
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fischer–Tropsch synthesis: effect of ammonia on product selectivities for a Pt promoted Co/alumina catalyst
Venkat Ramana Rao Pendyala,
Wilson D. Shafer,
Gary Jacobs,
Michela Martinelli,
Dennis E. Sparks and
Burtron H. Davis *
*
Center for Applied Energy Research, University of Kentucky, 2540 Research Park Drive, Lexington, KY-40511, USA. E-mail: burtron.davis@uky.edu; Tel: +1 859 257 0251
First published on 24th January 2017
Abstract
The effects of co-fed ammonia in synthesis gas on the activity and product selectivities of a typical cobalt catalyst (0.5% Pt–25% Co/Al2O3) were investigated during the Fischer–Tropsch synthesis using a continuously stirred tank reactor (CSTR). The product selectivities were compared at a similar CO conversion level for various concentrations (10–1000 ppmv) of ammonia, as well as clean (un-poisoned) conditions. The addition of 10–1000 ppmv ammonia (concentration of ammonia with respect to the syngas feed) significantly decreased activity; the percentage of deactivation was similar (∼40%) for the various concentrations of ammonia used. At similar CO conversions, the addition of ammonia caused an increase in olefin selectivity and the corresponding paraffin and alcohol selectivities were decreased compared to the ammonia free synthesis conditions. Olefin selectivity increased with increasing concentration of ammonia, and the paraffin and alcohol selectivities were decreased with increasing ammonia concentration. At similar CO conversions, ammonia addition exhibited a positive effect on hydrocarbon selectivity (i.e., lower light gas products and higher C5+) and also light gas product selectivities (C1–C4) were decreased and C5+ selectivity increased with increasing concentration of ammonia compared to ammonia free conditions.
1. Introduction
Fischer–Tropsch Synthesis (FTS) is a promising route for the transformation of synthesis gas (CO + H2, or syngas) into valuable hydrocarbons (gasoline, diesel, chemicals, etc.). It is suitable for the production of synthetic fuels from both fossil (e.g., coal, natural gas) and renewable (e.g., biomass) resources.1–3 This reaction produces a wide variety of products, from gases to heavy waxes. FTS is catalyzed by several transition metals including Fe, Co, and Ru, whereas only Co and Fe are commonly employed as reasonable commercial catalysts for the process. However, cobalt catalysts may feature high stability and activity, along with a lower deactivation rate, compared to iron catalysts,4 but require a narrower range of operating temperatures and pressures, because any overheat shifts the reaction towards methane formation and decreases the desired liquid product selectivity.5,6A gasification process is used to convert coal or biomass feedstocks into synthesis gas (CO and H2 mixture), which undergoes the Fischer–Tropsch reaction after H2/CO ratio adjustment and CO2 removal. However synthesis gas also contains various impurities that must be removed in order to prevent FT catalyst poisoning. Biomass-derived synthesis gas can contain both organic and inorganic impurities such as tars, benzene, toluene, xylene, NH3, HCN, H2S, COS, HCl, volatile metals, dust, and soot.7 Coal, which originates from biomass, contains many of the same inorganic impurities as found in biomass.8 Due to the variety of feedstocks that can be processed, significant variations in the composition of synthesis gas can be expected. This affects the nature of the impurities that are present (element, speciation), as well as their relative contents. Moreover, due to the high sensitivity of FT catalysts, severe specifications regarding syngas purity are required. Iron and cobalt catalysts share the sensitivity toward some, but not all, of the impurities commonly found in coal and biomass-derived syngas.
Among these impurities, high concentrations of sulfur compounds (≥500 ppbv) may cause irreversible deactivation of cobalt and iron-based catalysts during FT synthesis.9–12 The effect of ammonia on FTS has been reported in a few studies over iron and cobalt catalysts and the reported results, however, have been rather contradictory.13–18 Claeys et al.13 reported that co-feeding of up to 25% NH3 in syngas did not affect FT activity; similarly Borg et al.14 reported that 4.2 ppmv of ammonia does not have an impact for a CoRe/Al2O3 catalyst. An Exxon patent15 claims that a combined concentration of 100 ppb of NH3 and HCN in syngas will result in a catalyst half-life of only 4 days for a supported cobalt catalyst in a slurry reactor. However, the patent also indicates that hydrogen treatment of the catalyst can restore the initial activity. Van Berge16 reported that cobalt catalysts were rapidly but reversibly deactivated by HCN and NH3. In a recent investigation, Rausch et al.17 indicated that ammonia had a significant effect on the cobalt catalyst. Ordomsky et al.18 reported that the addition of NH3 and CH3CN during FT synthesis has a significant irreversible deactivation on supported cobalt catalysts, whereas no noticeable deactivation was observed with supported iron catalysts.
In our previous investigations,19–22 we reported that an iron catalyst was quite resistant to high levels (200 ppmw) of ammonia,19 whereas a cobalt catalyst underwent significant deactivation with parts per million levels of ammonia,20–22 regardless of whether ammonia gas or ammonium hydroxide was used. The addition of ammonia caused significant deactivation for alumina, titania and silica supported cobalt catalysts, but the rate of deactivation was higher for the silica-supported catalyst compared to the alumina and titania supported catalysts.20 In most studies reported in the open literature, the effect of ammonia on product selectivities over cobalt catalysts has not been reported at similar CO conversion levels. The aim of the present study is to compare the product selectivities at similar CO conversion levels for various concentrations of co-fed ammonia (10–1000 ppmv) as well as under clean conditions for a Pt-promoted Co/alumina catalyst.
2. Experimental
2.1 Catalyst preparation
Sasol/Catalox alumina (high purity γ-alumina, 140 m2 g−1) was used as the support for the cobalt catalyst. The catalyst was prepared by a slurry impregnation method, and cobalt nitrate was the precursor. In this method, which follows a Sasol patent,23 the ratio of the volume of solution used to the weight of alumina was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, such that the volume of solution was approximately 2.5 times the pore volume of the catalyst. Two impregnation steps were used, each to load 12.5% of Co by weight. After the second impregnation/drying step, the catalyst was calcined under air flow at 350 °C. The promoter was added by incipient wetness impregnation (IWI), and the precursor utilized for noble metal addition was tetraammineplatinum(II) nitrate. After Pt addition, the sample was dried and calcined again using the same conditions as used previously.
