
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDipolar vinyl sulfur fluorescent dyes. Synthesis and photophysics of sulfide, sulfoxide and sulfone based D–π–A compounds†
Matias Monçalvesa,
Gabriel M. Zanottod,
Josene M. Toldod,
Daniel S. Ramponc,
Paulo H. Schneiderb,
Paulo F. B. Gonçalvesd,
Fabiano S. Rodembusch *b and
Claudio C. Silveira*a
*b and
Claudio C. Silveira*a
aDepartamento de Química, Universidade Federal de Santa Maria, CEP 97105-900, Santa Maria-RS, Brazil. E-mail: silveira@quimica.ufsm.br; Fax: +55 55 3220-8754
bGrupo de Pesquisa em Fotoquímica Orgânica Aplicada, Universidade Federal do Rio Grande do Sul – Instituto de Química, Avenida Bento Gonçalves 9500. CEP 91501-970, Porto Alegre-RS, Brazil. E-mail: fabiano.rodembusch@ufrgs.br; Fax: +55 51 3308 7304; Tel: +55 51 33087204
cUniversidade Federal do Paraná, Departamento de Química, Laboratório de Polímeros e Catálise, CEP 81531-980, Curitiba, PR, Brazil
dGrupo de Química Teórica, Universidade Federal do Rio Grande do Sul – Instituto de Química, Avenida Bento Gonçalves, 9500, CP 15003, CEP 91501-970, Porto Alegre-RS, Brazil
First published on 27th January 2017
Abstract
New vinyl sulfides and sulfoxides were obtained in good yields using the Horner–Wadsworth–Emmons (HWE) reaction. The vinyl sulfides and sulfoxides presented preferentially the E-stereochemistry. The respective vinyl sulfones were obtained in good yields from the vinyl sulfides by a simple oxidation step using mCPBA, solely as the E isomers. These derivatives presented absorption maxima in the UV-A region with molar absorptivity coefficients and radiative rate constant values ascribed to spin and symmetry-allowed 1π–π* electronic transitions. Experimentally, a small solvatochromic effect indicated an almost absent charge transfer character in the ground-state. A fluorescence emission in the violet-green region with a significative redshift on the maxima with increasing solvent polarity was observed, which could be related to a better electron delocalization in the excited state, as a result of an intramolecular charge transfer state. DFT and TD-DFT calculations were performed at the PBE1PBE and CAM-B3LYP levels of theory. No solvatochromic effect was observed in the ground state, but a bathochromic effect was detected in the emission maxima. Starting from sulfides to sulfones, the addition of oxygen atoms improved the charge separation, enabling an ICT state, mainly in P3 and P5. This charge separation was more efficient in the triphenylamino derivatives than among the pyrene derivatives since the first one has a better electron-donating character.
Introduction
High-efficiency organic luminescent materials are critically important in a variety of high-tech innovations.1–5 Due to the rapid growth of this field, the development of novel and efficient emitters is a very important task. In the past few decades, push–pull structures composed of a π-conjugated core substituted by electron-donating (D) and electron-attracting (A) (D–π–A) groups have been extensively studied in materials chemistry.6–11 These molecular systems have also been extended to quadrupolar (D–π–A–π–D or A–π–D–π–A) compounds, which also have promising technological applications.12–17 In general, optical materials take important roles in optoelectronics, as part of transistors, OLEDs, solar cells, lightening agents and light modulators.18,19 These usual D–π–A molecular architectures play an important role in their photophysics because they not only show high fluorescent quantum yields owing to the effective radiative decay of their excited intramolecular charge-transfer (ICT) state but also possess impressive bipolar charge-transporting properties for their constitutive hole- and electron-transporting moieties,20–25 that can be modulated through tuning the groups electron donor and/or acceptor strengths.26–30 Concerning those functional groups, the organosulfides allow a tuneable electronic behavior that can be easily modulated through a single step oxidation to the respective sulfoxides or sulfones, which are effective electron acceptor groups.31–38In this regard, our continuous interests in the synthesis of organochalcogen compounds prompted us to access symmetrical and unsymmetrical vinyl sulfides by a straightforward approach, using a Wittig–Horner reaction, as precursors of products carrying a higher oxidation state on sulfur.39,40 In this sense, photoactive divinyl sulfides and sulfones have been obtained.31,41
However, there is a lack of information on the rationalization of comparative electronic effects exerted by organosulfides, sulfoxides and sulfones on excited and ground states of push–pull π-conjugated structures. Therefore, herein we describe the synthesis of novel vinyl sulfides, sulfoxides and sulfones containing pyrene and triphenylamine as end-capped electron donor groups. In addition, the electronic, photophysical and theoretical properties of these compounds were extensively explored, and the effect of the oxidation state of the sulfur on the electronic properties of push–pull molecular systems were evaluated, comparing the experimental results with the theoretical properties, predicted with TD-DFT calculations.
Experimental
General information
All reactions were performed in flame-dried glassware under ambient conditions unless otherwise stated. All solvents were purified according to the literature and transferred via syringe.42 Tetrahydrofuran (THF) was refluxed under Na/benzophenone and distilled immediately before use. Unless otherwise stated, commercial reagents were used without additional purification. The column chromatographic purification of the products was performed on silica gel (230–400 mesh). The thin-layer chromatography (TLC) plates were visualized by exposure to UV radiation (365 nm), iodine vapours and/or acid solution of vanillin.The 1H NMR spectra were recorded at 200/400 MHz and the 13C NMR spectra were measured at 50/100 MHz, in CDCl3 as solvent with TMS as reference. Coupling constants (J) are reported in Hertz. Abbreviations to denote the multiplicity of a particular signal are s (singlet), d (doublet), dd (doublet of doublets), t (triplet) and m (multiplet). A Perkin-Elmer CHN 2400 analyzer was used for elemental analyses. The infrared spectra were recorded on a FTIR spectrometer, with the samples dispersed in KBr pellets. The UV-Vis absorption spectra were recorded on a Shimadzu UV-2450 spectrophotometer in a concentration range of 10−4 to 10−5 M. The steady state fluorescence spectra were measured using a Shimadzu RF5301PC spectrofluorometer. The maximum absorption wavelength was used as excitation wavelength for fluorescence measurements. The quantum yield of fluorescence (ΦFL) was determined by applying the dilute optical method. Quinine sulphate (QS) in H2SO4 (0.5 mol L−1) (ΦFL = 0.55) was used as the quantum yield standard.43 All measurements were performed at room temperature (25 °C).
Theoretical calculations
In parallel to the experimental studies, theoretical calculations were carried out using the Gaussian 09 package44 for molecules P1–P6. For geometry optimizations and vibrational analysis in the ground state, the Density Functional Theory (DFT) was used with two hybrid exchange–correlation functionals: PBE1PBE, a global hybrid parameter free with 25% exact exchange functional45 and CAM-B3LYP, the long range corrected version of B3LYP using the Coulomb-attenuating method,46 both with the cc-pVDZ basis set. These functionals were chosen for this study since they have presented good results and have been extensively tested in different systems for a large range of properties, particularly for absorption and emission spectral simulations and electronic properties.47–50 However, in systems where it is possible to observe charge transfer, it is recommended to use the range-separated hybrid functionals, like the CAM-B3LYP.48,50 Geometry optimizations and vibrational analysis on the first excited state were performed using TD-DFT (Time Dependent Density Functional Theory) with the cc-pVDZ basis set. Population analysis and vertical transitions were performed using jun-cc-pVTZ basis sets. The jun-basis set, named calendar basis set, is recommended by Truhlar et al.51 as a better option to the default augmented (aug) basis sets, showing a lower computational cost. These basis sets are constructed by removing diffuse functions from the augmented (aug-) basis sets. The jun-cc-pV*Z basis sets remove the diffuse function from H and He and also remove the highest angular momentum diffuse function from all other atoms. All equilibrium geometries were characterized by the absence of imaginary frequencies. The influence of the solvent was considered in all calculations (geometry optimization and vertical transitions) using PCM (Polarizable Continuum Model)52 and the solvents used were 1,4-dioxane, dichloromethane, acetonitrile and ethanol. Charges from electrostatic potential using a grid based method (ChelpG) were used to obtain the electrostatic potential surface and dipole momenta. All images of the geometry and molecular orbital were generated using the ChemCraft program.53Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 to 50:50, v/v, EtOAc/hexanes) gave the product as a colourless oil (4.68 g, 90%). 1H NMR (200 MHz, CDCl3) δ (ppm): 7.47–7.42 (m, 2H), 7.33–7.21 (m, 3H), 4.21–4.06 (m, 4H), 3.24 (d, J = 14.0 Hz, 2H), 1.32 (t, 6H).:2 mixture of THF/H2O (15 mL) and cooled to 0 °C. A solution of 1 (1.507 g, 5.79 mmol) in THF (5 mL) was added dropwise. The mixture was stirred at room temperature for 93 h. The solvent was removed under reduced pressure and the resulting crude mixture was diluted with EtOAc, washed with saturated aqueous NH4Cl, dried over MgSO4 and concentrated under vacuum. The crude product was purified by flash chromatography (EtOAc/hexanes, 7:3, v/v) to yield 4 (1.03 g, 65%) as a light yellow oil. 1H NMR (200 MHz, CDCl3) δ (ppm): 7.77–7.65 (m, 2H), 7.60–7.51 (m, 3H), 4.25–4.03 (m, 4H), 3.50–3.22 (m, 2H), 1.36–1.21 (m, 6H).:9, v/v).:9, v/v).:8, v/v).
