
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual enhancement in the electroreduction of oxygen by NiCoPt by surface tunability through potential cycling†
Moorthi Lokanathanab,
Indrajit M. Patilac,
Alhasan Kabiru Usmanc,
Anita Swamic,
Pravin Walked,
M. Navaneethane and
Bhalchandra Kakade *ac
*ac
aSRM Research Institute, SRM University, Kattankulathur, 603 203, Chennai, India. E-mail: bhalchandrakakade.a@res.srmuniv.ac.in; Fax: +91 44 2745 6702; Tel: +91 44 2741 7920
bDepartment of Physics and Nanotechnology, SRM University, Kattankulathur, 603 203, Chennai, India
cDepartment of Chemistry, SRM University, Kattankulathur, 603 203, Chennai, India
dNational Centre for Nanosciences and Nanotechnology, University of Mumbai, Mumbai, 400098, India
eResearch Institute of Electronics, Shizuoka University, 3-5-1 Johoku, Naka-Ku, Hamamatsu, Shizuoka 432-8011, Japan
First published on 16th February 2017
Abstract
Chemically ordered interconnected nanostructures of NiCoPt alloy have been prepared using a simple solvothermal process and studied for oxygen reduction reaction (ORR) kinetics. NiCoPt/C catalyst has demonstrated an interesting trend of enhancement in the ORR activity along with long-term durability. The specific activity of 0.744 mA cm−2 for NCP10/C (NiCoPt/C prepared at reaction time of 10 h) is ∼3.7 times higher than that of Pt/C (0.2 mA cm−2). The durability of the catalyst was evaluated over 30k potential cycles in the lifetime regime. More significantly, a novel trend in the enhancement in the ORR activity during stability cycles has been observed for the first time, where a remarkable enhancement of 82% in the specific activity has been observed after 30k potential cycles. Thus, ∼7-fold higher activity of NCP10/C@30k over initial activity of commercial Pt/C would make a tremendous impact on fuel cell technology. Systematic X-ray diffraction studies were performed to supplement subsequent improvement in the ORR activity during potential cycling, where structural changes due to alloying and de-alloying taking place with formation of tetrahexahedron-like surfaces after 15k cycles. Furthermore, transmission electron microscopy (TEM) analysis after 30k durability cycles reveals better stability of NCP10/C nanostructure signifying the retention of Ni and Co due to the chemically ordered structures of NiCoPt alloy catalyst. The observed enhancement in durability might be due to the ordered arrangement of Pt and Ni/Co within the alloy.
1. Introduction
The great challenge in commercialization of clean energy, environmentally friendliness and high energy conversion efficiency of low temperature fuel cells, is severely hindered because of sluggish ORR kinetics,1,2 specifically for residential and automotive applications. A platinum (Pt) based catalyst of suitable composition with sustained catalytic activity of at least 2–4 times higher than that of commercial Pt and minimum poisoning could solve the disadvantage of using expensive Pt to achieve proton exchange membrane fuel cells (PEMFCs) as a viable technology with tremendous societal impact.3 A substantial breakthrough has been made on Pt based ternary electrocatalyst, such as alloying Pt with 3d-transition metals, including Pt1−x−yMxNy (M, N = Mn, Cr, Fe, V, Cu, Co, Ni etc.).4–22 Besides, in the past few decades synthetic methods such as sputtering,4 incipient wetness,5 electrodeposition,6 impregnation,7 solution route,8 dealloying,9 solvent co-reduction,10 surfactant-directed,11 hydrothermal,12 thermal decomposition,13 and multi-step processing14 have been immensely established. Such Pt-based catalysts with trimetalic compositions have shown extraordinary promise with regard to improved catalytic activity due to various aspects, including the composition effects,15 Pt-rich skin or layer,16 chemically ordered intermettalic,17 elemental rearrangement,18 annealing,19 exposed facets,20 surface structure,21 effect of nanoscale strain, metal–metal coordination number22 and so on. Further, Pt-rich trimetalic compositions like Pt3M0.5N0.5 (M, N: transition metals) have garnered substantial interest among researchers, compared with other trimetalic combinations, from the perspectives of both fundamental research and energy technology development, due to their unique ORR activity.19Specifically, higher activity of single crystalline Pt3Ni thin films has acquired intensive attention towards Pt–Ni structures with (111) surfaces and the specific activities are known to increase in the order of Pt (100) ≪ Pt (111) ≈ Pt (110), while the order changes to (100) < (110) ≪ (111) for spherical Pt–Ni alloy nanoparticles in aqueous HClO4 electrolyte.23 In addition, recently a successful synthesis of Pt3Ni nanocubes and octahedra along with their comparative ORR activities are reported in terms of (110) and (111) surface facets.24 Whereas, more recently, the correlation between the compositional segregation and element specific anisotropic growth of Pt–Ni nanocrystals and their ORR activity has been introduced.25,26 However, substantial confusion is still remaining due to the loss of shape and size after certain number of electrochemical cycles revealing a limited long-term stability of Pt–Ni particles. An electrocatalyst, which furnishes an initial high activity and corresponding shape stability with a small activity drop during the lifetime of the catalyst, is mandatory for commercial applications. However, only rare efforts have been made to overcome this problem. Recently, a potential class of stable and active Pt based trimetallic Pd@Pt–Ni octahedral nanoparticles brings the next level ORR activity performance.27 Further, Fe-doped Pt3Ni nanocrystals show a stability of octahedral shape even after 16k potential cycles in acidic media with only 25% loss in its mass activity.28 The problem of limited stability was overcome by Zhang et al. by introducing Cu into the Pt–Ni alloy and remarkable activity of 2.35 A mgPt−1 with only 19% of loss after 4k cycles along with retention of shape in acidic media has been achieved.29 More recently, Yamaguchi and co-workers have reported fct-PtFeCu and fct-PtFeCo nanoparticles as stable electrocatalysts for the ORR with the mass-activity loss of only 24% and 33.3% after 10k cycles in acidic media respectively.30 An activity comparison with disordered versus ordered structure of the similar system has already been reported.31 However, despite its importance, a continuous enhancement in the ORR activity due to surface changes (especially in case of ordered nanostructures) has not been noticed seriously during stability tests, though few recent reports find initial activity enhancement up to 4–5k cycles followed by continuous activity loss at higher potential cycles.32,33
Herein, we emphasize on an interesting and unusual ORR activity trend of a ternary alloy ordered nanocrystals of NiCoPt prepared in a simple solvothermal method followed by post-annealing treatment. This method is believed to be suitable for large-scale synthesis of ordered interconnected trimetallic nanocrystals with high Pt surface area. Among various catalysts NCP10/C (NiCoPt/C prepared at reaction time of 10 h) catalyst has shown excellent ORR activity and interesting trend in increase in activity while increasing cycles, perhaps because of improved synergistic effects between Pt, Co and Ni due to suppressed oxidation of Ni and Co in the fct structures. A systematic positive shift in the half wave potential (E1/2) of ORR polarization curve with respect to number of durability cycles indicates surface reconstruction of interconnected NiCoPt nanoparticles, providing more (111) facets in turn leading to the increase in the ORR activity. Structural changes have been studied after various durability cycles. We believe that such unique, yet simple synthetic methodology for preparation of NiCoPt/C electrocatalyst with sustained ORR activity could be a promising approach to reduce the activity loss in low temperature fuel cells.
2. Experimental section
2.1. Materials
Platinum(II) acetylacetonate (Pt(acac)2) was purchased from Sigma-Aldrich chemicals. Cobalt(II) acetate tetrahydrate [(CH3COO)2Co·4H2O], nickel(II) acetate tetrahydrate [(CH3COO)2Ni·4H2O] and N,N-dimethylformamide (C3H7NO) were purchased from SRL chemicals, Pvt. Ltd India. Polyvinylpyrrolidone (PVP; mol. weight ∼ 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Alfa Aesar. Potassium iodide (KI) and perchloric acid (HClO4) was purchased from Rankem chemicals, India Pvt. Ltd. Commercial Pt/C catalyst (20 wt% of Pt on carbon support) was purchased from Iron Power Ltd. Millipore water (18.2 MΩ) was used throughout all electrochemical measurements. All the chemicals were used as received without further purification.
000) was purchased from Alfa Aesar. Potassium iodide (KI) and perchloric acid (HClO4) was purchased from Rankem chemicals, India Pvt. Ltd. Commercial Pt/C catalyst (20 wt% of Pt on carbon support) was purchased from Iron Power Ltd. Millipore water (18.2 MΩ) was used throughout all electrochemical measurements. All the chemicals were used as received without further purification.
