DOI:
10.1039/C6RA28129J
(Paper)
RSC Adv., 2017,
7, 16341-16347
Preparation and dispersity of carbon nanospheres by carbonizing polyacrylonitrile microspheres
Received
13th December 2016
, Accepted 2nd March 2017
First published on 15th March 2017
Abstract
Polyacrylonitrile microspheres of about 240 nm were synthesized and used as a precursor for preparing carbon nanospheres (CNs) by oxidation and sequential carbonization. The relationship between preparation conditions and sphere properties (dispersity and yield) were investigated. For oxidation, a slow heating rate resulted in good dispersity and high final temperature led to high yield of carbon. For carbonization, a slow heating rate and a fast flow rate of protection gas were good for the high dispersity of CNs. Under optimized conditions of 0.1 °C min−1 to 300 °C for oxidation and 3 °C min−1 to 1000 °C for carbonization under N2 protection of 1.8 L min−1, CNs with 180 nm diameter, 4.51% nitrogen content, high purity, and good dispersity were fabricated. Moreover, after being further treated at 2800 °C, graphitized CNs with high crystal structures were obtained.
1. Introduction
Carbon materials have been widely used in various practical fields;1,2 moreover, carbon spheres have attracted significant attention in catalysis, adsorption, drug delivery, and energy conversion due to their unique sphere structure, high mechanical stability, and good surface permeability and accessibility.3–7 They have been prepared by various routes such as arc discharge, laser ablation, chemical vapor deposition, carbonization of polymers, etc.8–10 Among these methods, the carbonization of polymers has received significant attention because polymers made of carbon can have a unique π-conjugated system and high carbon content. Deshmukh et al.10 have reviewed carbonization of polymers as a carbonization route to prepare carbon spheres. A number of polymers including poly(divinylbenzene) colloidal particles, the copolymer of vinylidene chloride and acrylonitrile, and iron-doped polypyrrole nanoparticles have been reviewed as carbonization precursors. Bazargan et al.11 have, in particular, reviewed the carbonization of polymers to fabricate carbon spheres under high temperature and high pressure conditions. Many polymers such as polyethylene, polyvinylchloride, and polypropylene have been cited in the review.
For the abovementioned method of carbonizing polymers, possibilities of various polymers fabricated from carbon are endless. For other methods, apart from spherical products, some other by-products with different morphologies are often obtained, and the diameters of carbon spheres are not usually uniform. If polymers are first prepared as spheres and then carbonized, the products may have the advantages of good sphere size uniformity and almost no by-products because of the high equality and conversion for polymerization. Moreover, nitrogen-doped carbon materials have many potential uses such as practical environmental applications12 and efficient electrode material for supercapacitors13 and catalytic materials.14 PAN is a type of organic polymer material with high nitrogen content and the content of nitrogen can be controlled by the carbonization temperature; therefore, PAN can be used as a precursor for carbon materials with nitrogen groups. As a good carbon source, polyacrylonitrile (PAN) has long been used as a precursor for carbon fibers and it has also been polymerized as spheres and then carbonized for carbon nanospheres (CNs) of high purity and good uniformity.15 After further activation, the porous CNs derived from PAN-based CNs were found to have high-performance as supercapacitors because of their porous carbon structure and presence of a nitrogen group.7
However, because PAN has a melting point of 317 °C and decomposes when heated, deformation of sphere structures and low carbon yield will be inevitable if PAN spheres are directly subjected to carbonization. Furthermore, due to the small sizes of PAN nanospheres, they may aggregate during heat-treatment, making it a challenge to prepare CNs by the carbonization of PAN spheres. Yang et al.15 developed a method for coating PAN nanospheres with titanium phosphate, which acts as a protector to separate PAN nanospheres, thus protecting them from aggregation. Yao et al.16 reported a homogenous thermal stabilization method to prepare thermally stabilized nanospheres of PAN using an ionic liquid solution. They found that the extent of aggregation of the prepared spheres was closely related to the stabilization conditions.
In this study, PAN spheres were polymerized and carbonized to prepare CNs. Before carbonization, stabilization by oxidation in air was carried out to avoid the deformation of spheres and increase the carbon yield. The relationships between the preparation conditions and sphere properties (dispersity and yield) were mainly investigated. The preferred oxidation and carbonization conditions were proposed and CNs with good dispersity were prepared. The morphology and structure of CNs were also characterized.
