
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceREBaCo2O5+δ (RE = Pr, Nd, and Gd) as promising oxygen electrodes for intermediate-temperature solid oxide electrolysis cells
Shaoming Liu,
Wenqiang Zhang,
Yifeng Li and
Bo Yu *
*
Institute of Nuclear and New Energy Technology, Collaborative Innovation Center of Advanced Nuclear Energy Technology, Tsinghua University, Beijing 100084, China. E-mail: cassy_yu@tsinghua.edu.cn; Fax: +86-10-6277-1740; Tel: +86-10-8019-4039
First published on 14th March 2017
Abstract
Double-perovskite REBaCo2O5+δ (RE = Pr, Nd, and Gd) oxides were synthesized and evaluated as oxygen electrodes for intermediate-temperature solid oxide electrolysis cells (IT-SOECs). The crystal structures, thermal stability properties, electrical conductivities, and electrochemical performances of the REBaCo2O5+δ materials were investigated systematically. Among the three oxides, PrBaCo2O5+δ (PBCO) has the highest electrical conductivity of 645 S cm−1 at 600 °C. The electrical conductivity decreases as the ionic radius of the RE decreases, which is related to the increase in oxygen vacancy concentration. NdBaCo2O5+δ (NBCO) has the lowest polarization resistance of 0.95 Ω cm2 among the three oxides at 650 °C, which is mainly because NBCO has a better balance between thermal expansion compatibility and conductivity than PBCO and GdBaCo2O5+δ (GBCO). For electrochemical performance in SOECs, NBCO has the smallest current/voltage loss; at a voltage of 1.2 V, the power density of NBCO is 1.58 W cm−2 at 800 °C. Even at 700 °C, a maximum power density of 0.857 W cm−2 can still be achieved by NBCO. These results suggest that NBCO is a novel and promising candidate oxygen electrode material for IT-SOECs.
1 Introduction
A solid oxide electrolysis cell (SOEC) is a highly efficient energy converter that can generate hydrogen fuel by the electrolysis of steam using electricity and heat. If coupled to nuclear or renewable energy (such as solar energy, geothermal energy and wind power), it can achieve a high overall electrical-to-hydrogen energy conversion efficiency with low or zero CO2 emissions.1,2 An additional advantage of the SOEC is that it can be used in the power-to-gas process, especially in transforming redundant renewable energy into hydrogen gas.3 Moreover, it can be operated in fuel cell mode, generating energy when electricity demand is high.4Elevated temperature is favorable for high-efficiency hydrogen production by SOECs.5 However, high operating temperatures (900–1000 °C) also bring many serious problems, including interfacial reactions, delamination and high-cost interconnect materials. A way to solve these problems is by lowering the operating temperature of the SOEC into the intermediate temperature (IT, 600–800 °C) range, which can suppress the solid-state reactions between the cell components and use low-cost metallic interconnecting materials, consequently increasing the reliability and durability of the SOEC materials and components.6 Although the IT range is more suitable for SOFCs, the increase in ohmic resistance from the electrolyte and polarization resistance from the electrodes becomes the main barrier as the temperature decreases. The ohmic resistance can be reduced with a thin-film electrolyte. The conventional materials for SOECs include dense YSZ electrolytes, porous Ni-YSZ hydrogen electrodes, and perovskite-based La0.6Sr0.4MnO3−δ YSZ oxygen electrodes.7 When operating in the IT range, the polarization losses of the oxygen electrodes become the major source of performance degradation in SOECs.8–10 Therefore, much research has been devoted to finding candidates to replace La0.6Sr0.4MnO3−δ YSZ oxygen electrodes. Although SOECs can be seen as the inverse of SOFCs in principle, because of the existence of a large amount of water in SOECs, delamination phenomenon is more serious.
In recent years, mixed ionic–electronic conductors (MIECs) have attracted more attention. This is because MIECs can accelerate the oxygen evolution reaction (OER) in the oxygen electrodes of the SOECs, further increasing the active area.11–13 In addition, research on MIEC oxygen electrode materials with perovskite-type structures such as La1−xSrxCo1−yFeyO3−δ (LSCF),14–16 Ba1−xSrxCo1−yFeyO3−δ (BSCF)17,18 and SrCo0.7Fe0.2Nb0.1O3−δ (SCFN),19 and K2NiF4−δ-type structures (first member of the so-called Ruddlesden–Popper series) such as La2NiO4+δ,20,21 Pr2NiO4+δ (ref. 22–24) and Nd2NiO4+δ (ref. 25 and 26) has been carried out. One of the main advantages of these materials is high OER activity and oxygen reduction reaction (ORR) activity. Recently, the double perovskite-type structure (AA′B2O5+δ) has attracted much attention due to the particularity of the structure, where alkali-earth ions and lanthanides take up the A-site sub-lattice, and oxygen vacancies are localized into layers.27–30 Actually, the ordered double-perovskite structure provides channels for the oxygen ions to move more easily,27 and the difference in the ionic radii in the A and A′ sites has been found to be the driving force for producing the desired ordered double-perovskite structure. Rare earth elements are often used to occupy the A-site ion. At the A′ site, Ba or Sr, which has a larger ionic radius compared to rare earth elements, is more suitable. At the B site, transition metals such as Co, Fe, Cu and Mn are the most commonly used, and Co-based double perovskites were found to offer the highest electrochemical performance.31 Among these different double-perovskite materials, REBaCo2O5+δ (RE = La, Pr, Nd, Sm, Gd and Y) have been investigated as promising oxygen electrode materials for IT-SOFCs because of their high electronic conductivity,32 rapid oxygen ion diffusion, surface exchange kinetics,33–35 and excellent electrochemical performance.36–38 However, research on SOECs still focuses on traditional high-temperature SOFC materials, and few studies have addressed the differences among these double perovskite-type oxides and the feasibility of their application in SOECs.