:95 to 50:50, v/v, EtOAc/hexanes) gave the product as a colourless oil (4.68 g, 90%). 1H NMR (200 MHz, CDCl3) δ (ppm): 7.47–7.42 (m, 2H), 7.33–7.21 (m, 3H), 4.21–4.06 (m, 4H), 3.24 (d, J = 14.0 Hz, 2H), 1.32 (t, 6H).:2 mixture of THF/H2O (15 mL) and cooled to 0 °C. A solution of 1 (1.507 g, 5.79 mmol) in THF (5 mL) was added dropwise. The mixture was stirred at room temperature for 93 h. The solvent was removed under reduced pressure and the resulting crude mixture was diluted with EtOAc, washed with saturated aqueous NH4Cl, dried over MgSO4 and concentrated under vacuum. The crude product was purified by flash chromatography (EtOAc/hexanes, 7:3, v/v) to yield 4 (1.03 g, 65%) as a light yellow oil. 1H NMR (200 MHz, CDCl3) δ (ppm): 7.77–7.65 (m, 2H), 7.60–7.51 (m, 3H), 4.25–4.03 (m, 4H), 3.50–3.22 (m, 2H), 1.36–1.21 (m, 6H).:9, v/v).:9, v/v).:8, v/v).Results and discussion
Synthesis
The precursors, the phosphonates 1, 4 and 4-diphenylamino benzaldehyde (2) were prepared following reported procedures. The syntheses of the sulfur-based chromophores P1–P6, via the Horner–Wadsworth–Emmons reaction, are presented in Scheme 1. | ||
| Scheme 1 Synthesis of the sulfur-based dyes P1–P6. | ||
The synthesis of the vinyl sulfides and sulfoxides was carried out by treatment of the appropriate aldehyde with the phosphonates 1 or 4 using NaH as base, in THF at 60 °C for 24 h. The sulfides P1 and P2 were obtained in excellent yields, 93% and 89%, respectively, with a clear preference for the E isomers. The vinyl sulfoxides P3 and P4 were obtained in 71% and 68% yield, respectively. As expected, the stereoselectivity of the HWE reaction was very high, yielding P3 with clear preference for the E isomer and P4 solely as the E isomer. On the other hand, the vinyl sulfones P5 and P6 were successfully synthesized in good yields (78% and 75%, respectively) by oxidation of the vinyl sulfides P1 and P2 with mCPBA. As observed in a previous work,31,40 the oxidation reaction caused the isomerization of the Z isomers, yielding the sulfones as single E isomers. The dyes P1–P6 were fully characterized by 1H and 13C NMR, FTIR, high-resolution mass spectrometry (HRMS) or elemental analysis. The stereochemistry of the products was established on the basis of the coupling constants of the vinylic protons in their 1H NMR spectra (Jtrans ∼ 15 Hz and Jcis ∼ 10 Hz).
Photophysical characterization
The absorption spectra of the sulfur based compounds P1–P6 in different organic solvents are shown in Fig. 1 and 2. The relevant data from UV-Vis absorption spectroscopy are presented in Table 1. The UV-Vis absorption curves allowed to relate the experimental extinction coefficients ε with the calculated radiative rate constants k0e and the oscillator strengths (fe), using the Strickler–Berg relation presented in eqn (1),55
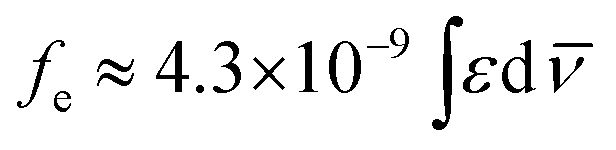 | (1) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) (cm−1) corresponding to a single electron oscillator. The rate constant (k0e) for emission can be related to the extinction coefficient for absorption using the eqn (2), where the pure radiative lifetime, τ0 is defined as 1/k0e.56
(cm−1) corresponding to a single electron oscillator. The rate constant (k0e) for emission can be related to the extinction coefficient for absorption using the eqn (2), where the pure radiative lifetime, τ0 is defined as 1/k0e.56
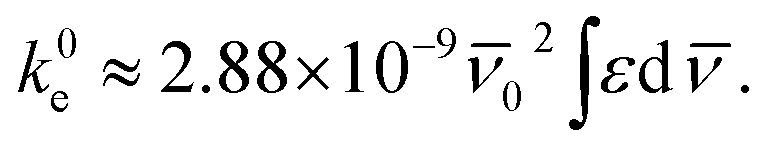 | (2) |
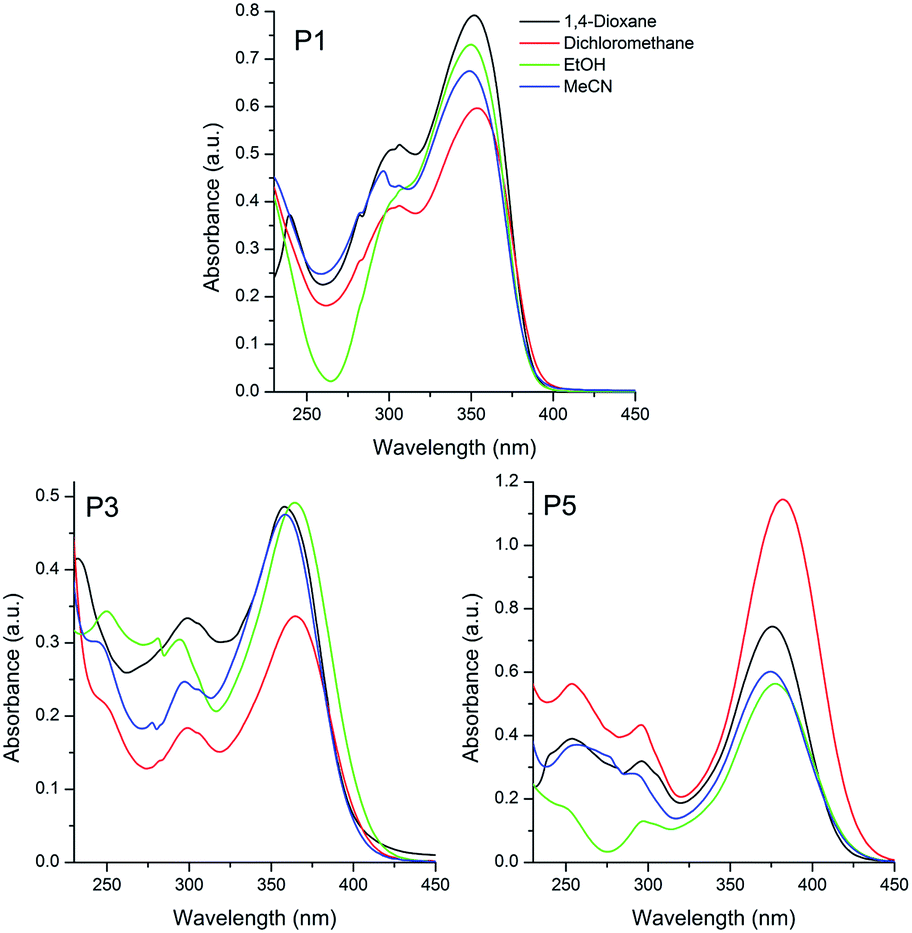 | ||
| Fig. 1 UV-Vis absorption spectra of the sulfide P1, sulfoxide P3 and sulfone P5 based triphenylamino compounds in different organic solvents [10−5 M]. | ||
| Dye | Solventa | Conc. | λabs | ε | fe | k0e | τ0 |
|---|---|---|---|---|---|---|---|
| a DIO = 1,4-dioxane, DCM = dichloromethane, EtOH = ethanol and MeCN = acetonitrile. | |||||||
| P1 | DIO | 2.21 | 352 | 3.59 | 0.96 | 7.71 | 1.30 |
| DCM | 2.89 | 354 | 2.07 | 0.47 | 3.75 | 2.67 | |
| EtOH | 2.21 | 350 | 3.31 | 0.83 | 6.78 | 1.48 | |
| MeCN | 2.21 | 348 | 3.06 | 0.85 | 7.02 | 1.43 | |
| P3 | DIO | 2.28 | 359 | 2.96 | 0.48 | 3.72 | 2.69 |
| DCM | 1.79 | 364 | 1.88 | 0.39 | 2.91 | 3.43 | |
| EtOH | 1.79 | 365 | 2.75 | 0.61 | 4.57 | 2.19 | |
| MeCN | 1.79 | 359 | 2.66 | 0.51 | 3.98 | 2.51 | |
| P5 | DIO | 2.19 | 376 | 3.39 | 0.62 | 4.42 | 2.26 |
| DCM | 1.41 | 382 | 3.30 | 0.93 | 6.35 | 1.58 | |
| EtOH | 1.72 | 378 | 3.28 | 0.66 | 4.65 | 2.15 | |
| MeCN | 1.72 | 375 | 3.49 | 0.61 | 4.31 | 2.32 | |
| P2 | DIO | 2.11 | 371 | 4.38 | 0.81 | 5.87 | 1.70 |
| DCM | 2.49 | 370 | 2.87 | 0.57 | 4.18 | 2.39 | |
| EtOH | 1.63 | 367 | 5.52 | 0.97 | 7.19 | 1.39 | |
| MeCN | 2.03 | 368 | 4.91 | 0.79 | 5.84 | 1.71 | |
| P4 | DIO | 2.19 | 362 | 3.91 | 0.69 | 5.26 | 1.90 |
| DCM | 2.56 | 369 | 2.79 | 0.52 | 3.83 | 2.61 | |
| EtOH | 2.21 | 369 | 4.12 | 0.77 | 5.66 | 1.77 | |
| MeCN | 2.21 | 360 | 4.08 | 0.81 | 6.24 | 1.60 | |
| P6 | DIO | 2.28 | 371 | 3.58 | 0.62 | 4.52 | 2.21 |
| DCM | 2.10 | 372 | 2.57 | 0.52 | 3.77 | 2.65 | |
| EtOH | 2.28 | 370 | 3.88 | 0.64 | 4.69 | 2.13 | |
| MeCN | 2.28 | 370 | 3.51 | 0.76 | 5.55 | 1.80 | |
The sulphur-based compounds containing the triphenylamino moiety presented absorption maxima located in the UV-A region (350, 362 and 378 nm for P1, P3 and P5, respectively) (Fig. 1). The molar absorptivity values (ε, ∼104 M−1 cm−1) and the calculated radiative rate constants (k0e, ∼108 s−1) indicated spin and symmetry allowed electronic transitions, related to 1π–π* transitions. Although no relation was detected between the location of the absorption maxima and the solvent polarity, it was observed for all compounds a small solvatochromic effect (Δλabs ∼ 450 cm−1), indicating an almost absent charge transfer character in the ground-state.