:1, such that the volume of solution was approximately 2.5 times the pore volume of the catalyst. Two impregnation steps were used, each to load 12.5% of Co by weight. After the second impregnation/drying step, the catalyst was calcined under air flow at 350 °C. The promoter was added by incipient wetness impregnation (IWI), and the precursor utilized for noble metal addition was tetraammineplatinum(II) nitrate. After Pt addition, the sample was dried and calcined again using the same conditions as used previously.
2.2 Catalyst characterization
BET surface area and porosity measurements of the calcined and ammonia exposed catalysts were conducted using a Micromeritics 3-Flex unit. Before performing the test, the temperature was gradually ramped to 160 °C and the sample was evacuated for at least 12 h to approximately 50 mTorr.
For the ammonia exposed catalyst, the catalyst was initially activated in a flow of 10 cm3 min−1 of H2 mixed with 20 cm3 min−1 of argon at 350 °C for 10 h, and then cooled to 220 °C under flowing Ar, after which ammonia gas (1% NH3 balanced nitrogen, 50 cm3 min−1) was flowed for an hour. The sample was then cooled to 85 °C under flowing of Ar. A gas mixture of 10 cm3 min−1 H2 and 20 cm3 min−1 Ar was flowed at 85 °C for 15 min to chemisorb hydrogen to the catalyst surface. The sample was then held at 85 °C under flowing Ar to remove and/or prevent adsorption of weakly bound species prior to increasing the temperature slowly to 350 °C, the reduction temperature of the catalyst.
2.3 Catalytic activity testing
FTS reaction experiments were conducted using a 1 L continuously stirred tank reactor (CSTR) equipped with a magnetically driven stirrer with turbine impeller, a gas-inlet line, and a vapor outlet line with a stainless steel (SS) fritted filter (2 μm) placed external to the reactor. A tube fitted with a SS fritted filter (0.5 μm opening) extending below the liquid level of the reactor was used to withdraw reactor wax (i.e., rewax, which is solid at room temperature), thereby maintaining a relatively constant liquid level in the reactor. Separate mass flow controllers were used to control the flow rates of hydrogen and carbon monoxide. Carbon monoxide, prior to use, was passed through a vessel containing lead oxide on alumina to remove traces of iron carbonyls. The gases were premixed in an equalization vessel and fed to the CSTR below the stirrer, which was operated at 750 rpm. The reactor temperature was maintained constant (±1 °C) using a temperature controller.The catalyst (∼12.0 g) was ground and sieved to 63–106 μm before loading into a fixed-bed reactor for 12 h of ex situ reduction at 350 °C and atmospheric pressure using a gas mixture of H2/He (60 NL per h) with a molar ratio of 1:3. The reduced catalyst (∼10.0 g) was transferred to a 1 L CSTR containing 310 g of melted Polywax 3000 under the protection of inert nitrogen gas. The reactor used for the reduction was weighed before and after reduction and after catalyst transfer in order to obtain an accurate amount of catalyst that was added. The transferred catalyst was further reduced in situ at 230 °C at atmospheric pressure using pure hydrogen (20 NL per h) for another 24 h before starting the FT reaction. In this study, the FTS conditions used were 220 °C, 1.99 MPa, H2/CO = 2, and a stirrer speed of 750 rpm.
Gas, water, oil, light wax, and heavy wax samples were collected daily and analyzed. Heavy wax samples were collected in a 200 °C hot trap connected to the filter containing dip tube. The vapor phase in the region above the reactor slurry passed continuously to the warm (100 °C) and then the cold (0 °C) traps located external to the reactor. The light wax and water mixture were collected daily from the warm trap and an oil plus water sample from the cold trap. Tail-gas from the cold trap was analyzed with an online HP Quad Series Micro GC, providing molar compositions of C1–C5 olefins and paraffins, as well as H2, CO and CO2. The analysis of the aqueous phase used an SRI 8610C GC with a thermal conductivity detector. The organic products were analyzed with an Agilent 6890 GC with a flame ionization detector and a 60 m DB-5 column. Hydrogen and carbon monoxide conversions were calculated based on GC analysis of the gas products, the gas feed rate, and the gas flow that was measured at the outlet of the reactor.
3. Results and discussion
To maintain experimental control, similar activation and reaction conditions (partial pressures of CO and H2) were used at various concentrations of ammonia exposure for a Pt–Co/alumina catalyst. After attaining steady state CO conversion, ammonia gas (10 to 1000 ppmv) was added to the syngas feed. The addition of ammonia (10–1000 ppmv) caused a significant initial drop in CO conversion and then the CO conversion remained relatively constant with addition of ammonia following the initial drop. More importantly, CO conversion did not recover to the expected value, even after termination of ammonia addition under normal FTS conditions, indicating that the catalyst had deactivated (i.e., NHx species are strongly adsorbed to deactivate cobalt active sites).20 The relative CO conversions for a Pt-promoted Co/alumina catalyst at various concentrations of ammonia compared to the pre-exposure conditions are shown in Fig. 1. This figure shows that the extent of deactivation is similar (±3%) for the various concentrations of ammonia used (10–1000 ppmv). The similar extents of poisoning are consistent with the chemisorption of the same amount of ammonia at the pressure represented by 10–1000 ppmv of ammonia.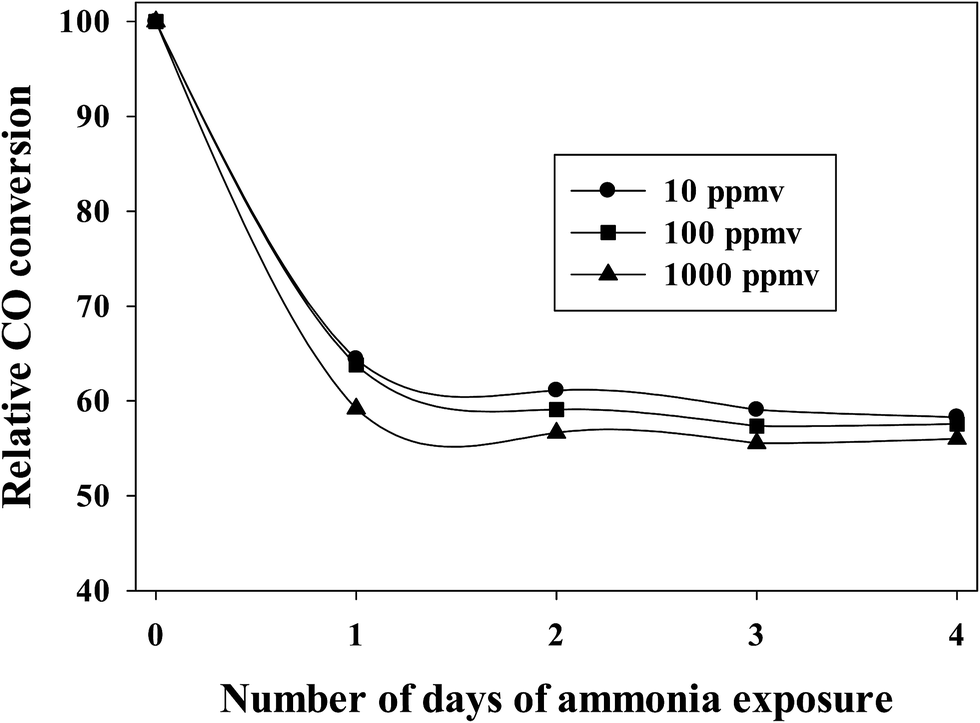 | ||
