
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of size-tunable superparamagnetic/polymeric core/shell nanoparticles by metal-free atom transfer radical polymerization at ambient temperature†
Xiaobing Wang‡
,
Ning You‡,
Fuqiang Lan,
Peng Fu*,
Zhe Cui*,
Xinchang Pang *,
Minying Liu* and
Qingxiang Zhao*
*,
Minying Liu* and
Qingxiang Zhao*
School of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, China. E-mail: zhaoqingxiang1@126.com; pangxinchang1980@163.com; lmy@zzu.edu.cn; fupeng@zzu.edu.cn; cuizhezzu@126.com; Fax: +86 371 67781596; Tel: +86 371 67781591
First published on 23rd January 2017
Abstract
A facile route to fabricate size-tunable superparamagnetic/polymeric core/shell nanoparticles with uniform distribution was introduced based on metal-free atom transfer radical polymerization at ambient temperature.
Core/shell nanostructures,1,2 in which an inorganic nanoparticle as the core was surrounded by a layer of inorganic or organic material, have been widely investigated both as a method to improve the stability and surface chemistry of the core materials and as a means of accessing unique physical and chemical properties that are not possible from one nanostructure alone.3 For example, the surfaces of metal or semiconductor nanoparticles are isolated by silica shells from interfacial chemistry;4,5 semiconductor shells increase the quantum yield of semiconductor nanoparticle cores;6 Au nanoparticles allow covalent attachment of organic ligands with thiol groups to generate unique optical properties;3,7 and polymeric shells aid the compatibility of inorganic nanoparticles as cores in polymer hosts.8,9 Whereas inorganic shells are typically grown from the surface of the core, many different methods have been described for fabricating organic polymer shells, for example, in situ polymerization from particle-bound initiators,10 direct attachment of functionalized polymers to surfaces of inorganic nanoparticles,2,11 layer-by-layer deposition,12,13 and synthesis of the nanoparticles in the presence of polymeric ligands.14 In all of these cases, the specific chemical interaction of the inorganic nanoparticle surface or the surface-bound polymers must be explicitly tailored in order to form the organic polymeric shell.15
Controlled free radical polymerizations (CRPs),16,17 as one of the most far reaching developments in polymer design and synthesis,18 allow facile access to functionalized polymers with controlled and well-defined structures.17 Among the controlled free radical polymerization techniques, atom transfer radical polymerization (ATRP) is the most utilized and operates via a redox equilibrium process mediated by a ligated metal catalyst [i.e., Cu(I), Ru(II), Fe(II), etc.] to make all the initiating sites growing with same polymerization speeds for the polymers with well-defined structures.19 However, for a variety of applications, such as microelectronics, biomaterials, functional organic–inorganic hybrid nanocomposites, etc., a key limiting factor in using ATRP is metal catalyst purification and contamination.20,21 Although a significant focus for the ATRP field since the initial discovery has therefore been directed toward lowering catalyst loadings22 or removal of residual metals,21 it is still difficult to widely utilize traditional ATRP technique for the fabrication of functional organic–inorganic hybrid nanocomposites,23 and we envisaged that a much more viable and ambitious solution to this grand challenge would be the development of a metal-free catalyst system for ATRP in the fabrication of organic–inorganic hybrid nanocomposites.18,24,25
Here we report an unconventional but robust strategy for the fabrication of size-tunable superparamagnetic/polymeric core/shell nanoparticles with uniform distribution by metal-free ATRP at ambient temperature. Firstly, the hydroxyl group of 12-hydroxydodecanoic acid was modified by the reaction between the hydroxyl group and 2-bromophenylacetyl bromide to prepare bi-functional ligands as the metal-free ATRP initiators, and then the bi-functional ligands reacted with iron(III) oxide to prepare precursors of superparamagnetic Fe3O4 nanoparticles. Superparamagnetic Fe3O4 nanoparticles with different sizes can be fabricated by in situ decomposition of precursors by using different organic solvents with different boiling points, and the bi-functional ligands as the metal-free ATRP initiators capped on the surface of Fe3O4 nanoparticles. Then MMA monomers were initiated to grow PMMA polymeric chains from the initiating sites on the surface of superparamagnetic Fe3O4 nanoparticles by the metal-free ATRP technique for the fabrication of core/shell superparamagnetic Fe3O4/PMMA hybrid nanoparticles, composed of superparamagnetic Fe3O4 nanoparticles as core and PMMA chains as shell with different sizes, 10-phenylphenothiazine as the photocatalyst under 380 nm LED irradiation at room temperature. The dimensions of Fe3O4 nanoparticles can be adjusted by changing the different organic solvents with different boiling points: higher boiling point for larger size of Fe3O4 nanoparticles, and the thickness of PMMA polymeric shell can be tuned by adjusting the LED irradiation time during the polymerization process.