A similar photophysical behaviour could be observed for the pyrene derivatives P2, P4 and P6, where 1π–π* electronic transitions were ascribed to the absorption maxima located at 369, 365 and 371 nm, respectively (Fig. 2). In addition, despite the presence of electron withdrawing and electron donating groups (EWG and EDG, respectively) in the π-conjugated system, the small value for the Δλabs (∼370 cm−1), also indicated the absence of charge transfer state in these compounds in the ground-state. It is worth mentioning that the studied sulfur derivatives containing the triphenylamino moiety P1, P3 and P5 presented a redshift on the location of the absorption maxima with increase of the electron withdrawing effect, from sulfide to sulfone (24–28 nm). Since this behavior is not observed in P2, P4 and P6, the redshift cannot be related only to changes in the withdrawing effect of the chromophore (sulfide–sulfoxide–sulfone). Thus, it is believed that the donating effect is also playing an important role on the observed photophysical behaviour, because the pyrene presents lower donor ability than the triphenylamino group. An almost constant radiative lifetime (τ0 ∼ 2 ns) was calculated for the compounds in all studied solvents, indicating that after radiation absorption, the dyes populate the same excited state.
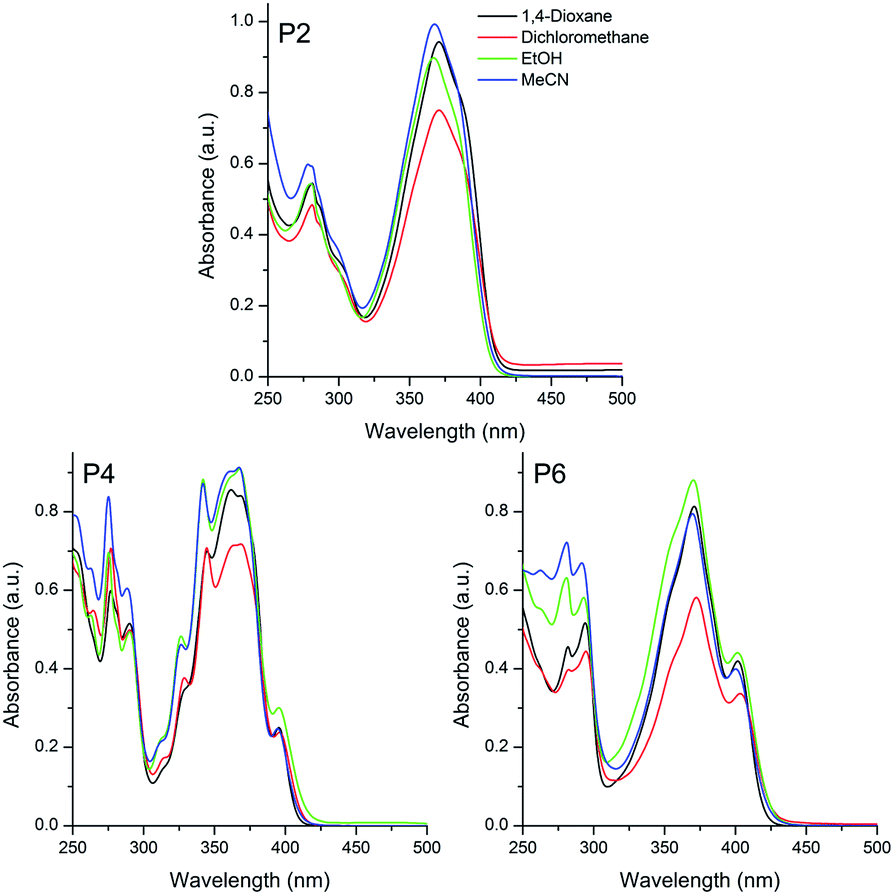 | ||
| Fig. 2 UV-Vis absorption spectra of the sulphide (P2), sulfoxide (P4) and sulfone (P6) based pyrene compounds in different organic solvents [10−5 M]. | ||
The fluorescence emission spectra of compounds P1–P6 are shown in Fig. 3 and 4. The relevant data from the steady-state fluorescence spectroscopy are presented in Table 2. The emission curves were obtained by exciting the compounds at the absorption maxima wavelength. The sulfur based compounds containing the triphenylamino moiety presented a main emission band located in the violet-green region, which can be related to a vertical transition from the lowest excited state with the largest oscillator strength, as observed in Table 1 for the almost unitary values of fe. For all compounds a significant redshift (Δλem) on the emission maxima is observed with increasing the solvent polarity. Values from 36 to 85 nm (1915 to 3531 cm−1) were calculated for sulfide to sulfone compounds.