| Fig. 1 Effect of ammonia addition on relative CO conversion, related to the pre-exposure conditions. | ||
The effect of ammonia on product selectivities over a Pt-promoted Co/alumina catalyst at various concentrations of ammonia is shown in Table 1. In FT synthesis, it is well known that the conversion level usually influences the selectivity, in part due to increasing partial pressure of water and by increasing the partial pressure of reactants.25,26 It is therefore important to compare the catalysts at a similar CO conversion level. Experiments were conducted for the un-poisoned and ammonia exposed catalyst at various concentrations. The addition of 10 ppmv ammonia caused an increase in olefin selectivity from 16.7 to 21.3%, while paraffin selectivity slightly decreased to 77.5% from 78.3% and alcohol selectivity decreased to 1.2% from 5.0%. For 100 ppmv addition of ammonia, olefin selectivity increased to 23.3% from 16.7%, paraffin selectivity decreased to 74.7 from 78.3% and alcohol selectivity decreased to 2.0% from 5.0%. Similarly, the addition of 1000 ppmv ammonia caused an increase in olefin selectivity to 26.8% from 16.7%, while paraffin selectivity decreased to 72.7% from 80.4% and alcohol selectivity decreased to 0.5% from 2.7%. At similar CO conversions, the addition of ammonia caused an increase in olefin selectivity and corresponding decreases in alcohol and paraffin selectivities compared to the ammonia free synthesis condition. With increasing concentration of ammonia, the extents were increased with similar trends (i.e., increase in olefin selectivity, decreases in paraffin and alcohol selectivities) compared to the ammonia free synthesis condition.
| Ammonia concentration (ppmv) | ||||||
|---|---|---|---|---|---|---|
| 10 | 100 | 1000 | ||||
| 0 | 10 | 0 | 100 | 0 | 1000 | |
| a Reaction conditions: T = 220 °C; P = 1.99 MPa; H2/CO = 2; 10% N2; S.V. = 4–5.2 slph per gcatalyst. | ||||||
| CO conversion | 36.3 | 36.6 | 36.3 | 36.5 | 28.5 | 28.9 |
| Paraffin | 78.3 | 77.5 | 78.3 | 74.7 | 80.4 | 72.7 |
| Alcohol | 5.0 | 1.2 | 5.0 | 2.0 | 2.7 | 0.5 |
| Olefin | 16.7 | 21.3 | 16.7 | 23.3 | 16.9 | 26.8 |
The effect of ammonia on hydrocarbon selectivities at similar CO conversions were compared for the unpoisoned and ammonia poisoned catalysts, which are shown in Table 2. The addition of 10 ppmv ammonia decreased the methane selectivity to 7.6% from 8.7% and the corresponding C5+ selectivity increased to 83.9% from 81.5%. For 100 ppmv addition of ammonia conditions, methane selectivity dropped to 6.7% from 8.7% and the C5+ selectivity was enhanced to 85.9% from 81.5%. Similarly, the addition of 1000 ppmv ammonia caused a significant decrease in methane selectivity (6.5% from 9.3%) and the corresponding C5+ selectivity increased to 86.2% from 80.2%. These results clearly indicate that addition of ammonia significantly decreases the lower hydrocarbons selectivity (C1–C4) and increases the corresponding higher hydrocarbon (C5+) selectivity. With increasing ammonia concentrations (10 to 1000 ppmv), lower hydrocarbon selectivities decreased and higher hydrocarbons selectivity increased.
| Ammonia concentration (ppmv) | ||||||
|---|---|---|---|---|---|---|
| 0 | 10 | 0 | 100 | 0 | 1000 | |
| a Reaction conditions: T = 220 °C; P = 1.99 MPa; H2/CO = 2; 10% N2; S.V. = 4–5.2 slph per gcatalyst. | ||||||
| CO conversion | 36.3 | 36.6 | 36.3 | 36.5 | 28.5 | 28.9 |
| C1 | 8.7 | 7.6 | 8.7 | 6.7 | 9.3 | 6.5 |
| C2–C4 | 9.8 | 8.5 | 9.8 | 7.4 | 10.5 | 7.3 |
| C5+ | 81.5 | 83.9 | 81.5 | 85.9 | 80.2 | 86.2 |
A loading of 0.5% Pt is typical of 25% Co/Al2O3 research catalysts for facilitating the reduction of cobalt oxides that strongly interact with the support.24,27 EXAFS experiments have revealed that Pt is in contact with Co at the atomic level, with no Pt–Pt coordination observed;28 moreover, comparing selectivity at constant conversion, Pt did not significantly alter the selectivity of FTS compared to an un-promoted catalyst.29 That is, the role of Pt is primarily to increase the extent of cobalt reduction to, in turn, increase the surface Co metal active site density. In our earlier study,22 the effect of ammonia over Pt promoted and un-promoted 20% Co/SiO2 catalysts was investigated. Similar trends were observed for the un-promoted and Pt-promoted silica supported cobalt catalysts, and the Pt promoted catalyst deactivated more than the un-promoted catalyst during the ammonia exposure. Regarding the conversion and selectivity trends, Pt promoted Co/SiO2 did not display any additional benefits or drawbacks relative to the un-promoted silica supported cobalt catalyst.