In the literature,38–40 it has been demonstrated that the best SOFC performance can be obtained by middle-sized ions such as Pr, Nd, and Gd occupying the A site, which can produce a good balance between electrochemical performance and thermal expansion coefficient (TEC). In this paper, three double-perovskite oxides, PrBaCo2O5+δ, NdBaCo2O5+δ, and GdBaCo2O5+δ, have been prepared and characterized. The effects of the RE3+ ions on the crystal structure, thermal stability properties, concentration of oxygen vacancies, electrical conductivity, and catalytic activity for the OER in SOECs are presented. The electrochemical performances of the REBaCo2O5+δ series of oxides with RE = Pr, Nd, and Gd used as oxygen electrodes for IT-SOECs were systematically investigated.
2 Experimental
2.1 Materials synthesis and cell fabrication
REBaCo2O5+δ (RE = Pr, Nd, and Gd) powders were synthesized by the EDTA–citrate method, as described by Zhou et al.41 The procedure was as follows: Pr2O3, Nd2O3, and Gd2O3 were initially dissolved separately in HNO3 (≈30%) to form the metal nitrate solutions, and precise concentrations were titrated using the EDTA method. The starting precursor was prepared with stoichiometric amounts of Pr(NO3)3/(Nd(NO3)3/Gd(NO3)3), Ba(NO3)2 and Co(NO3)2. Given amounts of EDTA and citric acid were added to the starting precursor (total metal cation![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric:EDTA acid = 1:1.5:1), and the pH values were controlled by NH3·H2O. When place in an oil bath at 90 °C, the solutions changed into transparent purple gels. After evaporation for 24 h at 120 °C in a drying oven, the gels were pre-fired for 5 h at 250 °C and calcined for 5 h at 1000 °C to obtain the desired oxide powders.
:citric:EDTA acid = 1:1.5:1), and the pH values were controlled by NH3·H2O. When place in an oil bath at 90 °C, the solutions changed into transparent purple gels. After evaporation for 24 h at 120 °C in a drying oven, the gels were pre-fired for 5 h at 250 °C and calcined for 5 h at 1000 °C to obtain the desired oxide powders.
Powders of Gd0.2Ce0.8O1.9 (GDC) oxides were synthesized using the citrate–nitrate combustion method.42 Solutions of Gd(NO3)3 and Ce(NO3)3 (1 M) were prepared, and their concentrations were titrated with EDTA. A stoichiometric amount of Gd(NO3)3 and Ce(NO3)3 were previously mixed into a solution under mechanical stirring. The necessary amount of citric acid (citric acid:total metal ions = 1.5:1) was then added to the solution. After continuous evaporation and stirring at 80 °C for 3 h, the solutions turned into gels, which were heated in a resistance furnace until spontaneous combustion occurred. Finally, the collected powders were sintered at 1000 °C for 2 h.
Some of the calcined REBaCo2O5+δ (RE = Pr, Nd, and Gd) powders were reground and pressed into bars (5 mm × 30 mm × 3 mm) with 150 MPa hydraulic pressure. After calcining for 5 h at 1150 °C, dense samples were prepared for electrical conductivity measurement. The electrochemical performances of the oxygen electrodes were measured using symmetrical cells with configurations of REBaCo2O5+δ|GDC|REBaCo2O5+δ (RE = Pr, Nd, and Gd), as shown in Fig. 2(g). Pressed pellets of GDC were calcined at 1500 °C for 2 h in air. The REBaCo2O5+δ (RE = Pr, Nd, and Gd) powders were prepared into slurries with ethyl cellulose and terpineol as the binders and coated on the surface of the GDC pellets. Finally, the symmetrical cells were calcined at 1000 °C for 2 h.
2.2 Characterization
The crystal structures of the obtained products were characterized by X-ray diffraction (XRD; BRUKER, Germany) using Cu Kα radiation with a step size of 0.02°. The scanning range was 10° to 80°. The microstructures of the powders were confirmed by scanning electron microscopy (SEM; LEO 1530).The electrical conductivities were studied using a four-probe DC method from 350 °C to 1000 °C at intervals of 50 °C under air atmosphere. The TECs were examined with a dilatometer from 150 °C to 900 °C at a heating rate of 5 °C min−1.
Electrochemical impedance spectroscopy was carried out using a Zahner IM6 electrochemical workstation over the frequency range of 100 kHz to 100 mHz. The signal voltage amplitude was 10 mV, and the operating temperature range was 650 °C to 800 °C.
The configuration of a single SOEC was Ni-YSZ|YSZ|GDC|oxygen electrode. The electrochemical measurements were performed in the temperature range of 650 °C to 800 °C. The electrochemical performances of the single-button cells were evaluated based on voltage–current (V–I) curves.