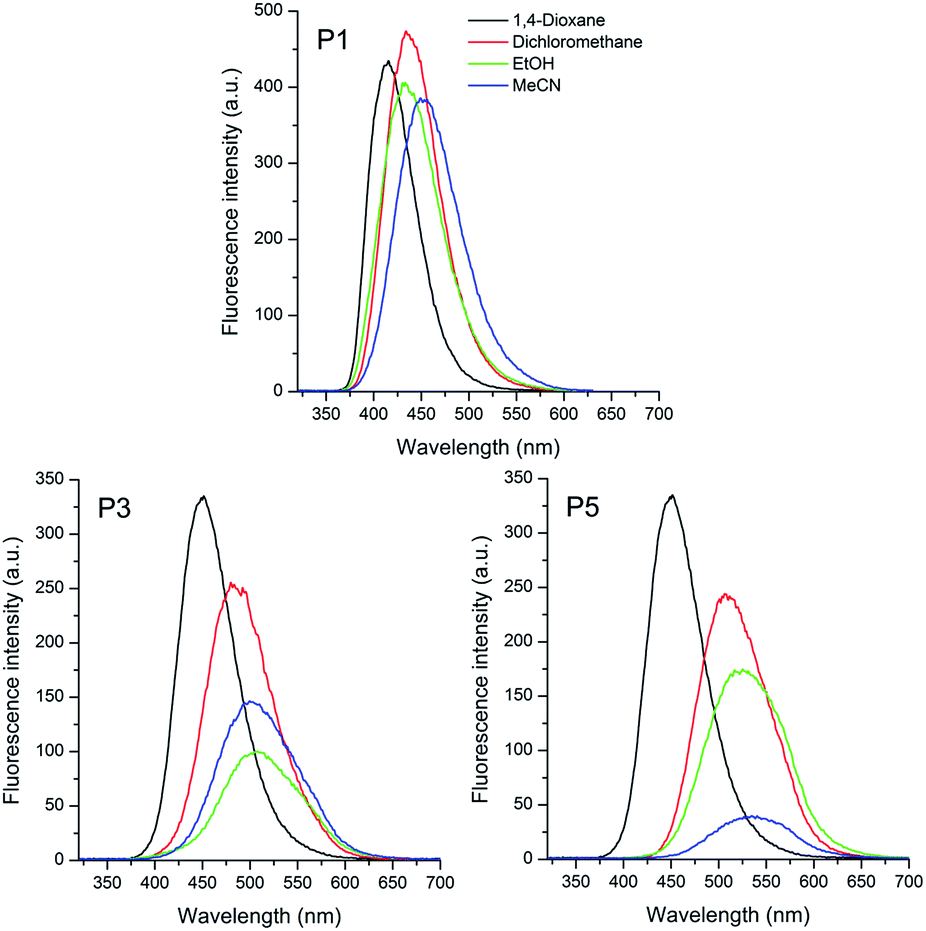 | ||
| Fig. 3 Fluorescence spectra of the sulphide (P1), sulfoxide (P3) and sulfone (P5) based triphenylamino compounds in different organic solvents [10−5 M]. | ||
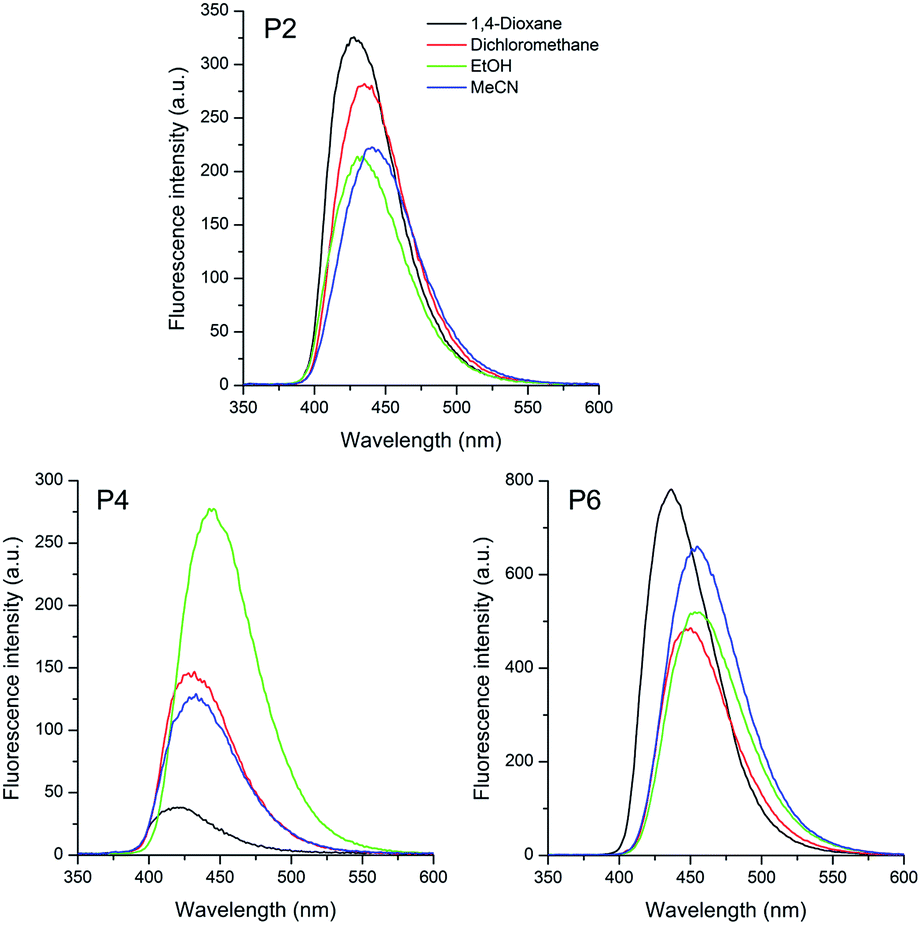 | ||
| Fig. 4 Fluorescence emission spectra of the sulfide P2, sulfoxide P4 and sulfone P6 based pyrene compounds in different organic solvents [10−5 M]. | ||
| Dye | Solventa | Solvent | λem | Δλem | ΔλST | ΦFL |
|---|---|---|---|---|---|---|
| a DIO = 1,4-dioxane, DCM = dichloromethane, EtOH = ethanol and MeCN = acetonitrile. | ||||||
| P1 | DIO | Hexane | 416 | 36/1915 | 4371 | 0.34 |
| DCM | Toluene | 437 | 5365 | 0.22 | ||
| EtOH | 1,4-Dioxane | 434 | 5694 | 0.32 | ||
| MeCN | Dichloromethane | 452 | 6612 | 0.46 | ||
| P3 | DIO | Hexane | 450 | 51/2262 | 5633 | 0.27 |
| DCM | Toluene | 483 | 6683 | 0.27 | ||
| EtOH | 1,4-Dioxane | 506 | 7634 | 0.45 | ||
| MeCN | Dichloromethane | 501 | 7895 | 0.35 | ||
| P5 | DIO | Hexane | 450 | 85/3531 | 6533 | 0.29 |
| DCM | Toluene | 508 | 6493 | 0.33 | ||
| EtOH | 1,4-Dioxane | 524 | 7371 | 0.45 | ||
| MeCN | Dichloromethane | 535 | 7975 | 0.20 | ||
| P2 | DIO | Hexane | 428 | 13/689 | 3590 | 0.26 |
| DCM | Toluene | 435 | 4039 | 0.23 | ||
| EtOH | 1,4-Dioxane | 433 | 4153 | 0.25 | ||
| MeCN | Dichloromethane | 441 | 4498 | 0.25 | ||
| P4 | DIO | Hexane | 417 | 16/886 | 3643 | 0.44 |
| DCM | Toluene | 432 | 3952 | 0.48 | ||
| EtOH | 1,4-Dioxane | 444 | 4578 | 0.32 | ||
| MeCN | Dichloromethane | 433 | 4683 | 0.16 | ||
| P6 | DIO | Hexane | 430 | 25/1278 | 3698 | 0.47 |
| DCM | Toluene | 449 | 4610 | 0.25 | ||
| EtOH | 1,4-Dioxane | 455 | 5049 | 0.36 | ||
| MeCN | Dichloromethane | 455 | 5049 | 0.45 | ||
The positive solvatochromism can be related to a better electron delocalization in the excited state afforded by an intramolecular charge transfer state in these compounds.
The increase of the calculated Stokes' shift with increase of the dielectric constant of the solvent (1,4-dioxane to acetonitrile) is consistent with the stabilization of the charge transfer state by the polar solvents (Table 2).
Moreover, this effect is greater in the sulfone derivative P5 due to higher electronic withdrawing character of this group relative to sulfoxide P3 and sulfide P1.
This behaviour indicates that the excited state of these compounds is more polar than the ground state one, i.e. μe > μg. In this scenario, the relaxed excited state seems to be more stabilized relative to the ground state by increasing the polarity of the solvent.
As already observed for compounds P1, P3 and P5, the pyrene based sulfur compounds also present a single emission band located in the visible region of the spectra (violet to blue). Values from the calculated oscillator strength also allowed to relate the observed deactivation pathway with the lowest excited state. Although much less intense than the triphenylamino analogues, a redshift on the emission maxima (Δλem) was also observed with changes in the solvent polarity. Compounds P2, P4 and P6 presented values from 13 to 25 nm (689 to 1278 cm−1), increasing from sulfide to sulfone derivatives, which can also be related to a ICT state. This difference is probably due to the less donating character of the pyrene group with regard to the triphenylamino moiety. Once again the sulfone derivative P6 presented the highest redshift due to higher electronic withdrawing character of this group compared to sulfoxide P4 and sulfide P2. In these compounds, the Stokes' shift values also relates to an ICT state in the excited state. It is worth mentioning that in ethanol some compounds presented an unexpected behaviour due to a specific solvent–fluorophore interaction, such as hydrogen bonding.57 The calculated fluorescence quantum yields (ΦFL) were in agreement with the values reported in the literature for push–pull vinylic compounds58 or sulfur compounds containing EDG or EWG moieties.20,37,59,60 The almost similar ΦFL calculated for all compounds indicate that different electron donating groups, as well as the oxidation state of the sulfur atom do not affect significantly the excited singlet state deactivation. Similar sulfoxide derivatives bearing a pyrene moiety were reported in fluorescence chemosensor applications.61 The very low fluorescence quantum yields were related to excited-state pyramidal inversion of the sulfoxide-appended fluorophores,62 which were suppressed after metal coordination. Besides the extension of the π-conjugation provided by the vinyl moiety, this group also seems to play an interesting role in the photophysics of the studied compounds. The observation of higher fluorescence quantum yields suggests that minimization of the pyramidal inversion of the sulfoxide is operative, as an excited state deactivation channel. On the other side, the shape of the absorption and emission spectra of the pyrene derivatives are different from those observed in similar compounds.37,61
To better understand the extend of the intramolecular charge transfer character in these compounds, the difference in the dipole moment between the excited and ground states (Δμeg) were obtained for dyes P1, P3 and P5 using the Lippert–Mataga eqn (3):63
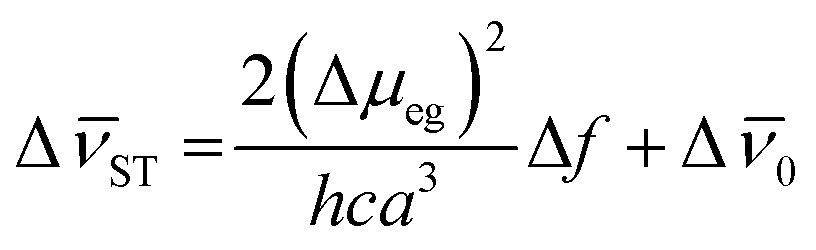 | (3) |
This equation compares the solvatochromic shifts from the absorption and emission maxima, as well as the Stokes' shift versus the orientation polarization function (Δf),56,57 were a linear correlation indicates intramolecular charge transfer character.64 In this equation, h is the Planck's constant, c is the speed of light, a is the Onsager cavity radius, which was obtained in gas phase using CAM-B3LYP/jun-cc-pVTZ level of theory. For these calculations were used values 0.5 angstrom smaller than those calculated (5.44, 5.36 and 5.12 angstrom for P1, P3 and P5, respectively).
Fig. 5 presents the Lippert plot of the absorption, fluorescence emission maxima and Stokes' shift (ΔST = abs − em) versus the orientation polarisability (Δf) for P1, P3 and P5. Any significant solvatochromic effect was observed taking the absorption maxima into account (Fig. 5a), which can be related to absence of charge transfer in the ground state. On the other hand, the negative slope on the emission maxima, as well as the increasing slope on the Stokes' shift (Fig. 5b and c, respectively) indicate intramolecular charge transfer (ICT) mechanism in the excited state. From the slope of the Stokes' shift versus the orientation polarisability plot, the change in the dipole moment (Δμeg) of the fluorophores upon excitation was estimated as 15.40 D for P1, 14.76 D for P3 and 14.43 D for P5. It is worth to mention that the ICT process, as well as the significative difference in the dipole moment between the excited and ground states were already predicted by theoretical calculations obtained in this work.
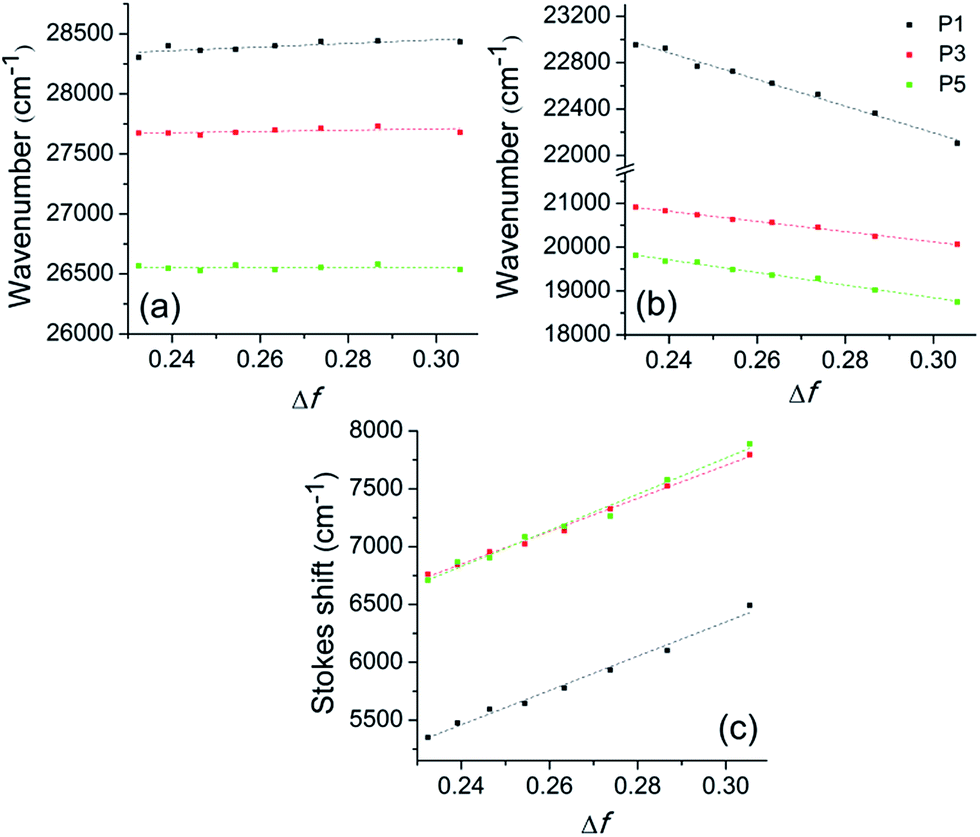 | ||
| Fig. 5 Solvent effect on the (a) absorption maxima, (b) fluorescence emission and (c) Stokes' shift of the sulfide P1, sulfoxide P3 and sulfone P5. Δf is given as Δf = [(ε − 1)/(2ε + 1)] − [(n2 − 1)/(2n2 + 1)], where ε is dielectric constant and n is the refractive index for a mixture of solvents.56 | ||
Theoretical calculations
The wavelengths of the absorption and emission maxima obtained with the CAM-B3LYP functional are shown in Table 3. A comparison between these results and those obtained with PBE1PBE is shown in the ESI (Table ESI1†). It is known from the literature that CAM-B3LYP generally provides a better description of the excited states with charge transfer and Rydberg states than hybrid functionals, like PBE1PBE.46,48,50 However, the latter is well known for furnishing good results in the absorption and emission calculated spectra, and exhibiting mean absolute errors about 0.30 eV on determinations of wavelength maxima.48,50,65–67 CAM-B3LYP is a range separated exchange and LYP correlation, which allows an improvement on the description of charge transfer states.50 This is an important fact to be taken into account when studying molecules which could have a strong ICT character, like those with a D–π–A structure. ICT plays a key role in solar energy conversion devices, as it impacts directly into the dye efficiency as sensitizer.68,69| Dye | Solventa | S0 | S1 | ||||
|---|---|---|---|---|---|---|---|
| λabs | fe | μ | λem | fe | μ | ||
| a DIO = 1,4-dioxane, DCM = dichloromethane, EtOH = ethanol and MeCN = acetonitrile.b Calculated with PBE1PBE/jun-cc-pVTZ//PBE1PBE/cc-pVDZ. The remaining data were calculated with CAM-B3LYP/jun-cc-pVTZ//CAM-B3LYP/cc-pVDZ. | |||||||
| P1 | DIOb | 368 | 0.853 | 1.85 | 451 | 0.785 | 10.98 |
| DIO | 329 | 0.938 | 1.83 | 412 | 0.999 | 4.19 | |
| DCM | 330 | 0.945 | 2.09 | 427 | 1.140 | 4.98 | |
| EtOH | 329 | 0.930 | 2.17 | 431 | 1.174 | 5.18 | |
| MeCN | 329 | 0.926 | 2.18 | 432 | 1.180 | 5.22 | |
| P3 | DIOb | 374 | 0.891 | 5.19 | 486 | 0.206 | 25.82 |
| DIO | 331 | 0.990 | 5.18 | 385 | 1.007 | 12.40 | |
| DCM | 332 | 1.006 | 5.98 | 407 | 1.194 | 13.46 | |
| EtOH | 331 | 0.996 | 6.23 | 412 | 1.235 | 13.79 | |
| MeCN | 331 | 0.993 | 6.27 | 414 | 1.243 | 13.87 | |
| P5 | DIOb | 395 | 0.923 | 7.98 | 531 | 0.120 | 29.72 |
| DIO | 343 | 1.073 | 7.83 | 399 | 0.965 | 18.28 | |
| DCM | 346 | 1.101 | 8.86 | 419 | 1.181 | 19.03 | |
| EtOH | 346 | 1.097 | 9.17 | 425 | 1.236 | 19.33 | |
| MeCN | 346 | 1.094 | 9.23 | 427 | 1.246 | 19.39 | |
| P2 | DIOb | 396 | 0.848 | 1.77 | 485 | 0.987 | 4.41 |
| DIO | 354 | 0.922 | 1.73 | 447 | 1.115 | 2.14 | |
| DCM | 354 | 0.929 | 1.94 | 471 | 1.366 | 2.47 | |
| EtOH | 353 | 0.904 | 2.00 | 479 | 1.437 | 2.56 | |
| MeCN | 353 | 0.897 | 2.01 | 480 | 1.450 | 2.58 | |
| P4 | DIOb | 384 | 0.915 | 4.24 | 444 | 1.068 | 4.76 |
| DIO | 350 | 0.984 | 4.36 | 420 | 1.155 | 4.84 | |
| DCM | 350 | 0.986 | 5.15 | 445 | 1.392 | 5.66 | |
| EtOH | 349 | 0.960 | 5.41 | 453 | 1.454 | 5.92 | |
| MeCN | 349 | 0.953 | 5.45 | 454 | 1.465 | 5.96 | |
| P6 | DIOb | 403 | 0.898 | 6.64 | 452 | 1.026 | 12.94 |
| DIO | 362 | 0.998 | 6.61 | 428 | 1.154 | 10.49 | |
| DCM | 365 | 1.004 | 7.59 | 458 | 1.385 | 11.82 | |
| EtOH | 364 | 0.979 | 7.88 | 468 | 1.445 | 12.21 | |
| MeCN | 364 | 0.972 | 7.94 | 469 | 1.456 | 12.29 | |
In the present work, CAM-B3LYP provided a better description of the absorption and emission spectra, presenting a lower mean deviation on absorption and emission maxima compared to the experimental data (Table ESI1†). Thus, the geometries and electronic properties of the compounds will be discussed based only on CAM-B3LYP calculations.