2.2. Synthesis of interconnected ordered NiCoPt alloy nanostructures
A simple solvothermal method has been employed to synthesize the interconnected PtNiCo nanoparticles. Typically, 30 mg of Pt(acac)2, 14.97 mg of cobalt(II) acetate, 14.88 mg of nickel(II) acetate, 160 mg of PVP and 0.166 g of KI were mixed with 15 mL of DMF. The whole mixture was sonicated for 30 minutes to obtain a homogeneous mixture. A transparent yellow color solution was transferred into 25 mL Teflon-lined stainless steel autoclave and kept at 150 °C for 10 h. The mixture was cooled at room temperature and the resultant nanostructures were supported on carbon black (Ketjen black carbon, ∼20 wt% metal loading) followed by ultrasonication and refluxing at 150 °C for 2 h. The sample was collected after centrifuge and washed with acetone and water. Finally, the obtained black powder was heat treated at 700 °C under N2/H2 atmosphere to bring structurally ordered interconnected nanostructure and obtained sample was designated as NCP10/C (NiCoPt/C prepared at reaction time of 10 h). For comparison purpose, similar experiments were repeated for 6 h, 8 h, 16 h and the samples were denoted as NCP6/C, NCP8/C, NCP16/C respectively.2.3. Characterization
The powder X-ray diffraction (XRD) was used for the structural analysis of Pt alloy nanoparticles. XRD measurements were performed on an X'pert pro diffractometer, PANanalytical using the CuKα line (λ = 1.5406 Å, 40 kV, 40 mA) in the 2θ range of 5–100° with a scan rate of 2° min−1 the FEI Tecnai T-20 electron microscope, operating at 200 kV was used for TEM analysis. EDX studies have been employed to quantify the metal loading by ‘Quanta 200 FEG FE-SEM’. The X-ray photoelectron spectroscopic (XPS) analysis was performed using a Shimadzu ESCA 3400 instrument.2.4. Electrochemical measurements
The electrochemical measurements were carried out in a typical three electrode system. A platinum (Pt) wire and reversible hydrogen electrode (RHE) were used as a counter and a reference electrode respectively. All electrochemical measurements were carried on a CHI 760E bipotentiostat (CH Instruments, Inc., USA). The glassy carbon (GC) electrode (geometric surface area: 0.0707 cm2) was polished with 0.05 mm alumina slurry, followed by careful rinsing and ultrasonication for 4 min to absolute cleaning, using Milli-Q water. The active catalyst was drop-casted on cleaned GC electrode using a slight modification in our previous recipe.34 Briefly, 2 mg catalyst was dissolved in 1 mL solution containing 200 μL IPA, 796 μL DI water and 4 μL Nafion (5 wt%, Aldrich). Further, sonication for 1 h in ice cold water to make homogeneous slurry followed by a 4 μL of this catalyst ink was drop casted onto the polished and cleaned GC electrode surface. The electrode was dried over night with slow rate of evaporation. The Pt loading has been maintained as 20 μgPt cm−2 for both NCP10/C and commercial Pt/C for better comparison of activities (similar approach was employed for other catalysts too). Then the cyclic voltammograms (CVs) were carried out in N2 saturated 0.1 M HClO4 solution to clean electrochemically by 100 cycles between 0.05 and 1.2 V vs. RHE with a scanning rate of 100 mV s−1 until getting constant curve. The electrochemical active surface area (ECSA) was determined under similar conditions with a scanning rate of 20 mV s−1. Hupd region was used to obtain charge required to form a monolayer of hydrogen on the catalyst surface to evaluate ECSA of the catalyst.For rotating ring-disk electrode (RRDE) measurements, catalyst ink and electrodes were prepared by a similar method to that of the rotating-disk electrode (RDE) measurements and 7 μL of the ink drop-coated onto 4 mm diameter electrode (geometric surface area of 0.1256 cm2). The disk electrode was scanned at a rate of 10 mV s−1 and the ring potential was kept constant at 1.3 V vs. RHE.