3 Results and discussion
3.1 Powder characterization
Samples of the REBaCo2O5+δ (RE = Pr, Nd, and Gd) particles were obtained from the precursors and calcined at 1000 °C for 5 h under an air atmosphere, and GDC powder was calcined at 1000 °C for 2 h. The XRD patterns of PBCO, NBCO, GBCO and GDC at room temperature are shown in Fig. 1(a) and (b). These samples clearly consist of only single phases, in agreement with the previously reported results.30,43,44 Ref. 39 and 45 indicated that the double perovskite can have one of two different crystal structures depending on the temperature and oxygen content. Chavez et al.39 found that cooling rate plays an important role in the crystal structure. The crystal structures of REBaCo2O5+δ (RE = Pr, Nd, and Gd) powders are listed in Table 1. PBCO and NBCO crystallize in a tetragonal structure with P4/mmm, while GBCO crystallizes in an orthorhombic structure with Pmmm; the change of the structure from tetragonal to orthorhombic is related to the splitting of the peak around 2θ = 22.7°. This distortion may be due to the difference in the oxygen content. Table 1 also illustrates the associated lattice parameters and unit cell volumes. The increasing trend in the lattice volumes of REBaCo2O5+δ (RE = Pr, Nd and Gd) powders follows the increasing trend in ionic radii (Pr > Nd > Gd). The chemical compatibilities between an oxygen electrode and electrolyte were also examined. The REBaCo2O5+δ oxygen electrode and GDC electrolyte were mixed with a mass ratio of 1:1 and then calcined at 1000 °C for 5 h in air atmosphere. Fig. 1(b) displays the XRD patterns of the PBCO–GDC, NBCO–GDC and GBCO–GDC mixtures after heat treatment. The XRD patterns of GDC powders are also shown in Fig. 1(b). No structural changes or new phases are indicated in these patterns, indicating that no chemical reactions occurred between REBaCo2O5+δ (RE = Pr, Nd, and Gd) and GDC. Fig. 2(a)–(c) show the SEM images of PBCO, GBCO and NBCO powders calcined at 1000 °C for 5 h under air atmosphere. The samples are seen to possess similar particle morphologies and accompany with agglomerate phenomenon, and the average particle sizes are less than 1 μm.
 | ||
| Fig. 1 XRD patterns of (a) double-perovskite REBaCo2O5+δ (RE = Pr, Nd, and Gd) and (b) REBaCo2O5+δ (RE = Pr, Nd, and Gd)–GDC mixtures sintered at 1000 °C for 5 h. | ||
| Material | Space group | A (Å) | b (Å) | c (Å) | Lattice volume (Å3) | SBET (m2 g−1) | Mean pore diameter (nm) |
|---|---|---|---|---|---|---|---|
| PBCO | P4/mmm | 3.90672 | 3.90672 | 7.65202 | 116.79 | 11.756 | 3.4 |
| NBCO | P4/mmm | 3.89406 | 3.89406 | 7.60997 | 115.4 | 9.779 | 3.39 |
| GBCO | Pmmm | 3.90899 | 3.87454 | 7.53093 | 114.06 | 7.098 | 3.41 |
| GDC | Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
5.41673 | 5.41673 | 5.41673 | 158.93 | — | — |
 | ||
| Fig. 2 SEM images of the various REBaCo2O5+δ samples: (a) PBCO, (b) GBCO and (c) NBCO. Cross-sectional images of the symmetrical cells made from (d) PBCO, (e) GBCO and (f) NBCO and (g) the single cell. | ||
The microstructures of the three-phase boundaries, which are related to the active areas of the oxygen electrode, have a great influence on the electrochemical performance. Therefore, the electrochemical performance of the oxygen electrode depends strongly on the pore size and surface area of the oxygen electrode powders. Smaller particles with the larger surface areas have more contact points between particles and electrolyte, allowing for the growth and migration of the active sites.46,47 However, oxygen electrodes consisting of smaller particles are more fragile, and delaminating phenomena become more serious at the operating temperature. Larger particles with the smaller surface areas have larger contact points, facilitating gas diffusion but limiting the formation of oxygen vacancies and oxygen ions. The surface area (SBET) and pore size distributions of REBaCo2O5+δ (RE = Pr, Nd, and Gd) samples were measured based on N2 sorption curves using the BET and BJH methods, respectively (as shown in Table 1). The analysis of experimental data reveals that surface area decreases with decreasing RE ionic radius: SBET(PBCO) > SBET(NBCO) > SBET(GBCO). Moreover, the pore size for all three samples is about 3.4 nm.