As illustrated in Scheme 1, in order to synthesize bi-functional ligands as the metal-free ATRP initiators, the hydroxyl group of 12-hydroxydodecanoic acid was modified by the reaction between the hydroxyl group and 2-bromophenylacetyl bromide (Scheme S1†). Subsequently, the iron-carboxylate complex with the metal-free ATRP initiating sites was prepared by reacting iron(III) oxide and bi-functional ligands for the precursors of Fe3O4 nanocrystals (Scheme S2†). The iron-carboxylate complex was slowly heated to 320 °C in 1-octadecene solution, and then kept at that temperature for 30 min for the synthesis of Fe3O4 nanocrystals under argon.26–28 When the reaction temperature reached 320 °C, a severe reaction occurred, at the same time, and the original transparent solution became turbid and brownish black. The nanocrystals can be easily re-dispersed in various organic solvents, such as toluene, chloroform, hexane and so on. During the forming of Fe3O4 nanocrystals, the surface of nanoparticles was in situ capped by bi-functional ligands with metal-free ATRP initiating sites as end functional groups. In order to investigate the architectures of Fe3O4 nanocrystals, TEM characterization was carried out. The dark spherical structures in the TEM micrographs corresponded to Fe3O4 nanocrystals (Fig. 1), and the average diameter of nanoparticles was 12.1 ± 1.1 nm based on TEM images. X-ray diffraction (XRD) measurement also confirms the crystal structure of Fe3O4 nanocrystals, and the diffraction pattern of Fe3O4 nanoparticles was shown in Fig. S1.† In addition, energy dispersive spectroscopy (EDS) microanalysis was also used to confirm the composition of Fe3O4 nanocrystals by combining the XRD characterization, as shown in Fig. S2.† In order to determine the existence of organic initiators on the surface of Fe3O4 nanocrystals, FT-IR and TGA were used to characterize the metal-free ATRP initiators on the surface of Fe3O4 nanocrystals (Fig. S3 and S4†). The weight fraction of organic initiators on the surface of Fe3O4 nanoparticles can be determined by TGA (4.8%). Based on TGA result, the degradation temperature is about 368 °C, it is critical to ensure the existence of organic initiators after the preparation of Fe3O4 nanocrystals. The particle size of Fe3O4 nanocrystals can be controlled by using different solvents with different boiling points. For example, when reaction solvent was changed from 1-octadecene to trioctylamine (b.p. 365 °C), the particle size of the Fe3O4 nanocrystals can be changed from 12.1 ± 1.1 nm to 22.3 ± 2.9 nm, and the TEM images was shown in Fig. S5.† The results of tuning the sizes of Fe3O4 nanocrystals by using different solvents with different boiling points can be explained by the higher reactivity of iron-carboxylate complex in the solvent with higher boiling point.26
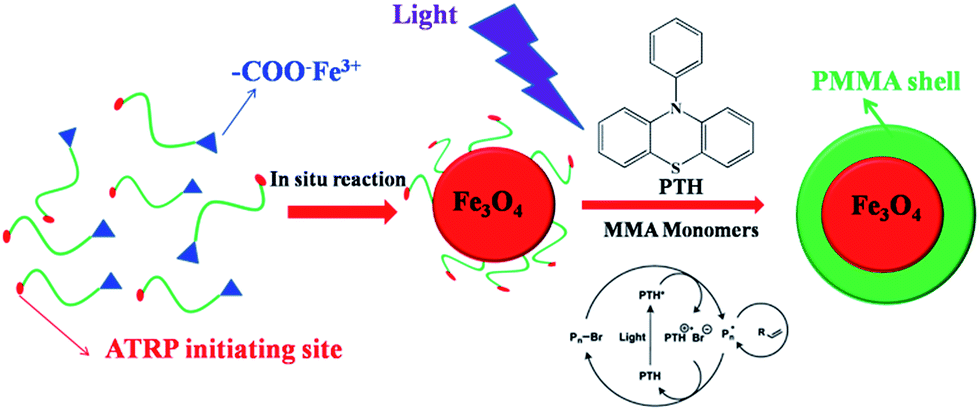 | ||
| Scheme 1 Schematic stepwise representation of the synthetic route to core/shell PMMA-capped Fe3O4 nanoparticles by metal-free ATRP based on PTH as photocatalyst. | ||
 | ||
| Fig. 1 Representative TEM images of Fe3O4 nanoparticles capped with metal-free ATRP initiating sites as ligands. (A) 50 nm; (B) 10 nm; (C) 5 nm; (D) 2 nm. | ||