3. Results and discussion
Fig. 1 illustrates a schematic demonstration of formation of NiCoPt nanoparticles as well as the disordered to ordered structure transformation during post annealing treatment. A simple solvothermal method has been employed to prepare NiCoPt alloy nanostructures followed by annealing to form ordered structures. Here PVP acts as a capping agent and KI has been employed as an additive (to control the facets) and further to avoid oxidation of Co and Ni in presence of N-dimethyl formamide (DMF). Fig. 2A shows comparative XRD patterns of NCP10/C and standard Pt (ICSD: 03-065-2868). The powder XRD pattern of NCP10/C shows classic Pt like face-centered cubic (fcc) features, corresponding to the (111), (200), (220) and (311) planes. The peak positions are shifted to higher 2θ angles, specifying the presence of Co and Ni into the Pt lattice to form an ordered L10 phase with a reduced Pt–Pt distance. Further, the presence of ordered phase of face centered tetragonal (fct) of NCP nanostructure is clearly confirmed by superlattice peaks of (001) and (110) indicating an intermetallic Pt2CoNi phase.33 Importantly, the degree of ordering is observed to be 84%, which is evaluated using XRD analysis, which is also in agreement with previous report.35 The composition of NCP10 has also been observed as Pt43Ni42Co15 using EDX analysis shown in Fig. S1A.†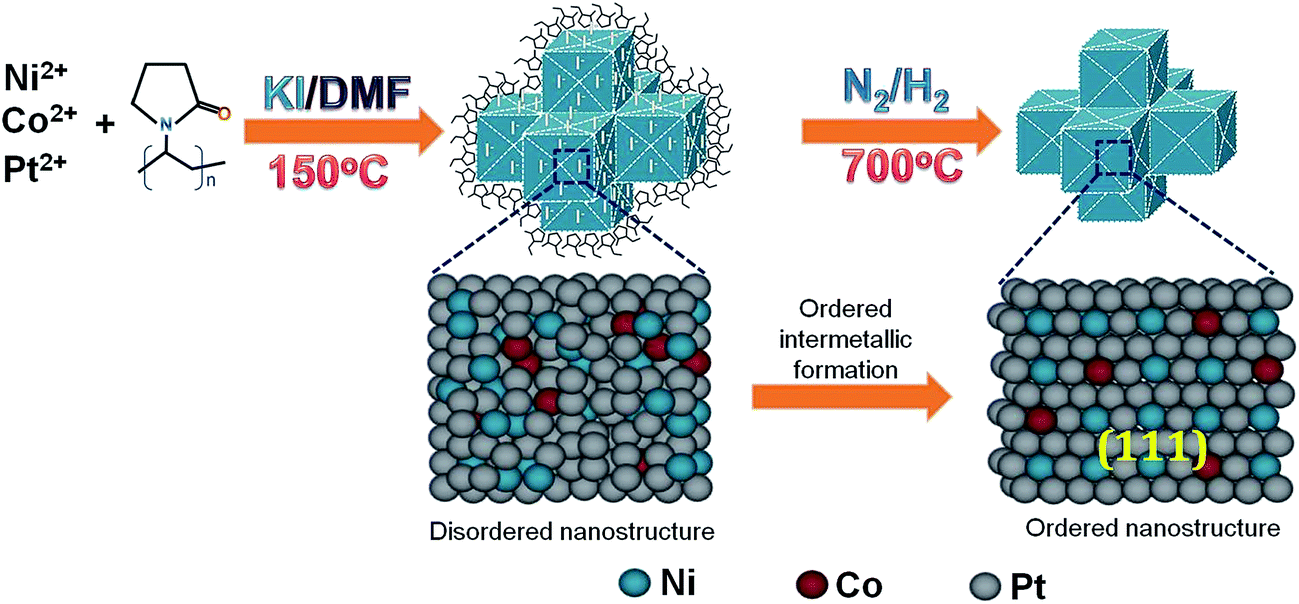 | ||
| Fig. 1 Schematic illustration of formation of NCP/C electrocatalyst. | ||
 | ||
| Fig. 2 (A) XRD pattern of NCP10/C catalyst; (B and C) low resolution TEM images of NCP10/C; and (D and E) HR-TEM images of NCP10/C showing (111) facet (red circle), steps (yellow circle) and twin boundaries (blue circle); inset showing the FFT pattern of twin boundaries. | ||
Fig. 2B and C reveal low-resolution TEM images of NCP10/C to investigate the particle size and particle distribution on the carbon support. It is evident from the Fig. 2C that the most of the NCP particles are interconnected with exposed concave surfaces and truncated edges. Additionally, all nanoparticles are interconnected and form very strong network on the carbon support with the average particle size of ∼20 nm (of course a large heterogeneity in the particles size has been observed). The HRTEM images of NCP10/C catalyst exhibiting atomic fringes with d-spacing of 0.221 nm corresponding to (111) facets that are clearly seen in Fig. 2D and E. Inset of Fig. 2E represents the Fast Fourier Transform (FFT) pattern corresponds to twin boundaries, indicating a clear evidence for defect sites at edges. Whereas, yellow and blue dotted circles indicate steps and twin boundaries respectively (defects), which have been seen in Fig. 2E. Also, Fig. 2E confirms a maximum coverage of catalytic active (111) facets, which have been seen everywhere in the sample, whereas steps and twin boundaries (defects) are observed at the edges and corners only. More significantly, Fig. S2A (ESI†) shows HAADF-STEM images of the NCP10/C with elemental mapping for Pt, Co and Ni, clearly exhibiting the homogeneous distribution of these elements throughout the structures.
Electrochemical properties of NCP10/C have been studied in comparison with that of commercial Pt/C electrocatalyst and shown in Fig. 3. Accordingly, inset of Fig. 3A shows superimposed cyclic voltammograms for NCP10/C and Pt/C electrocatalysts, recorded in presence of N2-saturated 0.1 M HClO4 solution at scan rate of 20 mV s−1 between 0.05 to 1.2 V vs. RHE. Two distinct potential regions were observed corresponding to hydrogen under potential adsorption/desorption (H+ + e− = Hupd) processes on the nanocrystal surface, similar to a polycrystalline Pt surface. The second potential region beyond ∼0.6 V indicates the formation of a hydroxide layer (2H2O = OHad + H3O+ + e−) on the catalyst surface. The electrochemical active surface area per unit weight (ECSA; m2 g−1) was determined as 68.1 and 65.4 m2 g−1 for NCP10/C and Pt/C respectively, from the Hupd region after double-layer correction and normalization to a value of 0.21 mC cm−2, corresponding to the monolayer of hydrogen on a clean polycrystalline Pt surface.36 Higher value of ECSA has been observed for NCP10/C catalyst due to branched surfaces indicating higher active sites on NiCoPt ordered interconnected nanostructures (also incident in the HR-TEM images). However, the composition and electronic structure (prominent factors) of the alloy particles comprising three different metals/elements would have more effect on their surface properties rather than the size and shape of the particles.
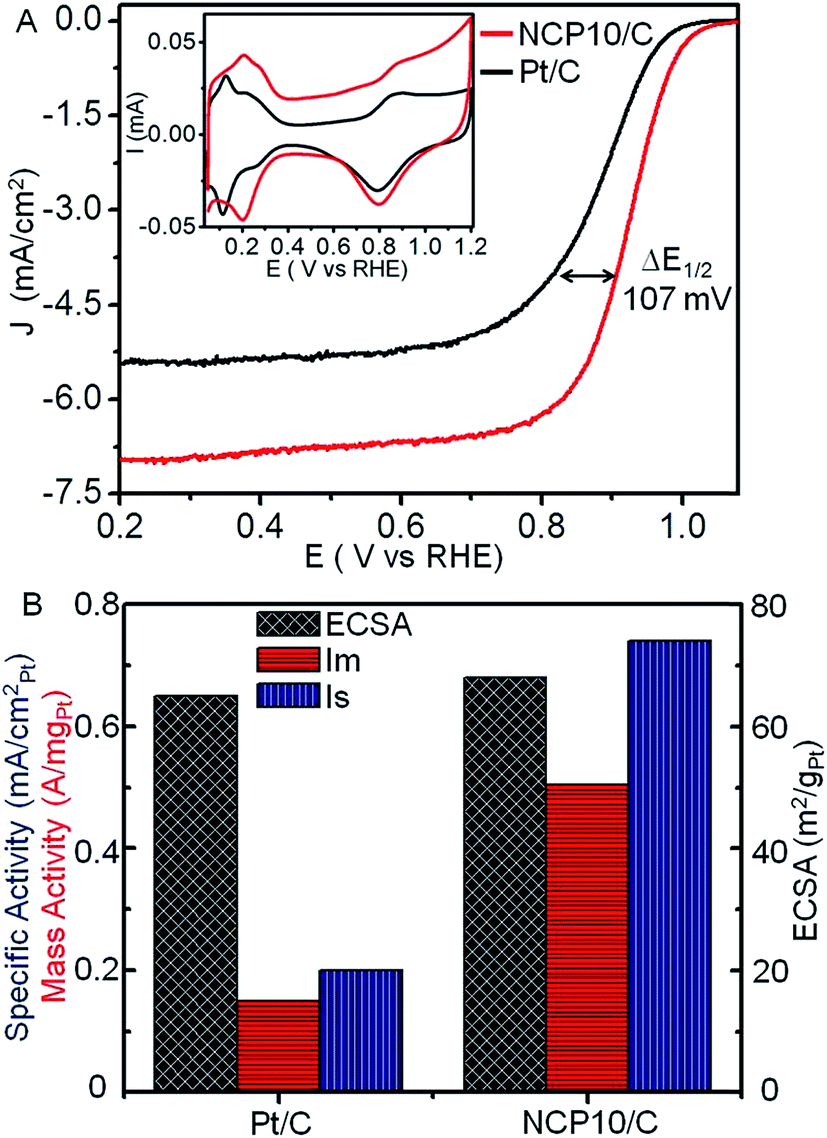 | ||
| Fig. 3 (A) ORR polarization curves recorded in the presence of O2-saturated 0.1 M HClO4 at 25 °C with a sweep and rotation rate of 10 mV s−1 1600 rpm respectively; inset shows CVs of NCP10/C and commercial Pt/C catalyst, recorded in the presence of N2-saturated 0.1 M HClO4 at a sweep rate of 20 mV s−1; (B) histogram of comparative Is and Im values calculated at 0.9 V and ECSA values of NCP10/C and commercial Pt/C. | ||
Fig. 3A shows the comparative ORR polarization curves for NCP10/C and Pt/C catalysts recorded in the presence of O2-saturated 0.1 M HClO4 at 25 °C with a scan rate of 10 mV s−1. ORR activities have been quantified at E = 0.9 V vs. RHE to avoid the interferences from mass-transport losses at higher current densities.3 As expected, NCP10/C shows a single reduction wave with a mixed kinetic-diffusion control region between 1.0 and 0.75 V vs. RHE, followed by a flat diffusion-limited current region in the potential range 0.7 to 0.2 V, as shown in Fig. 3A. The kinetic