Calculated vertical absorption and emission wavelength maxima for the molecules P1–P6 can be seen in Table 3. Changes in the absorption and emission wavelengths were observed by changing the donor and acceptor groups attached to the central part of the molecule. In P1, P3, and P5 the presence of the donor group triphenylamino leads to absorption maxima located around 329, 331, and 345 nm, respectively. By changing the donor group to pyrene (P2, P4, and P6), the mean values of absorption wavelength maxima are shifted to 353, 350 and 363 nm, respectively. In general, the oxidation of vinyl sulfides, producing the respective vinyl sulfoxides and sulfones, causes deviations to red in the absorption wavelength maxima. This tendency is the same observed in the experimental results and can be related to the increase of the electron withdrawing effect from sulfide to sulfone and to the nature of the electron-donating group. Since the triphenylamino group has greater donor ability than pyrene, it is possible to see higher charge separation in P1, P3, but mainly in P5, as observed in the HOMO–LUMO plots (Fig. 7). In addition, the high oscillator strength in all cases indicates a high probability of transition associated to HOMO–LUMO, that can be assigned to 1π–π* electronic transitions.
The increase in solvent polarity did not present a significant effect in the absorption maxima, indicating the lack of solvatochromic effects in the ground state. In contrast, the main emission bands showed a significant bathochromic effect when the dielectric constant of the solvent was increased. This effect was higher when the solvent was changed from 1,4-dioxane to dichloromethane and can be assigned to a greater stabilization of the molecules by the solvent in the excited state than in the ground state. Since the molecules are more polar in S1, an increase in the solvent polarity will stabilize the first excited state more than the ground state.70 It has also been noted that changes in dipole moment from S0 to S1 are more significant in sulfones than in sulfides and sulfoxides. This can be attributed to the higher electron-withdraw character of the former group. In addition, the higher electron-donating character of the triphenylamino group helps to increase the difference in dipole moment from S0 to S1, improving the charge separation and enabling the existence of an intramolecular charge transfer state in the molecule (that aspect will be subsequently discussed).
Fig. 6 shows the optimized geometries calculated for the ground and first excited states using CAM-B3LYP and 1,4-dioxane as solvent. The main differences in bond lengths and angles for the structures are summarized in Tables ESI2 and ESI3.† Since the E isomer was the major product in all cases, only the geometries of these isomers were calculated. For P1, P3, and P5, in the ground state, it can be seen that both external phenyl rings are out-of-plane, presenting a dihedral angle d1 of approximately 45°. The central phenyl ring is in the same plane of the double bond, showing a dihedral angle d2 around 0°. In excited state, d1 comes close to 35°, while d2 undergoes a small variation, assuming a planar structure along the π-bridge part of the conjugated system, in agreement with previous reports in the literature.71 It is interesting to note that the phenyl ring attached to sulfur is not in the same plane of the π bridge, being almost perpendicular to it (d3). Upon excitation, there is a rotation of this ring in P1, while no significant rotation is observed in P3 and P5. Compounds P2, P4, and P6 have a pyrene motif as donor. In these molecules, the dihedral angle between the pyrene moiety and the double bond is in the range of 16–30° in the ground state, and becomes almost planar in the excited state. In the same way as in P1, P3 and P5, the phenyl ring bonded to the sulfur is not in the same plane of the remainder part of the molecule, being almost perpendicular to it, and rotating in P2 from S0 to S1. Comparing the geometries of the ground and excited states, the attachment of oxygen atoms to the sulfur does not lead to significant changes in the structures. This is reflected in a small geometrical reorganization after the electronic transition. By changing the solvent, a very small shift is observed in the geometries, as can be seen in Table ESI2.†
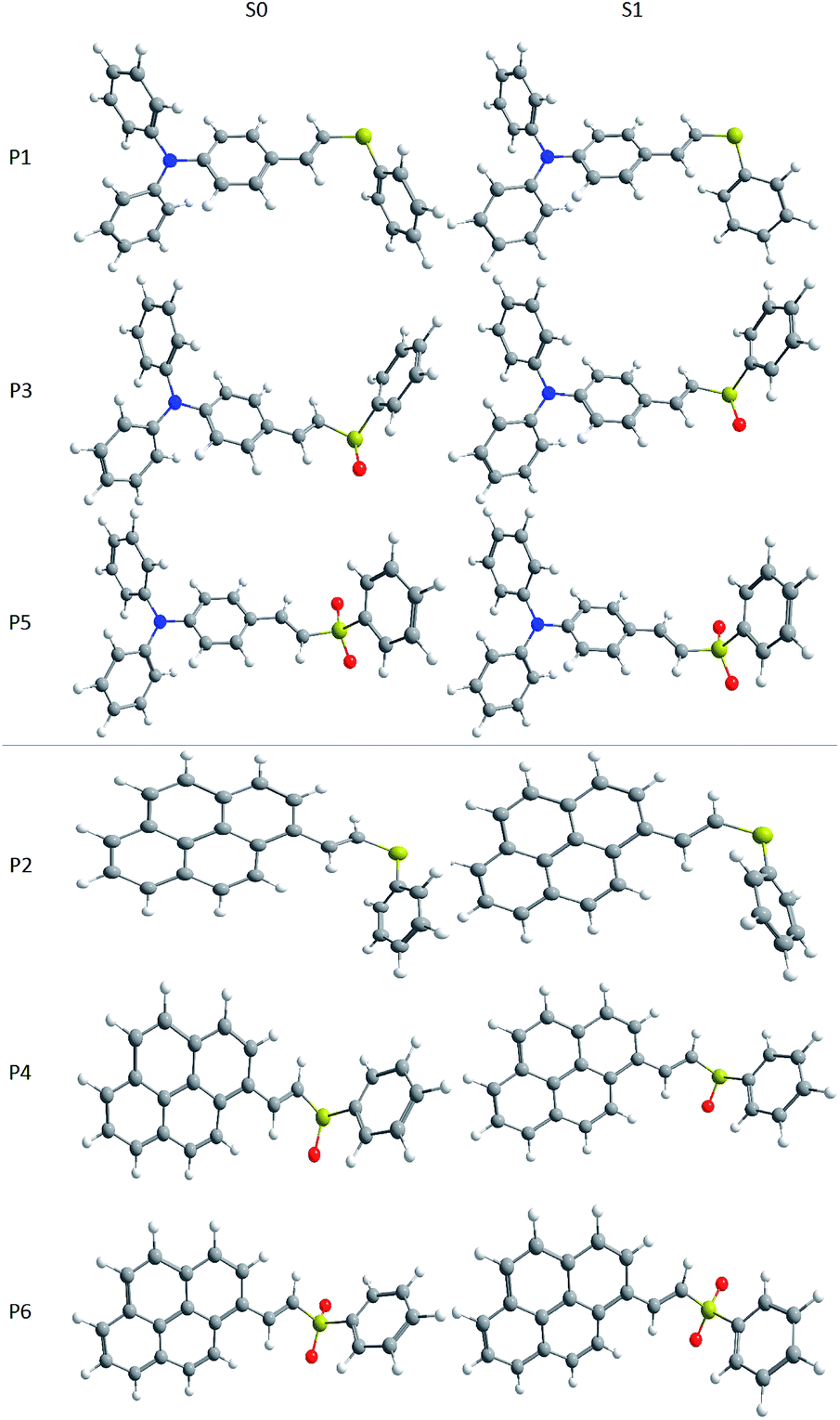 | ||
| Fig. 6 Molecular geometries of the P1–P6 dyes in the ground (left) and first excited (right) states, calculated at the CAM-B3LYP level in 1,4-dioxane. | ||
The geometry of the molecules has an important role in systems that can present intramolecular charge transfer. For example, it is expected that the planarity of the donor moiety and the whole conjugated system (donor–π-bridge–acceptor) improves the mobility of electrons, resulting in a better charge transfer.68 The molecules under study have a D–π–A structure, which is desirable for a sensitizer, as it helps to separate spatially the HOMO and LUMO molecular orbitals.69 In fact, despite pyrene derivatives are more planar than the triphenylamino derivatives, this does not improve the charge separation in the molecules, as can be seen in Fig. 7. As the change in the dielectric constant of the solvent has no considerable effect in molecular orbitals, these plots are representative for all studied solvents (Fig. ESI1 and ESI2†).