3.2 Thermal stability properties
TGA curves of the samples were measured to confirm the formation temperature of double perovskite. Fig. 3(a) shows that weight loss occurs with increasing temperature. The TGA curves consist of three parts. First, the change from room temperature to about 285 °C is mainly due to the evaporation of adsorbed water. Second, the change from 285 °C to 800 °C is attributed to the decomposition of organics. In the last part from 800 °C to 1000 °C, there is almost no weight loss on the TG curves. Therefore, to obtain the stable double-perovskite phase, the sintering temperature must be higher than 800 °C.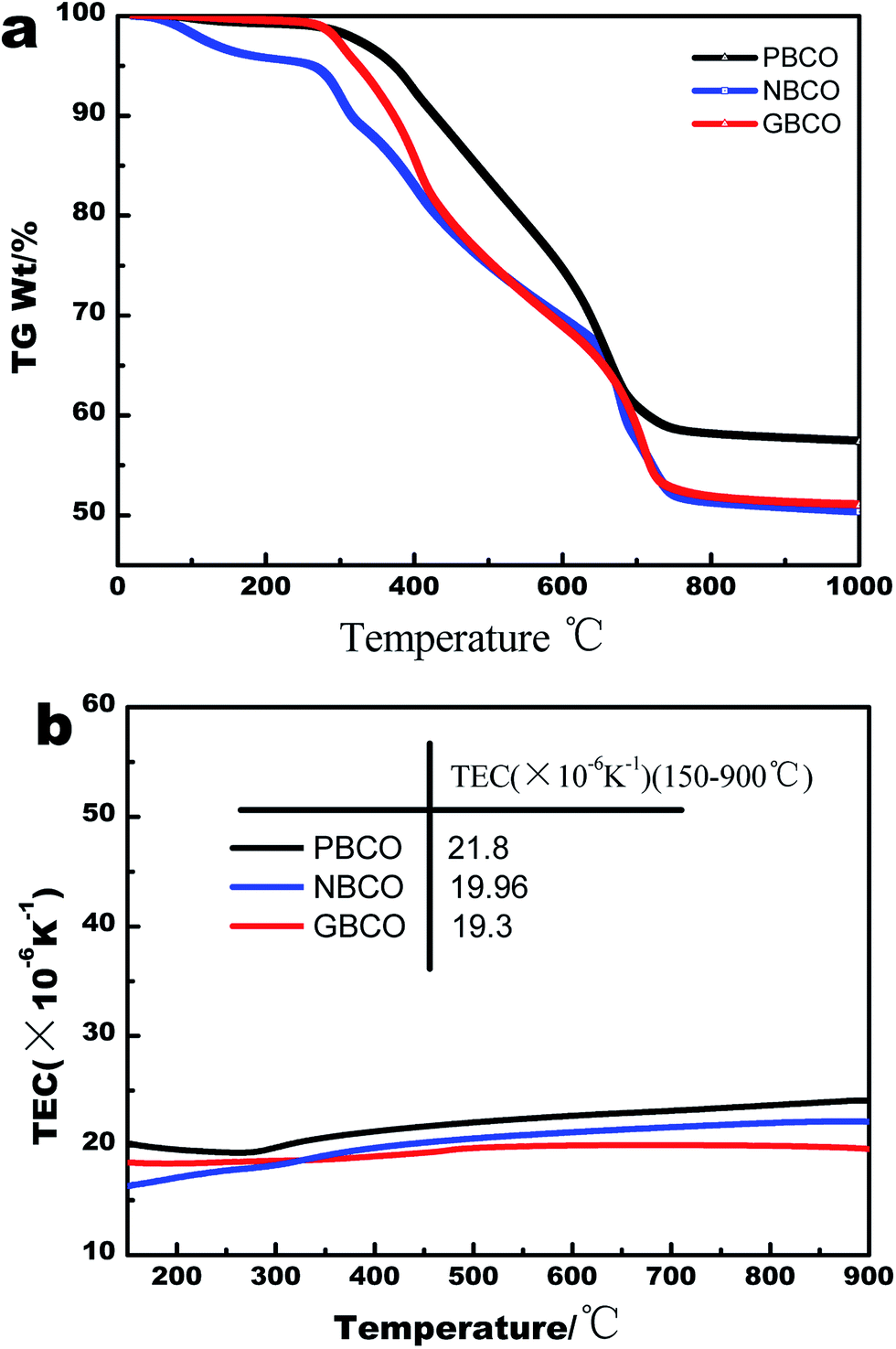 | ||
| Fig. 3 (a) TGA plots of the REBaCo2O5+δ (RE = Pr, Nd, and Gd) samples recorded in the air with a heating/cooling rate of 10 °C min−1. (b) Thermal expansion curves of PBCO, NBCO and GBCO from 150 °C to 900 °C in air with a heating rate of 5 °C min−1. | ||
The TECs were examined by a dilatometer from 150 °C to 900 °C at a heating rate of 5 °C min−1. Fig. 3(b) shows the thermal expansion curves of PBCO, NBCO, and GBCO. The left side of the Y-axis represents the TECs of PBCO, NBCO, and GBCO. Clearly, the average TECs could be acquired by fitting the thermal expansion curves with a quartic polynomial. The TECs of PBCO, NBCO, and GBCO are much higher than that of GDC, mainly because Co3+ cations often present in the form of high-spin states (HS, t42ge2g) at high temperature; these states have larger ionic radii compared to low-spin (LS, t62ge0g) or intermediate-spin (IS, t52ge1g) states.48,49 The mismatch between the oxygen electrode and electrolyte in TEC may reduce the long-term stability and electrochemical performance of single cells due to the stress formed at the boundary between the electrode material and electrolyte material. The average TECs in Fig. 3(b) decrease upon changing the RE from Pr to Gd, which is consistent with the decreasing size of the RE3+ ion.