Subsequently, the metal-free ATRP initiating sites on the surface of Fe3O4 nanocrystals was used to initiate the polymerization of methyl methacrylate (MMA) under 380 nm LED irradiation at room temperature for the fabrication of Fe3O4/PMMA core/shell nanostructures. In the metal-free ATRP system, 10-phenylphenothiazine (PTH) was used as photocatalyst that was synthesized from phenothiazine and chlorobenzene using C–N cross-coupling chemistry (Ered(PTH˙+/PTH*) = −2.1 V vs. SCE).18 The proposed mechanism of initiating MMA polymerization is reversible electron transfer from photoexcited PTH in order to reversibly activate the alkyl bromide initiator. After the alkyl bromide initiator was reduced by photoexcited photocatalyst, the radical was formed to initiate MMA polymerization, at the same time, PTH as photocatalyst was oxidized to stable radical cation species. Then the propagating polymer radical was oxidized to form dormant species. The whole process was cycled to form polymer chains (Scheme 1).18,24 Even if PTH as photocatalyst has been used for the metal-free ATRP,18 for the first time, metal-free ATRP by using PTH as photocatalyst was utilized for the preparation of size-tunable superparamagnetic/polymeric core/shell nanoparticles. In the traditional ATRP process, a ligated metal catalyst [i.e., Cu(I), Ru(II), Fe(II), etc.] was usually used to mediate the redox equilibrium process. However, for functional organic–inorganic hybrid nanocomposites, a key limiting factor in using ATRP is metal catalyst purification and contamination. In our fabrication system, the thickness of PMMA polymeric shell can be tuned by adjusting the LED irradiation time during the polymerization process. Firstly, the Fe3O4 nanocrystals with the average diameter of 12.1 ± 1.1 nm was used as example, PMMA polymeric chains can grow on the surface of Fe3O4 nanocrystals, PTH as the photocatalyst under 380 nm LED irradiation for 5 h at room temperature. In order to further investigate the architectures of core/shell Fe3O4/PMMA nanostructures by TEM characterization, PMMA polymeric shell was stained by ruthenium tetroxide (RuO4).29 The clear ∼4 nm shell structures in the TEM micrographs corresponded to PMMA domain as shell (Fig. 2). In addition, in order to obtain real information about PMMA grafting chains, the detachment of PMMA chains from the surface of Fe3O4 nanocrystals was carried out by dissolving Fe3O4 nanocrystals in acid condition. A monomodal peak with narrow distribution (PDI = 1.20) can be obtained for the detached PMMA grafting chains from GPC (Fig. S6†), and the molecular weight is 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1, which is close to 13050 g mol−1 (PMMA chain obtained from free initiators). The existence of Fe3O4 core was almost no effect on initiating MMA polymerization. In addition, the existence of capped PMMA chains on the surface of Fe3O4 core was also confirmed by 1H-NMR, FT-IR and TGA (Fig. S7–S9†). The weight fraction of PMMA in core/shell Fe3O4/PMMA nanoparticles was determined by TGA (16.2%). The thickness of PMMA polymeric shell can be tuned by adjusting the LED irradiation time during the polymerization process, and all results were demonstrated in Table S1.† For example, when the LED irradiation time increased to 10 h, the thickness of PMMA polymeric shell also increased to ∼6 nm (Fig. S10†).
100 g mol−1, which is close to 13050 g mol−1 (PMMA chain obtained from free initiators). The existence of Fe3O4 core was almost no effect on initiating MMA polymerization. In addition, the existence of capped PMMA chains on the surface of Fe3O4 core was also confirmed by 1H-NMR, FT-IR and TGA (Fig. S7–S9†). The weight fraction of PMMA in core/shell Fe3O4/PMMA nanoparticles was determined by TGA (16.2%). The thickness of PMMA polymeric shell can be tuned by adjusting the LED irradiation time during the polymerization process, and all results were demonstrated in Table S1.† For example, when the LED irradiation time increased to 10 h, the thickness of PMMA polymeric shell also increased to ∼6 nm (Fig. S10†).
 | ||
| Fig. 2 Representative TEM characterization results of core/shell Fe3O4/PMMA with different scale bars. (A) 20 nm; (B) 2 nm. Inset: representative configuration comprising reaction vial surrounded by 380 nm LEDs with a tube blowing compressed air for cooling. | ||
Core/shell Fe3O4/PMMA nanoparticles with different sizes show superparamagnetic behavior at room temperature. The hysteresis loops of two samples with different sizes were measured at 300 K, as shown in Fig. 3(A), while keeping the LED irradiation time fixed (5 h). Both of two samples show no remanence or coercivity at 300 K, that is, superparamagnetic behavior.30 In order to quantitatively evaluate the magnetic response of core/shell Fe3O4/PMMA nanoparticles to the applied magnetic field, the mass magnetization was characterized at 300 K. The saturation magnetization of Fe3O4 colloidal nanocrystals capped with PMMA polymeric shell was determined to be 20.9 and 30.2 emu g−1 (dimensions (D) of Fe3O4 nanocrystals: ∼12 nm and ∼22 nm). The increasing saturation magnetization should be due to the increasing size of Fe3O4 nanocrystals.30
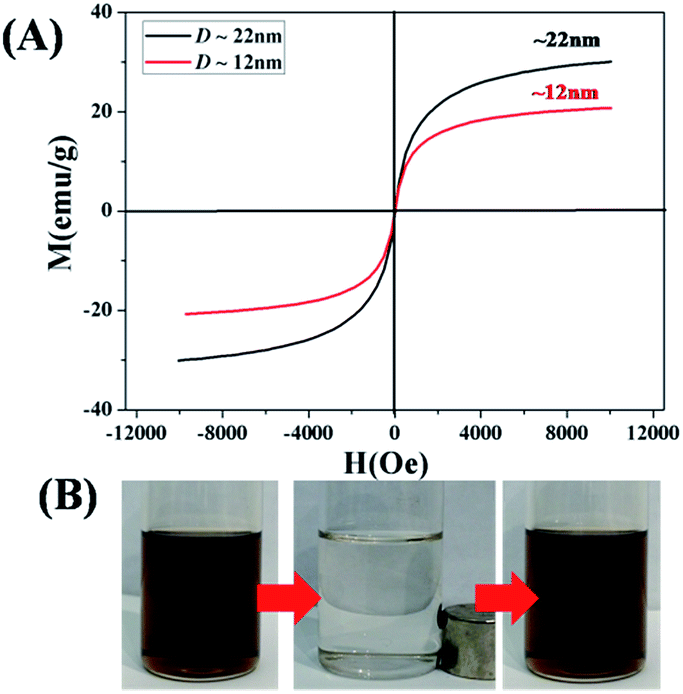 | ||
| Fig. 3 Superparamagnetic behavior of core/shell Fe3O4/PMMA nanoparticles with different sizes. (A) Mass magnetization M as a function of applied external field H at 300 K when Fe3O4 nanocrystals as core with different sizes while keeping the LED irradiation time fixed (5 h). (B) Digital images of a toluene Fe3O4 colloidal nanocrystals shown in Fig. 2. Without magnetic field, with magnetic field for 10 min, and after the magnetic field was removed (from left to right). | ||
Owing to surface-tethered hydrophobic PMMA polymeric chains as shell, the resulting hybrid inorganic–organic core/shell Fe3O4/PMMA nanoparticles by metal-free ATRP process are highly organic solvent-dispersed. The external magnetic responses were observed by dispersing core/shell Fe3O4/PMMA nanoparticles into toluene. As shown in Fig. 3(B), Fe3O4 colloidal nanocrystals deposited on the wall of the vial under the influence of external magnetic field within several minutes. When external magnet was removed, the aggregated Fe3O4 colloidal nanocrystals can be back into the original toluene solution.
In conclusion, we have developed an unconventional but robust strategy for the fabrication of size-tunable superparamagnetic/polymeric core/shell nanoparticles with uniform distribution by metal-free ATRP at ambient temperature. The bi-functional ligands as the metal-free ATRP initiators were firstly prepared by reacting the hydroxyl group of 12-hydroxydodecanoic acid and 2-bromophenylacetyl bromide, and then the bi-functional ligands reacted with iron(III) oxide to prepare precursors of superparamagnetic Fe3O4 nanoparticles. Superparamagnetic Fe3O4 nanoparticles with different sizes can be fabricated by in situ decomposition of precursors by using different organic solvents with different boiling points, and the bi-functional ligands as the metal-free ATRP initiators capped on the surface of Fe3O4 nanoparticles. Then MMA monomers were initiated to grow PMMA polymeric chains from the initiating sites on the surface of superparamagnetic Fe3O4 nanoparticles by the metal-free ATRP technique for the fabrication of core/shell superparamagnetic Fe3O4/PMMA hybrid nanoparticles, composed of superparamagnetic Fe3O4 nanoparticles as core and PMMA chains as polymeric shell with different sizes, PTH as the catalyst under 380 nm LED irradiation at room temperature. The dimensions of Fe3O4 nanoparticles as core can be adjusted by changing the different organic solvents with different boiling points: higher boiling point for larger size of Fe3O4 nanoparticles, and the thickness of PMMA polymeric shell can be tuned by adjusting the LED irradiation time during the polymerization process. Hence, this metal-free ATRP strategy may stand out as an emerging methodology to fabricate a myriad of other functional organic–inorganic hybrid nanocomposites with novel and tailorable physical properties for a range of intriguing applications, such as magnetic devices, nanoelectrics, sensors, catalysis and other fields.
Acknowledgements
The work was financially supported by the National High Technology Research and Development Program of China (863 Program) (No. 2011AA02A204-03), the Science and Technology Innovation Talents plans of Henan Province of China (No. 124200510011), 1000 Young Talent (to Xinchang Pang), Postdoctoral Research Sponsorship in Henan Province (2014004, to Zhe Cui), Key Science and Technology Development Program of Henan Province (152102310070, to Zhe Cui) and Outstanding Young Talent Research Fund of Zhengzhou University (1421320045, to Peng Fu).Notes and references
- Y. Kang and T. A. Taton, Angew. Chem., 2005, 44, 409–412 CrossRef CAS PubMed
.
- Z. Li, J. Zhang, P. Fu, Z. Cui, X. Pang, M. Liu and Q. Zhao, RSC Adv., 2016, 6, 79263–79267 RSC
- T. K. Mandal, M. S. Fleming and D. R. Walt, Nano Lett., 2002, 2, 3–7 CrossRef
- K. Kamata, Y. Lu and Y. Xia, J. Am. Chem. Soc., 2003, 125, 2384–2385 CrossRef CAS PubMed
- S. P. Mulvaney, M. D. Musick, C. D. Keating and M. J. Natan, Langmuir, 2003, 19, 4784–4790 CrossRef CAS
- K. Wu, G. Liang, D. Kong, J. Chen, Z. Chen, X. Shan, J. R. Mcbride and T. Lian, Chem. Sci., 2015, 7, 1238–1244 RSC
- Y. Cao, R. Jin and C. A. Mirkin, J. Am. Chem. Soc., 2001, 123, 7961–7962 CrossRef CAS PubMed
- M. K. Corbierre, N. S. Cameron, M. Sutton, S. G. J. Mochrie, L. B. Lurio, A. Rühm and R. B. Lennox, J. Am. Chem. Soc., 2001, 123, 10411–10412 CrossRef CAS PubMed
- S. Kim and M. G. Bawendi, J. Am. Chem. Soc., 2003, 125, 14652–14653 CrossRef CAS PubMed
- J. K. Oh, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6983–7001 CrossRef CAS
- S. J. Jeon, S. M. Yang, B. J. Kim, J. D. Petrie, S. G. Jang, E. J. Kramer, D. J. Pine and G. R. Yi, Chem. Mater., 2009, 3739–3741 CrossRef CAS
- L. Shen, J. Funct. Biomater., 2011, 2, 355–372 CrossRef CAS PubMed
- D. I. Gittins and F. Caruso, J. Phys. Chem. B, 2001, 105, 6846–6852 CrossRef CAS
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS PubMed
- M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS
- M. Ouchi, T. Terashima and M. Sawamoto, ChemInform, 2010, 41, 1–2 Search PubMed
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS PubMed
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, d. A. J. Read, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed
- C. Cheng, E. Khoshdel and K. L. Wooley, Nano Lett., 2006, 6, 1741–1746 CrossRef CAS PubMed
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed
- A. J. D. Magenau, N. C. Strandwitz, G. Armando and M. Krzysztof, Science, 2011, 332, 81–84 CrossRef CAS PubMed
- X. Pang, Y. He, J. Jung and Z. Lin, Science, 2016, 353, 1268–1272 CrossRef CAS PubMed
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed
- X. Pan, M. Lamson, J. Yan and K. Matyjaszewski, ACS Macro Lett., 2015, 4, 192–196 CrossRef CAS
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342–4345 CrossRef CAS PubMed
- J. Bai, X. Wang, P. Fu, Z. Cui, Q. Zhao, X. Pang and M. Liu, RSC Adv., 2016, 6, 9429–9435 RSC
- T. M. Chou, P. Prayoonthong, A. Aitouchen and M. Libera, Polymer, 2002, 43, 2085–2088 CrossRef CAS
- S. Xuan, Y.-X. J. Wang, J. C. Yu and K. Cham-Fai Leung, Chem. Mater., 2009, 21, 5079–5087 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26290b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |