
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC-7 modified flavonoids as novel tyrosyl-tRNA synthetase inhibitors†
Zhu-Ping Xiao *a,
Wei Weia,
Qi Liua,
Peng-Fei Wangb,
Xing Luoa,
Fang-Yuan Chena,
Yang Caoa,
Hong-Xia Huanga,
Mi-Mi Liua and
Hai-Liang Zhu*ab
*a,
Wei Weia,
Qi Liua,
Peng-Fei Wangb,
Xing Luoa,
Fang-Yuan Chena,
Yang Caoa,
Hong-Xia Huanga,
Mi-Mi Liua and
Hai-Liang Zhu*ab
aCollege of Chemistry and Chemical Engineering, Hunan Engineering Laboratory for Analyse and Drugs Development of Ethnomedicine in Wuling Mountains, Jishou University, Jishou 416000, P. R. China. E-mail: xiaozhuping2005@163.com
bState Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing 210093, P. R. China. E-mail: zhuhl@nju.edu.cn
First published on 18th January 2017
Abstract
Twenty C-7 modified flavonoids were designed and synthesized. Biological evaluation in vitro indicated that compounds generated by SYBYL-X with high scores also showed good inhibitory activities against TyrRS. Compounds containing the nargenin core exhibit better enzyme inhibitory activities than other flavonoid cores, with (S)-5-hydroxy-4′-hydroxy-7-(2-morpholino-2-oxoethoxy)-2,3-dihydroflavone (b1) being the most active (IC50 = 0.10 ± 0.01 μM) in all assayed compounds. All compounds were also assayed for antimicrobial activities against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa, and b1 also displayed excellent activity, showing 6-fold more potent than the marketed antibiotic ciprofloxacin. In comparison with Gram-positive organism, all these derivatives exhibited better activity against Gram-negative organism, and did not displayed significant differences between the two assayed Gram-negative strains (E. coli ATCC 8739 and P. aeruginosa ATCC 9027).
1. Introduction
Since the first therapeutic use of penicillin in 1941, each newly marketed antibiotic has invariably resulted in the emergence of drug-resistant pathogens,1–3 which has rendered marketed antibiotics less effective.4–6 One particularly troubling event is the widespread emergence of MRSA (methicillin resistant Staphylococcus aureus) strains, which are resistant to many major classes of drugs.7,8 Therefore, new classes of antibacterial agents are urgently needed to combat drug-resistant bacteria.Flavonoids, natural products of the benzopyran class heterocycles, ubiquitously present in fruits and vegetables.9 In the past decades, a wide range of important biological activities have also been described, such as antioxidant, antibacterial, selective anticancer, anti-inflammatory and antiallergic properties.10–16 However, most flavonoids were not developed as clinical drugs because of poor bioavailability (less than 5%) and induction or inhibition of some metabolic enzymes.17 Therefore, great interest has been attracted in the field of structural modifications of the flavonoid skeleton for improving bioavailability and toxicity profiles.18
AaRSs (aminoacyl tRNA synthetases) play a crucial role in the process of protein synthesis, and are believed to be novel targets of antibacterial, antifungal and antiparasitic drugs.19–21 Recently, our efforts have focused on the development of novel TyrRS inhibitors based on modifications of 3-arylfuran-2(5H)-one core.20,22,23 4-Hydroxyphenyl, a moiety of the substance of TyrRS, binds at the bottom of the active pocket,24 which is often observed in flavonoid molecules, such as naringenin from pomelo, apigenin from celery and daidzein from soybean (Fig. 1). Based on this consideration, modifications of flavonoid skeleton were here performed to mimic the substance of TyrRS (Tyrosyl-tRNA Synthetase), tyrosyl adenylate, as possible TyrRS inhibitors. Therefore, a series of novel flavonoid derivatives were designed and synthesized, and were subsequently evaluated for biological activities against a representative Gram-positive organism (Staphylococcus aureus ATCC 6538) and two Gram-negative organisms (Escherichia coli ATCC 8739; Pseudomonas aeruginosa ATCC 9027). The results indicated that some of the synthesized compounds show very good antibacterial activities.
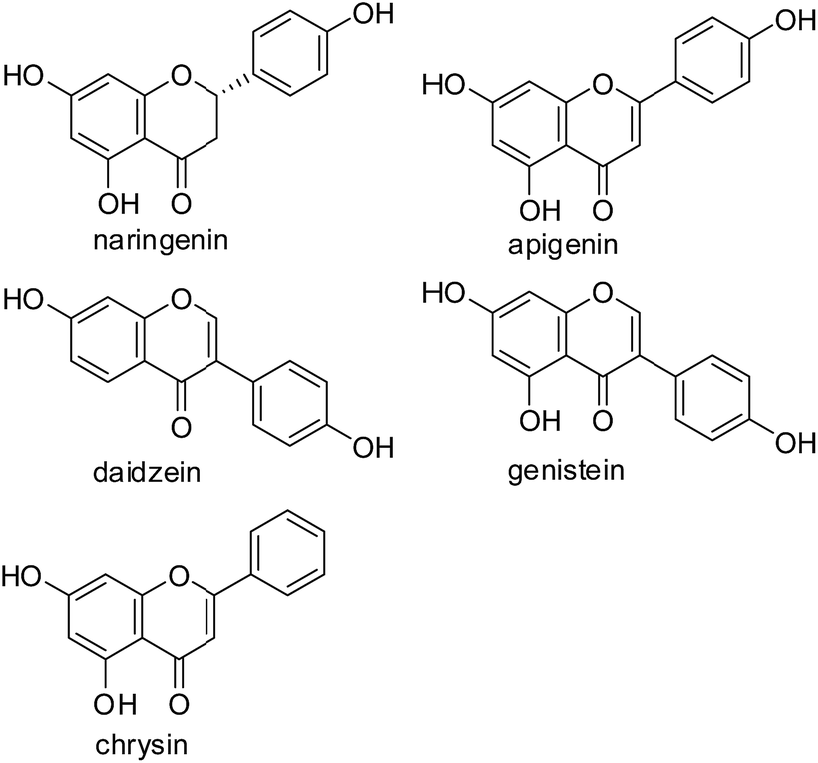 | ||
| Fig. 1 Structures of representative flavonoids. | ||
2. Results and discussion
2.1 Design and virtual screen
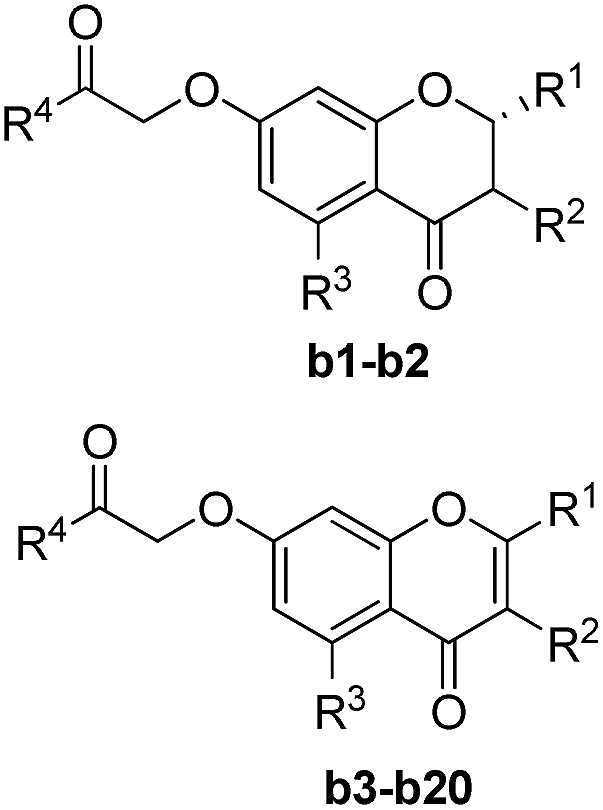
Based on scaffold hopping drug design, our laboratory recently developed several novel TyrRS inhibitors.25,26 As a continuation of this drug discovery program, scaffold hopping of tyrosyl adenylate was performed by Surflex Sim suite of SYBYL-X 2.1.1 (ref. 27) according to the hypothesis mentioned above. In details, tyrosyl adenylate were selected as the template for superposing with a series of typical O-graft flavonoids based on shape matching method (Fig. 2). Results revealed that 7-O-graft flavonoid was able to be the scaffold hopping structure of tyrosyl adenylate. To further determine the possibility of developing TyrRS inhibitors from this scaffold, compound library with C-7 side chain modifications were generated using the Library Design software module within the SYBYL-X environment. Subsequently, virtual screening was performed using docking mode of Surflex-Dock software module, where 20 possible conformations (poses) for each docked ligand were produced. The docked conformations of ligands were evaluated and ranked using Total_Score and Cscore (Concensus score) modules. Compounds were selected according to synthesis possibility and values of Total_Score and Cscore, and the obtained top 20 compounds (b1–b20) were listed in Table 1.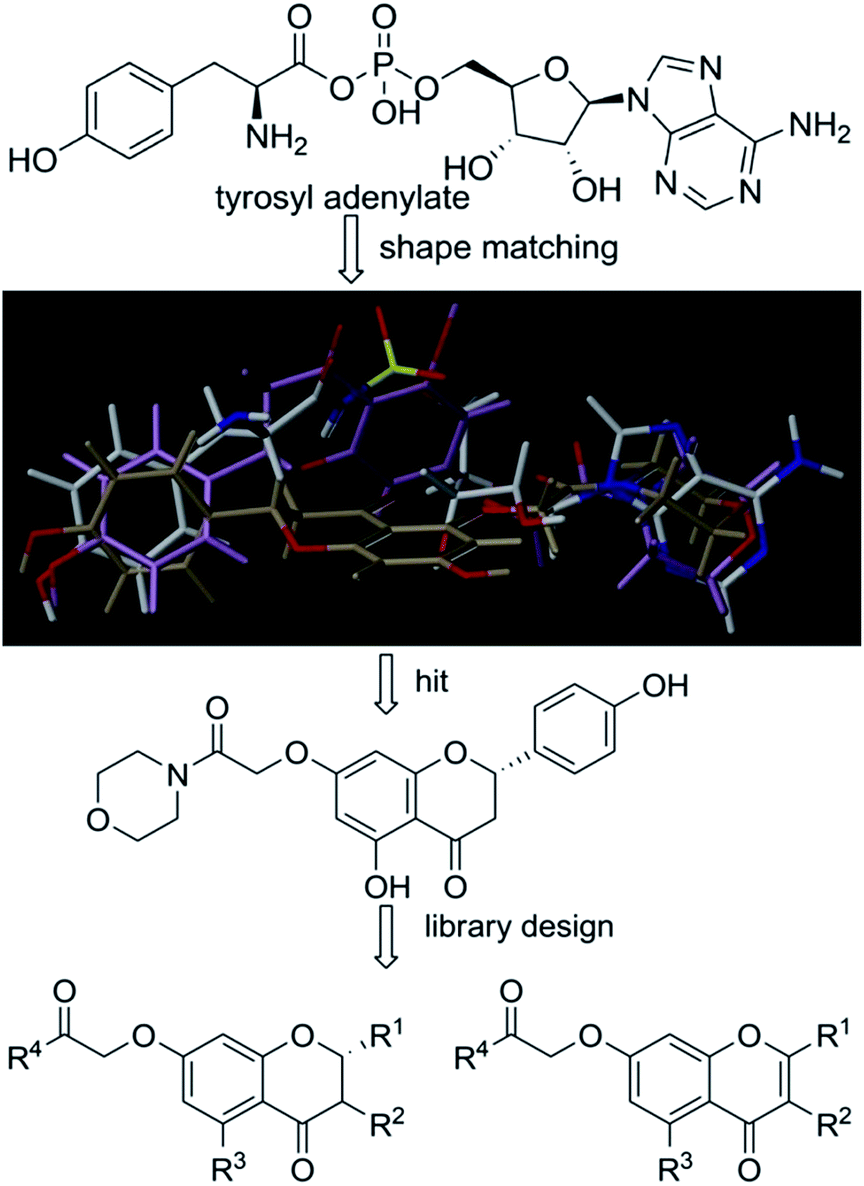 | ||
| Fig. 2 Scaffold hopping and library design. Superpositions of tyrosyl adenylate (white) with a 7-O-graft flavonoid derivative (light purple) and 5-O-graft flavonoid derivative (light brown) generated by Surflex Sim suite of SYBYL-X 2.1.1. | ||
| Entry |
|
Score | IC50 (μM) | |||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | |||
| b1 |  |
H | OH |  |
10.20 | 0.10 ± 0.01 |
| b2 | 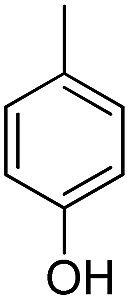 |
H | OH | 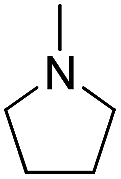 |
8.65 | 2.7 ± 0.1 |
| b3 | H | 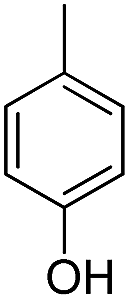 |
OH | 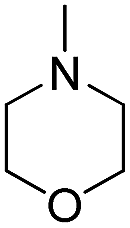 |
9.26 | 0.20 ± 0.01 |
| b4 | H |  |
OH |  |
7.74 | 17 ± 0.9 |
| b5 | H |  |
OH |  |
9.15 | 0.60 ± 0.02 |
| b6 | H |  |
OH |  |
9.26 | 1.10 ± 0.07 |
| b7 | H |  |
H |  |
7.21 | >100 |
| b8 | H | 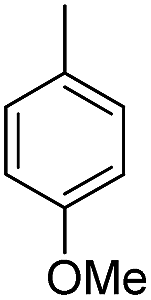 |
H | 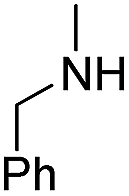 |
6.99 | >100 |
| b9 | H | 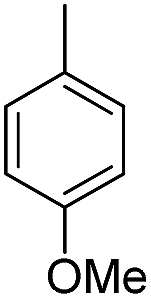 |
H | 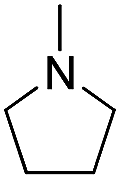 |
7.99 | 16 ± 1.0 |
| b10 | H | 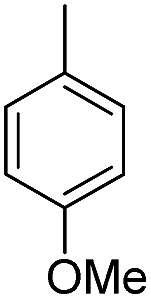 |
H | 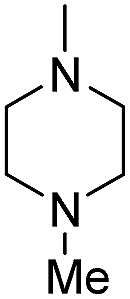 |
8.82 | 7.7 ± 0.3 |
| b11 | H | 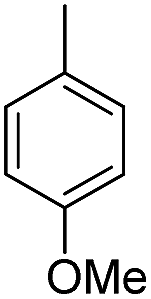 |
H | 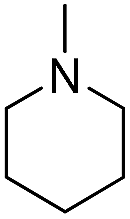 |
8.54 | 5.2 ± 0.2 |
| b12 | H | 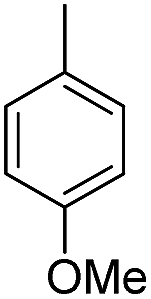 |
H | 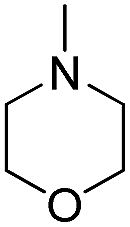 |
8.62 | 2.0 ± 0.1 |
| b13 | H | 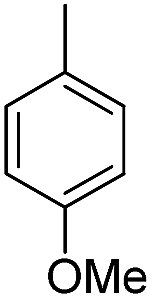 |
H | 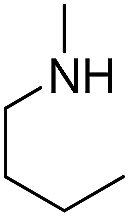 |
7.42 | 34 ± 1.8 |
| b14 | H | 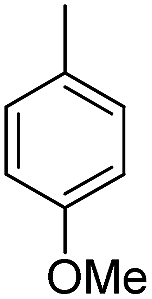 |
H | 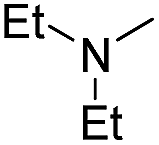 |
7.21 | 41 ± 2.2 |
| b15 | H | 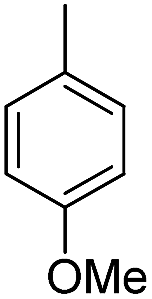 |
H | 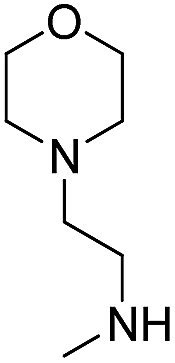 |
8.22 | 6.7 ± 0.3 |
| b16 | 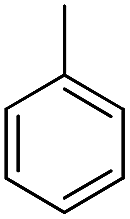 |
H | OH | 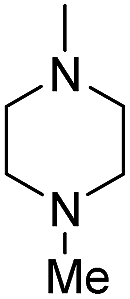 |
8.34 | 20 ± 1.1 |
| b17 | 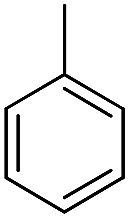 |
H | OH | 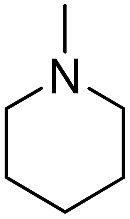 |
8.57 | 13 ± 0.8 |
| b18 |  |
H | OH |  |
7.56 | 86 ± 6.2 |
| b19 |  |
H | OH |  |
8.66 | 6.5 ± 0.4 |
| b20 | 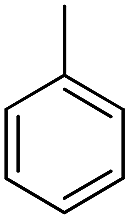 |
H | OH | 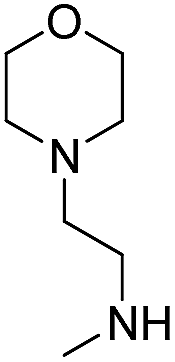 |
7.74 | 28 ± 1.6 |
2.2 Synthesis and characterization
The synthetic route illustrated in Schemes 1 and 2 was developed according to the reported methods with some modification,28–31 and the prepared compounds are depicted in Table 1. In general, deprotonation and alkylation of the 5-hydroxyl group is hardly since the hydrogen atom of the 5-hydroxyl group is attracted to the oxygen atom of the 4-carbonyl group forming intramolecular hydrogen bond.32 Chrysin therefore shows excellent O-alkylation selectivity at 7-OH in an alkaline environment, which followed by the production of compounds b16–b20. This was supported the signal at about δ 12.8 in 1H NMRs of b16–b20, which was assigned to the hydrogen atom of 5-OH. Similarly, 5-OH in b1–b6 leaves intact in alkylation of their precursors naringenin and genistein. In comparison with 4′-OH, 7-OH shows 2 orders of magnitude decrease in pKa values of naringenin and genistein,32–34 and alkylation of the hydroxyl groups occurs following a specific sequential position order: 7 > 4′.35,36 The signals at about δ 12.8 and 9.6 in b1–b6 were assigned to 5-OH and 4′-OH, respectively.37–39 These facts suggested the selective O-alkylation of 7-OH.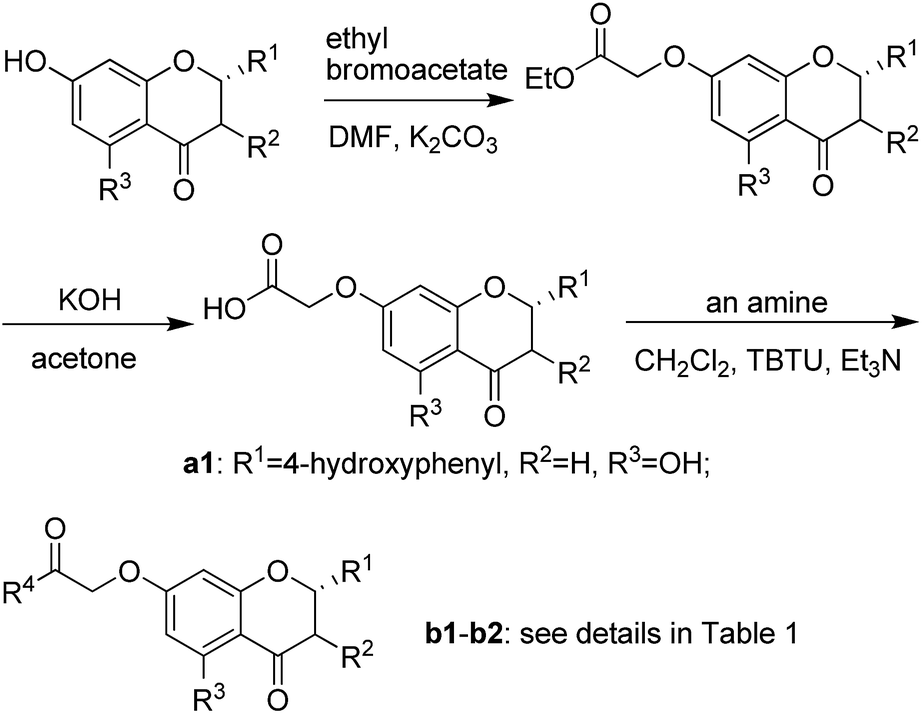 | ||
| Scheme 1 Synthetic route of compounds b1–b2. | ||
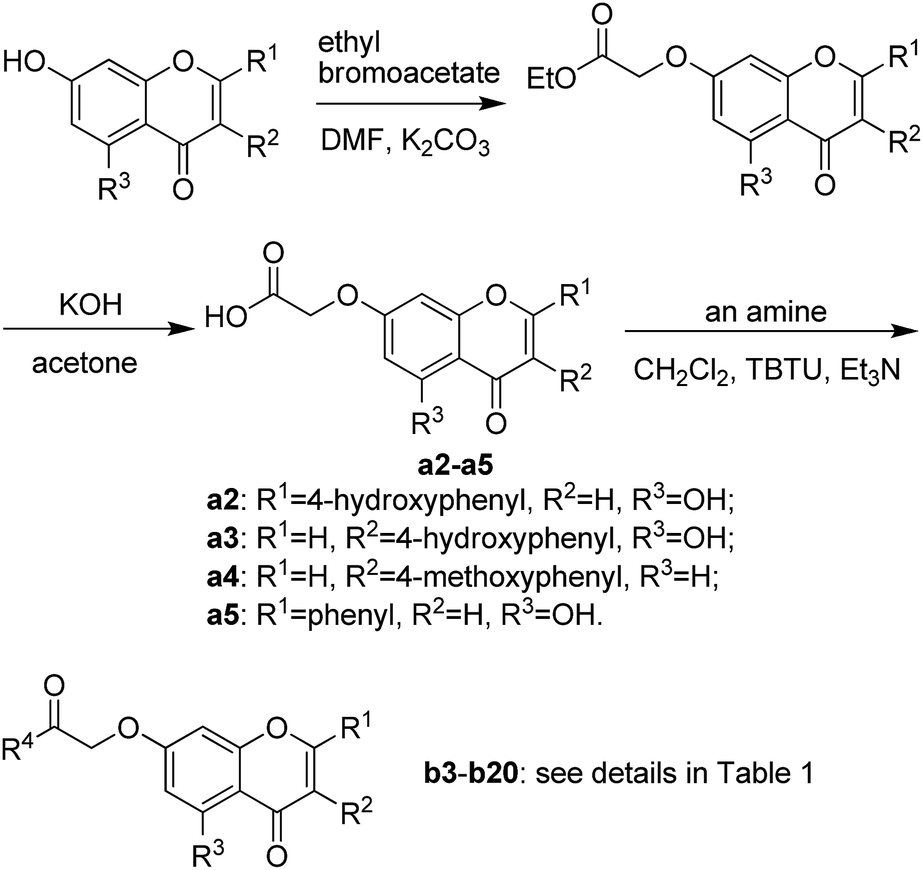 | ||
| Scheme 2 Synthetic route of compounds b3–b20. | ||
2.3 Inhibitory activities of flavonoid derivatives against TyrRS
Compounds b1–b20 were synthesized (Schemes 1 and 2) to test for inhibitory activity against TyrRS from E. coli, and IC50s of these compounds are presented in Table 1. The main flavonoids involving in modifications are naringenin, genistein, 4′-O-methyldaidzein and chrysin. In these four series, derivatives from naringenin were determined to be the most potent, and compound b1 with a morpholine ring in the 7-side chain displayed IC50 of 0.10 ± 0.01 μM against TyrRS, being the most active. Substitution of the genistein core (b3) for naringenin core (b1) led to a slight decrease in potency. It was changed into daidzein or chrysin core, however, resulting in a significant decrease of potency with IC50 values of 6- to 70-fold increase (b1 vs. b12 or b19, b2 vs. b9 or b18). It can be seen that R4 at the C-7 side chain is also a major influence of molecule potency, and variation of the group at this position may result in large changes of IC50 values. N-Containing heterocyclic compounds were more active than alkyl or aromatic amino containing analogues, with evidences of anilino and benzylamino containing compounds (b7 and b8) being completely loss of activity against this enzyme. Replacement of morpholino with piperidino or N-methylpiperazino caused 2 to 5-fold decreases in potency, while a dramatic decrease was observed when it was changed into pyrrolidino group (b1 vs. b2, b12 vs. b9 and b19 vs. b18).2.4 Antibacterial activity
All compounds were tested against representative Gram-positive organism (S. aureus ATCC 6538) and two Gram-negative organisms (E. coli ATCC 8739 and P. aeruginosa ATCC 9027), which are presented in Table 2. The results revealed that compounds with outstanding inhibition against TyrRS also exhibit strong antibacterial activities, which shows that their inhibition of TyrRS may lead to the antibacterial activity. In comparison with Gram-positive organism, all these derivatives exhibited better activity against Gram-negative organism, and significant differences were not observed between the two assayed Gram-negative strains. Only one compound b1 was proved to exhibit activity comparable to that of the marketed antibiotic penicillin G against S. aureus ATCC 6538. While three compounds, b1, b3 and b5, showed better activities than marketed antibiotic ciprofloxacin against Gram-negative organisms, together with two compounds (b6 and b10) being compared to ciprofloxacin. Out of these compounds, b1 was the most active with MIC50 of 0.11 μg mL−1, showing 6-fold more potent than ciprofloxacin.| Entry | MIC50 (μg mL−1) | ||
|---|---|---|---|
| A | B | C | |
| a (A) E. coli ATCC 8739; (B) P. aeruginosa ATCC 9027; (C) S. aureus ATCC 6538. | |||
| b1 | 0.11 | 0.29 | 0.59 |
| b2 | 1.8 | 2.7 | 14.8 |
| b3 | 0.37 | 0.55 | 1.6 |
| b4 | 44.3 | 56.1 | 47.8 |
| b5 | 0.55 | 0.71 | 9.3 |
| b6 | 0.67 | 1.2 | 10.9 |
| b7 | >100 | >100 | >100 |
| b8 | >100 | >100 | >100 |
| b9 | 30.2 | 42.4 | 35.5 |
| b10 | 1.0 | 2.2 | 12.9 |
| b11 | 2.7 | 3.0 | 6.8 |
| b12 | 15.3 | 18.7 | 29.4 |
| b13 | >100 | >100 | >100 |
| b14 | >100 | >100 | >100 |
| b15 | 10.2 | 14.2 | 25.6 |
| b16 | 70.4 | 94.9 | 82.7 |
| b17 | 18.5 | 22.7 | 31.3 |
| b18 | >100 | >100 | >100 |
| b19 | 6.7 | 8.5 | 17.8 |
| b20 | >100 | >100 | 98 |
| Ciprofloxacin | 0.67 | 0.81 | — |
| Penicillin G | — | — | 0.54 |
2.5 In vitro toxicity
As these derivatives were inhibitors against TyrRS, we studied the toxicity of active compounds measuring cytotoxicity (EC50) against mammalian cells (HeLa). None of the derivatives showed significant toxicity up to 100 μM concentration (Table 3), which indicates assayed compounds showed selectivity >100.| Entry | EC50 (μM) |
|---|---|
| b1 | >100 |
| b3 | >100 |
| b5 | >100 |
| b6 | >100 |
| b10 | >100 |
2.6 Docking studies
Molecular dockings of the most active compound b1 were performed to illustrate the binding mode and to provide an evidence for the potency of inhibition. Initially sketched b1 was subjected to energy minimization (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iterations) by the Powell minimization standard method. The final b1 conformational coordinates were stored as a mol2 file. Accuracy docking was performed using the Surflex-Dock GeomX software module within the SYBYL-X 2.1.1 environment. Molecular docking results revealed that b1 docks favorably into the active site of TyrRS, and adopts a binding pose as that of co-crystallized ligand Tyr-AMS (5-O-[N-(L-tyrosyl)sulfamoyl]adenosine) (Fig. 3). Along the active site, the morpholino moiety packs in the upper cleft of the binding pocket, and forms hydrophobic interactions with Val 53, Leu 227 and Ile 228. Oxygen atom at C-7 as a hydrogen bond acceptor receives two strong hydrogen bond interactions from Gly 197 and Asp 200 with H⋯O distances of 2.185 and 2.868 Å, while Asp 200 in turn as a acceptor builds another O–H⋯O hydrogen bond with 5-OH. On the contrary, the B-ring of the naringenin core drops completely into the bottom of the binding pocket, with OH as the hydrogen bond acceptor as well as donor forming two hydrogen bonds (O–H⋯O) with Tyr 37 and Asp 182, respectively. In addition, three non-classical hydrogen bonds (C–H⋯O) were observed between B-ring and the enzyme backbone with H⋯O distances in the range of 2.3 to 3.2 Å. Hydrogen bonds built between 4-C
000 iterations) by the Powell minimization standard method. The final b1 conformational coordinates were stored as a mol2 file. Accuracy docking was performed using the Surflex-Dock GeomX software module within the SYBYL-X 2.1.1 environment. Molecular docking results revealed that b1 docks favorably into the active site of TyrRS, and adopts a binding pose as that of co-crystallized ligand Tyr-AMS (5-O-[N-(L-tyrosyl)sulfamoyl]adenosine) (Fig. 3). Along the active site, the morpholino moiety packs in the upper cleft of the binding pocket, and forms hydrophobic interactions with Val 53, Leu 227 and Ile 228. Oxygen atom at C-7 as a hydrogen bond acceptor receives two strong hydrogen bond interactions from Gly 197 and Asp 200 with H⋯O distances of 2.185 and 2.868 Å, while Asp 200 in turn as a acceptor builds another O–H⋯O hydrogen bond with 5-OH. On the contrary, the B-ring of the naringenin core drops completely into the bottom of the binding pocket, with OH as the hydrogen bond acceptor as well as donor forming two hydrogen bonds (O–H⋯O) with Tyr 37 and Asp 182, respectively. In addition, three non-classical hydrogen bonds (C–H⋯O) were observed between B-ring and the enzyme backbone with H⋯O distances in the range of 2.3 to 3.2 Å. Hydrogen bonds built between 4-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Arg 89, 3-CH2 and Tyr 175 also furnish important contributions to binding affinities (Fig. 4). The docking studies demonstrate that b1 strongly binds with TyrRS, powerfully supporting its excellent inhibitory potency in enzyme assays.
O and Arg 89, 3-CH2 and Tyr 175 also furnish important contributions to binding affinities (Fig. 4). The docking studies demonstrate that b1 strongly binds with TyrRS, powerfully supporting its excellent inhibitory potency in enzyme assays.
 | ||
| Fig. 3 Binding pose of compound b1 with TyrRS (shown as gray) and the original binding pose of co-crystal ligand Tyr-AMS (shown as red). | ||
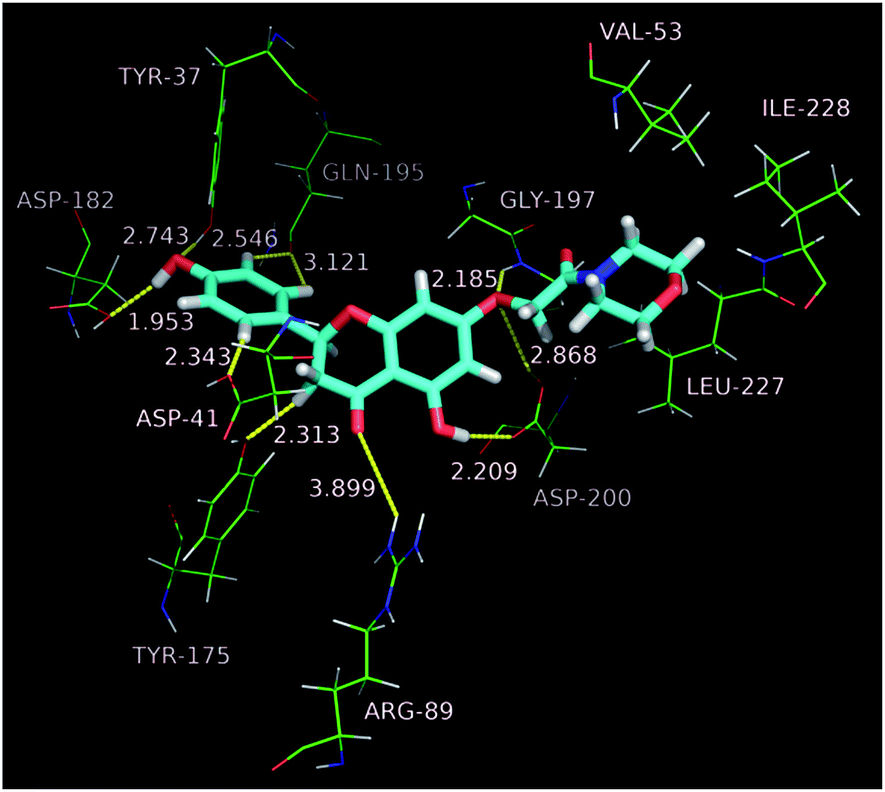 | ||
| Fig. 4 Binding mode of compound b1 with TyrRS. | ||
3. Experimental
3.1 Chemistry
All chemicals (reagent grade) used were purchased from Aldrich (U.S.A) and Sinopharm Chemical Reagent Co., Ltd (China). Separation of the compounds by column chromatography was carried out with silica gel 60 (200–300 mesh ASTM, E. Merck). The quantity of silica gel used was 30–70 times the weight charged on the column. Then, the eluates were monitored using TLC (thin-layer chromatography). Melting points (uncorrected) were determined on a XT4 MP apparatus (Taike Corp., Beijing, China). ESI mass spectra were obtained on a Mariner System 5304 mass spectrometer, and 1H NMR and 13C NMR spectra were recorded on Bruker AV-300 or 400 spectrometer at 25 °C with TMS and solvent signals allotted as internal standards. Chemical shifts were reported in ppm (δ). Elemental analyses were performed on a CHN-O-Rapid instrument and were within ±0.4% of the theoretical values.3.1.2.1 (S)-5-Hydroxy-4′-hydroxy-7-(2-morpholino-2-oxoethoxy)-2,3-dihydroflavone (b1). Light yellow powder, 57.4%, mp 161–162 °C; 1H NMR (DMSO): 2.60 (dd, J = 17.2 Hz, J = 3.0 Hz, 1H); 3.29–3.32 (m, 1H); 3.42 (t, J = 6.8 Hz, 4H); 4.33 (t, J = 7.2 Hz, 2H); 4.82 (s, 2H); 5.49 (dd, J = 13.4 Hz, J = 2.9 Hz, 1H); 6.07 (dd, J = 7.8 Hz, J = 2.3 Hz, 2H); 6.79 (s, 1H); 6.80 (s, 1H); 7.33 (d, J = 8.5 Hz, 2H); 9.62 (s, 1H); 12.07 (s, 1H); EIMS m/z 399 (M+). Anal. calcd for C21H21NO7: C, 63.15; H, 5.30; N, 3.51; found: C, 63.09; H, 5.31; N, 3.51.
3.1.2.2 (S)-5-Hydroxy-4′-hydroxy-7-(2-(pyrrolidin-1-yl)-2-oxoethoxy)-2,3-dihydrofla-vone (b2). Light yellow powder, 53.9%, mp 260–262 °C; 1H NMR (DMSO): 1.72–1.79 (m, 2H); 1.84–1.91 (m, 2H); 2.71 (dd, J = 17.1 Hz, J = 3.0 Hz, 1H); 3.33–3.43 (m, 5H); 4.81 (s, 2H); 5.48 (dd, J = 11.1 Hz, J = 2.9 Hz, 1H); 6.07 (dd, J = 7.8 Hz, J = 2.3 Hz, 2H); 6.79 (s, 1H); 6.81 (s, 1H); 7.33 (d, J = 8.5 Hz, 2H); 9.61 (s, 1H); 12.08 (s, 1H); EIMS m/z 383 (M+). Anal. calcd for C21H21NO6: C, 65.79; H, 5.52; N, 3.65; found: C, 65.83; H, 5.51; N, 3.65.
3.1.2.3 5-Hydroxy-4′-hydroxy-7-(2-morpholino-2-oxoethoxy)isoflavone (b3). White powder, 82.5%, mp 235–237 °C; 1H NMR (DMSO): 3.46 (t, J = 4.7 Hz, 4H); 3.59 (t, J = 4.8 Hz, 2H); 3.64 (t, J = 4.8 Hz, 2H); 5.03 (s, 2H); 6.44 (d, J = 2.5 Hz, 1H); 6.67 (d, J = 2.3 Hz, 1H); 6.84 (d, J = 8.4 Hz, 2H); 7.40 (d, J = 8.3 Hz, 2H); 8.42 (s, 1H); 9.63 (s, 1H); 12.96 (s, 1H); EIMS m/z 397 (M+). Anal. calcd for C21H19NO7: C, 63.47; H, 4.82; N, 3.52; found: C, 63.50; H, 4.81; N, 3.52.
3.1.2.4 5-Hydroxy-4′-hydroxy-7-(2-butylamino-2-oxoethoxy)isoflavone (b4). White powder, 64.8%, mp 203–205 °C; 1H NMR (DMSO): 0.85–0.897 (t, J = 7.3 Hz, 3H); 1.23–1.32 (m, 2H); 1.34–1.55 (m, 2H); 3.14 (q, J = 6.7 Hz, 2H); 4.62 (s, 2H); 6.46 (d, J = 2.3 Hz, 1H); 6.66 (d, J = 2.3 Hz, 1H); 6.84 (d, J = 8.6 Hz, 2H); 7.40 (d, J = 8.6 Hz, 2H); 8.17 (t, J = 5.8 Hz, 1H); 8.43 (s, 1H); 9.63 (s, 1H); 12.96 (s, 1H); EIMS m/z 383 (M+). Anal. calcd for C21H21NO6: C, 65.79; H, 5.52; N, 3.65; found: C, 65.74; H, 5.53; N, 3.65.
3.1.2.5 5-Hydroxy-4′-hydroxy-7-(2-piperidino-2-oxoethoxy)isoflavone (b5). White powder, 65.5%, mp 235–237 °C; 1H NMR (DMSO): 1.44–1.47 (m, 2H); 1.52–1.77 (m, 4H); 3.38–3.45 (m, 4H); 4.99 (s, 2H); 6.42 (d, J = 2.2 Hz, 1H); 6.65 (d, J = 2.2 Hz, 1H); 6.83 (d, J = 8.6 Hz, 2H); 7.40 (d, J = 8.5 Hz, 2H); 8.42 (s, 1H); 9.63 (s, 1H); 12.96 (s, 1H); EIMS m/z 395 (M+). Anal. calcd for C22H21NO6: C, 66.83; H, 5.35; N, 3.54; found: C, 66.87; H, 5.31; N, 3.54.
3.1.2.6 5-Hydroxy-4′-hydroxy-7-(2-(4-methylpiperazino)-2-oxoethoxy)isoflavone (b6). White powder, 63.2%, mp 193–195 °C; 1H NMR (DMSO): 3.51 (s, 3H); 3.85–3.94 (m, 4H); 4.08–4.23 (m, 4H); 4.62 (s, 2H); 4.64 (d, J = 2.2 Hz, 1H); 6.66 (d, J = 2.3 Hz, 1H); 6.84 (d, J = 8.6 Hz, 2H); 7.40 (d, J = 8.6 Hz, 2H); 8.44 (s, 1H); 9.64 (s, 1H); 12.96 (s, 1H); EIMS m/z 410 (M+). Anal. calcd for C22H22N2O6: C, 64.38; H, 5.40; N, 6.83; found: C, 64.34; H, 5.41; N, 6.84.
3.1.2.7 4′-Methoxy-7-(2-(3,5-dichlorophenylamino)-2-oxoethoxy)isoflavone (b7). Light yellow powder, 82.4%, mp 208–210 °C; 1H NMR (DMSO): 3.79 (s, 3H); 4.95 (s, 2H); 7.00 (d, J = 8.8 Hz, 2H); 7.14–7.30 (m, 2H); 7.35 (t, J = 1.9 Hz, 1H); 7.53 (d, J = 8.8 Hz, 2H); 7.75 (d, J = 1.9 Hz, 2H); 8.09 (d, J = 8.7 Hz, 1H); 8.45 (s, 1H); 10.51 (s, 1H); EIMS m/z 469 (M+). Anal. calcd for C24H17Cl2NO5: C, 61.29; H, 3.64; Cl, 15.08; N, 2.98; found: C, 61.23; H, 3.64; Cl, 15.09; N, 2.98.
3.1.2.8 4′-Methoxy-7-(2-benzylamino-2-oxoethoxy)isoflavone (b8). White powder, 57.8%, mp 195–197 °C; 1H NMR (DMSO): 3.79 (s, 3H); 4.37 (d, J = 6.1 Hz, 2H); 4.76 (s, 2H); 7.01 (d, J = 8.8 Hz, 2H); 7.13–7.37 (m, 7H); 7.54 (d, J = 8.7 Hz, 2H); 8.07 (d, J = 9.6 Hz, 1H); 8.44 (s, 1H); 8.78 (t, J = 6.1 Hz, 1H); EIMS m/z 415 (M+). Anal. calcd for C25H21NO5: C, 72.28; H, 5.10; N, 3.37; found: C, 72.23; H, 5.11; N, 3.37.
3.1.2.9 4′-Methoxy-7-(2-pyrrolidino-2-oxoethoxy)isoflavone (b9). Light yellow powder, 70.3%, mp 139–140 °C; 1H NMR (DMSO): 1.74–1.81 (m, 2H); 1.88–1.94 (m, 2H); 3.32 (t, J = 6.9 Hz, 2H); 3.45 (t, J = 6.8 Hz, 2H); 3.73 (s, 3H); 4.85 (s, 2H); 6.45 (d, J = 2.5 Hz, 1H); 6.54 (dd, J = 9.0 Hz, J = 2.5 Hz, 1H); 6.89 (d, J = 8.6 Hz, 2H); 7.22 (d, J = 8.6 Hz, 2H); 8.01 (d, J = 9.0 Hz, 1H); 8.45 (s, 1H); EIMS m/z 379 (M+). Anal. calcd for C22H21NO5: C, 69.64; H, 5.58; N, 3.69; found: C, 69.68; H, 5.57; N, 3.69.
3.1.2.10 4′-Methoxy-7-(2-(4-methylpiperazino)-2-oxoethoxy)isoflavone (b10). White powder, 76.8%, mp 178–180 °C; 1H NMR (DMSO): 2.23 (s, 3H); 2.27–2.34 (m, 2H); 2.37–2.43 (m, 2H); 3.45–3.52 (m, 4H); 3.80 (s, 3H); 5.05 (s, 2H); 7.00 (d, J = 8.8 Hz, 2H); 7.10 (dd, J = 8.9 Hz, J = 2.4 Hz, 1H); 7.15 (d, J = 2.4 Hz, 1H); 7.54 (d, J = 8.7 Hz, 2H); 8.04 (d, J = 8.9 Hz, 1H); 8.43 (s, 1H); 13C NMR (DMSO): 44.26, 46.01, 54.94, 55.60, 66.56, 102.02, 114.07, 115.49, 118.23, 123.79, 124.51, 127.30, 130.53; EIMS m/z 408 (M+). Anal. calcd for C23H24N2O5: C, 67.63; H, 5.92; N, 6.86; found: C, 67.67; H, 5.91; N, 6.85.
3.1.2.11 4′-Methoxy-7-(2-piperidino-2-oxoethoxy)isoflavone (b11). White powder, 59.8%, mp 146–148 °C; 1H NMR (DMSO): 1.41–1.47 (m, 2H); 1.51–1.65 (m, 4H); 3.38–3.46 (m, 4H); 3.80 (s, 3H); 5.04 (s, 2H); 7.01 (d, J = 8.7 Hz, 2H); 7.10 (dd, J = 8.9 Hz, J = 2.4 Hz, 1H); 7.14 (d, J = 2.3 Hz, 1H); 7.54 (d, J = 8.7 Hz, 2H); 8.04 (d, J = 8.8 Hz, 1H); 8.44 (s, 1H); EIMS m/z 393 (M+). Anal. calcd for C23H23NO5: C, 70.21; H, 5.89; N, 3.56; found: C, 70.17; H, 5.90; N, 3.56.
3.1.2.12 4′-Methoxy-7-(2-morpholino-2-oxoethoxy)isoflavone (b12). White powder, 84.6%, mp 186–188 °C; 1H NMR (DMSO): 3.45–3.51 (m, 4H); 3.57–3.64 (m, 2H); 3.64–3.68 (m, 2H); 3.80 (s, 3H); 5.07 (s, 2H); 7.01 (d, J = 8.7 Hz, 2H); 7.11 (dd, J = 8.9 Hz, J = 2.4 Hz, 1H); 7.17 (d, J = 2.4 Hz, 1H); 7.54 (d, J = 8.7 Hz, 2H); 8.04 (d, J = 8.9 Hz, 1H); 8.43 (s, 1H); EIMS m/z 395 (M+). Anal. calcd for C22H21NO6: C, 66.83; H, 5.35; N, 3.54; found: C, 66.88; H, 5.31; N, 3.54.
3.1.2.13 4′-Methoxy-7-(2-butylamino-2-oxoethoxy)isoflavone (b13). White crystal, 60.5%, mp 148–152 °C; 1H NMR (DMSO): 0.86 (t, J = 7.3 Hz, 3H); 1.28 (p, J = 7.3 Hz, 2H); 1.42 (p, J = 7.1 Hz, 2H); 3.14 (q, J = 6.7 Hz, 2H); 3.79 (s, 3H); 4.66 (s, 2H); 7.00 (d, J = 8.7 Hz, 2H); 7.14 (d, J = 1.9 Hz, 1H); 7.14 (dd, J = 8.7 Hz, J = 2.0 Hz, 1H); 7.53 (d, J = 8.7 Hz, 2H); 8.06 (d, J = 8.6 Hz, 1H); 8.21 (t, J = 5.9 Hz, 1H); 8.44 (s, 1H); EIMS m/z 381 (M+). Anal. calcd for C22H23NO5: C, 69.28; H, 6.08; N, 3.67; found: C, 69.23; H, 6.09; N, 3.67.
3.1.2.14 4′-Methoxy-7-(2-diethylamino-2-oxoethoxy)isoflavone (b14). Pink powder, 71.3%, mp 124–126 °C; 1H NMR (DMSO): 1.05 (t, J = 7.1 Hz, 6H); 3.30 (q, J = 6.9 Hz, 4H); 3.79 (s, 3H); 5.01 (s, 2H); 7.00 (d, J = 8.8 Hz, 2H); 7.09 (dd, J = 8.4 Hz, J = 2.4 Hz, 1H); 7.10 (d, J = 2.4 Hz, 1H); 7.53 (d, J = 8.7 Hz, 2H); 8.04 (d, J = 9.1 Hz, 1H); 8.43 (s, 1H); EIMS m/z 381 (M+). Anal. calcd for C22H23NO5: C, 69.28; H, 6.08; N, 3.67; found: C, 69.33; H, 6.07; N, 3.67.
3.1.2.15 4′-Methoxy-7-(2-(2-morpholino)ethylamino-2-oxoethoxy)isoflavone (b15). White powder, 75.5%, mp 178–180 °C; 1H NMR (DMSO): 2.30–2.44 (m, 6H); 3.28 (q, J = 6.5 Hz, 2H); 3.55 (t, J = 4.6 Hz, 4H); 3.80 (s, 3H); 4.69 (s, 2H); 7.01 (d, J = 8.7 Hz, 2H); 7.15 (d, J = 2.3 Hz, 1H); 7.16 (dd, J = 8.6 Hz, J = 2.4 Hz, 1H); 7.54 (d, J = 8.8 Hz, 2H); 8.08 (d, J = 9.6 Hz, 1H); 8.12 (t, J = 5.7 Hz, 1H); 8.44 (s, 1H); EIMS m/z 438 (M+). Anal. calcd for C24H26N2O6: C, 65.74; H, 5.98; N, 6.39; found: C, 65.71; H, 5.99; N, 6.40.
3.1.2.16 5-Hydroxy-7-(2-(4-methyl)piperazino-2-oxoethoxy)flavone (b16). Light yellow powder, 77.8%, mp 144–146 °C; 1H NMR (DMSO): 2.85 (s, 3H); 2.95–3.20 (m, 4H); 3.35–3.60 (m, 4H); 5.09 (s, 2H); 6.47 (d, J = 2.2 Hz, 1H); 6.85 (d, J = 2.2 Hz, 1H); 7.06 (s, 1H); 7.58–7.67 (m, 3H); 8.10 (d, J = 8.2 Hz, 2H); 12.85 (s, 1H); EIMS m/z 394 (M+). Anal. calcd for C22H22N2O5: C, 66.99; H, 5.62; N, 7.10; found: C, 66.94; H, 5.61; N, 7.11.
3.1.2.17 5-Hydroxy-7-(2-piperidino-2-oxoethoxy)flavone (b17). Yellow powder, 61.7%, mp 181–183 °C; 1H NMR (DMSO): 1.40–1.47 (m, 2H); 1.57–1.62 (m, 4H); 3.39–3.46 (m, 4H); 5.00 (s, 2H); 6.42 (d, J = 2.2 Hz, 1H); 6.82 (d, J = 2.2 Hz, 1H); 7.06 (s, 1H); 7.57–7.66 (m, 3H); 8.11 (d, J = 6.6 Hz, 2H); 12.82 (s, 1H); EIMS m/z 379 (M+). Anal. calcd for C22H21NO5: C, 69.64; H, 5.58; N, 3.69; found: C, 69.59; H, 5.59; N, 3.69.
3.1.2.18 5-Hydroxy-7-(2-pyrrolidino-2-oxoethoxy)flavone (b18). Light yellow powder, 80.6%, mp 159–161 °C; 1H NMR (DMSO): 1.79 (p, J = 6.8 Hz, 2H); 1.92 (p, J = 6.8 Hz, 2H); 3.34 (t, J = 6.8 Hz, 2H, overlapping with signals of water); 3.48 (t, J = 6.8 Hz, 2H); 4.92 (s, 2H); 6.42 (d, J = 2.2 Hz, 1H); 6.82 (d, J = 2.2 Hz, 1H); 7.06 (s, 1H); 7.57–7.66 (m, 3H); 8.11 (d, J = 6.6 Hz, 2H); 12.82 (s, 1H); EIMS m/z 365 (M+). Anal. calcd for C21H19NO5: C, 69.03; H, 5.24; N, 3.83; found: C, 69.08; H, 5.23; N, 3.83.
3.1.2.19 5-Hydroxy-7-(2-morpholino-2-oxoethoxy)flavone (b19). Yellow powder, 58.7%, mp 212–213 °C; 1H NMR (DMSO): 3.42–3.49 (m, 4H); 3.53–3.61 (m, 2H); 3.61–3.68 (m, 2H); 5.03 (s, 2H); 6.44 (d, J = 2.3 Hz, 1H); 6.82 (d, J = 2.3 Hz, 1H); 7.06 (s, 1H); 7.57–7.66 (m, 3H); 8.10 (d, J = 6.6 Hz, 2H); 12.82 (s, 1H); EIMS m/z 381 (M+). Anal. calcd for C21H19NO6: C, 66.13; H, 5.02; N, 3.67; found: C, 66.09; H, 5.03; N, 3.67.
3.1.2.20 5-Hydroxy-7-(2-(2-morpholino)ethylamino-2-oxoethoxy)flavone (b20)42. Yellow powder, 63.4%, mp 216–217 °C; 1H NMR (DMSO): 2.30–2.42 (m, 4H); 3.20–3.33 (m, 4H); 3.45–3.60 (m, 4H); 4.67 (s, 2H); 6.46 (d, J = 2.2 Hz, 1H); 6.85 (d, J = 2.3 Hz, 1H); 7.08 (s, 1H); 7.58–7.67 (m, 3H); 8.11 (d, J = 6.6 Hz, 2H); 8.85 (bs, 1H); 12.83 (s, 1H); EIMS m/z 424 (M+). Anal. calcd for C23H24N2O6: C, 65.08; H, 5.70; N, 6.60; found: C, 65.11; H, 5.69; N, 6.59.
3.2 Extraction of the TyrRS and enzyme assay
TyrRS was over-expressed in E. coli and purified to near homogeneity (∼98% as judged by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis)) using standard purification procedures. TyrRS activity was measured by aminoacylation using modifications to previously described methods.43 The assays were performed at 37 °C in a mixture containing (final concentrations) 100 mM Tris/Cl pH 7.9, 50 mM KCl, 16 mM MgCl2, 5 mM ATP (adenosine triphosphate), 3 mM DTT (DL-dithiothreitol), 4 mg mL−1 E. coli MRE600 tRNA (Roche) and 10 μM L-tyrosine (0.3 μM L-[ring-3,5-3H] tyrosine (PerkinElmer, specific activity: 1.48–2.22 TBq mmol−1), 10 μM carrier). TyrRS (0.2 nM) was preincubated with a range of inhibitor concentrations for 10 min at room temperature followed by the addition of pre-warmed mixture at 37 °C. After specific intervals, the reaction was terminated by adding aliquots of the reaction mix into ice-cold 7% trichloroacetic acid and harvesting onto 0.45 mm hydrophilic Durapore filters (Millipore Multiscreen 96-well plates) and counted by liquid scintillation. The rate of reaction in the experiments was linear with respect to protein and time with less than 50% total tRNA acylation. IC50s correspond to the concentration at which half of the enzyme activity is inhibited by the compound. The results are presented in Table 1.3.3 Antimicrobial activity
The antibacterial activities of the synthesized compounds were tested against Gram-positive bacterial strain (Staphylococcus aureus ATCC 6538, penicillin G sodium as positive control) and two Gram-negative bacterial strains (Escherichia coli ATCC 8739 and Pseudomonas aeruginosa ATCC 9027, ciprofloxacin as positive control). The MTT (methylthiazolyldiphenyl-tetrazolium bromide) proliferation assay was used to measure the MIC50s of the test compounds by determining the absorbance of the cells in culture.44 A stock solution of the synthesized compound (1000 μg mL−1) in DMSO was prepared with different concentrations using sterilized liquid medium (50% (v/v) of DMSO in PBS (phosphate-buffered saline)). A specified quantity of the medium containing the test compound was added into 96-well plates, which was replaced by the sterilized liquid medium as blank control. Suspension of the microorganism was prepared to contain approximate 105 cfu mL−1 and applied to 96-well plates with serially diluted compounds (or blank control) to be tested and incubated at 37 °C. In the case of fungi, plates were incubated at 28 °C. Fifty μL of PBS containing 3 mg of MTT per mL was added to each well. Incubation was continued for 4–5 h, then 100 μL of 10% sodium dodecyl sulfate containing 5% isopropanol and 10 mol mL−1 HCl was added to extract the dye. After 8–10 h of incubation, the control OD (optical density) value (at 570 nm) should be controlled under 1.0–2.0 to achieve reliable results. The observed MIC50s were presented in Table 2.3.4 Cytotoxicity assay45
Cytotoxicity of selected compounds was assessed against an HeLa cell line. The cells were grown in a 96-well plate in DMEM media supplemented with 10% fetal bovine serum and 5% penicillin–streptomycin until they reached 70–80% confluency. The cells were then treated with serially diluted compounds. Two controls were made, one containing no cells (blank control) and the other control containing cells with no compounds (control). Cells were incubated for 24 h at 37 °C under 5% CO2 atmosphere. After 24 h, the supernatant was carefully removed and 100 μL of 5 mg mL−1 concentration MTT solution was added to each well. The plate was incubated for 3 h at 37 °C under 5% CO2 atmosphere. The cells were then treated with 100 μL of DMSO to solubilize formazan crystals. The plate was then read at 570 nm. The quantity of viable cells after treatment with each compound was expressed as a percentage of the control.3.5 Protocol of docking study
The X-ray structure of TyrRS from Bacillus stearothermophilus was downloaded from the Protein Data Bank (PDB code: 3ts1)24 and was modified by adding hydrogen atoms and removing water as well as cocrystallized substrate (tyrosinyl adenylate) using SYBYL-X version 2.1.1 software suite (Tripos, Inc., St. Louis, MO).27 The active site was defined as all the amino acid residues confined within a 5 Å radius sphere centered about tyrosinyl adenylate, and the composite structure without original ligand was utilized as the in silico model for docking studies. Default parameters and values within the minimization dialogue were used except where otherwise mentioned. The docked conformations of ligands were evaluated and ranked using Surflex-Dock and four scoring functions implemented in the Cscore software module within the SYBYL-X environment. The Cscore module allowed consensus scoring that integrated multiple well-known scoring functions such as ChemScore, D-Score, G-Score and PMF-Score to evaluate docked ligand conformations.464. Conclusions
A series of flavonoid derivatives was designed as TyrRS inhibitors based on scaffold hopping drug design. They were generated by Library Design software module within the SYBYL-X environment. Twenty compounds with top scores were obtained, and nineteen of them were reported for the first time. Enzyme assays identified three compounds showing excellent inhibitory activities against TyrRS with IC50 lower than 1 μM. In general, all the assayed compounds exhibited better activity against Gram-negative organism than Gram-positive organism. Furthermore, compounds with good inhibition against TyrRS also exhibit good antibacterial activities. Out of these compounds, b1 is the most active, which structurally containing a nargenin core with MIC50 of 0.11 μg mL−1 against E. coli ATCC 8739 shows 6-fold more potent than marketed antibiotic ciprofloxacin, and further investigations are being carried out in our laboratory. These findings together with molecular docking studies revealed that antibacterial activities of these flavonoid based amides may be caused by their inhibition of TyrRS, and compounds with a naringenin core are worth to be modified for discovery of novel antibacterial agents.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
The work was financed by grants from National Natural Science Foundation of China (grant No. 21262013 and 81273382), by a Project supported by Hunan Provincial Natural Science Foundation of China (grant No. 2015JJ2116).Notes and references
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- P. Fernandes, Nat. Biotechnol., 2006, 24(12), 1497–1503 CrossRef CAS PubMed.
- F. Fontaine, A. Hequet, A. S. Voisin-Chiret, B. Alexandre, L. Aurélien, C. Thierry, J. Claude and R. Sylvain, J. Med. Chem., 2014, 57(6), 2536–2548 CrossRef CAS PubMed.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6(1), 29–40 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325(5944), 1089–1093 CrossRef CAS PubMed.
- S. P. Kawatkar, T. A. Keating, N. B. Olivier, J. N. Breen, O. M. Green, S. Y. Guler, M. F. Hentemann, J. T. Loch, A. R. McKenzie, J. V. Newman, L. J. Otterson and G. Martinez-Botella, J. Med. Chem., 2014, 57(11), 4584–4597 CrossRef CAS PubMed.
- E. Klein, D. L. Smith and R. Laxminarayan, Emerging Infect. Dis., 2007, 13(12), 1840–1846 CrossRef PubMed.
- T. Lam, M. T. Hilgers, M. L. Cunningham, B. P. Kwan, K. J. Nelson, V. Brow-Driver, V. Ong, M. Trzoss, G. Hough, K. G. Shaw and J. Finn, J. Med. Chem., 2014, 57(3), 651–668 CrossRef CAS PubMed.
- A. K. Verma and R. Pratap, Tetrahedron, 2012, 68(41), 8523–8538 CrossRef CAS.
- R. Landolfi, R. L. Mower and M. Steiner, Biochem. Pharmacol., 1984, 33(9), 1525–1530 CrossRef CAS PubMed.
- P. A. Berg and P. T. Daniel, Prog. Clin. Biol. Res., 1988, 280, 157–171 CAS.
- S. Gupta, F. Afaq and H. Mukhtar, Biochem. Biophys. Res. Commun., 2001, 287(4), 914–920 CrossRef CAS PubMed.
- L. C. Chiang, L. T. Ng, I. C. Lin, P. L. Kuo and C. C. Lin, Cancer Lett., 2006, 237(2), 207–214 CrossRef CAS PubMed.
- P. W. Snijman, E. Joubert, D. Ferreira, X. C. Li, Y. Q. Ding, I. R. Green and W. C. A. Gelderblom, J. Agric. Food Chem., 2009, 57(15), 6678–6684 CrossRef CAS PubMed.
- M. Dueñas, S. González-Manzano, A. González-Paramás and C. Santos-Buelga, J. Pharm. Biomed. Anal., 2010, 51(2), 443–449 CrossRef PubMed.
- H. Gao, H. Wang and J. Peng, Cell Biochem. Biophys., 2014, 69(1), 27–34 CrossRef CAS PubMed.
- L. Tang, Q. Feng, J. Zhao, L. N. Dong, W. Liu, C. H. Yang and Z. Q. Liu, Food Chem. Toxicol., 2012, 50(5), 1460–1467 CrossRef CAS PubMed.
- M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed.
- T. J. Bullwinkle and M. Ibba, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(8), 2252–2257 CrossRef CAS PubMed.
- Z. P. Xiao, W. Wei, P. F Wang, W. K. Shi, N. Zhu, M. Q. Xie, Y. W. Sun, L. X. Li, Y. X. Xie, L. S. Zhu, N. Tang, H. OuYang, X. H. Li, G. C. Wang and H. L. Zhu, Eur. J. Med. Chem., 2015, 102, 631–638 CrossRef CAS PubMed.
- P. V. S. Mohana, R. Sankaranarayanan, M. Udayakumar and S. Thamotharan, Res. J. Pharm., Biol. Chem. Sci., 2015, 6(3), 1737–1743 Search PubMed.
- X. D. Wang, R. C. Deng, J. J. Dong, Z. Y. Peng, X. M. Gao, S. T. Li, W. Q. Lin, C. L. Lu, Z. P. Xiao and H. L. Zhu, Bioorg. Med. Chem., 2013, 21(17), 4914–4922 CrossRef CAS PubMed.
- X. D. Wang, W. Wei, P. F. Wang, Y. T. Tang, R. C. Deng, B. Li, S. S. Zhou, J. W. Zhang, L. Zhang, Z. P. Xiao, H. Ou-Yang and H. L. Zhu, Bioorg. Med. Chem., 2014, 22(14), 3620–3628 CrossRef CAS PubMed.
- P. Brick, T. N. Bhat and D. M. Blow, J. Mol. Biol., 1989, 208(1), 83–98 CrossRef CAS PubMed.
- Z. P. Xiao, X. B. He, Z. Y. Peng, T. J. Xiong, J. Peng, L. H. Chen and H. L. Zhu, Bioorg. Med. Chem., 2011, 19(5), 1571–1579 CrossRef CAS PubMed.
- W. Wei, W. K. Shi, P. F. Wang, X. T. Zeng, P. Li, J. R. Zhang, Q. Li, Z. P. Tang, J. Peng, L. Z. Wu, M. Q. Xie, C. Liu, X. H. Li, Y. C. Wang, Z. P. Xiao and H. L. Zhu, Bioorg. Med. Chem., 2015, 23(20), 6602–6611 CrossRef CAS PubMed.
- SYBYL Molecular Modeling Software, version SYBYL-X 2.1, Tripos Inc., St. Louis, MO, 2015 Search PubMed.
- E. R. Lee, Y. J. Kang, H. Y. Choi, G. H. Kang, J. H. Kim, B. W. Kim, Y. S. Han, S. Y. Nah and H. D. Paik, Biol. Pharm. Bull., 2007, 30, 2394–2398 CAS.
- M. Pal, R. Dakarapu, K. Parasuraman, V. Subramanian and K. R. Yeleswarapu, J. Org. Chem., 2005, 70, 7179–7187 CrossRef CAS PubMed.
- K. Dahlén, M. Grøtli and K. Luthman, Synlett, 2006, 897–900 Search PubMed.
- M. Pal, V. Subramanian, K. Parasuraman and K. R. Yeleswarapu, Tetrahedron, 2003, 59, 9563–9570 CrossRef CAS.
- G. J. Nan, J. Shi, Y. R. Huang, J. Sun, J. H. Lv, G. D. Yang and Y. P. Li, J. Chem. Eng. Data, 2014, 59, 1304–1311 CrossRef CAS.
- I. Kron, Z. Pudychová-Chovanová, B. Veliká, J. Guzy and P. Perjési, Monatsh. Chem., 2012, 143, 13–17 CrossRef CAS.
- A. Dhainaut, G. Lewin, E. Canet, M. Lonchampt and Y. Rolland, US 5889003, 1999.
- M. Bouktaib, S. Lebrun, A. Atmani and C. Rolando, Tetrahedron, 2002, 58, 10001–10009 CrossRef CAS.
- Q. G. Mei, C. Wang, W. C. Yuan and G. L. Zhang, Beilstein J. Org. Chem., 2015, 11, 288–293 CrossRef CAS PubMed.
- N. Ohta, G. Kuwata, H. Akahori and T. Watanabe, Agric. Biol. Chem., 1979, 43(7), 1415–1419 CAS.
- J. M. Hastings, M. K. Hadden and B. S. J. Blagg, J. Org. Chem., 2008, 73, 369–373 CrossRef CAS PubMed.
- M. F. Oldfield, L. Chen and N. P. Botting, J. Labelled Compd. Radiopharm., 2007, 50, 1266–1271 CrossRef CAS.
- S.-T. Chung, Y.-T. Huang, H.-Y. Hsiung, W.-H. Huang, C.-W. Yao and A.-R. Lee, Chem. Biodiversity, 2015, 12(4), 685–696 CAS.
- J. D. Williams, M. C. Torhan, V. R. Neelagiri, C. Brown, N. O. Bowlin, M. Di, C. T. McCarthy, D. Aiello, N. P. Peet, T. L. Bowlin and D. T. Moir, Bioorg. Med. Chem., 2015, 23(5), 1027–1043 CrossRef CAS PubMed.
- H. Choe, J. Kim and S. Hong, Bioorg. Med. Chem. Lett., 2013, 23, 4324–4327 CrossRef CAS PubMed.
- G. P. Sang, P. Schimmel and S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11043–11049 CrossRef PubMed.
- X.-D. Wang, R.-C. Deng, Y. Liu, B. Li, S. Huang, H. Ouyang and Z.-P. Xiao, Asian J. Chem., 2014, 26, 8015–8018 CrossRef.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS PubMed.
- M. Hassani, W. Cai, K. H. Koelsch, D. C. Holley, A. S. Rose, F. Olang, J. P. Lineswala, W. G. Holloway, J. M. Gerdes, M. Behforouz and H. D. Beall, J. Med. Chem., 2008, 51, 3104–3115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28061g |
| This journal is © The Royal Society of Chemistry 2017 |