The 1-olefin contents in total hydrocarbons as a function of carbon number at similar CO conversions for the un-poisoned and 1000 ppmv ammonia poisoned catalysts are shown in Fig. 2. At similar CO conversion level, the 1-olefin content (selectivity) of the ammonia exposed catalyst is higher than for the unpoisoned catalyst at all carbon numbers (C2–C17). Ethylene is more reactive than other low molecular weight 1-olefins, and thus its selectivity is correspondingly lower. The low ratio of ethylene to ethane was probably due to the rapid readsorption30 and hydrogenation and/or incorporation of ethylene for chain propagation.31 Linear 1-olefins are known to be the main primary organic products of FTS. Once formed, they can readsorb from the catalyst surface and undergo secondary reactions: hydrogenation, isomerization, re-insertion, hydrogenolysis and hydroformylation. The observed product can be impacted by secondary reactions. Olefin secondary reactions strongly depend on reaction conditions (temperature, partial pressures of H2, CO and water and residence time), the catalyst used and the olefin chain length. The olefin content (from C3 onwards) decreased with increasing carbon number for both unpoisoned and ammonia poisoned catalysts. The decrease in olefin content with increasing carbon number (for C3+ hydrocarbons) has been attributed to differences in reactivity, intraparticle diffusional effects, residence time differences due to vapor pressure differences and solubility variation with molecular weight.6,32–36 Diffusivity decreases with increases in molecular weights of the species, and higher molecular weight 1-olefins have longer residence times within catalyst pores, thus increasing the probability for secondary reactions.32–34 Increases in solubility with increases in carbon number also result in increased residence times for higher molecular weight hydrocarbons in the reactor as well as their higher liquid phase concentrations, resulting in a greater extent of secondary reactions.6,34–36 Due to diffusional restrictions, larger α-olefins spend longer times in a pore, which increases the probability of their readsorption on FTS active sites before exiting the pore. In a typical liquid product produced from cobalt catalyst, olefins will appear around up to carbon number 25, whereas paraffins will appear above about carbon number 35. Olefins are not observed above carbon number ∼25 in the liquid products because their long pore residence time ensures that olefins will readsorb many times; only chains that terminate as unreactive paraffins exit the catalyst. Thus the higher paraffinic content of larger hydrocarbons is not the result of direct hydrogenation of α-olefins to the corresponding paraffin of equal size,32,33 but reflects instead the enhanced readsorption of α-olefins and surface chain initiation steps that lead to their ultimate desorption as paraffins.
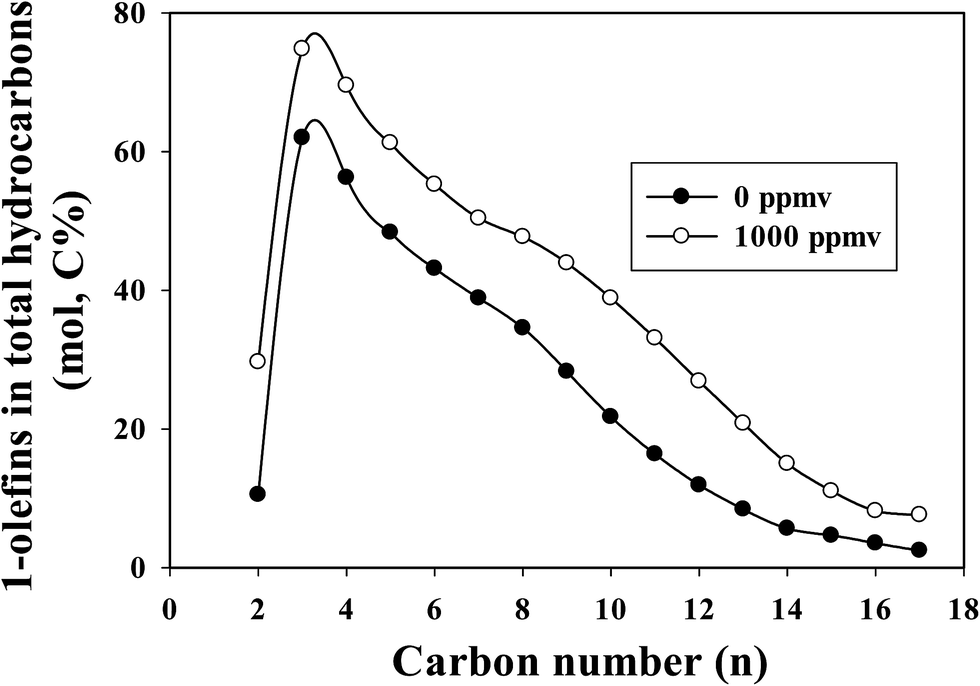 | ||
| Fig. 2 The effect of ammonia on 1-olefin content in total hydrocarbons at similar CO conversion level (∼28.5%) for a Pt-promoted Co/alumina catalyst. | ||
The decrease in methane selectivity along with higher 1-olefin content of the ammonia exposed catalysts might be due to the suppression of secondary reactions of olefins (such as hydrogenation, isomerization, re-insertion and hydroformylation). Two additional experiments were carried out to further confirm the suppression of secondary hydrogenation reaction of olefins, by co-feeding 1-decene to the unpoisoned and ammonia poisoned catalysts during FT synthesis. Gas chromatographic results and 1-decene conversion of the un-poisoned and 1000 ppmv ammonia exposed conditions are shown in Fig. 3. The conversion of 1-decene was calculated from its rate of consumption assuming that the FT product formation rate was not affected by 1-decene addition. The added decene is primarily converted to the corresponding linear paraffin (decane). The activity and selectivity results of 1-decene under FT conditions for the unpoisoned and ammonia exposed catalysts are shown in Table 3. The 1-decene conversion of the ammonia exposed (1000 ppmv) catalyst was found to be 46.0%. The selectivity of the hydrogenation product (decane) was found to be 90.0% and the remaining 10.0% selectivity was to isomerization products (cis and trans isomers of 2-decene). For the clean catalyst, 85.4% 1-decene conversion was observed, and the selectivities of the hydrogenation and isomerization products were found to be 91.0% and 9.0%, respectively. These results clearly reveal that addition of ammonia caused a significant suppression of secondary reactions of olefins such as hydrogenation and isomerization for the supported cobalt catalysts. The 1-decene addition during FT over unpoisoned and ammonia exposed catalysts does not have any significant impact on methane selectivity. This suggests that hydrogenolysis leading to methane from added 1-decene and the olefins formed during synthesis played a negligible role.