3.3 Electrical conductivity
The electrical conductivities of the REBaCo2O5+δ materials were measured using the four-probe DC method under air atmosphere. The traditional oxygen electrode material La0.8Sr0.2MnO3−δ (LSM) was used as the reference material. Fig. 4 shows the electrical conductivities of REBaCo2O5+δ and LSM. It can be seen that the conductivities of REBaCo2O5+δ are higher than those of LSM at temperatures below 800 °C, and PBCO has a higher conductivity than NBCO and GBCO. For all samples, the electrical conductivity decreases with increasing temperature, and the conductivity decreases from Pr to Ga. While exposed to air atmosphere, the conductivities of REBaCo2O5+δ oxides decrease with increasing temperature. These results suggest metallic behavior in the oxides, which agrees with previous reports.30 When the temperature reaches a certain value, the conductivity of REBaCo2O5+δ decreases dramatically. This is caused by the evolution of lattice oxygen, which not only forms many oxygen vacancies, but also causes the reduction of Co(IV) to Co(III). The high concentration of oxygen vacancies decreases the covalence of the O–Co–O bonds and leads to a decline in the concentration of mobile charge carriers, resulting in a dramatic drop in conductivity.35,50–52 The maximum electrical conductivities of PBCO, NBCO and GBCO are 775, 361, and 328 S cm−1 at 350 °C, respectively. When the temperature reached 600 °C, the electrical conductivities of the samples reaches 645 S cm−1 (PBCO), 308 S cm−1 (NBCO) and 318 S cm−1 (GBCO). The conductivity differences in reported studies for PBCO, NBCO and GBCO are quite high.37,53–56 This is mainly due to the different synthesis methods used to make the powders. The highest reported conductivity values are 844 S cm−1 for PBCO,57 952 S cm−1 for NBCO and 512 S cm−1 for GBCO38 at 600 °C. The lowest conductivity values are 161 S cm−1 for PBCO, 192 S cm−1 for NBCO and 105 S cm−1 for GBCO58 at 600 °C. Throughout the IT range, the electrical conductivity decreases with decreasing RE ionic radius: σPBCO > σNBCO > σGBCO. We can explain this trend by ionic polarization; an RE3+ with a positive charge typically has a stronger attraction to the O2− electron clouds and deforms them towards the RE3+ ion, thus improving the covalent interactions between RE3+ and O2−. We take PBCO and GBCO as examples. In these two mixed oxides with the same double-perovskite crystal structure, Pr3+ and Gd3+ have the same charges. In this situation, Gd3+ with a smaller radius deforms the electron cloud of O2− more severely and strengthens the covalent interactions of the RE–O bond. As a result, the O 2p electrons are more likely to transfer to the Gd 5d states rather than to the Co 3d states (Fig. 5(a)).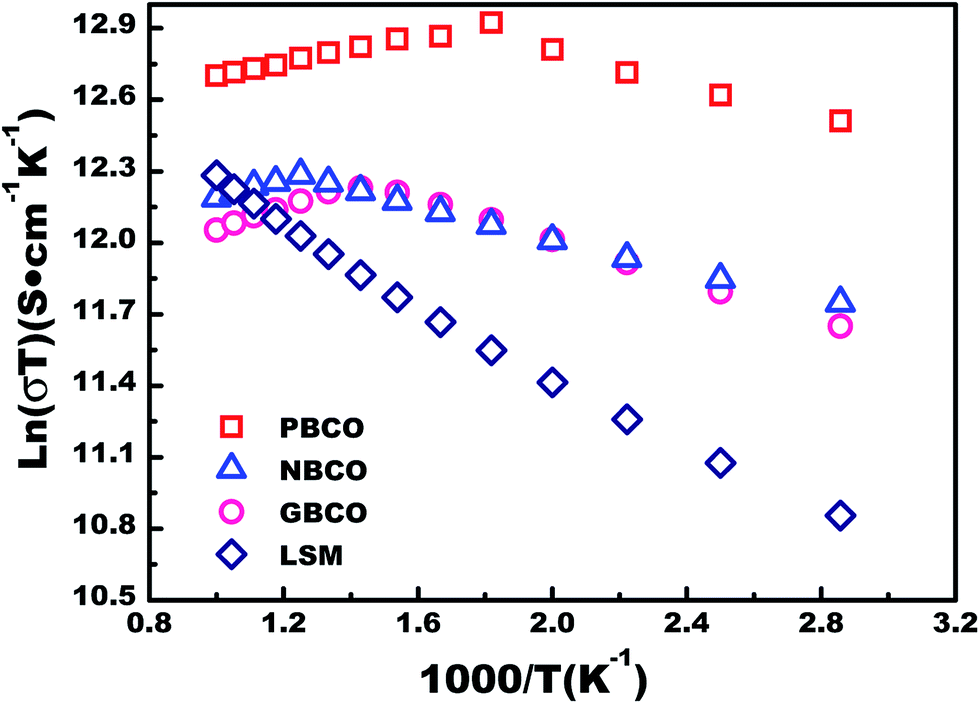 | ||
| Fig. 4 Arrhenius plots of ln(σT) vs. 1000/T for REBaCo2O5+δ (RE = Pr, Nd, and Gd) and La0.8Sr0.2MnO3−δ (LSM) in air. | ||
 | ||
| Fig. 5 (a) The contradistinctive electron distributions in PBCO and GBCO. In the GBCO system, the stronger polarization interaction between Gd3+ and O2− decreases the overlap between Co and O and causes electron transformation towards Gd 5d states. (b) Band structures of PBCO and GBCO. The electron transformation from O 2p states to RE 5d states causes the decline of the O 2p center about the Fermi level (especially in GBCO), resulting in a decrease in electrical conductivity. | ||
As shown in Fig. 5(b), the electron transformation from O 2p to RE 5d will cause the decline of the O 2p center about the Fermi level, resulting in a decrease in electrical conductivity.38,59 Therefore, more significant ionic polarization leads to poorer electronic conductivity in these three oxides.
3.4 Electrochemical performance
Fig. 6 shows the EIS spectra of symmetrical cells employing PBCO, NBCO, and GBCO as oxygen electrodes. All the impedance spectra were fitted by Z-view software, and the equivalent circuit is shown in Fig. 6, wherein L1 is the inductance. CPE1 and CPE2 represent constant phase elements. The intercept of the high-frequency impedance arc on the real axis is attributed to the ohmic resistance (Rohm), which comes from the electrolyte, electrodes, collectors and lead wires. Two electrode processes corresponding to two impedance arcs were the main factors limiting the ORR. The polarization resistance at high frequency (R1) is related to the migration of the oxygen ions from the three-phase boundary into the electrolyte, while the polarization resistance at low frequency (R2) is related to molecular oxygen diffusion, adsorption, and dissociation processes. The total polarization resistance (Rp) corresponds to the sum of R1 and R2. Table 2 shows the total polarization resistance (Rp) for PBCO, NBCO, and GBCO used as oxygen electrodes. The total polarization resistance (Rp) decreases with increasing temperature due to the enhancement in the catalytic activity of the oxygen electrode and the rate of OER, which consequently accelerates oxygen diffusion, adsorption and dissociation processes. Therefore, the increased oxygen ion mobility rate could facilitate oxygen transport then leading to the decrease of the polarization resistance. For example, the Rp values of PBCO decrease from 1.83 Ω cm2 at 650 °C to 0.13 Ω cm2 at 800 °C, which agrees with the results of previous reports.39,60 Fig. 6 also shows that the charge transfer reaction is the rate-limiting step in the OER for the REBaCo2O5+δ oxygen electrode. Therefore, to improve the performances of IT-SOECs, a lower polarization impedance oxygen electrode material is necessary. According to our results, NBCO has the lowest polarization resistance; the polarization resistance Rp was only 0.95 Ω cm2 at 650 °C. When compared with the literature, large differences in the area-specific polarization resistance (ASR) values of PBCO, NBCO and GBCO can be found. For example, the highest ASR values are 0.75 Ω cm2 for PBCO,57 1.18 Ω cm2 for NBCO37 and 5.77 Ω cm2 for GBCO61 at 700 °C. The lowest ASR values are 0.02 Ω cm2 for PBCO,58 0.04 Ω cm2 for NBCO58 and 0.03 Ω cm2 for GBCO58 at 700 °C. This is attributed to many factors, such as physical and microstructural parameters, that affect the ASR values measured by impedance spectroscopy. | ||
| Fig. 6 Impedance spectra of the symmetric cells with PBCO, NBCO and GBCO used as oxygen electrodes at different temperatures. | ||
| T (°C) | The total cathodic polarization resistance Rp (Ω cm2) | |||
|---|---|---|---|---|
| 650 | 700 | 750 | 800 | |
| PBCO | 1.83 | 0.69 | 0.27 | 0.13 |
| NBCO | 0.95 | 0.14 | 0.38 | 0.07 |
| GBCO | 1.55 | 0.66 | 0.31 | 0.14 |
The electrical conductivity of NBCO is lower than that of PBCO, whereas the polarization resistance of NBCO is the lowest. This is because the smaller radius of Nd3+ facilitates the migration of O2− in the lattice. As an opposite extreme, the relatively large Pr3+ inhibits the relaxation of the lattice and retains a rigid structure, which is unfavorable for the migration of O2−. According to this analysis, the ionic conductivity increases with decreasing RE ionic radius: σPBCO < σNBCO < σGBCO.
Combined with the preceding analysis in Section 3.3, PBCO has the highest electrical conductivity but the lowest ionic conductivity, and the ion-conductive process restricts its performance; GBCO has the highest ionic conductivity but the lowest electrical conductivity, and the electronically conductive process limits its performance. In the case of NBCO, its relatively high mixed ionic–electronic conductivity leads to relatively good electrical properties. On the other hand, NBCO has the lowest TEC and may have the lowest contact resistance with the electrolyte. These factors combine to produce the relatively good performance of NBCO.
Fig. 7(a)–(c) are the V–I curves of the single solid-oxide electrolysis cells in SOEC mode with PBCO, NBCO and GBCO used as the oxygen electrodes of the single cells. It can be seen that the electrochemical performances of these materials decrease with decreasing temperature. For a voltage of 1.2 V, the power density values for PBCO, NBCO and GBCO at 800 °C are 0.65, 1.58 and 0.4 W cm−2, respectively; at 700 °C, the power density values for PBCO, NBCO and GBCO are 0.13, 0.857 and 0.208 W cm−2, respectively. NBCO has the highest power density, which is higher than that of SrCo0.8Fe0.1Ga0.1O3−δ (ref. 62) (for a voltage of 1.5 V, the power density is 0.873 W cm−2 at 750 °C) and close to that of La0.58Sr0.4Co0.2Fe0.8O3 (ref. 63) (for a voltage of 1.02 V, the power density is 0.75 W cm−2 at 777–780 °C) at even higher temperature. Fig. 7(d) shows the ASR values for REBaCo2O5+δ (RE = Pr, Nd, and Gd), which can be calculated from the slopes of the V–I curves. The ASR is represented by the total resistance of the single cell. It can be seen that NBCO has the least ASR, which may be because NBCO, which has a relatively low TEC and a relatively high electric conductivity, has the lowest contact resistance. The results indicate that the double-perovskite NBCO oxygen electrode with GDC electrolyte exhibits excellent electrochemical performance, further suggesting that NBCO is suitable for use as an IT-SOEC oxygen electrode.