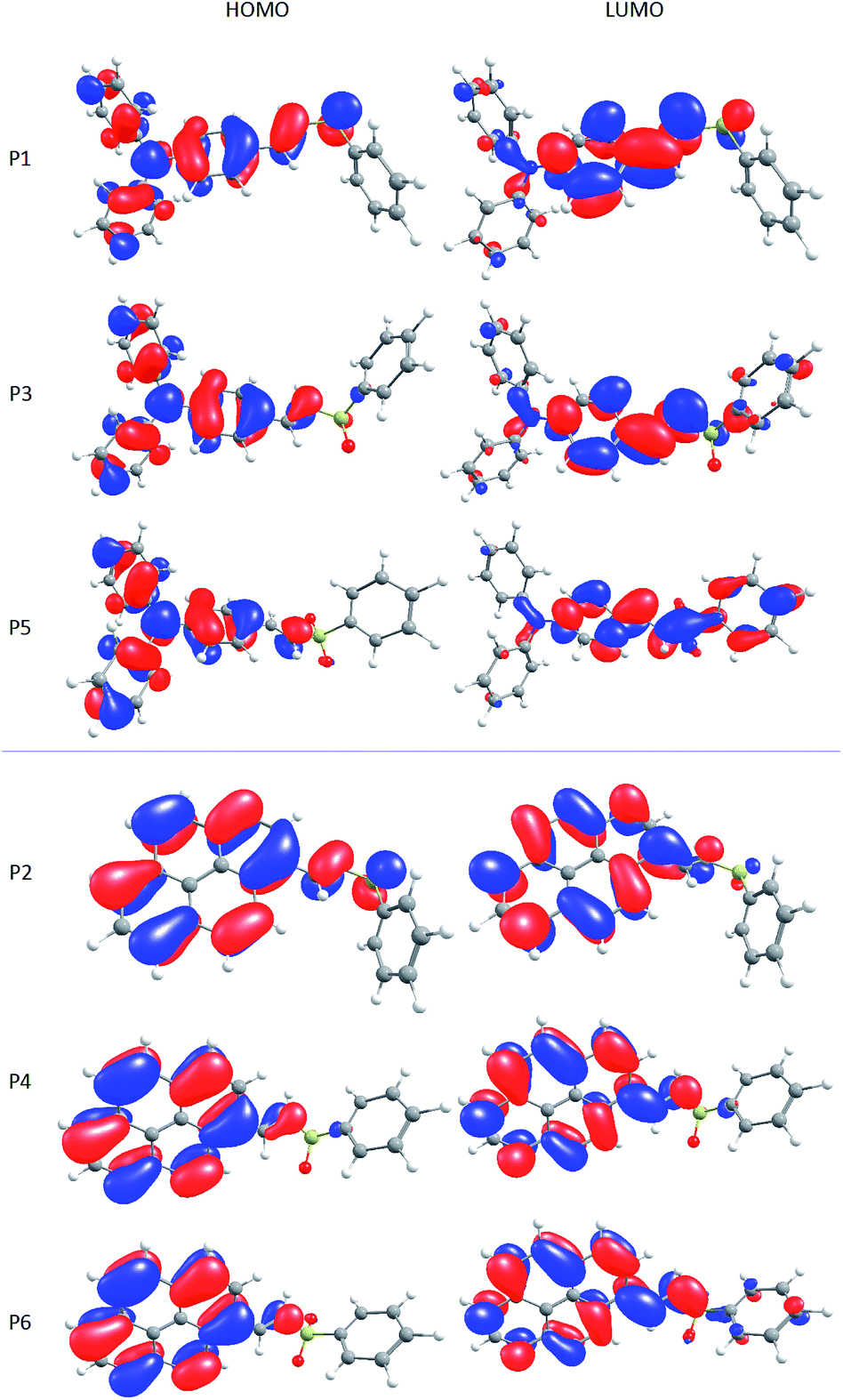 | ||
| Fig. 7 HOMO and LUMO of the P1–P6 dyes, calculated at the CAM-B3LYP level in 1,4-dioxane. | ||
It is possible to observe that in P1, the HOMO is localized at the donor group (triphenylamino or pyrene), but the LUMO is in the same group. This means that there is not an efficient charge separation, but the addition of oxygen atoms can help to improve this behaviour. Mainly in P5, a clear spatial separation of HOMO and LUMO can be observed, which means that P5 presents an ICT character. Furthermore, the large redshift in the maxima emission wavelength and the large dipole moment changes between the ground and first excited states are evidences of the ICT character in P5.
On the other hand, P2, P4 and P6 undergo a different pathway, since pyrene has a small electron-withdrawing character. The attachment of oxygen atoms to sulfur still increases the molecular orbital separation (even poorly from P2 to P4 and a little bit better from P4 to P6). However, the HOMO and LUMO are virtually in the same portion of the molecule, indicating a local excitation, leading to a high electron–hole recombination rate.69,72
The charge transfer character could be evaluated using descriptors based in TD-DFT, like the Δr-index. This descriptor was proposed in 2013 by Guido et al. and is based on the distances between the centroids of the orbitals involved in the excitations and interpreted in terms of the hole–electron distance.72 This distance is related to the nature of the electronic transition since, often, local excitations are characterized by short distances, while larger distances are usually found for charge transfer excitations. Using this descriptor, available in Multiwfn, a multifunctional wavefunction analyzer,73 we have obtained values equal to 1.8, 3.2, 4.1 Å for P1, P3 and P5, respectively and values equal to 0.9, 0.9 and 1.5 Å for P2, P4 and P6, respectively. According to the authors, these values indicate that charge transfer excitations are expected only for P3 and P5, in agreement with our previous discussion based on HOMO–LUMO separations and dipole momenta variations.
Conclusions
In summary, new vinyl sulfides and sulfoxides were successfully synthesized by reaction of appropriate aldehydes (triphenylamine and pyrene derivatives) and phosphonates, using NaH as base. These compounds were obtained in excellent yields with a clear preference for the E isomers. It could also be observed that the stereoselectivity of the Horner–Wadsworth–Emmons reaction was significantly high. In addition, the related vinyl sulfones were also accessed in good yields by a simple oxidation step of the respective vinyl sulfides using mCPBA. The oxidation reaction took place with isomerization of the Z isomers, yielding the sulfones as single E isomers. The introduction of sulfoxide and sulfones as electron-withdrawing groups resulted in a series of small dyes with the D–π–A structure. All studied compounds presented absorption maxima located in the UV-A region with molar absorptivity coefficient ε values (∼104 M−1 cm−1) and calculated radiative rate constants k0e (∼108 s−1) related to spin and symmetry allowed 1π–π* electronic transitions. For both sets of compounds, despite the presence of electron withdrawing and electron donating groups in the vinylic conjugated system, a small solvatochromic effect was observed, indicating an almost absent charge transfer character in the ground-state. The sulfur based compounds containing the triphenylamino moiety present a main emission band located in the violet-green region. These compounds show a significant redshift on the emission maxima with increasing solvent polarity (1915 to 3531 cm−1, from sulfide to sulfone, respectively). The positive solvatochromism can be related to a better electron delocalization in the excited state resulting from an intramolecular charge transfer state in these compounds. Moreover, this effect is greater in the sulfone derivative due to the higher electronic withdrawing character of this group relative to sulfoxides and sulfides. The pyrene based sulfur compounds also present an emission band in the violet-blue region. A less intense redshift in the emission maxima with increasing solvent polarity was observed in the pyrene derivatives due to the less donating character of the pyrene group relative to the triphenylamino moiety. DFT and TD-DFT calculations were performed at the PBE1PBE and CAM-B3LYP levels. The CAM-B3LYP functional presented a better agreement with the experimental photophysical data. In the ground state no solvatochromic effect was observed. On the other hand, a bathochromic effect was detected in the emission maxima when the solvent dielectric constant was increased. This behavior indicates a higher stabilization of S1 compared to S0. All the vertical electronic transitions can be assigned to a 1π–π* character. Starting from sulfides to sulfones, the addition of oxygen atoms improves the charge separation, enabling an ICT state mostly in P3 and P5. This charge separation is more evident among the triphenylamino derivatives than in the pyrene derivatives, since the first one has a better electron-donating character.Acknowledgements
The authors thank CNPq, CAPES and Instituto Nacional de Inovação em Diagnósticos para a Saúde Pública (INDI-Saúde) for financial support and scholarships. Theoretical calculations were carried out with the support of the Centro Nacional de Supercomputação (CESUP), from the Universidade Federal do Rio Grande do Sul.Notes and references
- M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485 RSC.
- C. Z. Zhang, C. Lu, J. Zhu, G. Y. Lu, X. Wang, Z. W. Shi, F. Liu and Y. Cui, Chem. Mater., 2006, 18, 6091 CrossRef CAS.
- J. J. Kim, J. Lee, S. P. Yang, H. G. Kim, H. S. Kweon, S. Yoo and K. H. Jeong, Nano Lett., 2016, 16, 2994 CrossRef CAS PubMed.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231 CrossRef CAS PubMed.
- B. E. Hardin, H. J. Snaith and M. D. McGeheem, Nat. Photonics, 2012, 6, 162 CrossRef CAS.
- A. Osuka and T. Tanaka, Chem. Soc. Rev., 2015, 44, 943 RSC.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171 CrossRef CAS PubMed.