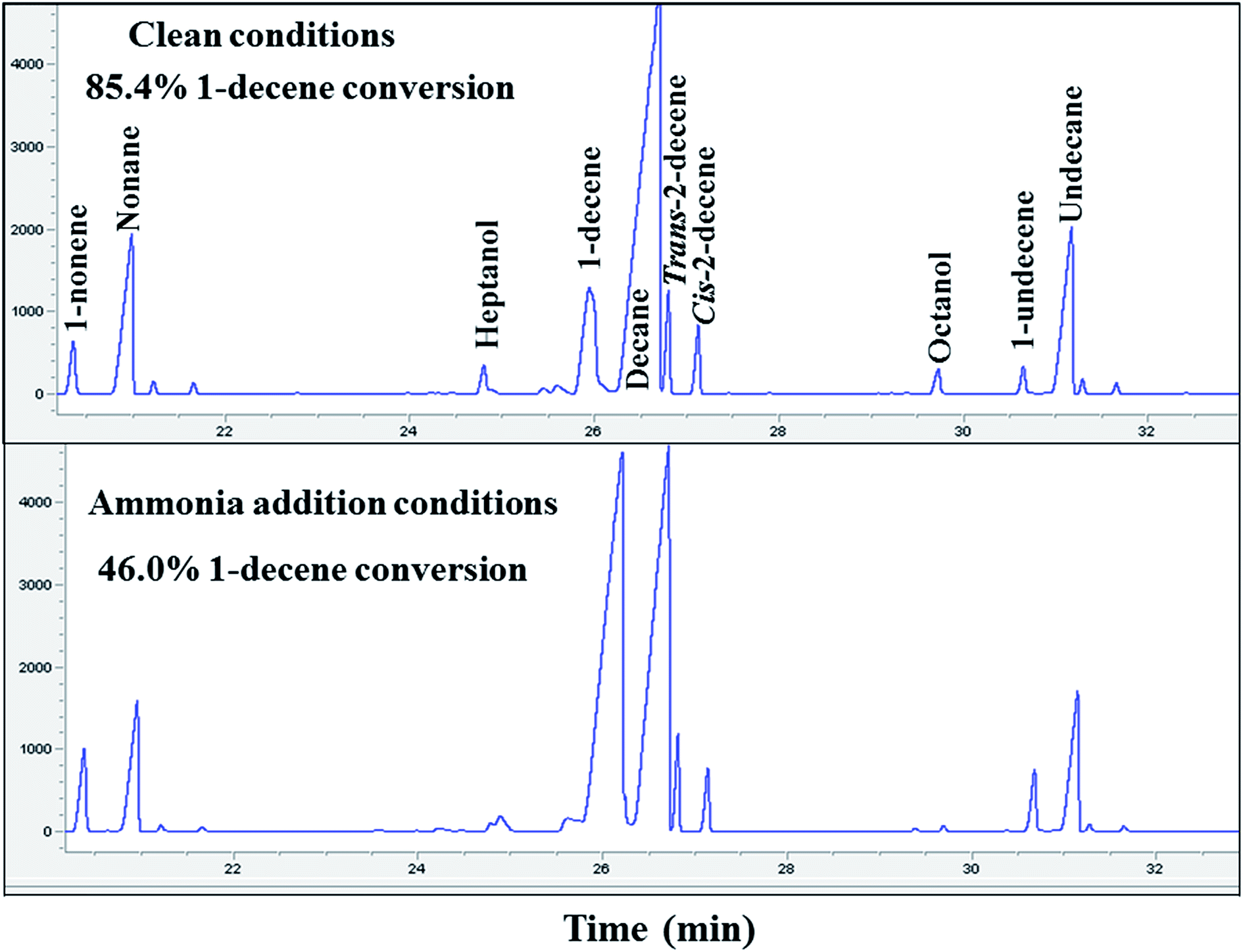 | ||
| Fig. 3 Gas chromatographic results of 1-decene conversion at clean and 1000 ppmv ammonia addition conditions. | ||
| Conversion/selectivity (%) | Unpoisoned run | Ammonia exposed run (1000 ppmv) |
|---|---|---|
| a FT reaction conditions: T = 220 °C; P = 1.99 MPa; H2/CO = 2; 10% N2; S.V. = 3–6 slph per gcatalyst, 1-decene (1 mL h−1). | ||
| 1-Decene conversion | 85.4 | 46.0 |
| Selectivity to hydrogenated product (decane) | 91.0 | 90 |
| Selectivity to isomerization products (cis and trans 2-decene) | 9.0 | 10.0 |
The effect of ammonia on FTS activity and selectivity of cobalt catalysts is unclear; some researchers have reported no effect,13,14 while others have reported that ammonia had a significant impact.15–18,20,21 In the open literature, most of the studies have not reported the product selectivities at similar CO conversion level. Recently, Rausch et al.17 reported that ammonia addition had a significant impact on activity and product selectivity as well. The activity of the catalyst was decreased with increasing the concentration of ammonia. The addition of ammonia resulted in an increase in olefin selectivity and decreases in paraffin and alcohol selectivities. Ordomsky et al.18 also reported that small amounts of acetonitrile and ammonia affected the FTS activity and selectivity of the cobalt catalysts. The deactivation of the cobalt catalyst was explained by possible formation of cobalt nitride, which may be responsible for the selective blocking of methanation sites. Lower methane and higher C5+ selectivity of the cobalt catalysts was explained by a decrease in intrinsic hydrogenation activity of cobalt sites after the formation of cobalt nitride. The effect of ammonia on product selectivity of Pt–Co/alumina catalyst is similar to the effect of potassium promoter to an iron-based FT catalysts. As we mentioned in our earlier studies,37,38 the addition of potassium to the iron catalysts increases the strength of CO adsorption and suppresses H2. This results in a high CO/H concentration ratio on the catalyst surface and consequently leads to a lower hydrogenation activity. A lower hydrogenation activity would suppress the hydrogenation of surface CHx species and also secondary hydrogenation of olefins. As a result, low selectivity to methane and high selectivity to higher hydrocarbons and olefins was observed.