 | ||
| Fig. 7 I–V curves recorded in SOEC mode for single cells in which the oxygen electrode is (a) PBCO, (b) NBCO and (c) GBCO. (d) Area-specific polarization resistance (ASR) values for REBaCo2O5+δ (RE = Pr, Nd, and Gd) at different temperatures. | ||
4 Conclusions
REBaCo2O5+δ (RE = Pr, Nd, and Gd) oxides with one single phase structure were synthesized using an EDTA–citrate process. These oxides have been used as oxygen electrodes for practical IT-SOECs. The electrical conductivity values of the samples are 645 S cm−1 (PBCO), 308 S cm−1 (NBCO) and 318 S cm−1 (GBCO) at 600 °C. The conductivities of REBaCo2O5+δ are higher than those of LSM at temperatures below 800 °C, and PBCO has a higher conductivity than NBCO and GBCO. At a given temperature, the electrical conductivity decreases as the ionic radius of the RE decreases: σPBCO > σNBCO > σGBCO. This trend is related to the increasing concentration of oxygen vacancies. In these oxides, NBCO has the lowest polarization resistance. The polarization resistance Rp of this material at 650 °C is 0.95 Ω cm2, which is mainly because NBCO could has a better balance between TEC and electrochemical performance than PBCO and GBCO. In single SOEC tests, at a voltage of 1.2 V, the power density values of NBCO, PBCO and GBCO at 800 °C are 1.58, 0.65 and 0.4 W cm−2, respectively. Even at 700 °C, the power density for NBCO is 0.857 W cm−2. Therefore, the double-perovskite NBCO oxygen electrode with GDC electrolyte exhibits an excellent electrochemical performance, indicating that NBCO is a promising oxygen electrode material for IT-SOECs in practical applications. In the next step, the performances of single cells with sizes of 10 cm × 10 cm operated in SOEC mode will be evaluated.Acknowledgements
This work was supported financially by the Natural Science Foundation of China (No. 91645126, 21273128 and 51202123) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13026).References
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed.
- E. Stefan and T. Norby, Chem. Commun., 2016, 52, 1759–1767 RSC.
- A. Varone and M. Ferrari, Renewable Sustainable Energy Rev., 2015, 45, 207–218 CrossRef.
- Y. Zheng, J. C. Wang, B. Yu, W. Q. Zhang, J. Chen, J. L. Qiao and J. J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- M. Y. Liu, B. Yu, J. M. Xu and J. Chen, At. Energy Sci. Technol., 2009, 43, 881–885 CAS.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- C. Stoots, J. O'Brien and J. Hartvigsen, Int. J. Hydrogen Energy, 2009, 34, 4208–4215 CrossRef CAS.
- S. P. Jiang, Asia-Pac. J. Chem. Eng., 2016, 11, 386–391 CrossRef CAS.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- P. Moçoteguy and A. Brisse, Int. J. Hydrogen Energy, 2013, 38, 15887–15902 CrossRef.
- S. B. Adler, ChemInform, 2004, 104, 4791–4843 CAS.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 CAS.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- E. P. Murray, M. J. Sever and S. A. Barnett, Solid State Ionics, 2002, 148, 27–34 CrossRef.
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Energy Environ. Sci., 2011, 4, 4984–4993 CAS.
- M.-B. Choi, B. Singh, E. D. Wachsman and S.-J. Song, J. Power Sources, 2013, 239, 361–373 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- W. Zhou, R. Ran and Z. Shao, J. Power Sources, 2009, 192, 231–246 CrossRef CAS.
- S. Lü, Y. Bo, X. Meng, X. Zhao, J. Yuan, C. Fu, Y. Zhang, L. Yang, H. Fan and J. Yang, J. Power Sources, 2015, 273, 244–254 CrossRef.
- Y. S. Yoo, M. Choi, J. H. Hwang, H. N. Im, B. Singh and S. J. Song, Ceram. Int., 2015, 41, 6448–6454 CrossRef CAS.
- G. T. Kim, S. Y. Wang, A. J. Jacobson, Z. Yuan and C. L. Chen, J. Mater. Chem., 2007, 17, 1316–1320 RSC.
- S. Miyoshi, T. Furuno, O. Sangoanruang, H. Matsumoto and T. Ishihara, J. Electrochem. Soc., 2007, 154, B57–B62 CrossRef CAS.
- E. Boehm, J. M. Bassat, P. Dordor, F. Mauvy, J. C. Grenier and P. Stevens, Solid State Ionics, 2005, 176, 2717–2725 CrossRef CAS.
- J. M. Bassat, M. Burriel, O. Wahyudi, R. Castaing, M. Ceretti, P. Veber, I. Weill, A. Villesuzanne, J. C. Grenier, W. Paulus and J. A. Kilner, J. Phys. Chem. C, 2013, 117, 26466–26472 CAS.
- M. Lagunabercero, A. Hanifi, H. Monzon, J. Cunningham, T. Etsell and P. Sarkar, J. Mater. Chem. A, 2014, 2, 9764–9770 CAS.
- F. Mauvy, J. M. Bassat, E. Boehm, J. P. Manaud, P. Dordor and J. C. Grenier, Solid State Ionics, 2003, 158, 17–28 CrossRef CAS.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Appl. Phys. Lett., 2005, 86, 091910 CrossRef.
- A. Tarancón, S. J. Skinner, R. J. Chater, F. Hernández-Ramírez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175 RSC.
- G. Kim, S. Wang, A. J. Jacobson, L. Reimus, P. Brodersen and C. A. Mims, J. Mater. Chem., 2007, 17, 2500–2505 RSC.
- K. Zhang, L. Ge, R. Ran, Z. Shao and S. Liu, Acta Mater., 2008, 56, 4876–4889 CrossRef CAS.
- A. Manthiram, J. H. Kim, Y. N. Kim and K. T. Lee, J. Electroceram., 2011, 27, 93–107 CrossRef CAS.
- A. Maignan, C. Martin, D. Pelloquin, N. Nguyen and B. Raveau, J. Solid State Chem., 1999, 142, 247–260 CrossRef CAS.
