multidrug-resistant K562 cells, which hints at the potential of mPEG–PCL NPs to reverse P-glycoprotein-mediated multidrug resistance. Zastre J. et al.26 reported the MePEG-b-PCL diblock copolymers increased the cellular accumulation of R-123 into caco-2 cells through several mechanisms, including the “depot” effect for free unimer partitioning into the membrane and modulate P-gp, the endocytic pathway, and the fluidization of membranes mediated by free amphiphilic unimer resulting in enhanced permeability via enhanced transcellular and paracellular diffusion. These studies may give the reason for our results that encapsulation of drugs into mPEG–PCL NPs enhanced the delivery of drugs into cells. Hence, our findings indicate that drug cocktails can be more efficiently deliver in to cells through mPEG–PCL NPs and possess exert synergistic effect relative to free drugs.
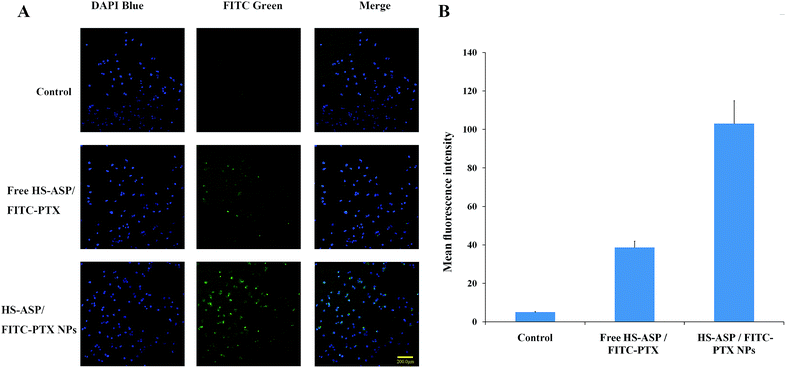 |
| | Fig. 7 Cellular uptake of different formulations containing HS-ASP and FITC-PTX against LL/2 cells. (A) The microscopic fluorescence images of LL/2 cells incubate with blank mPEG–PCL NPs, free HS-ASP and FITC-PTX solution and dual drug loaded NPs for 4 h at 37 °C. The nuclei are stained blue with DAPI and the cellular distribution of FITC-PTX is shown as green fluorescence in the cytosol. (B) The relative fluorescence intensity measured by ArrayScan® VTI HCS reader. | |
Affect of HS-ASP/PTX on tubulin evaluated by confocal microscopy
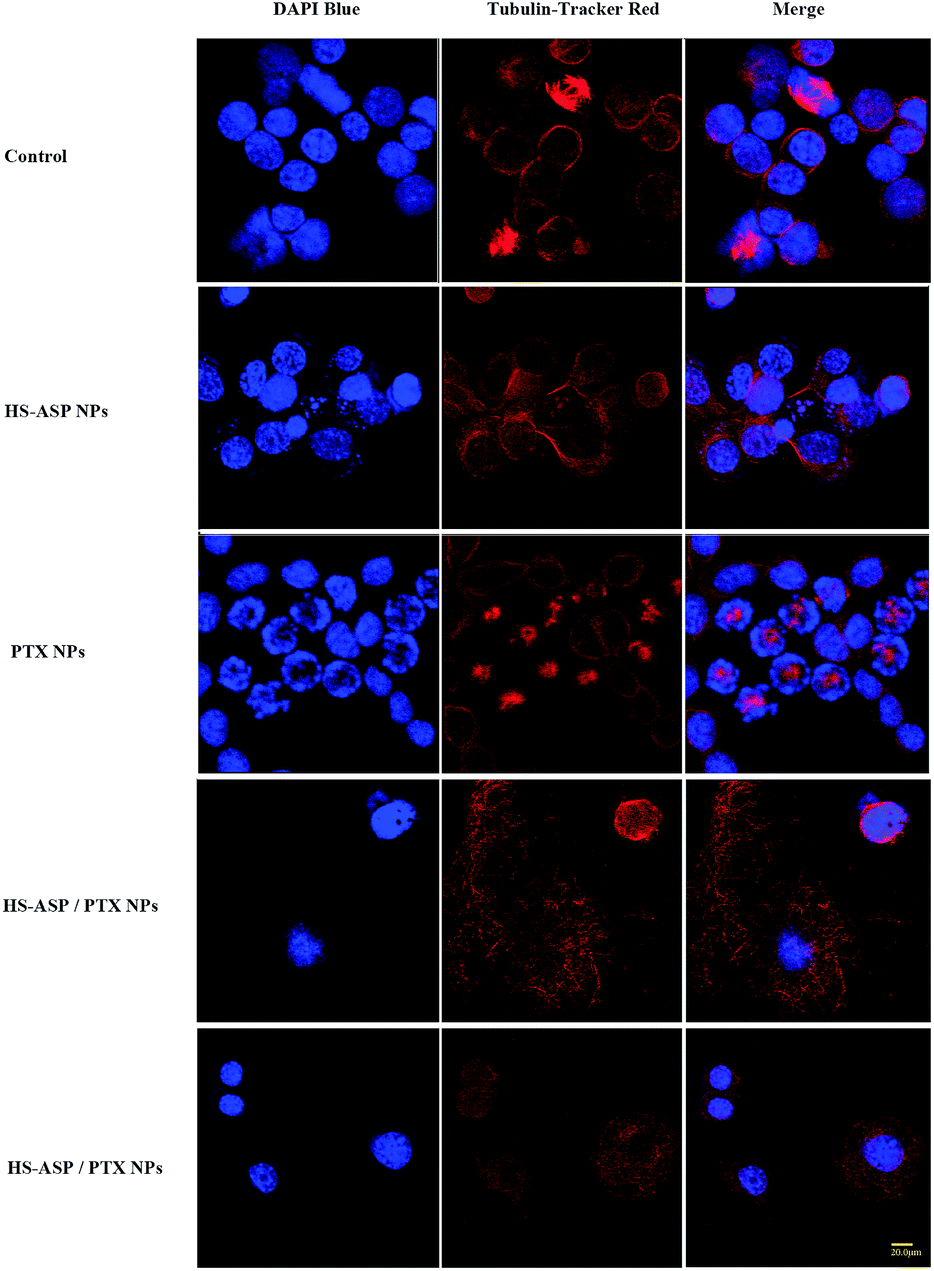
To further explore the possible mechanism for the synergistic therapeutic effect, confocal microscopy is employed to observe the morphology of tubulin (Fig. 8) by Tubulin Tracker Red in LL/2 cells treated with NPs containing HS-ASP 30 μg mL−1, PTX 3 μg mL−1, and HS-ASP 30 μg mL−1 with PTX 3 μg mL−1. Untreated cell shows an organized cytoplasmic microtubule complex (CMTC) that radiates from the centrosomal region and the mitotic spindle. The