To understand the reason for changes in activity and product selectivities following ammonia exposure, the catalysts were investigated by characterizing the freshly calcined and ammonia exposed catalysts by using BET surface area and hydrogen chemisorption techniques. BET surface area and pore size distribution results of the Pt–Co/alumina catalysts are presented in Table 4. The BET surface area and pore size distribution results of the freshly calcined, FT used, ammonia exposed during FT and the regenerated (after ammonia exposure mild in situ H2 treated) catalysts had similar values (±3%, within experimental error range). These values suggest that ammonia exposure during FTS does not cause any significant structural changes of the catalyst (e.g., pore collapse) as was the case with Co/SiO2 catalysts.20 The hydrogen chemisorption results of the freshly calcined catalyst are shown in Table 5. Ammonia addition may decrease the reactivity of hydrogen. For a hydrogen activated freshly calcined clean catalyst the H2-TPD up to 325 °C shows complete desorption of hydrogen (139 μmol g−1), while for the ammonia pretreated catalyst, only 88% of the H2 had desorbed (122 μmol g−1) by that temperature (325 °C). Both fresh and ammonia-exposed catalysts desorbed relatively similar amounts of hydrogen after a 30 min hold at 350 °C. During FT reaction, continuous addition of ammonia caused a 40% drop in activity, but in TPD, hydrogen desorbed was lowered by only 12%. Note that we flowed H2 at 85 °C for 15 min during TPD measurement, which may have removed a fraction of the NHx species from the surface prior to the TPD, so the result may not reflect the effect of constant NH3 addition during FT.
| Catalyst | BET surface area (m2 g−1) | Single point pore volume (cm3 g−1) | Single point pore diam. (nm) | BET SA ratio with fresh case |
|---|---|---|---|---|
| a Used for Fischer–Tropsch reaction (after extraction treated with 1% O2/N2 treated at 300 °C/4 h). | ||||
| γ-Al2O3 support (Catalox Sba 150) | 143.0 | 0.450 | 12.6 | — |
| Calcined 0.5% Pt–25% Co/Al2O3 | 98.2 | 0.245 | 10.1 | — |
| Unpoisoned 0.5% Pt–25% Co/Al2O3a | 95.6 | 0.248 | 10.4 | 0.97 |
| Ammonia exposed 0.5% Pt–25% Co/Al2O3a | 98.7 | 0.253 | 10.2 | 1.00 |
| After ammonia exposure mild in situ H2 treated 0.5% Pt–25% Co/Al2O3a | 96.6 | 0.250 | 10.3 | 0.98 |
The effect of ammonia on the catalytic activity can be explained by the adsorption of NHx species on the cobalt surface which may or may not form surface cobalt nitride and which severely decreases the ability to adsorb the reactants (hydrogen and CO). This changes the selectivity due to a coverage of NHx species on metal sites that normally adsorb hydrogen, creating a relatively hydrogen poor surface. Ordomsky et al.18 proposed that the formation of cobalt nitride might be responsible for the changes in activity and selectivity following ammonia exposure. There are two likely scenarios for the beneficial impacts on selectivity (lower methane and higher C5+) with ammonia addition for the alumina supported cobalt catalyst; one is that ammonia may inhibit the methanation sites responsible for the deviation of methane selectivity above that produced from ASF kinetics; another possibility is that ammonia inhibits termination of chain growth. The evidence for the former is a slight decrease in the deviation from the ASF distribution for methane and the evidence for the latter is an increase in the value of α. The formation of surface cobalt nitride or adsorbed NHx might block the methanation sites, which results in lower methane and higher C5+ selectivity. In earlier studies39,40 it was reported that cobalt nitride forms in the presence of ammonia over alumina and silica supported cobalt catalysts. In one of our very recent investigations,41 we studied the effect of COS on the activity and selectivity of a Pt–Co/alumina catalyst. The addition of COS caused a significant irreversible deactivation, and lower hydrocarbon (C1–C4) selectivities increased and higher hydrocarbon (C5+) selectivity decreased compared with unpoisoned catalyst at similar CO conversion level. S K-edge XANES analysis of the COS poisoned cobalt catalyst indicated that the formation of cobalt sulfide (Co–S) was responsible for the decrease in CO conversion and change in product selectivities. Similarly, in the present work, the addition of ammonia over cobalt catalyst might cause the formation of cobalt nitride, which might be responsible for the changes in activity and selectivity.
In our previous investigations,20,21 we reported that the addition of ammonia caused a significant decrease in CO conversion over supported cobalt catalysts and that catalyst activity did not recover even after termination of ammonia addition under normal FT conditions. However, the results for this study show that the activity of the Pt–Co/alumina catalyst virtually recovered to the initial values after the catalyst had been subjected to a mild in situ hydrogen treatment (i.e., similar FTS conditions but the carbon monoxide was switched off) for a 24 h period (Fig. 4). The addition of ammonia caused a significant decrease in activity and also altered the product selectivity. After termination of ammonia addition, the activity and selectivities did not recover to the initial values but remained at the poisoned level; this may be explained by blocking of active sites by chemisorbed surface NHx species, thereby restricting the adsorption of reactants (H2 and CO). These surface NHx species are not removed after the termination of the ammonia feeding. However, mild in situ hydrogen treatment removed the surface NHx species and the activity and product selectivity recovered to the initial values. The olefin/paraffin ratio of the Pt–Co/alumina prior to, during and after addition of ammonia and after hydrogen treatment conditions are shown in Fig. 5. As we discussed earlier, the addition of ammonia resulted in an increase in the olefin/paraffin ratio or olefin content relative to ammonia free synthesis conditions. After the termination of ammonia addition, the catalyst did not recover the activity and product selectivities (hydrocarbons, and olefin/paraffin ratio) as well. However, after a mild in situ hydrogen treatment, the activity and product selectivities recovered almost to their initial values (Fig. 4 and 5, respectively). These results indicate that the effect of ammonia is able to be reversed (activity and product selectivity) with mild in situ hydrogen treatment over a Pt–Co/alumina catalyst.