- L. Zhao, B. B. He, B. Lin, H. P. Ding, S. L. Wang, Y. H. Ling, R. R. Peng, G. Y. Meng and X. Q. Liu, J. Power Sources, 2009, 194, 835–837 CrossRef CAS.
- C. Zhu, X. Liu, C. Yi, L. Pei, D. Wang, D. Yan, K. Yao, T. Lü and W. Su, J. Power Sources, 2010, 195, 3504–3507 CrossRef CAS.
- Q. Zhou, F. Wang, Y. Shen and T. He, J. Power Sources, 2010, 195, 2174–2181 CrossRef CAS.
- J. H. Kim, J. Bae, M. Cassidy, P. A. Connor, W. Zhou and J. Irvine, ECS Trans., 2009, 25, 2707–2715 CAS.
- W. Wang, S. P. Teng, S. H. Chan and T. S. Zhang, ECS Trans., 2009, 25, 2277–2281 CAS.
- J. H. Kim and A. Manthiram, J. Electrochem. Soc., 2008, 155, 7551–7557 Search PubMed.
- E. Chavez, M. Mueller, L. Mogni and A. Caneiro, in Xix Latin American Symposium on Solid State Physics, ed. A. Serquis, C. Balseiro and P. Bolcatto, 2009, vol. 167 Search PubMed.
- R. Pelosato, G. Cordaro, D. Stucchi, C. Cristiani and G. Dotelli, J. Power Sources, 2015, 298, 46–67 CrossRef CAS.
- W. Zhou, Z. Shao and W. Jin, J. Alloys Compd., 2006, 426, 368–374 CrossRef CAS.
- B. Yu, W. Zhang, J. Xu and J. Chen, Int. J. Hydrogen Energy, 2008, 33, 6873–6877 CrossRef CAS.
- C. J. Zhu, X. M. Liu, C. S. Yi, D. Yan and W. H. Su, J. Power Sources, 2008, 185, 193–196 CrossRef CAS.
- T. Chen, H. L. Zhao, Z. X. Xie, Y. Lu and N. S. Xu, Int. J. Hydrogen Energy, 2012, 37, 19133–19137 CrossRef CAS.
- P. S. Anderson, C. A. Kirk, J. Knudsen, I. M. Reaney and A. R. West, Solid State Sci., 2005, 7, 1149–1156 CrossRef CAS.
- H. C. Jin, J. H. Jang, H. R. Ji and S. M. Oh, J. Power Sources, 2000, 87, 92–100 CrossRef.
- S. Wang, Y. Jiang, Y. Zhang, J. Yan and W. Li, Solid State Ionics, 1998, 113, 291–303 CrossRef.
- L. Ruiz-González, K. Boulahya, M. Parras, J. Alonso and J. M. González-Calbet, Chem.–Eur. J., 2002, 8, 5694–5700 CrossRef.
- I. O. Troyanchuk, N. V. Kasper, D. D. Khalyavin, H. Szymczak, R. Szymczak and M. Baran, Phys. Rev. B, 1998, 58, 2418–2421 CrossRef CAS.
- N. E. Volkova, L. Y. Gavrilova, V. A. Cherepanov, T. V. Aksenova, V. A. Kolotygin and V. V. Kharton, J. Solid State Chem., 2013, 204, 219–223 CrossRef CAS.
- S. Pang, X. Jiang, X. Li, Z. Su, H. Xu, Q. Xu and C. Chen, Int. J. Hydrogen Energy, 2012, 37, 6836–6843 CrossRef CAS.
- M. B. Choi, S. Y. Jeon, J. S. Lee, H. J. Hwang and S. J. Song, J. Power Sources, 2010, 195, 1059–1064 CrossRef CAS.
- S. Park, S. Choi, J. Kim, J. Shin and G. Kim, ECS Electrochem. Lett., 2012, 1, F29–F32 CrossRef CAS.
- L. Zhao, J. Shen, B. He, F. Chen and C. Xia, Int. J. Hydrogen Energy, 2011, 36, 3658–3665 CrossRef CAS.
- S. Yoo, S. Choi, J. Kim, J. Shin and G. Kim, Electrochim. Acta, 2013, 100, 44–50 CrossRef CAS.
- H. Zhao, Y. Zheng, C. Yang, Y. Shen, Z. Du and K. Świerczek, Int. J. Hydrogen Energy, 2013, 38, 16365–16372 CrossRef CAS.
- L. Jiang, F. Li, T. Wei, R. Zeng and Y. Huang, Electrochim. Acta, 2014, 133, 364–372 CrossRef CAS.
- K. Zhang, L. Ge, R. Ran, Z. Shao and S. Liu, Acta Mater., 2008, 56, 4876–4889 CrossRef CAS.
- K. T. Lee and A. Manthiram, J. Electrochem. Soc., 2006, 153, A794–A798 CrossRef CAS.
- J. Zou, J. Park, B. Kwak, H. Yoon and J. Chung, Solid State Ionics, 2012, 206, 112–119 CrossRef CAS.
- E. Chavez, M. Mueller, L. Mogni and A. Caneiro, J. Phys.: Conf. Ser., 2009, 167, 012043 CrossRef.
- X. Meng, Y. Shen, M. Xie, Y. Yin, N. Yang, Z. F. Ma, J. C. D. D. Costa and S. Liu, J. Power Sources, 2016, 306, 226–232 CrossRef CAS.
- J. Schefold and A. Brisse, ECS Trans., 2013, 53, 53–61 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |