
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoreductive synthesis of monodispersed Au nanoparticles with citric acid as reductant and surface stabilizing reagent†
Yasuhiro Shiraishi *ab,
Haruki Tanakaa,
Hirokatsu Sakamotoa,
Satoshi Ichikawac and
Takayuki Hiraia
*ab,
Haruki Tanakaa,
Hirokatsu Sakamotoa,
Satoshi Ichikawac and
Takayuki Hiraia
aResearch Center for Solar Energy Chemistry and Division of Chemical Engineering, Graduate School of Engineering Science, Osaka University, Toyonaka 560-8531, Japan. E-mail: shiraish@cheng.es.osaka-u.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Saitama 332-0012, Japan
cInstitute for NanoScience Design, Osaka University, Toyonaka 560-8531, Japan
First published on 18th January 2017
Abstract
Colloidal suspensions of gold nanoparticles (AuNPs) in water can be prepared by heating water containing HAuCl4 with citric acid as reductant and surface stabilizing reagent. This thermal reduction method, however, promotes aggregation of the formed AuNPs, resulting in bimodal distribution of AuNPs. Here we report that UV (254 nm) irradiation of water containing HAuCl4 with citric acid at room temperature successfully reduces HAuCl4 and produces monodispersed AuNPs, while suppressing aggregation of the formed AuNPs. Changing the intensity of UV light or the amount of citric acid added successfully produces AuNPs with tunable sizes and narrow size distributions.
Introduction
Gold nanoparticles (AuNPs) have attracted much attention due to their unique optical,1 electronic,2 and chemical properties3 that differ significantly from those of bulk Au. Of particular interest is focused on application to cancer diagnosis, therapy, and bioimaging because of their localized surface plasmon resonance (LSPR) enhanced light scattering and absorption properties.4 In addition, due to the high surface-to-volume ratio, high surface energy, and strong LSPR absorption in the visible wavelength region, AuNPs often exhibit unusual catalytic,5 photocatalytic,6,7 or electronic properties.8 These properties strongly depend on the size and dispersibility of AuNPs.9 Preparation of monodispersed AuNPs and their accurate size control, therefore, poses a challenge for advanced processing.Thermal reduction of Au3+ is a common approach for the preparation of colloidal suspension of AuNPs. Ascorbic acid10 and NaBH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11–13 have often been used as a reductant, but these methods need surface stabilizing reagent. Thermal reduction with trisodium citrate, the so-called Turkevich method,14–19 is the simplest and most popular procedure for the preparation of AuNPs in water because trisodium citrate act as both reductant and surface stabilizing reagent. The Na+ cations in the molecule, however, contaminate the formed AuNPs.20 The use of metal-free reductant is therefore necessary. Citric acid is one potential reductant for this purpose:21 simple stirring of water containing HAuCl4 with citric acid at elevated temperature successfully produces AuNPs. This method, however, inevitably promotes thermal aggregation of the formed AuNPs due to the weak adsorption of citric acid onto the AuNPs surface.21 Reduction of HAuCl4 at relatively low temperature is therefore necessary for the preparation of monodispersed AuNPs.
11–13 have often been used as a reductant, but these methods need surface stabilizing reagent. Thermal reduction with trisodium citrate, the so-called Turkevich method,14–19 is the simplest and most popular procedure for the preparation of AuNPs in water because trisodium citrate act as both reductant and surface stabilizing reagent. The Na+ cations in the molecule, however, contaminate the formed AuNPs.20 The use of metal-free reductant is therefore necessary. Citric acid is one potential reductant for this purpose:21 simple stirring of water containing HAuCl4 with citric acid at elevated temperature successfully produces AuNPs. This method, however, inevitably promotes thermal aggregation of the formed AuNPs due to the weak adsorption of citric acid onto the AuNPs surface.21 Reduction of HAuCl4 at relatively low temperature is therefore necessary for the preparation of monodispersed AuNPs.
Recently, photoreduction of Au3+ has also received much attention as an alternative method for AuNPs synthesis because it can be operated at room temperature by UV irradiation. Many of the reported photoreduction methods need both reductants and surface stabilizing reagents.22–29 Although some systems can use single chemicals behaving as both reductant and surface stabilizing reagent such as ionic liquids, dendrimers, polyvinylpyrrolidone, and trisodium citrate, they are relatively expensive or contain Na+ cation contaminant.30–33 Yang et al.34 used citric acid for photoreductive synthesis of AuNPs. They found that UV irradiation of water containing HAuCl4 and citric acid successfully reduces Au3+ and produces AuNPs. They also studied the pH effects on the rate of AuNPs formation by UV-vis absorption spectroscopy and the size of AuNPs by transmission electron microscopy (TEM) observations, but did not clarify the dispersibility of the formed AuNPs.
In the present work, we studied photoreductive synthesis of AuNPs in pure water with citric acid by UV (254 nm) irradiation at room temperature. The size and dispersibility of AuNPs were studied by TEM and dynamic laser scattering (DLS) analysis and were compared with those obtained by thermal reduction. We found that this photoreduction method successfully produces monodispersed AuNPs, while suppressing subsequent aggregation of the formed AuNPs. We also report here that changing the intensity of UV light and the amount of citric acid successfully produces monodispersed AuNPs with tunable sizes and narrow size distributions.