cells are generally uniform in size and have single nuclei that are round in shape (Fig. 8A). Upon treating with HS-ASP NPs, there is no obvious change of CMTC, but no mitotic spindle is found in cells (Fig. 8B). In contrast, the CMTC disappears and microtubule assembly is limited to the formation of asters radiating from the centrosomes in cells when treated with PTX NPs, and hence there are many mitotic cells with multiple asters (Fig. 8C), which leads to asymmetrical cell division and formation of cells with an abnormal size and DNA content. PTX NPs induces the shape change and multinucleation of cells. Furthermore, many of these cells displays the morphological characteristics of apoptosis: cell shrinkage, vacuolated cytoplasm, and regions of intense chromatin staining around the nuclear periphery. It can be concluded that, when the cells are treated with HS-ASP/PTX NPs (Fig. 8D and E), most of them are degraded, of which the CMTC is completely lost and replaced by bundles of free microtubules. Cell polarity is lost through the appearance of undulating membranes all around the cell periphery. The cytoplasm is accumulated in a central dome around the nucleus. The cell periphery consisted of very thin lamellae, from which microtubules are usually excluded. A possible mechanism for this phenomenon is that abnormally excess microtubules are degraded owing to PTX treatment, and meanwhile a large number of new tubulin are simultaneously synthesized to support normal metabolism in cells. However, when concurrently treats with HS-ASP NPs, HS-ASP inducts cell cycle arrest and apoptosis, resulting in completely losing cell organized CMTC and containing only short microtubules arranged in various disorganized ways. These results show that HS-ASP has no significant effect on the CMTC but might inhibit mitosis of LL/2 cells. On the other hand, it might sensitize the cells to the tubulin disruption effect of paclitaxel.