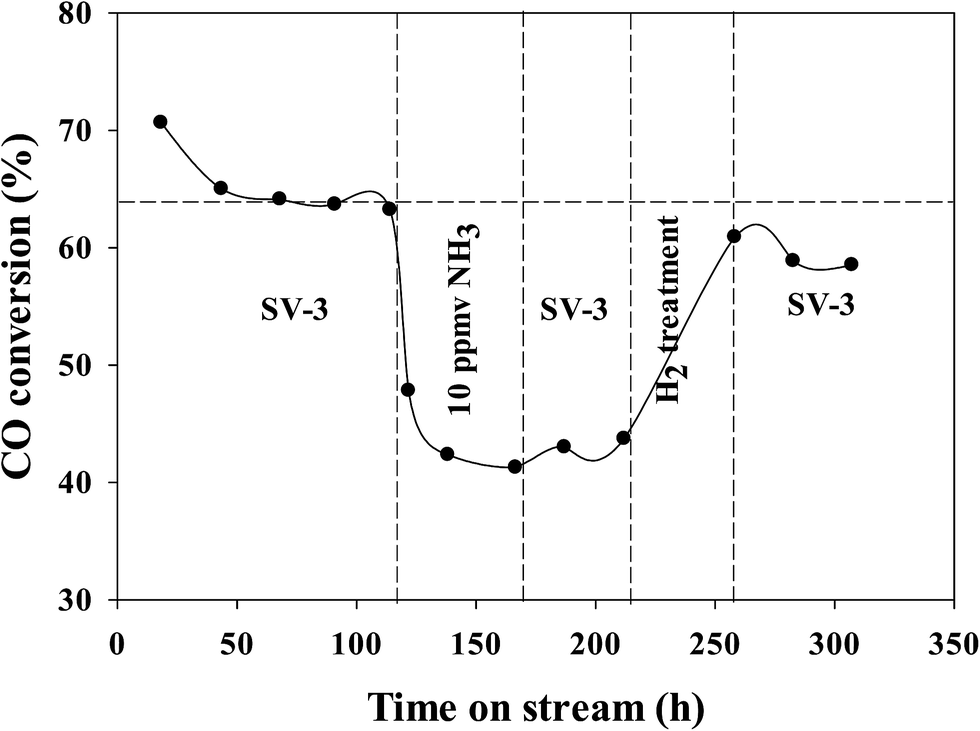 | ||
| Fig. 4 Run including ammonia addition and hydrogen treatment for the Pt–Co/alumina catalyst. Replotted from ref. 20. | ||
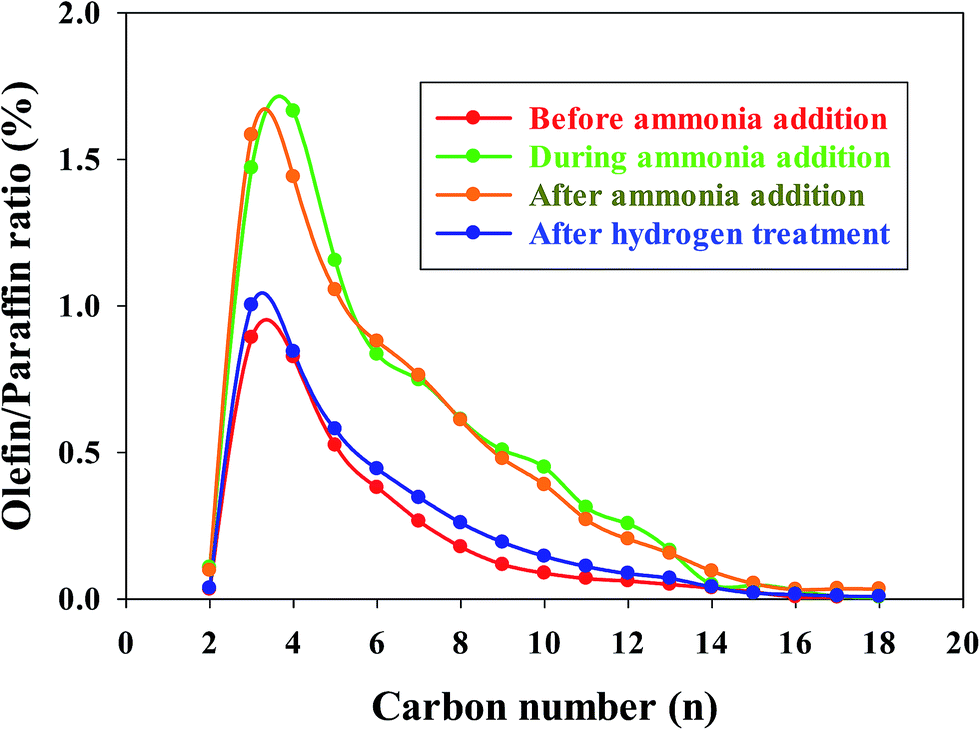 | ||
| Fig. 5 Olefin/paraffin ratio of before, during, after the addition of ammonia (10 ppmv) and also after hydrogen treatment for the Pt–Co/alumina catalyst. (Note: After hydrogen treatment means hydrogen treated after ammonia addition.) | ||
4. Conclusions
The effects of ammonia on activity and product selectivities were investigated by varying the concentration of ammonia during Fischer–Tropsch synthesis. The addition of ammonia caused a significant step drop in CO conversion, and the extent of decrease in CO conversion was similar for all concentrations (10 to 1000 ppmv) tested in this study. The product selectivities were compared at similar CO conversion level for unpoisoned and ammonia poisoned catalysts. At similar CO conversion, the addition of ammonia caused an increase in olefin selectivity and decreased the corresponding paraffin and alcohol selectivities. With increasing concentration of ammonia, the formation of olefins increased and the formation of paraffins and alcohols decreased. The hydrocarbon selectivity was also significantly impacted by the addition of ammonia; lower hydrocarbon selectivities (C1–C4) decreased and the corresponding higher hydrocarbon selectivity (C5+) increased for the ammonia exposed catalysts compared to under clean conditions at similar CO conversions. The decrease in methane selectivity accompanied by the higher olefin content of the ammonia poisoned catalyst is due to the suppression of secondary reactions of olefins (i.e., hydrogenation and isomerization). The suppression of secondary reactions with ammonia addition might be due to the formation of cobalt nitride. The effects of ammonia on activity and product selectivities were able to be reversed (virtually recovered to the initial values) with mild in situ hydrogen treatment over an ammonia exposed Pt–Co/alumina catalyst.Acknowledgements
This work carried out at the CAER was supported by the Commonwealth of Kentucky and DOE grant (DE-FC26-08-NT05988).References
- E. van Steen and M. Claeys, Chem. Eng. Technol., 2008, 31, 655 CrossRef CAS.
- A. de Klerk, Green Chem., 2008, 10, 1249 RSC.
- H. Jahangiri, J. Bennett, P. Mahjoubi, K. Wilson and S. Gu, Catal. Sci. Technol., 2014, 4, 2210 CAS.
- P. M. Maitlis and A. de Klerk, Greener Fischer–Tropsch Processes, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- R. Philippe, M. Lacroix, L. Dreibine, C. Pham-Huu, D. Edouard, S. Savin, F. Luck and D. Schweich, Catal. Today, 2009, 147, S305 CrossRef CAS.
- R. A. Dictor and A. T. Bell, J. Catal., 1986, 97, 121 CrossRef CAS.
- C. N. Hamelinck, A. P. C. Faaij, H. den Uil and H. Boerrigter, Energy, 2004, 29, 1743 CrossRef CAS.
- F. N. Cayan, M. J. Zhi, S. R. Pakalapati, I. Celik, N. Q. Wu and R. Gemmen, J. Power Sources, 2008, 185, 595 CrossRef CAS.