2. Experimental
Materials and preparation processes
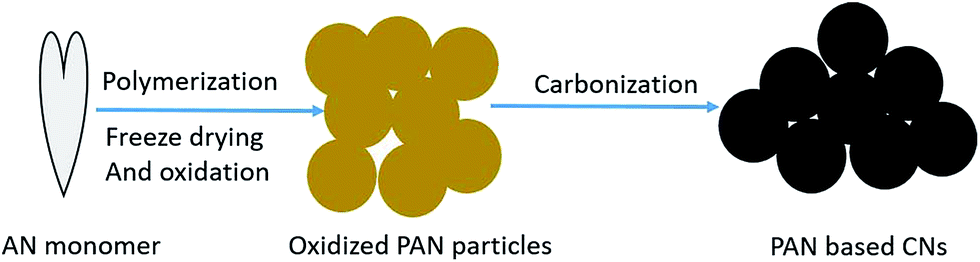
Acrylonitrile (AN) as the monomer and potassium persulfate (KPS) as the initiator were provided by China National Pharmaceutical Group Corporation. The preparation processes of CNs are shown in Fig. 1. PAN microspheres were prepared by the soapless emulsion polymerization of 30 mL AN and 30 mg KPS in 300 mL deionized water. The reaction was held at 65 °C for 6 h under the protection of N2 with intensive stirring. The PAN latex was then freeze-dried and stabilized in air under certain conditions of temperature and heating rate. Finally, the oxidized PAN spheres were subjected to carbonization at 1000 °C for 1 h under a fixed heating rate and N2 flow rate.
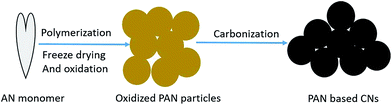 |
| | Fig. 1 Preparation processes for CNs. | |
Characterization
The morphologies of the spheres were investigated via FEI/Philips XL30 ESEM FEG field emission scanning electron microscopy (SEM). Transmission electron microscopy (TEM) images were obtained via a Hitachi H-8100 TEM system with an accelerating voltage of 200 kV. The size distribution and dispersity of the spheres were characterized using a laser diffraction particle size analyzer, the Mastersizer 2000, from Malvern Instruments Ltd., using water as the dispersant. The thermal behavior of PAN spheres was studied by thermogravimetric analysis (TGA, PyrisIand) and differential scanning calorimetry (DSC, using the DSC 8000 from PerkinElmer Company). X-ray diffraction (XRD) patterns of the samples were obtained in the 2-theta range of 10–70° using a Bruker D8 ADVANCE diffractometer with Cu Kα radiation. Infrared spectra were acquired using a VERTEX 70 Fourier transform infrared (FTIR) spectrometer (Bruker). Elemental analysis was conducted using the EA2400-II organic element analyzer. The C, H, and N contents were analyzed and the O element content was calculated.
3. Results and discussion
Morphology and thermal behavior of PAN microspheres
The morphology of the prepared PAN microspheres was characterized by SEM and is shown in Fig. 2. It can be seen that the products are all perfect microspheres with smooth surfaces and they have almost the same diameter, ranging from 230 to 250 nm. The microspheres are also well dispersed, with no aggregation i.e. PAN microsphere structures with good dispersity and uniform size can be obtained by the polymerization of AN in water.
 |
| | Fig. 2 SEM images of PAN microspheres. | |
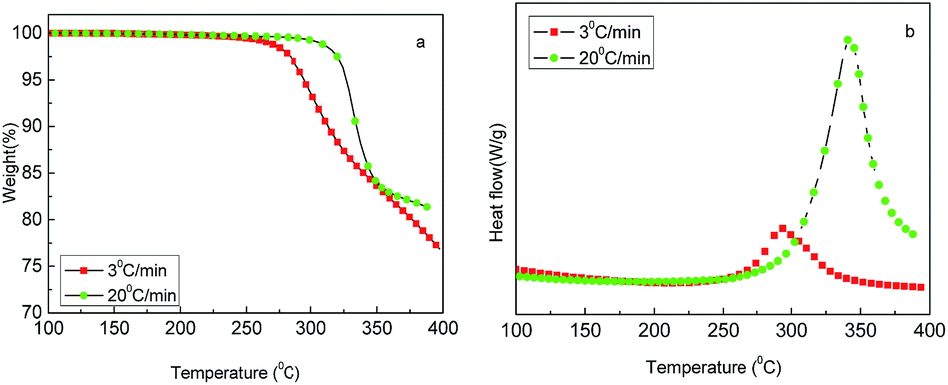
The thermal behavior of freeze-dried PAN powders was investigated using TGA-DSC in the temperature range of 100–400 °C under air atmosphere with the heating rates of 3 and 20 °C min−1. As shown in Fig. 3(a), an obvious weight loss occurred at 250 °C. It can be seen that the decomposition of PAN happened early, but slowly at 3 °C min−1, and later, but fast at 20 °C min−1. Fig. 3(b) shows DSC curves with a distinct exothermic peak in the temperature range of 250–400 °C, which is late but strong at 20 °C min−1 and early but relatively weak at 3 °C min−1; moreover, the weight loss varied according to the heating rate. In theory, the weight loss should be less at a low heating rate, whereas in this experiment, the result was contrary; the reason may be that at 20 °C min−1, the time was quite short and decomposition was not completely carried out. However, according to TGA-DSC, it can be inferred that the heat treatment of PAN in the temperature range of 200–400 °C greatly affected the stabilization reaction intensity and the yield of CNs. In fact, for the preparation of carbon materials from the PAN precursor, the oxidative stabilization of PAN at temperatures between 200 and 300 °C is the most critical step of the entire process, which is well known in the field of PAN-based carbon fiber.17