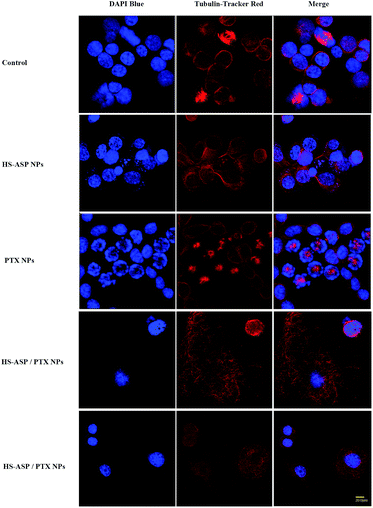 |
| | Fig. 8 Confocal laser scanning microscopy images of tubulin in LL/2 cells. The cells are cultured on 35 mm glass bottom dishes for 24 h. After culture with blank NPs, HS-ASP NPs, PTX NPs and HS-ASP/PTX NPs for 4 h, the cells are incubated with Tubulin Tracker Red for 1 h at 37 °C and subsequently examined by confocal microscopy. | |
Generally, synergistic combination of two or more drugs is a promising strategy to overcome undesirable toxicity and other side effects that limit the utility of many potential drugs by countering biological compensation, allowing reduced dosage of each agent or accessing context-specific multiple targets. Herein, LL/2 cells are applied to test the synergistic toxicity of two drugs and the enhanced toxicity after co-loaded in mPEG–PCL NPs. Dual-drug treatment shows synergistic effect stemmed from better proliferation inhibition effect over single-drug solutions observed. Much impressively, when both drugs are loaded in a single NP, this effect is further augmented, and the IC50 value is also reduced much more. HS-ASP is able to play the multiple roles to change the cellular metabolism and pH homeostasis through releasing the H2S.9,27 These contributes to sensitize the cells to PTX treatment. Moreover, HS-ASP is a low toxic derivative of aspirin that is one of the most widely used chemotherapeutic drugs in the clinic.19 As a result, the mPEG–PCL NPs co-loaded with PTX and HS-ASP posed by different action mechanism are expected to have good synergism effect against a wide range of cancer cell lines while showing low toxicity to normal cells.
Conclusions
In summary, we developed a novel mPEG–PCL nanocarrier system for simultaneous co-delivery of HS-ASP and PTX. The favourable size distribution, high drug encapsulation capacity, simultaneous releasing profile of mPEG–PCL NPs demonstrated its great potential for cocktail administration of combination drugs via intravenous injection for anticancer treatments. HS-ASP and PTX co-loaded mPEG–PCL NPs could be feasibly internalized by LL/2 tumour cells, and showed significant synergistic effect in suppression of cell growth by disrupting the normal microtubule network, inducing the cell cycle arrest and promoting the apoptotic response. The present studies indicates that the co-delivery system provides a promising platform for the cocktail treatment of non-small cell lung cancer, and possible of other types of cancer as well. Further studies are required to investigate the anticancer effect in vivo, the optimal doses of both anticancer drugs with maximal anticancer efficacy but fewest side effects, and the application of this strategy to treat other different tumours.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (81402500), China Postdoctoral Science Foundation Funded Project (2014M560720), National Key Research and Development Plan (2016YFC0303700), Youth Science and Technology Foundation of Science and Technology Department of Sichuan Province (2016JQ0031) and Foundation of Science and Technology Department of Sichuan Province (2015SZ0182). The authors report no conflicts of interest in this work.
Notes and references
- C. Jiang, H. Wang, X. Zhang, Z. Sun, F. Wang, J. Cheng, H. Xie, B. Yu and L. Zhou, Int. J. Pharm., 2014, 475, 60–68 CrossRef CAS PubMed.
- X.-Y. Ke, V. W. L. Ng, S.-J. Gao, Y. W. Tong, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2014, 35, 1096–1108 CrossRef CAS PubMed.
- S. Lv, Z. Tang, M. Li, J. Lin, W. Song, H. Liu, Y. Huang, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 6118–6129 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, Y. Wu, Y.-l. Hu, K. Nan, G. Nie and H. Chen, Biomaterials, 2011, 32, 8281–8290 CrossRef CAS PubMed.
- M. Chattopadhyay, R. Kodela, N. Nath, A. Barsegian, D. Boring and K. Kashfi, Biochem. Pharmacol., 2012, 83, 723–732 CrossRef CAS PubMed.
- M. Chattopadhyay, R. Kodela, N. Nath, C. R. Street, C. A. Velázquez-Martínez, D. Boring and K. Kashfi, Biochem. Pharmacol., 2012, 83, 733–740 CrossRef CAS PubMed.
- X. Sun, Z. Pang, H. Ye, B. Qiu, L. Guo, J. Li, J. Ren, Y. Qian, Q. Zhang and J. Chen, Biomaterials, 2012, 33, 916–924 CAS.
- Y. H. Yu, E. Kim, D. E. Park, G. Shim, S. Lee, Y. B. Kim, C.-W. Kim and Y.-K. Oh, Eur. J. Pharm. Biopharm., 2012, 80, 268–273 CrossRef CAS PubMed.
- A. L. Lee, Y. Wang, H. Y. Cheng, S. Pervaiz and Y. Y. Yang, Biomaterials, 2009, 30, 919–927 CrossRef CAS PubMed.
- L. Cai, X. Wang, W. Wang, N. Qiu, J. Wen, X. Duan, X. Li, X. Chen, L. Yang and Z. Qian, Int. J. Nanomed., 2012, 7, 4499–4510 CAS.
- T. Fang, Y. Dong, X. Zhang, K. Xie, L. Lin and H. Wang, Int. J. Pharm., 2016, 512, 39–48 CrossRef CAS PubMed.
- H. Wang, H. Xie, J. Wang, J. Wu, X. Ma, L. Li, X. Wei, Q. Ling, P. Song and L. Zhou, Adv. Funct. Mater., 2015, 25, 4956–4965 CrossRef CAS.
- H. Wang, H. Xie, J. Wu, X. Wei, L. Zhou, X. Xu and S. Zheng, Angew. Chem., Int. Ed., 2014, 53, 11532–11537 CrossRef CAS PubMed.
- J. Wang, H. Wang, J. Li, Z. Liu, H. Xie, X. Wei, D. Lu, R. Zhuang, X. Xu and S. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 19228–19237 CAS.
- C. Gong, Y. Xie, Q. Wu, Y. Wang, S. Deng, D. Xiong, L. Liu, M. Xiang, Z. Qian and Y. Wei, Nanoscale, 2012, 4, 6004–6017 RSC.
- E. Markovsky, H. Baabur-Cohen and R. Satchi-Fainaro, J. Controlled Release, 2014, 187, 145–157 CrossRef CAS PubMed.
- D. J. Mc Carthy, M. Malhotra, A. M. O'Mahony, J. F. Cryan and C. M. O'Driscoll, Pharm. Res., 2015, 32, 1161–1185 CrossRef CAS PubMed.
- Y. Gao, M. Jiang, Y. Ma, S. Wu, W. Li, X. Yang, Y. Li, X. Jing and H. Jiang, Anticancer Drugs, 2016, 27, 77–83 CrossRef CAS PubMed.
- M. Chattopadhyay, R. Kodela, N. Nath, Y. M. Dastagirzada, C. A. Velázquez-Martínez, D. Boring and K. Kashfi, Biochem. Pharmacol., 2012, 83, 715–722 CrossRef CAS PubMed.
- T.-C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS PubMed.
- Y. Gao, L. Chen, W. Gu, Y. Xi, L. Lin and Y. Li, Mol. Pharm., 2008, 5, 1044–1054 CrossRef CAS PubMed.
- M. J. Santander-Ortega, N. Csaba, L. González, D. Bastos-González, J. L. Ortega-Vinuesa and M. J. Alonso, Colloid Polym. Sci., 2010, 288, 141–150 CAS.
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS PubMed.
- Z. Li, Y. Zhao, D. Zhang, S. Zhuang and Y. Yamaguchi, Sens. Actuators, B, 2016, 230, 779–784 CrossRef CAS.
- Y. Diao, H. Li, Y. Fu, M. Han, Y. Hu, H. Jiang, Y. Tsutsumi, Q. Wei, D. Chen and J. Gao, Int. J. Nanomed., 2011, 6, 1955–1962 CAS.
- J. Zastre, J. Jackson, M. Bajwa, R. Liggins, F. Iqbal and H. Burt, Eur. J. Pharm. Biopharm., 2002, 54, 299–309 CrossRef CAS PubMed.
- Q. Xu, Y. Yao, Z. Ma and Z. Xia, Sci. Adv. Mater., 2012, 4, 888–892 CrossRef CAS.
Footnote |
| † The authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ad
*ad