- W. Ma, G. Jacobs, D. E. Sparks, W. D. Shafer, H. H. Hamdeh, S. D. Hopps, V. R. R. Pendyala, Y. Hu, Q. Xiao and B. H. Davis, Appl. Catal., A, 2016, 513, 127 CrossRef CAS.
- W. Ma, G. Jacobs, W. D. Shafer, V. R. R. Pendyala, Q. Xiao, Y. Hu, B. H. Davis and B. H. Davis, Catal. Lett., 2016, 146, 1204 CrossRef CAS.
- D. E. Sparks, G. Jacobs, M. K. Gnanamani, V. R. R. Pendyala, W. Ma, J. Kang, W. D. Shafer, R. A. Keogh, U. M. Graham, P. Gao and B. H. Davis, Catal. Today, 2013, 215, 67 CrossRef CAS.
- S. S. Pansare and J. D. Allison, Appl. Catal., A, 2010, 387, 224 CrossRef CAS.
- M. Claeys, E. van Steen, T. Sango, C. de Vries, R. Henke, A. K. Rausch and F. Roessner, 9th Novel Gas Conversion Symposium, Lyon, France, 2010 Search PubMed.
- Ø. Borg, N. Hammer, B. C. Enger, R. Myrstad, O. A. Lindvåg, S. Eri, T. H. Skagseth and E. Rytter, J. Catal., 2011, 279, 163 CrossRef.
- S. C. LeViness, C. J. Mart, W. C. Behrmann, S. J. Hsia and D. R. Neskora, Slurry Hydrocarbon Synthesis Process with Increased Catalyst life. International: Patent WO 98/50487, 1998.
- P. J. van Berge and E. A. Caricato, Oral Presentation at Catalysis Society of South Africa (CATSA), Kruger National Park, South Africa, 2000 Search PubMed.
- A. K. Rausch, L. Schubert, R. Henkel, E. van Steen, M. Claeys and F. Roessner, Catal. Today, 2016, 275, 94 CrossRef CAS.
- V. V. Ordomsky, A. Carvalho, B. Legras, S. Paul, M. Virgine, V. L. Sushkevich and A. Y. Khodakov, Catal. Today, 2016, 275, 84 CrossRef CAS.
- W. Ma, G. Jacobs, D. E. Sparks, V. R. R. Pendyala, S. G. Hopps, G. A. Thomas, H. H. Hamdeh, A. MacLennan, Y. Hu and B. H. Davis, J. Catal., 2015, 326, 149 CrossRef CAS.
- V. R. R. Pendyala, G. Jacobs, C. Bertaux, S. Khalid and B. H. Davis, J. Catal., 2016, 337, 80 CrossRef CAS.
- V. R. R. Pendyala, M. K. Gnanamani, G. Jacobs, W. Ma, W. D. Shafer and B. H. Davis, Appl. Catal., A, 2013, 468, 38 CrossRef CAS.
- V. R. R. Pendyala, G. Jacobs, W. Ma and B. H. Davis, in Fischer–Tropsch Synthesis, Catalysts, and Catalysis: Advances and Applications, ed. B. H. Davis and M. L. Occelli, CRC Press, Boca Raton, 2015 Search PubMed.
- R. L. Espinoza, J. L. Visagie, P. J. van Berge and F. H. Bolder, US Pat. 5,733,839, 1998.
- G. Jacobs, T. K. Das, Y. Zhang, J. Li, G. Racoillet and B. H. Davis, Appl. Catal., A, 2002, 233, 263 CrossRef CAS.
- Ø. Borg, Z. Yu, D. Chen, E. A. Blekkan, E. Rytter and A. Holmen, Top. Catal., 2014, 57, 491 CrossRef.
- J. Li, X. Zhan, Y. Zhang, G. Jacobs, T. K. Das and B. H. Davis, Appl. Catal., A, 2002, 228, 203 CrossRef CAS.
- G. Jacobs, Y. Ji, B. H. Davis, D. C. Cronauer, A. J. Kropf and C. L. Marshall, Appl. Catal., A, 2007, 333, 177 CrossRef CAS.
- G. Jacobs, J. A. Chaney, P. M. Patterson, T. K. Das, J. C. Maillot and B. H. Davis, J. Synchrotron Radiat., 2004, 11, 414 CrossRef CAS PubMed.
- W. Ma, G. Jacobs, R. A. Keogh, D. B. Bukur and B. H. Davis, Appl. Catal., A, 2012, 437–438, 1 CrossRef CAS.
- T. Komaya and A. T. Bell, J. Catal., 1994, 146, 237 CrossRef CAS.
- G. P. Van der Laan and A. A. C. M. Beenackers, Catal. Rev.: Sci. Eng., 1999, 41, 255 CAS.
- R. J. Madon, S. C. Reyes and E. Iglesia, J. Phys. Chem., 1991, 95, 7795 CrossRef CAS.
- E. Iglesia, S. C. Reyes and R. J. Madon, J. Catal., 1991, 129, 238 CrossRef CAS.
- D. B. Bukur, X. Lang, A. Akgerman and Z. Feng, Ind. Eng. Chem. Res., 1997, 36, 2580 CrossRef CAS.
- H. Schulz and M. Claeys, Appl. Catal., A, 1999, 186, 71 CrossRef CAS.
- E. W. Kuipers, S. Scheper, J. H. Wilson, H. Vinkenburg and H. Oosterbeek, J. Catal., 1996, 158, 228 CrossRef.
- V. R. R. Pendyala, U. M. Graham, G. Jacobs, H. H. Hamdeh and B. H. Davis, ChemCatChem, 2014, 6, 1952 CrossRef CAS.
- V. R. R. Pendyala, U. M. Graham, G. Jacobs, H. H. Hamdeh and B. H. Davis, Catal. Lett., 2014, 144, 1704 CrossRef CAS.
- Z. Yao, A. Zhu, J. Chen, X. Wang, C. T. Au and C. Shi, J. Solid State Chem., 2007, 180, 2635 CrossRef CAS.
- Y. Shi, Y. Wan, R. Zhang and D. Zhao, Adv. Funct. Mater., 2008, 18, 2436 CrossRef CAS.
- V. R. R. Pendyala, G. Jacobs, W. Ma, W. D. Shafer, D. E. Sparks, A. MacLennan, Y. Hu and B. H. Davis, Catal. Today Search PubMed , submitted.
| This journal is © The Royal Society of Chemistry 2017 |