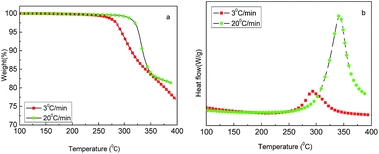 |
| | Fig. 3 TGA-DSC profiles of PAN microsphere powders: (a) TG and (b) DSC. | |
Oxidative stabilization of PAN microspheres
Because of the abovementioned importance of oxidative stabilization, the oxidation factors of heating rate and final temperature were investigated to determine their effects on the dispersity and carbon yield of PAN spheres. The dried PAN powders were subjected to stabilization in an oven with a pump to provide fresh air. As for the heating rate, the powders were heated to the same final temperature of 300 °C and held for 30 min at 0.1, 0.5, 1, and 3 °C min−1. The oxidized materials were correspondingly designated as OPAN01300, OPAN05300, OPAN1300, and OPAN5300, according to the heating rate.
Fig. 4 shows the SEM images of PAN spheres oxidized at different heating rates. It can be seen that at slow heating rates of 0.1 and 0.5 °C min−1 (Fig. 4(a) and (b)), the sphere structure was almost entirely retained after oxidation, whereas at fast heating rates of 1 and 3 °C min−1 (Fig. 4(c) and (d)), some large particles without spherical morphology were found, which may have been caused by the melting of the spheres. Moreover, aggregation occurred more easily at high heating rates, as seen from the images. Therefore, it can be regarded that a slow heating rate of oxidation is better for maintaining the sphere structure and preventing the melting of the spheres.
 |
| | Fig. 4 SEM images of PAN spheres oxidized at different heating rates: (a) 0.1, (b) 0.5, (c) 1, and (d) 3 °C min−1. | |
Since the morphology of the oxidized PAN particles should be spherical and any melting or aggregation may result in an increase in the particle size, a laser diffraction particle size analyzer was used to investigate the size distribution and then to estimate the dispersity of the spheres. Fig. 5 shows the particle size distribution of the oxidized spheres at different heating rates, and Table 1 presents the statistic diameters of different types. It can be seen from Fig. 5 that all the products have large distributions, up to 10 μm, indicating large melted particles or the existence of aggregation. In fact, even nanoparticles with good dispersity may also be found to have large size distribution due to the nanometer level sizes, which easily results in aggregation in the characterization process using the laser diffraction particle size analyzer (this is closely related to the dispersant used). However, it can still be seen that the materials with slow heating rate have greater distributions of small diameters. For OPAN01300 and OPAN05300, a distribution peak below 1 μm was found, and these two materials have obviously smaller diameters of D[3,2] and D(0.1) as compared to those of OPAN1300 and OPAN3300, which illustrates that a slow heating rate is better for high dispersity.
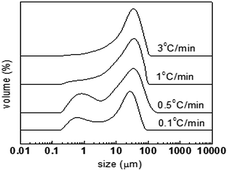 |
| | Fig. 5 Particle size distribution of PAN spheres after oxidation at different heating rates. | |
Table 1 Particle size diameters of PAN spheres after oxidation at different heating rates
| Samples |
D[3,2] (μm) |
D(0.1) (μm) |
D(0.5) (μm) |
D(0.9) (μm) |
| OPAN01300 |
2.27 |
0.64 |
15.39 |
45.54 |
| OPAN05300 |
2.46 |
0.72 |
15.70 |
64.04 |
| OPAN1300 |
5.68 |
2.78 |
22.79 |
60.89 |
| OPAN3300 |
8.72 |
4.67 |
26.97 |
63.62 |
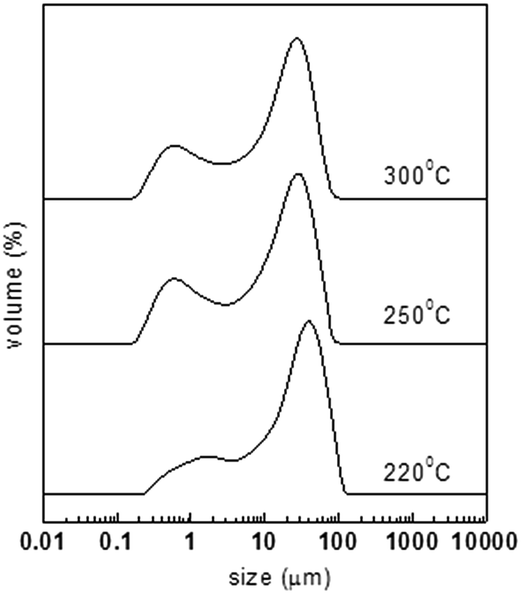
The powders were heated at the same rate of 0.1 °C min−1 to different final temperatures of 220, 250, and 300 °C. The oxidized materials were correspondingly designated as OPAN01220, OPAN01250, and OPAN01300 according to the final temperature. Fig. 6 shows the particle size distribution of the oxidized spheres at different final temperatures, and Table 2 presents the statistic diameters of different types. It can be seen that at the final temperature of 220 °C, the materials have more distributions of large diameter than at 250 and 330 °C. This should not be regarded as more melting or chemical cross-linking of spheres because at 220 °C, less heating was required. It should only be inferred that OPAN01220 with small sized spheres tended to physically aggregate during the characterization. From Table 2, it can be seen that the three materials have almost the same magnitude of D[3,2] and D(0.9), that is, the final temperature of oxidation had little effect on the dispersity of the spheres in the range from 220 to 300 °C.
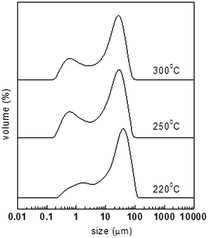 |
| | Fig. 6 Particle size distribution of PAN spheres after oxidation at different final temperatures. | |
Table 2 Particle size diameters of PAN spheres after oxidation at different final temperatures
| Samples |
D[3,2] (μm) |
D(0.1) (μm) |
D(0.5) (μm) |
D(0.9) (μm) |
| OPAN01220 |
4.40 |
1.42 |
25.86 |
68.43 |
| OPAN01250 |
2.12 |
0.60 |
14.46 |
46.77 |
| OPAN01300 |
2.27 |
0.64 |
15.39 |
45.54 |
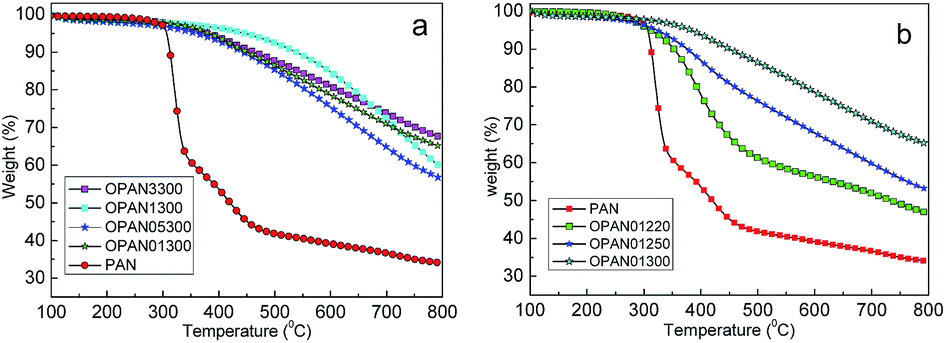
Carbon yields of different oxidized PAN spheres were approximately investigated by TGA (Fig. 7) at the heating rate of 5 °C min−1 to 800 °C under N2 protection. The yields for the different processes are shown in Table 3. It can be seen from Fig. 7(a) that there is little difference in the carbon yield for different oxidation heating rates, as observed via TGA, whereas in Fig. 7(b), the yields of different final temperatures exhibit large differences. As shown in Table 3, with the combination of oxidation and carbonization, the yields of materials oxidized at 300 °C (OPAN01300, OPAN05300, OPAN1300, and OPAN3300) were higher (46.5–56.8%) than those oxidized at 220 and 250 °C (37.8–39.8%), and the yield of PAN without oxidation was only 34.1%, which was very low.