Results and discussion
Thermal reduction
AuNPs were prepared with citric acid by conventional thermal reduction method. A water (2 mL) containing HAuCl4·4H2O (0.2 mM) and citric acid (0.5 mM) was stirred at 60 °C in a quartz cell within a Peltier temperature controller. Fig. 1a shows time-dependent change in absorption spectra of the solution. A distinctive LSPR band of AuNPs appears at 534 nm and increases with time. Simultaneously, red-shifted absorption band at λ > 600 nm, assigned to interparticle-coupled plasmon excitons of aggregated AuNPs, increases.35 This suggests that AuNPs formed by thermal reduction of HAuCl4 are subsequently aggregated.21,36 It is noted that the obtained solution maintain its transparency at least 2 days, where a precipitate formation scarcely occurs. Fig. 1b (blue) shows hydrodynamic diameter of AuNPs prepared at 60 °C by 25 min stirring, measured by DLS analysis. Bimodal particle distributions with average diameters ca. 40 nm and 150 nm were observed. This indicates that ca. 40 nm AuNPs formed by thermal reduction of HAuCl4 are subsequently aggregated and produce ca. 150 nm aggregates. Fig. 1c shows typical TEM images of the particles. The TEM samples were prepared by dipping a microgrid into the solution (2 s) followed by drying under air (10 s) and in vacuo (30 min). The sizes of AuNPs and their aggregates are ca. 40 nm and 150 nm, respectively, which agree with the DLS results (Fig. 1b).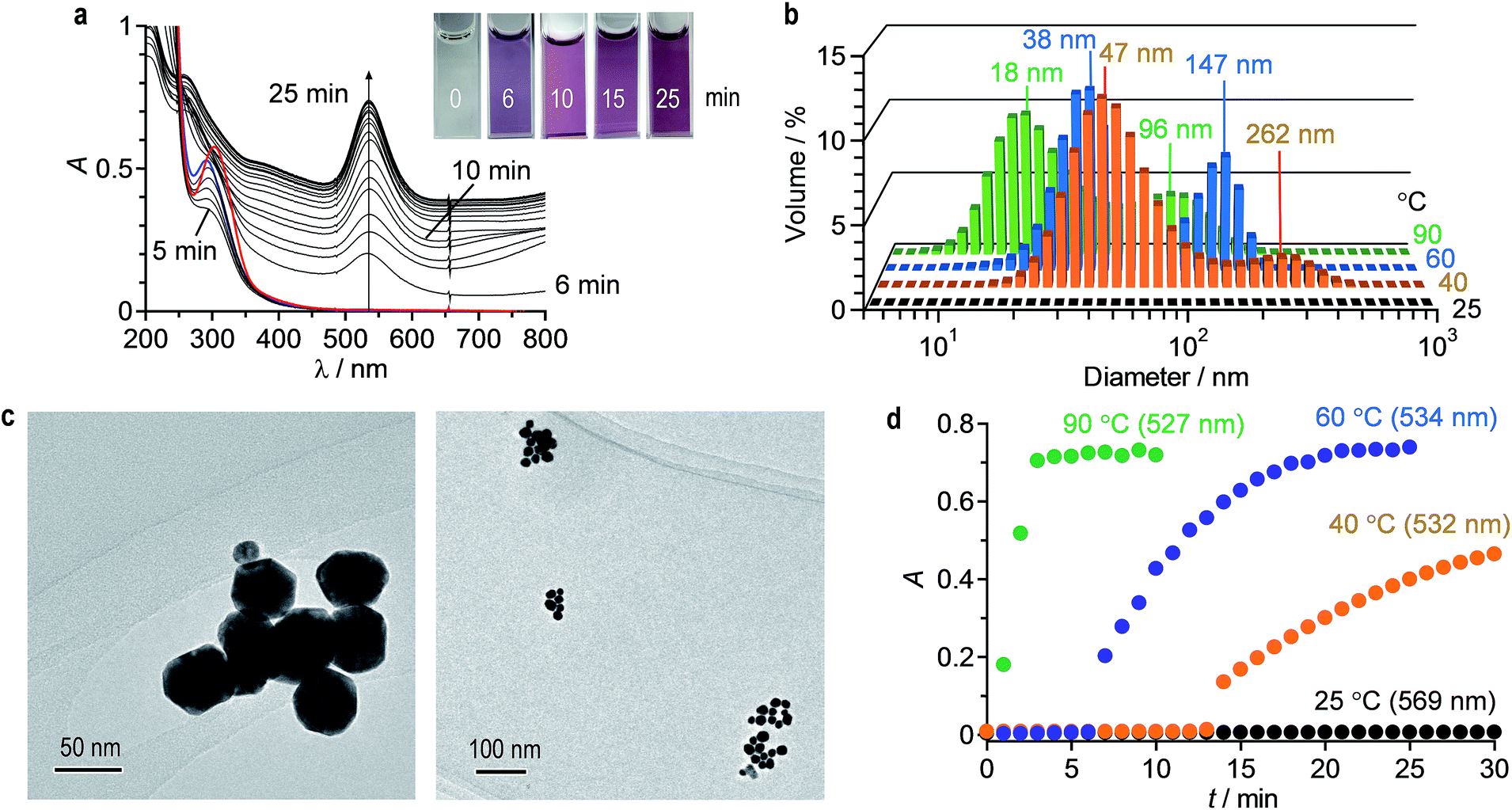 | ||
| Fig. 1 (a) Time-dependent change in absorption spectra of water containing HAuCl4·4H2O (0.2 mM) and citric acid (0.5 mM) during stirring at 60 °C in the dark. The red is the spectrum for the solution containing HAuCl4·4H2O at 25 °C, and the blue is the spectrum for the solution after addition of citric acid (1 min). (b) Hydrodynamic diameter of the solution obtained by stirring in the dark at 90 °C (10 min), 60 °C (25 min), 40 °C (30 min), and 25 °C (30 min), measured by DLS analysis. (c) Typical TEM images of the solution obtained by 25 min stirring at 60 °C in the dark. (d) Time-dependent change in the LSPR absorbance of the solutions during stirring at different temperatures in the dark. | ||
Thermal reduction of HAuCl4 was performed at 90 °C, 40 °C, and 25 °C, respectively. As shown in Fig. S1 (ESI†), stirring at 90 °C and 40 °C shows LSPR band of AuNPs as well as red-shifted band of the aggregates, as is the case at 60 °C (Fig. 1a). In addition, as shown in Fig. 1b, DLS analysis of these solutions also shows bimodal particle distributions, indicating that AuNPs undergo thermal aggregation even at 40 °C. However, as shown in Fig. S1 (ESI†), stirring at 25 °C scarcely increases the LSPR band. As shown in Fig. 1d (black), LSPR absorbance scarcely increases even after 30 min stirring, indicating that HAuCl4 is not reduced at 25 °C. These data indicate that the thermal reduction method is difficult to prepare monodispersed AuNPs while suppressing subsequent their aggregation.
Photoreduction
AuNPs were prepared by the photoreduction method. Water (2 mL) containing HAuCl4·4H2O (0.2 mM) and citric acid (0.5 mM) was stirred at 25 °C under irradiation of 254 nm light by a Xe lamp (light intensity: 150 mW m−2). Fig. 2a shows the time-dependent change in absorption spectra of the solution. As is the case for thermal reduction (Fig. 1a), LSPR absorption of AuNPs appears at 530 nm and increases with irradiation time. In this case, red-shifted absorption scarcely appears, indicating that the formed AuNPs scarcely aggregate. It must also be noted that the obtained solution maintain its transparency at least 2 days, where a precipitate formation scarcely occurs, as is the case for the thermal reduction. As shown in Fig. 2b, DLS analysis shows unimodal distribution of AuNPs with an average diameter ca. 50 nm, where aggregated AuNPs are scarcely observed. This suggests that photoreduction method scarcely promotes aggregation of the formed AuNPs. TEM observations confirm this. As show in Fig. 2c (right side), the TEM images obtained by 100 min stirring of the solution show well-dispersed ca. 50 nm AuNPs, where large aggregates are scarcely formed. As shown in Fig. S2 (ESI†), X-ray photoelectron spectroscopy (XPS) analysis of the AuNPs prepared by both thermal- and photoreduction methods exhibits characteristic Au 4f5/2 and 4f7/2 peaks at ca. 84.3 and 88.0 eV, respectively, assigned to Au0. In that, a component assigned to Au3+ or Au+ is scarcely observed at 85–86 eV,37,38 indicating that these cations are not contained in the obtained AuNPs.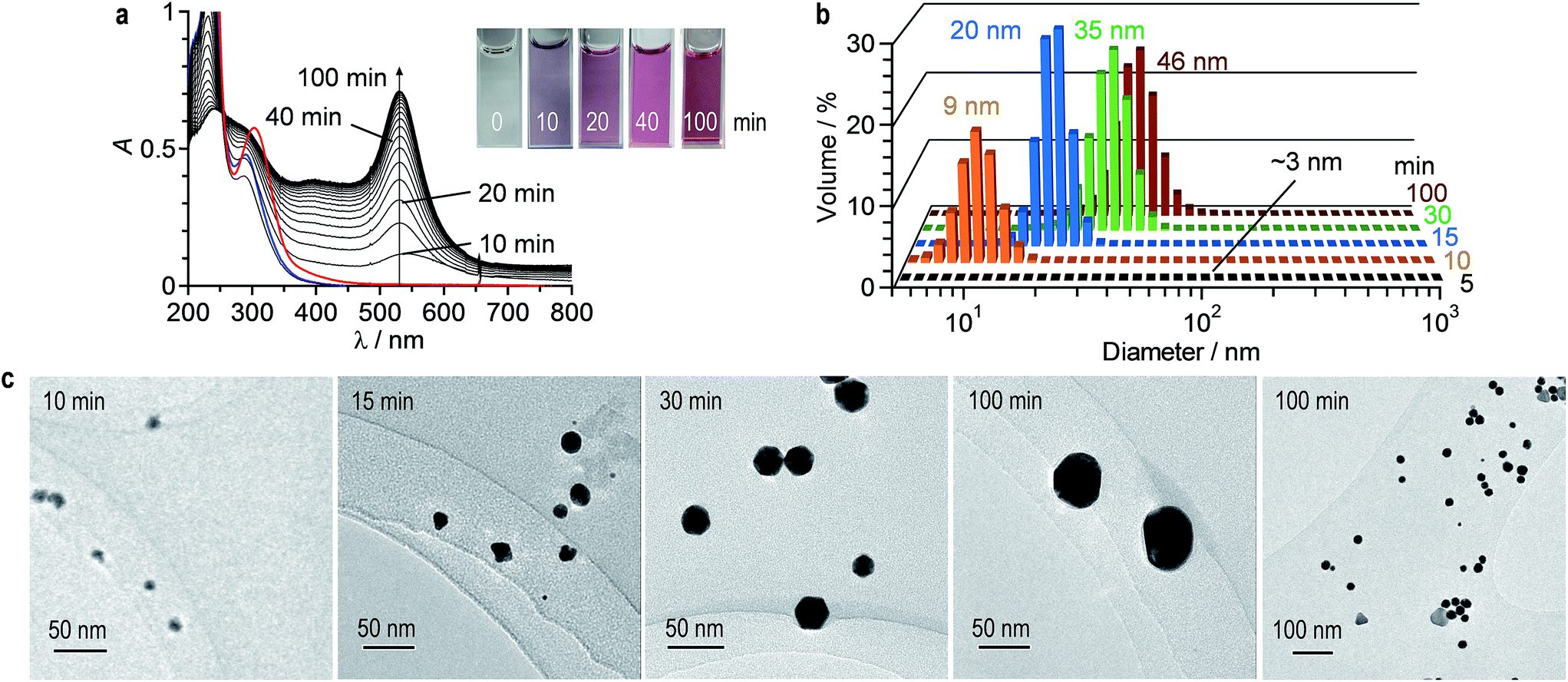 | ||
| Fig. 2 (a) Time-dependent change in absorption spectra of water containing HAuCl4·4H2O (0.2 mM) and citric acid (0.5 mM) during stirring at 25 °C under 254 nm irradiation (light intensity: 150 mW m−2). The red is the spectrum of a solution containing HAuCl4·4H2O at 25 °C, and the blue is the spectrum for the solution after addition of citric acid (1 min). (b) Hydrodynamic diameter and (c) TEM images of AuNPs obtained after different photoirradiation time. | ||
Mechanism for AuNPs formation by thermal reduction
It is well known that thermal reduction creates AuNPs by the nucleation and growth processes.34 As shown in Scheme 1, coordination of citric acid with Au3+Cl4− produces a citrate–Au3+Cl3− complex.14 Absorption spectra confirm this. As shown by the red line in Fig. 1a, the ligand-to-metal charge transfer (LMCT) band of AuCl4− appears at 303 nm.39 As shown by the blue line, addition of citric acid to this solution rapidly leads to blue shift of this band to 290 nm within 1 min due to the formation of a citrate–Au3+Cl3− complex.14 Heating the solution at 60 °C decreases this band, along with an increase in the LSPR band of AuNPs. This indicates that, as shown in Scheme 1, thermal activation of the citrate–Au3+Cl3− produces a reduced Au2+Clnm− species, along with a formation of oxidation products of citric acid such as dicarboxyacetone, acetoacetate, and CO2.40–44 Subsequent reduction of the formed citrate–Au2+Clnm− complex followed by the reduction of citrate–Au+Clnm− complex produce Au0, resulting in the formation of Au0 nuclei. Further adsorption of Au0 onto the nuclei leads to a growth of AuNPs.34 In that, the citrate anions are adsorbed onto the AuNPs surface and behave as a surface stabilizing reagent.45 Negative charge of these surface anions suppresses aggregation of AuNPs due to the electrostatic repulsion.46 A rise in temperature, however, weakens the adsorption of these anions, inevitably promoting aggregation of AuNPs.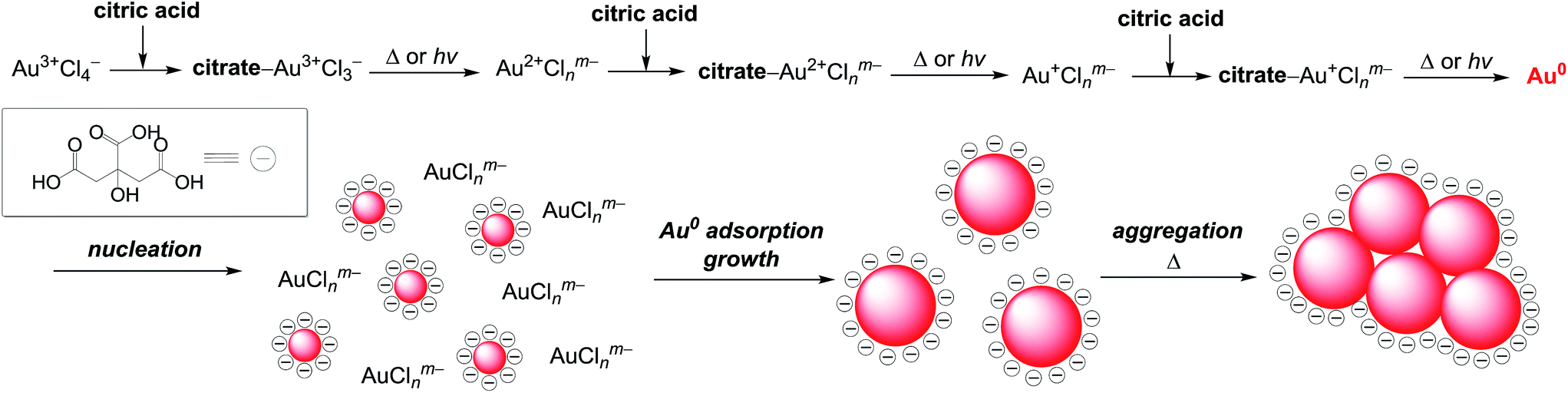 | ||
| Scheme 1 Mechanism for the formation of AuNPs by thermal reduction and photoreduction methods. | ||
Mechanism for AuNPs formation by photoreduction
In the photoreduction method, AuNPs are formed by the mechanism similar to that of thermal reduction (Scheme 1). As shown in Fig. 2a, UV irradiation of the citrate–Au3+Cl3− complex leads to a decrease in its LMCT band (blue line), along with a formation of LSPR absorption, as is the case for thermal reduction (Fig. 1a). GC analysis of the gas phase after 100 min photoirradiation detects the formation of CO2. The AuNPs in the solution were removed by centrifugation. The obtained solution was concentrated by evaporation and dissolved in DMSO-d6. As shown in Fig. S3 (ESI†), 1H NMR analysis of the solution shows a formation of acetoacetate and dicarboxyacetone as the oxidation products of citric acid. These findings indicate that, as shown in Scheme 1 (top), consecutive photoexcitation of the citrate–AuClnm− complexes reduces Au3+ to Au0 with the oxidation of citric acid, as is the case for thermal activation.40–44 Similar mechanism has been proposed in the photoreduction of some metal cations such as Ag+ and Fe3+ with citric acid by UV irradiation.47,48As shown in Scheme 1 (bottom), the formation of Au0 nuclei and their growth produce AuNPs, as is the case for thermal reduction. As shown in Fig. 2a, photoirradiation for 5 min scarcely leads to an increase in the LSPR absorption, where AuNPs are scarcely observed by DLS analysis (Fig. 2b; detection limit: 3 nm). Photoirradiation for 10 min creates a LSPR band, where small (∼10 nm) AuNPs are observed by DLS (Fig. 2b) and TEM (Fig. 2c, left). Further photoirradiation creates larger AuNPs; DLS and TEM confirm the growth of AuNPs. These data suggest that, as shown in Scheme 1, the photoreduction method produces AuNPs via the nucleation and growth mechanism in a manner similar to that of thermal reduction. Photoirradiation for 100 min finally produces ca. 50 nm AuNPs. At this temperature (25 °C), the adsorbed citric acid molecules sufficiently stabilize the particles and suppress their aggregation, resulting in the formation of monodispersed AuNPs.
Size control of AuNPs by photoreduction
It is well known that, in the thermal reduction, the size of AuNPs depends strongly on the rate of AuClnm− reduction.49 Increased rate of AuClnm− reduction creates a large number of Au0 nuclei and decreases the number of AuClnm− remaining in solution. This suppresses the growth of the particles and creates smaller AuNPs. As shown in Fig. 1d, stirring of a HAuCl4 solution with citric acid at higher temperature accelerates the increase in the LSPR absorption. In addition, as shown in Fig. 1b, the size of AuNPs becomes smaller at higher temperature. This is because, at higher temperature, enhanced reduction of AuClnm− creates larger number of Au0 nuclei and produces smaller AuNPs.50In the photoreduction, the rate of AuClnm− reduction can be controlled by the intensity of UV light and the amount of citric acid. Fig. 3a shows effect of the light intensity on the time-dependent change in the LSPR absorbance of the solution. The increased light intensity increases the absorbance more rapidly because it enhances photoexcitation of the citrate–AuClnm− complexes and promotes rapid reduction to Au0. Fig. 3b shows the size of AuNPs prepared at different light intensity. The increased light intensity indeed creates smaller AuNPs, where their aggregation does not occur. In this case, monodispersed 50–70 nm AuNPs can be created while maintaining narrow size distribution with <23% standard deviation.
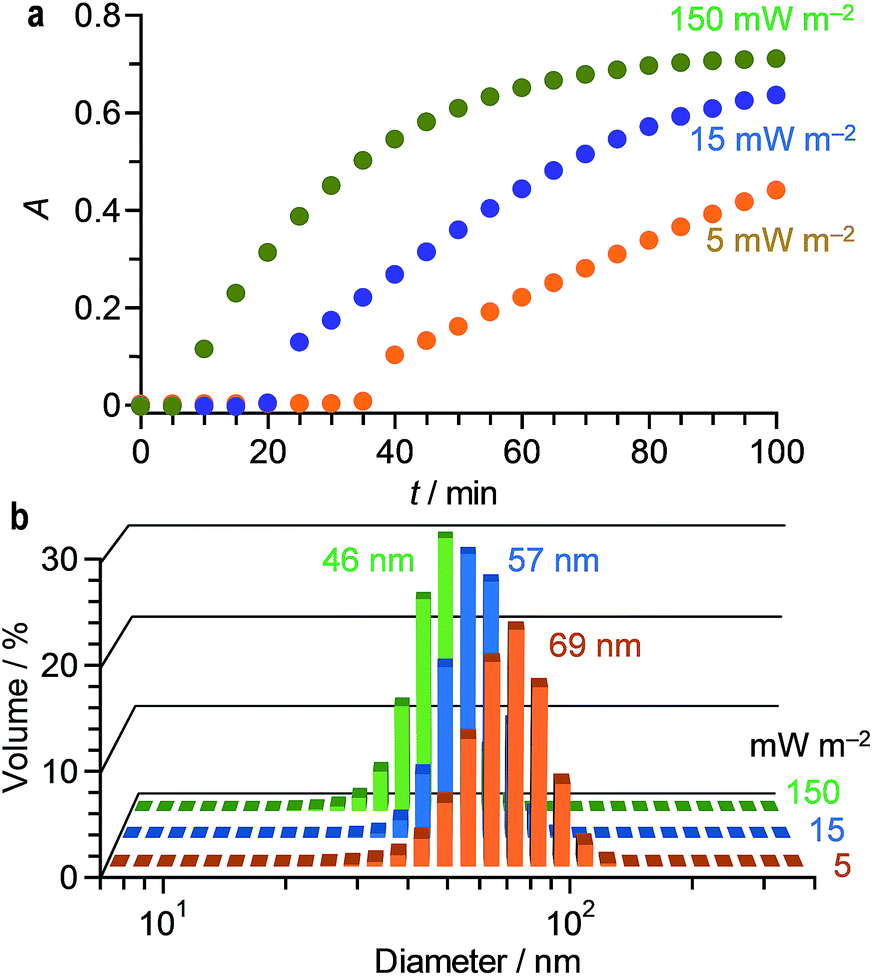 | ||
| Fig. 3 (a) Time-dependent change in the LSPR absorbance of water containing HAuCl4·4H2O (0.2 mM) and citric acid (0.5 mM) during stirring at 25 °C under 254 nm irradiation at different light intensity. Changes in absorption spectra are summarized in Fig. S4 (ESI†). (b) Hydrodynamic diameter of AuNPs measured by DLS analysis after photoirradiation at different light intensity for 100 min. | ||
It is also noted that the rate of photoreduction depends on the wavelength of incident light; UV irradiation is necessary. As shown in Fig. S6 (ESI†), the rate of LSPR absorption increase decreases by photoexcitation with longer wavelength light (254 > 360 > 510 nm), where the photoexcitation by 510 nm light scarcely increases the LSPR absorbance. This is because, as shown by the blue line in Fig. 2a, the absorbance of the citrate–AuClnm− complexes decreases at longer wavelengths and scarcely absorbs light in the visible region.
Fig. 4a shows effect of the citric acid amount on the time-dependent change in the LSPR absorbance of the solution. The increased amount of citric acid rapidly increases the LSPR absorbance because it produces a larger number of citrate–AuClnm− complexes and promotes rapid formation of larger number of Au0 nuclei. Therefore, as shown in Fig. 4b and c, the increased amount of citric acid produces smaller AuNPs, where their aggregation does not occur. Changing the citric acid amount successfully creates monodispersed AuNPs with 22–70 nm diameters, while maintaining narrow size distribution with <54% standard deviation.
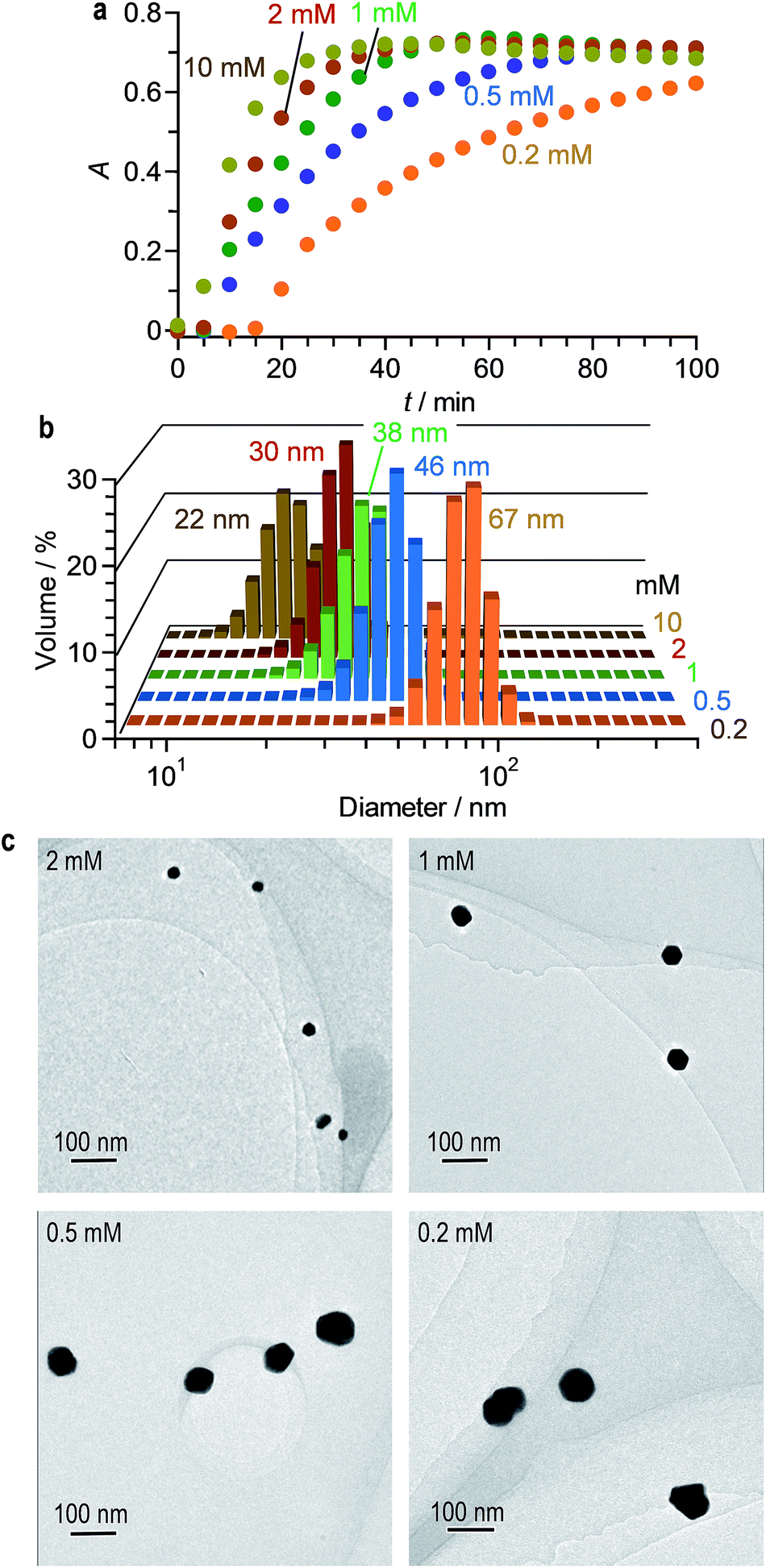 | ||
| Fig. 4 (a) Time-dependent change in absorption spectra of water containing HAuCl4·4H2O (0.2 mM) and different amount of citric acid during stirring at 25 °C under 254 nm irradiation (light intensity: 150 mW m−2). Changes in absorption spectra are summarized in Fig. S5 (ESI†). (b) Hydrodynamic diameter and (c) TEM images of AuNPs obtained after photoirradiation for 100 min with different citric acid amount. | ||
Conclusion
We found a simple method for preparation of monodispersed AuNPs. UV irradiation (254 nm) of water containing HAuCl4 and citric acid at room temperature successfully produces monodispersed AuNPs, while suppressing aggregation of the formed AuNPs. Changing the light intensity or the amount of citric acid successfully produces AuNPs with tunable sizes (20–70 nm) and narrow size distributions (<54% standard deviation). This simple but effective photo process may become one powerful method for the synthesis of monodispersed AuNPs. The results presented here may contribute to the design of simpler and more efficient method for metal nanoparticle processing.Experimental
Materials and methods
All reagents were supplied from Wako and used without further purification. Water was purified by the Milli-Q system. Thermal reduction was performed as follows: water (2 mL) containing HAuCl4·4H2O (0.2 mM) and citric acid (0.5 mM) was stirred at the designated temperature in the dark. Photoreduction was carried out as follows: water (2 mL) containing HAuCl4·4H2O (0.2 mM) and required amount of citric acid was stirred at 25 °C under 254 nm light irradiation. All reactions were performed on an UV-visible photodiode-array spectrophotometer (Shimadzu; Multispec-1500) equipped with a Peltier temperature controller (Shimadzu; S-1000) using a 10 mm path length quartz cell. The light irradiation was performed with a Xe lamp (300 W; Asahi Spectra Co. Ltd.; Max-302) equipped with 254, 360, or 510 nm band-pass filter.51 The full-width at half-maximum of the light (FWHM) was 12 nm. The photon number entered into the reaction vessel was determined with a spectroradiometer (USR-40, USHIO Inc.).52Analysis
1H NMR spectra were obtained on a JEOL JNM-AL400 Excalibur. Hydrodynamic diameter was measured on a dynamic laser scattering spectrometer (LB-500, HORIBA). TEM observations were performed on an FEI Tecnai G2 20ST analytical electron microscope (200 kV).53 XPS analysis was performed on a Kratos Axis Ultra spectrometer (Jasco Corp.).54Acknowledgements
This work was supported by the Grant-in-Aid for Challenging Exploratory Research (No. 15K14203) from the Japan Society for the Promotion of Science (JSPS).Notes and references
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
- B. Hvolbaek, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Nørskov, Nano Today, 2007, 2, 14–18 CrossRef.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- Y. Sugano, Y. Shiraishi, D. Tsukamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2013, 52, 5295–5299 CrossRef CAS PubMed.
- D. I. Gittins, D. Bethell, D. J. Schiffrin and R. J. A. Nichols, Nature, 2000, 408, 67–69 CrossRef CAS PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- B. Xavier, A. Ramanand and P. Sagayaraj, Pharma Chem., 2012, 4, 1467–1470 CAS.
- J.-W. Kim, J. H. Kim, S. J. Chung and B. H. Chung, Analyst, 2009, 134, 1291–1293 RSC.
- S. H. Lee, K. H. Bae, S. H. Kim, K. R. Lee and T. G. Park, Int. J. Pharm., 2008, 364, 94–101 CrossRef CAS PubMed.
- N. Zhou, J. Wang, T. Chen, Z. Yu and G. Li, Anal. Chem., 2006, 78, 5227–5230 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Frens, Nature (London), Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
- A. A. Volkert, V. Subramaniam and A. J. Haes, Chem. Commun., 2011, 47, 478–480 RSC.
- S. K. Sivaraman, S. Kumar and V. Santhanam, J. Colloid Interface Sci., 2011, 361, 543–547 CrossRef CAS PubMed.
- B. J. Luc and D. Bruno, Semicond. Sci. Technol., 2010, 57–78 Search PubMed.
- F. Shiba, CrystEngComm, 2013, 15, 8412–8415 RSC.
- M. Harada and S. Kizaki, Cryst. Growth Des., 2016, 16, 1200–1212 CAS.
- Y. Zhou, T. Ping, I. Maitlo, B. Wang, M. Y. Akram, J. Nie and X. Zhu, Nanotechnology, 2016, 27, 215301–215309 CrossRef PubMed.
- Y. L. Jung, J. H. Park, M. I. Kim and H. G. Park, Nanotechnology, 2016, 27, 055501–055507 CrossRef PubMed.
- H. Ma, D. Lin, H. Liu, L. Yang, L. Zhang and J. Liu, Mater. Chem. Phys., 2013, 138, 573–580 CrossRef CAS.
- F. H. Anka, S. D. Perera, C. Ratanatawanate and K. J. Balkus Jr, Mater. Lett., 2012, 75, 12–15 CrossRef CAS.
- F. B. Lollmahomed and R. Narain, Langmuir, 2011, 27, 12642–12649 CrossRef CAS PubMed.
- M. Harada and H. Einaga, Langmuir, 2007, 23, 6536–6543 CrossRef CAS PubMed.
- M. Y. Han and C. H. Quek, Langmuir, 2000, 16, 362–367 CrossRef CAS.
- Y. Nakazato, K. Taniguchi, S. Ono, T. Eitoku and K. Katayama, Phys. Chem. Chem. Phys., 2009, 11, 10064–10072 RSC.
- B. I. Zapadinskii, V. T. Shashkova, L. A. Pevtsova, A. V. Kotova, I. A. Matveeva and A. O. Stankevich, Polym. Sci., Ser. B, 2013, 55, 31–34 CrossRef CAS.
- K. Esumi, A. Suzuki, N. Aihara, K. Usui and K. Torigoe, Langmuir, 1998, 14, 3157–3159 CrossRef CAS.
- W.-E. Lu, M.-L. Zheng, W.-Q. Chen, Z.-S. Zhao and X.-M. Duan, Phys. Chem. Chem. Phys., 2012, 14, 11930–11936 RSC.
- S. Yang, Y. Wang, Q. Wang, R. Zhang and B. Ding, Colloids Surf., A, 2007, 301, 174–183 CrossRef CAS.
- P. K. Jain, W. Huang and M. A. EI-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
- Y. Shiraishi, H. Tanaka, H. Sakamoto, S. Ichikawa and T. Hirai, RSC Adv., 2015, 5, 77572–77580 RSC.
- D. C. Lim, R. Dietsche, G. Ganteför and Y. D. Kim, Appl. Surf. Sci., 2009, 256, 1148–1151 CrossRef CAS.
- Y. L. Mikhlin, A. S. Romanchenko and I. P. Asanov, Geochim. Cosmochim. Acta, 2006, 70, 4874–4888 CrossRef CAS.
- K. D. N. Vo, E. Guillon, L. Dupont, C. Kowandy and X. Coqueret, J. Phys. Chem. C, 2014, 118, 4465–4474 CAS.
- I. Ojea–Jimenez, F. M. Romero, N. G. Bastus and V. Puntes, J. Phys. Chem. C, 2010, 114, 1800–1804 Search PubMed.
- B. Rodorígez–González, P. Mulvaney and L. M. Z. Liz–Marzán, J. Phys. Chem. A, 2007, 221, 415–426 Search PubMed.
- X. Wu, P. L. Redmond, H. Liu, Y. Chen, M. Steigerwald and L. Brus, J. Am. Chem. Soc., 2008, 130, 9500–9506 CrossRef CAS PubMed.
- C. H. Munro, W. E. Smith, M. Garner, J. Clarkson and P. C. White, Langmuir, 1995, 11, 3712–3720 CrossRef CAS.
- C. Xue, G. S. Metraux, J. E. Millstone and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 8337–8344 CrossRef CAS PubMed.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- Y. Shiraishi, K. Tanaka, E. Shirakawa, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2013, 52, 8304–8308 CrossRef CAS PubMed.
- X. Li, X. Mao, X. Zhang, Y. Wang, Y. Wang, H. Zhang, X. Hao and C. Fan, Sci. China: Chem., 2015, 58, 457–466 CrossRef CAS.
- N. Seraghni, S. Belattar, Y. Mameri, N. Debbache and T. Sehili, Int. J. Photoenergy, 2012, 630425, 1–10 CrossRef.
- K. Okitsu, A. Yue, S. Tanaba, H. Matsumoto, Y. Yobiko and Y. Yoo, Bull. Chem. Soc. Jpn., 2002, 75, 2289–2296 CrossRef CAS.
- M. Tran, R. DePenning, M. Turner and S. Padalkar, Mater. Res. Express, 2016, 3, 105027–105036 CrossRef.
- Y. Shiraishi, S. Sumiya and T. Hirai, Chem. Commun., 2011, 47, 4953–4955 RSC.
- H. Koizumi, Y. Shiraishi, S. Tojo, M. Fujitsuka, T. Majima and T. Hirai, J. Am. Chem. Soc., 2006, 128, 8751–8753 CrossRef CAS PubMed.
- Y. Shiraishi, H. Sakamoto, Y. Sugano, S. Ichikawa and T. Hirai, ACS Nano, 2013, 7, 9287–9297 CrossRef CAS PubMed.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data (Fig. S1–S6). See DOI: 10.1039/c6ra27771c |
| This journal is © The Royal Society of Chemistry 2017 |