- J. A. Davies, A. Langovan, P. A. Sullivan, B. C. D. H. Olbricht, T. R. Ewy, C. M. Isborn, B. E. Eichinger, B. H. Robinson, P. J. Reid, X. Li and L. R. Dalton, J. Am. Chem. Soc., 2008, 130, 10565 CrossRef CAS PubMed.
- G. Xie, D. Chen, X. Li, X. Cai, Y. Li, D. Chen, K. Liu, Q. Zhang, Y. Cao and S. J. Su, ACS Appl. Mater. Interfaces, 2016, 8, 27920 CAS.
- S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923 CrossRef CAS.
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10 CrossRef CAS PubMed.
- L. Yan, X. Chen, Q. He, Y. Wang, X. Wang, Q. Guo, F. Bai, A. Xia, D. Aumiler, S. Vdović and S. Lin, J. Phys. Chem. A, 2012, 116, 8693 CrossRef CAS PubMed.
- C. Sissa, F. Terenziani, A. Painelli, R. Siram, K. Bhaskar and S. Patil, J. Phys. Chem. B, 2012, 116, 4959 CrossRef CAS PubMed.
- J. Z. Cheng, C. C. Lin, P. T. Chou, A. Chaskar and K. T. Wong, Tetrahedron, 2011, 67, 734 CrossRef CAS.
- E. Zojer, D. Beljonne, P. Pacher and J. L. Bredas, Chem.–Eur. J., 2004, 10, 2668 CrossRef CAS PubMed.
- P. Hrobárik, V. Hrobáriková, V. Semak, P. Kasák, E. Rakovský, I. Polyzos, M. Fakis and P. Persephonis, Org. Lett., 2014, 16, 6358 CrossRef PubMed.
- B. D. Zhao, G. L. Li, Y. Z. Shi, H. Q. Zhang and T. Wang, RSC Adv., 2015, 5, 54749 RSC.
- W. Hu, F. Bai, X. Gong, X. Zhan, H. Fu and T. Bjornholm, in Organic Optoelectronics, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- S. Kumar, P. Kumar, R. Srivastava, S. K. Pal and S. Ghosh, J. Phys. Chem. C, 2016, 120, 12723 CAS.
- T. H. Huang, J. T. Lin, L. Y. Chen, Y. T. Lin and C. C. Wu, Adv. Mater., 2006, 18, 602 CrossRef CAS.
- S. L. Lin, L. H. Chan, R. H. Lee, M. Y. Yen, W. J. Kuo, C. T. Chen and R. J. Jeng, Adv. Mater., 2008, 20, 3947 CrossRef CAS.
- Y. Zhang, S. L. Lai, Q. X. Tong, M. F. Lo, T. W. Ng, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61 CrossRef.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706 CrossRef CAS PubMed.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580 RSC.
- J. Ye, Z. Chen, M. K. Fung, C. Zheng, X. Ou, X. Zhang, Y. Yuan and C. S. Lee, Chem. Mater., 2013, 25, 2630 CrossRef CAS.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137 CrossRef CAS PubMed.
- A. R. Morales, A. Frazer, A. W. Woodward, H. Y. Ahn-White, A. Fonari, P. Tongwa, T. Timofeeva and K. D. Belfield, J. Org. Chem., 2013, 78, 1014 CrossRef CAS PubMed.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS PubMed.
- J. S. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS PubMed.
- H. Zhu, M. Li, J. Hu, X. Wang, J. Jie, Q. Guo, C. Chen and A. Xia, Sci. Rep., 2016, 6, 24313 CrossRef CAS PubMed.
- M. Monçalves, D. S. Rampon, P. H. Schneider, F. S. Rodembusch and C. C. Silveira, Dyes Pigm., 2014, 102, 71 CrossRef.
- Q. Verolet, A. Rosspeintner, S. Soleimanpour, N. Sakai, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2015, 137, 15644 CrossRef CAS PubMed.
- J. Míšek, A. V. Jentzsch, S. Sakurai, D. Emery, J. Mareda and S. Matile, Angew. Chem., Int. Ed., 2010, 49, 7680 CrossRef PubMed.
- S. O. Jeon, T. Earmme and S. A. Jenekhe, J. Mater. Chem. C, 2014, 2, 10129 RSC.
- M. Wang, X. Ma, J. Yu, X. Jia, D. Han, T. Zhou, J. Yang, J. Nie and T. Wang, Polym. Chem., 2015, 6, 4424 RSC.
- C. Nerungsi, P. Wanitchang, S. Sahasithiwat, K. Sadorn, T. Kerdcharoen and T. Thongpanchang, Tetrahedron Lett., 2010, 51, 6392 CrossRef CAS.
- S. Malashikhin and N. S. Finney, J. Am. Chem. Soc., 2008, 130, 12846 CrossRef CAS PubMed.
- T. H. Lee, K. L. Tong, S. K. So and L. M. Leung, Synth. Met., 2005, 155, 116 CrossRef CAS.
- C. C. Silveira, F. Rinaldi and R. C. Guadagnin, Eur. J. Org. Chem., 2007, 4935 CrossRef CAS.
- G. Padilha, T. S. Kaufman and C. C. Silveira, Tetrahedron Lett., 2016, 57, 3349 CrossRef CAS.
- M. M. Bassaco, M. Monçalves, F. Rinaldi, T. S. Kaufman and C. C. Silveira, J. Photochem. Photobiol., A, 2014, 290, 1 CrossRef CAS.
- W. L. F. Armarego, in Purification of Laboratory Chemicals, ed. W. L. F. Armarego and C. L. L. Chai, fifth edn, Elsevier Academic Press, Cornwall, UK, 2003 Search PubMed.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01,Gaussian, Inc., Wallingford CT, USA, 2013 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- D. M. P. Aroche, J. M. Toldo, R. R. Descalzo, P. F. B. Gonçalves and F. S. Rodembusch, New J. Chem., 2015, 39, 6987 RSC.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845 RSC.
- M. Pastore, E. Mosconi, F. Angelis and M. Grätzel, J. Phys. Chem. C, 2010, 114, 7205 CAS.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019 CrossRef CAS.
- E. Papajak, J. Zheng, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- G. A. Andrienko, CHEMCRAFT, http://www.chemcraftprog.com Search PubMed.
- K. Otrubova, B. F. Cravatt and D. L. Boger, J. Med. Chem., 2014, 57, 1079 CrossRef CAS PubMed.
- S. J. Strickler and R. A. Berg, J. Phys. Chem., 1962, 37, 814 CrossRef CAS.
- N. J. Turro, J. C. Scaiano and V. Ramamurthy, in Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, USA, 1st edn, 2008 Search PubMed.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006, ch. 6, pp. 205–235 Search PubMed.
- S. Achelle, A. Barsella, B. Caro and F. R. Guen, RSC Adv., 2015, 5, 39218 RSC.
- B. Huang, Q. Qi, W. Jiang, J. Tang, Y. Liu, W. Fan, Z. Yin, F. Shi, X. Ban, H. Xu and Y. Sun, Dyes Pigm., 2014, 111, 135 CrossRef CAS.
- B. Huang, Z. Yin, X. Ban, Z. Ma, W. Jiang, W. Tian, M. Yang, S. Ye, B. Lin and S. Yueming, J. Lumin., 2016, 172, 7 CrossRef CAS.
- R. S. Kathayat and N. S. Finney, J. Am. Chem. Soc., 2013, 135, 12612 CrossRef CAS PubMed.
- W. Lee and W. S. Jenks, J. Org. Chem., 2001, 66, 474 CrossRef CAS PubMed.
- S. A. El-Daly, A. M. Asiri and K. A. Alamry, J. Fluoresc., 2014, 24, 1307 CrossRef CAS PubMed.
- E. Lippert, W. Lüder, F. Moll, W. Nägele, H. Boos, H. Prigge and I. Seinbold-Blankenstein, Angew. Chem., 1961, 73, 695 CrossRef CAS.
- J. P. Cerón-Carrasco, A. Ripoche, F. Odobel and D. Jacquemin, Dyes Pigm., 2012, 92, 1144 CrossRef.
- H. Roohi, F. Hejazi, N. Mohtamedifar and M. Jahantab, Spectrochim. Acta, Part A, 2014, 118, 228 CrossRef CAS PubMed.
- R. S. Iglesias, L. F. Campo, F. S. Rodembusch and V. Stefani, Int. J. Quantum Chem., 2008, 108, 2334 CrossRef CAS.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC.
- S. Agrawal, N. J. English, K. R. Thampi and J. M. MacElroy, Phys. Chem. Chem. Phys., 2012, 14, 12044 RSC.
- Z. R. Grabowski and K. Rotkiewicz, Chem. Rev., 2003, 103, 3899 CrossRef PubMed.
- C. Bernini, J. Chem. Theory Comput., 2014, 10, 3925 CrossRef CAS PubMed.
- C. A. Guido, P. Cortona, B. Mennucci and C. Adamo, J. Chem. Theory Comput., 2013, 9, 3118 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterization of the compounds and additional results of the theoretical calculations. See DOI: 10.1039/c6ra27989a |
| This journal is © The Royal Society of Chemistry 2017 |