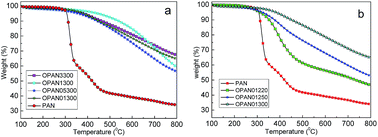 |
| | Fig. 7 TGA graphs of PAN spheres oxidized at different heating rates (a) and final temperatures (b). | |
Table 3 Yield of PAN spheres under different oxidation conditions
| Samples |
Yield of oxidation |
Yield of TG (wt%) |
Total yield (wt%) |
| PAN |
— |
34.1 |
34.1 |
| OPAN01300 |
79.5 |
65.2 |
51.8 |
| OPAN05300 |
82.0 |
56.7 |
46.5 |
| OPAN1300 |
83.7 |
59.9 |
50.13 |
| OPAN3300 |
84.1 |
67.6 |
56.8 |
| OPAN01220 |
84.9 |
46.9 |
39.8 |
| OPAN01250 |
81.3 |
53.2 |
37.8 |
Oxidation is very important for preparing PAN-based carbon materials. During oxidation, the structure of the isolated chain of PAN could transform into a type of cyclization structure via the oxidation, dehydrogenation, and cyclization reactions.13,14 The oxidation of PAN as a carbon fiber precursor has been widely investigated and is often performed in the temperature range of 200–300 °C. Sometimes higher temperatures up to 300 °C and even up to 400 °C are suggested for complete stabilization. If the temperature is too high, PAN polymers may be overheated and fuse or even burn. However, if the temperature is too low, the reactions are slow and the stabilization is incomplete, which may result in a low carbon yield.15 As for the oxidation of PAN spheres in this study, a slow heating rate can result in good dispersity and a high final temperature can result in a high yield of carbon. The oxidation conditions of OPAN01300 were chosen in the subsequent experiments.
Carbonization of oxidized PAN microspheres
Carbonization is the last step after polymerization and oxidation. It is often carried out by heat treatment at a high temperature for some time with a certain elevating procedure under inert gas protection. After carbonization, PAN polymers are often transformed into high carbon content, typically, 95%;18 as for CNs, dispersity and carbon yield are also important for this process. The materials of OPAN01300 were placed in a tubular furnace for carbonization; the heating rate and flow rate of N2 were mainly investigated. The powders were heated to 1000 °C and held for 60 min under N2 protection of 1.5 L min−1 at 3, 5, and 10 °C min−1. The carbonized materials were correspondingly designated as CPAN3-1.5, CPAN5-1.5, and CPAN10-1.5 according to the heating rate. As for the N2 flow rate, the powders were heated to 1000 °C at 5 °C min−1 and held for 60 min under N2 protection of 1.0, 1.5, and 1.8 L min−1. The carbonized materials were correspondingly designated as CPAN5-1.0, CPAN5-1.5, and CPAN5-1.8 according to the flow rate.
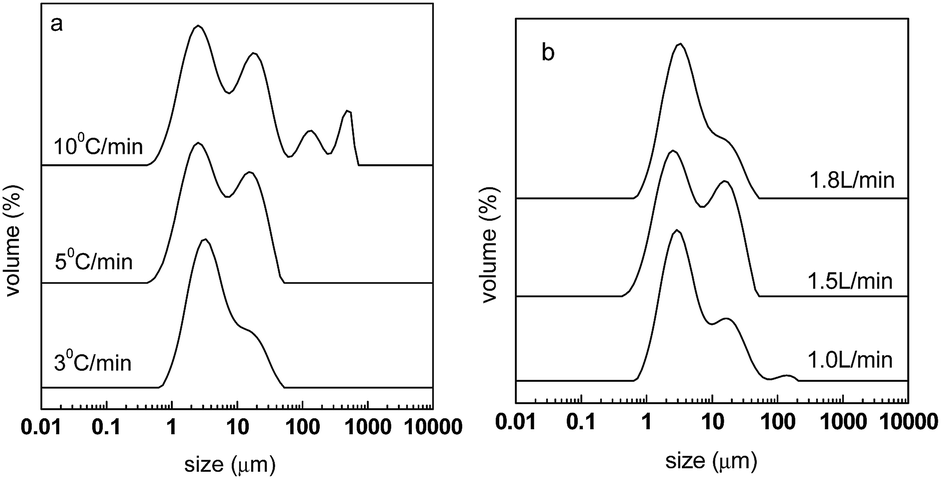
The particle size distribution and diameters of carbon spheres after carbonization are shown in Fig. 8 and Table 4, respectively. It can be seen from Fig. 8(a) that for the heating rates of 5 and 10 °C min−1, more distributions of large sizes were found, which correspond to large diameter sizes of D[3,2], D(0.9), as shown in Table 4. It can be regarded that a fast heating rate is not good for the dispersity of the spheres. Fig. 8(b) shows that for a N2 flow rate of 1.8 L min−1, less distributions of large sizes are found. This is consistent with Table 4, where the materials of CPAN5-1.8 have smaller diameters of D(0.5) and D(0.9). Therefore, it can be inferred that a large flow rate of N2 is good for high dispersity, and the reason may be that this could help the emission of decomposed gas and prevent it from participating in cross-linking reactions.
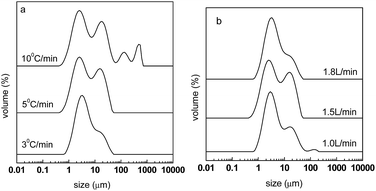 |
| | Fig. 8 Particle size distribution of carbon spheres after carbonization at different heating rates (a) and N2 flow rates (b). | |
Table 4 Particle size diameters of carbon spheres carbonized under different conditions
| Samples |
D[3,2] (μm) |
D(0.1) (μm) |
D(0.5) (μm) |
D(0.9) (μm) |
| CPAN3-1.5 |
2.66 |
1.19 |
3.43 |
12.91 |
| CPAN5-1.5 |
3.43 |
1.48 |
5.03 |
16.63 |
| CPAN10-1.5 |
4.07 |
1.59 |
7.66 |
31.70 |
| CPAN5-1.0 |
3.62 |
1.72 |
4.32 |
14.41 |
| CPAN5-1.8 |
3.62 |
1.77 |
4.38 |
11.38 |
The carbon yields under different carbonization conditions are shown in Table 5. It can be seen that the yields are not very different. The largest yield is 44.3% for CPAN3-1.5 and the smallest yield is 37.1% for CPAN5-1.8. It can be inferred that a fast heating rate and slow N2 flow rate are good for high carbon yield.
Table 5 Yield of carbon spheres carbonized under different conditions
| Samples |
CPAN3-1.5 |
CPAN5-1.5 |
CPAN10-1.5 |
CPAN5-1.0 |
CPAN5-1.8 |
| Yield of carbonization (%) |
44.3 |
45.4 |
46.7 |
44.6 |
37.1 |
Preparation and structure of CNs under preferred conditions
According to the abovementioned research, slow heating rate and high final temperature for oxidation and fast flow rate of protection gas for carbonization are good for high dispersity of PAN-based CNs. Therefore, the preferred conditions were controlled at 0.1 °C min−1 to 300 °C for oxidation and 3 °C min−1 to 1000 °C for carbonization under N2 flow of 1.8 L min−1.
The morphologies of the prepared CNs as direct powders and dispersed in ethanol were characterized by SEM, as shown in Fig. 9. It can be seen that the sphere morphology of PAN was well retained and the spheres had a good dispersity. After carbonization, the diameter of the spheres decreased from about 240 nm to about 180 nm.
 |
| | Fig. 9 SEM images of the CNs prepared under optimized conditions: (a) direct observation and (b) dispersed in ethanol. | |
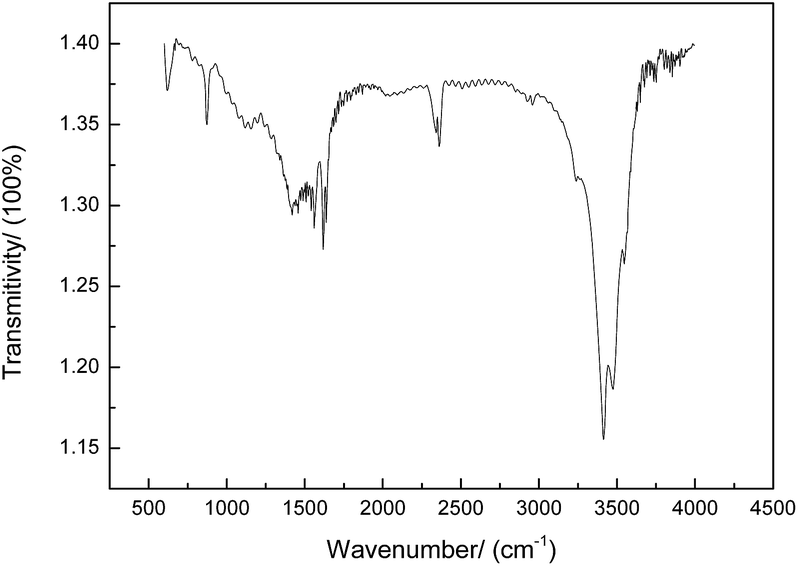
The FTIR spectra of the prepared CNs are shown in Fig. 10. The 3300–3500 cm−1 region of the infrared spectrum is sensitive to –OH, –NH2, and –NH vibrations. The absorption peak at 2240 cm−1 belongs to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond stretching vibration of PAN.19 The bands at 600–1680 cm−1 are attributed to the C
N bond stretching vibration of PAN.19 The bands at 600–1680 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond stretching of the CNs, demonstrating the aromatization during the carbonization process. Moreover, the peak at 1460 cm−1 is assigned to the saturated alkyl groups such as –CH3 and –CH2 and the peak at 890–910 cm−1 is due to CH2. The results of elemental analysis of various states of PAN are shown in Table 6, and it can be seen that the polymerized PAN only contains three elements, C, H, and N, with the content in good agreement with that in theory. With subsequent oxidation, oxygen is introduced with the content of about 22.95%. After carbonization, the carbon content tends to greatly increase, whereas the contents of other elements tend to largely decrease. Moreover, it was observed that the higher the temperature, the more obvious the trend. The carbon content is about 81.85% at 1000 °C and 95.41% at 1500 °C. The nitrogen content is about 4.51% at 1000 °C and 0.05% at 1500 °C. A cyclization reaction occurs when PAN is oxidized, and nitrogen plays a very important role in cyclization.20
C bond stretching of the CNs, demonstrating the aromatization during the carbonization process. Moreover, the peak at 1460 cm−1 is assigned to the saturated alkyl groups such as –CH3 and –CH2 and the peak at 890–910 cm−1 is due to CH2. The results of elemental analysis of various states of PAN are shown in Table 6, and it can be seen that the polymerized PAN only contains three elements, C, H, and N, with the content in good agreement with that in theory. With subsequent oxidation, oxygen is introduced with the content of about 22.95%. After carbonization, the carbon content tends to greatly increase, whereas the contents of other elements tend to largely decrease. Moreover, it was observed that the higher the temperature, the more obvious the trend. The carbon content is about 81.85% at 1000 °C and 95.41% at 1500 °C. The nitrogen content is about 4.51% at 1000 °C and 0.05% at 1500 °C. A cyclization reaction occurs when PAN is oxidized, and nitrogen plays a very important role in cyclization.20
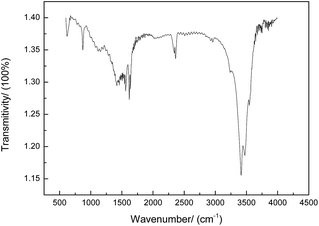 |
| | Fig. 10 FTIR spectra of CNs under preferred conditions. | |
Table 6 Elemental content for different processes
| Element |
PAN |
OPAN01300 |
CPAN3-1.8 |
CPAN(1500 °C) |
| C (wt%) |
67.42 |
52.62 |
81.85 |
95.41 |
| H (wt%) |
5.80 |
2.76 |
1.41 |
0.18 |
| N (wt%) |
26.78 |
21.67 |
4.51 |
0.05 |
| O (calculated, wt%) |
— |
22.95 |
12.23 |
4.36 |
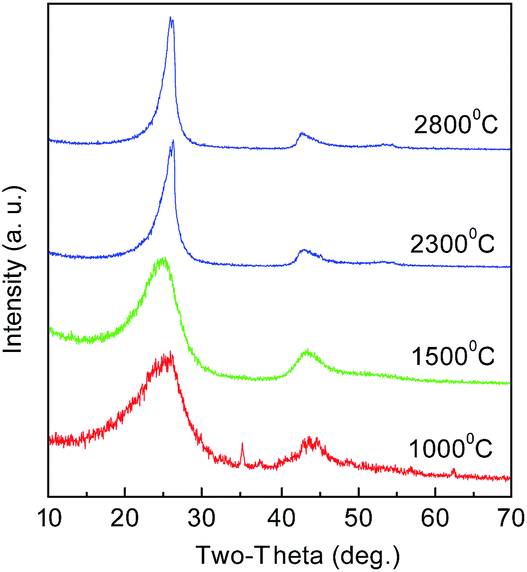
The CNs were also treated at high temperatures of 1500, 2300, and 2800 °C. The structures were analyzed by XRD, as shown in Fig. 11. It can be seen that all the materials have the characteristic diffraction profile of carbon at 2θ of about 260 for the (002) lattice plane and at about 430 for (100).21 The peak is broad at low temperature, suggesting a low graphitization degree and the possible presence of amorphous carbon. With the increase in temperature, the (002) peak becomes distinctly sharp and intense, indicating that PAN polymers tend to transform into the graphite crystal structure at high temperature. This trend seems more obvious at 2300 °C, indicating a decreased interlayer spacing of d002 and an increased stacking height of Lc. The TEM images of the products treated at 1000 and 2800 °C are shown in Fig. 12. When graphitized at 2800 °C, many distinct well-defined regions linking the graphitic layered domains with different orientations could be seen, indicating that many highly graphitized crystal structures were developed.
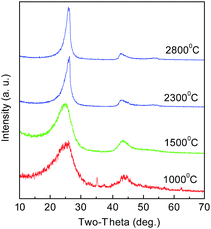 |
| | Fig. 11 XRD spectra of CNs after heat treatment at different temperatures. | |
 |
| | Fig. 12 TEM images of CNs: (a) carbonized at 1000 °C, (b), (c), and (d) graphitized at 2800 °C. | |
4. Conclusions
In this study, PAN microspheres of about 240 nm were polymerized and used as precursors for CNs preparation. The processes of oxidation and carbonization were mainly studied to determine their effects on the dispersity and carbon yield of the CNs prepared. For oxidation, it was found that a slow heating rate can result in a good dispersity and a high final temperature can result in a high yield of carbon. For carbonization, a slow heating rate and a fast flow rate of N2 were good for high dispersity of CNs. Under optimized conditions of 0.1 °C min−1 to 300 °C for oxidation and 3 °C min−1 to 1000 °C for carbonization under the N2 flow rate of 1.8 L min−1, CNs with diameter of 180 nm, high purity, and good dispersity were fabricated. After being further treated at 2800 °C, CNs with highly graphitized crystal structures were obtained.
Acknowledgements
We gratefully acknowledge the support of the National Science Foundation of China (51102169, 51272157, 51102168), Shanghai Nature Science Foundation (16ZR1423400), and Hujiang Foundation of China (B14006).
References
- Q. Liu, G. Qiu, Z. Zhou, J. Li, G. L. Amy, J. Xie and J. Y. Lee, Environ. Sci. Technol., 2016, 50(19), 10596–10605 CrossRef CAS PubMed.
- Y. Liu, J. Xie, C. N. Ong, C. D. Vecitis and Z. Zhou, Opt. Commun., 2015, 1, 769–778 CAS.
- J. L. Li, Z. L. Li, J. H. Tong, C. G. Xia and F. W. Li, RSC Adv., 2015, 5, 70010–70016 RSC.
- Z. G. Jia, Z. Y. Li, S. B. Li, Y. H. Li and R. S. Zhu, J. Mol. Liq., 2016, 220, 56–62 CrossRef CAS.
- T. Y. Wang, P. Zhao, Q. F. Zhao, B. Wang and S. L. Wang, Drug Delivery, 2016, 23, 420–428 CrossRef CAS PubMed.
- S. C. Ray, Z. N. Tetana, R. Erasmus, A. Mathur and N. J. Coville, Int. J. Energy Res., 2014, 38, 444–451 CrossRef CAS.
- L. Yao, G. Z. Yang and P. Han, RSC Adv., 2016, 6, 43748–43754 RSC.
- Y. J. Qi, M. Zhang, L. Qi and Y. Qi, RSC Adv., 2016, 6, 20814–20823 RSC.
- A. Boudjemaa, T. Mokrani, K. Bachari and N. J. Coville, Mater. Sci. Semicond. Process., 2015, 30, 456–461 CrossRef CAS.
- A. A. Deshmukh, S. D. Mhlanga and N. J. Coville, Food Mater. Sci. Eng., 2010, 70, 1–28 CrossRef.
- A. Bazargan, Y. Yan, C. W. Hui and G. McKay, Ind. Eng. Chem. Res., 2013, 52, 12689–12702 CrossRef CAS.
- Y. Liu, L. Yu, C. N. Ong and J. Xie, Nano Res., 2016, 9, 1983–1993 CrossRef CAS.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- L. C. Yang, Y. Shi, Q. S. Gao, B. Wang, Y. P. Wu and Y. Tang, Carbon, 2008, 46, 1816–1818 CrossRef CAS.
- Y. Yao, T. Zhou, T. Yang, R. Xiang and Y. Wu, Carbon, 2013, 58, 249–251 CrossRef CAS.
- A. Gupta, New aspects in the oxidative stabilization of polyacrylonitrile – based carbon fibers, The Pennsylvania State University, 1996 Search PubMed.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. T. Roth, Nature, 2007, 446, 60–63 CrossRef CAS PubMed.
- G. A. Cooper and R. M. Mayer, J. Mater. Sci., 1971, 6, 60–67 CrossRef CAS.
- Y. Z. Jin, Y. J. Kim, C. Gao, Y. Q. Zhu, A. Huczko, M. Endo and H. W. Kroto, Carbon, 2006, 44, 724–729 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yu Binbin,
Song Shen,
Tang Zhihong,
Yu Dengguang and
Yang Junhe*
*,
Yu Binbin,
Song Shen,
Tang Zhihong,
Yu Dengguang and
Yang Junhe*