current was determined after separating from diffusion part using Koutecky–Levich approach.3,37 The values of electrocatalytic activities of Pt/C, used for comparison, are in close agreement with the established benchmark activity of the Pt based catalysts reported by Gasteiger et al.3 The specific activity of 0.744 mA cmPt−2 has been achieved for NCP10/C which is ∼3.7 times higher than Pt/C (0.2 mA cmPt−2), demonstrating an excellent catalytic behavior of NCP10/C. Fig. 3B also reveals a comparison of ECSA, specific and mass activities of both NCP10/C and Pt/C, where NCP10/C shows a mass activity (Im) of 0.505 A mgPt−1, which is ∼3.4 times higher than that of Pt/C (0.15 A mgPt−1). In addition, the ORR activity of NCP10/C (ordered fct structure) was compared with disordered NCP10/C-fcc (prepared separately after heat treatment at 500 °C). Upon comparison, ordered NCP10/C (Im = 505.2 mA mgPt−1) exhibits better ORR activity than disordered NCP10/C-fcc (Im = 315 mA mgPt−1), due to lattice contraction in the former (Fig. S3;† comparative XRD and electrochemical studies have been shown), which is in close agreement with previous report.31 This higher activity of NCP10/C can be attributed mainly to; (1) the synergistic effects among Pt, Ni and Co, and (2) the ordered structure (fct) with L10 phase of NiCoPt. The following main effects may govern the electrochemical properties of NCP10/C. Firstly, the change in electronic structure of Pt by the incorporation of transition metal (Ni and Co) atoms into the Pt lattice, thus reducing the binding energy of oxygenated species on the surface and then producing more Pt sites available for incoming O2 or H2O molecules.38 Secondly, increased Pt (111) surface along with ordered structures and perhaps the high index facets are responsible to improve the electrocatalytic activity.26a,b,c
Further, Fig. S4† shows the rotating ring-disk electrode (RRDE) voltammograms for NCP10/C, clearly showing a four-electron transfer pathway and follows first order kinetics for ORR mechanism (yielding less than 1% peroxide intermediates in the range 0.05 to 1.0 V vs. RHE). Moreover, the detailed comparative electrochemical studies of all other trimetallic catalysts (viz. NCP6/C, NCP8/C and NCP16/C) prepared at various time periods are shown in Fig. S5 and S6 and Table S1 in ESI.† It is important to note that the higher non-faradic contribution in case of NCP10/C has been observed due to heterogeneity in the particle sizes of the nanostructures.
Usually, Pt-based catalyst face severe problems due to dissolution and agglomeration under alternating potentials during catalyst life time durability tests.39,40 Under acidic conditions, especially at fuel cell operating conditions, Pt-alloys exhibit transition metal dissolution forming a skeletal-like structure with enhanced surface area.41,42 Further, more severe treatment of voltage cycling between 0.6 and 1.4 V could be employed to mimic the shut down/start up cycles of a fuel cell to monitor stability of the carbon support.43 We evaluated the stability of the NCP10/C catalysts by measuring their ECSA loss over the course of 30k potential cycles (a triangular potential vs. time curves for an interval of 8 s) between 0.6 and 1.0 V in N2-saturated 0.1 M HClO4 solution, at a potential sweep rate of 100 mV s−1, where the initial stable CV was recorded after the activation of 100 cycles. Accordingly, comparative CVs after selective cycles have been shown in Fig. 4A, where a systematic change in the Hupd region has been seen. Fig. 4B shows the variation of ECSA with number of cycles, where NCP10/C and Pt/C respectively reveal ∼5% and ∼40% loss in ECSA after 5k cycles. The combined effects of dissolution/corrosion (seen in Fig. 4A) of the carbon support and/or hence detachment of metal nanoparticles can have serious effects on ECSA. Fig. 4B demonstrates only ∼22% and ∼24% loss in ECSA values for NCP10/C after 15k and 30k cycles, respectively. These losses in ECSA of NCP10/C are much lower than those obtained for PtIrCo/C catalyst,44 where almost 64% loss in ECSA could be attributed to a weaker Pt–Ir–Co bonding or dissolution effects, due to the wet chemical synthetic procedure. Surprisingly, excellent stability of similar trimetallic Rh–Pt–Ni catalyst has been reported by Strasser and co-workers.33 Interestingly, a recent study reveals tremendous stability of Pt monolayer on Pd–Au electrocatalyst due to inhibition of the oxidation or dissolution of Pd after alloying with Au.42 More interestingly, an initial increase in the activity of Pt monolayer on Pd–Au has been explained due to slight contraction of surface Pt owing to Pd dissolution and surface atom rearrangement into higher coordinated surfaces.41 Similarly, in present case, a continuous improvement in the ORR activity of NCP10/C (shown in polarization curves in Fig. 4C) with concomitant stability even up to 30k cycles indicates surface modification of interconnected NiCoPt particles. Further, durability tests indicate the better stability of NCP10/C with negligible deterioration of the nanocrystal surfaces (inset of Fig. 4C clearly indicating positive shift in polarization curves). The ORR activities are improved due to formation of spongy-like structures32 or relaxation followed by rearrangement of atoms46 on the ordered interconnected nanostructures. Additionally, inhibition of Ni or Co dissolution due to fct phase formation (alloying controls the redox properties of transition metals) could be one of the reasons for impressive stability with improved ORR activity of NCP10/C. Recently, Strasser et al., reported, an initial improvement in electrocatalytic activity due to doping of trace amount of Co on (111) surface of the octahedral Pt–Ni nanoparticles, wherein, Co leaching takes place on further cycling leading to decrease in the activity.45 On the other hand, in the present case, Co content on the surface as well as in the Pt lattice has been maintained due to single step solvothermal approach. Fig. 4D shows regular variation in mass and specific activity of NCP10/C during the stability test, where a continuous enhancement in the activity after various potential cycling has been observed. A remarkable gain of 82% in the specific activity has been observed after 30k cycles.
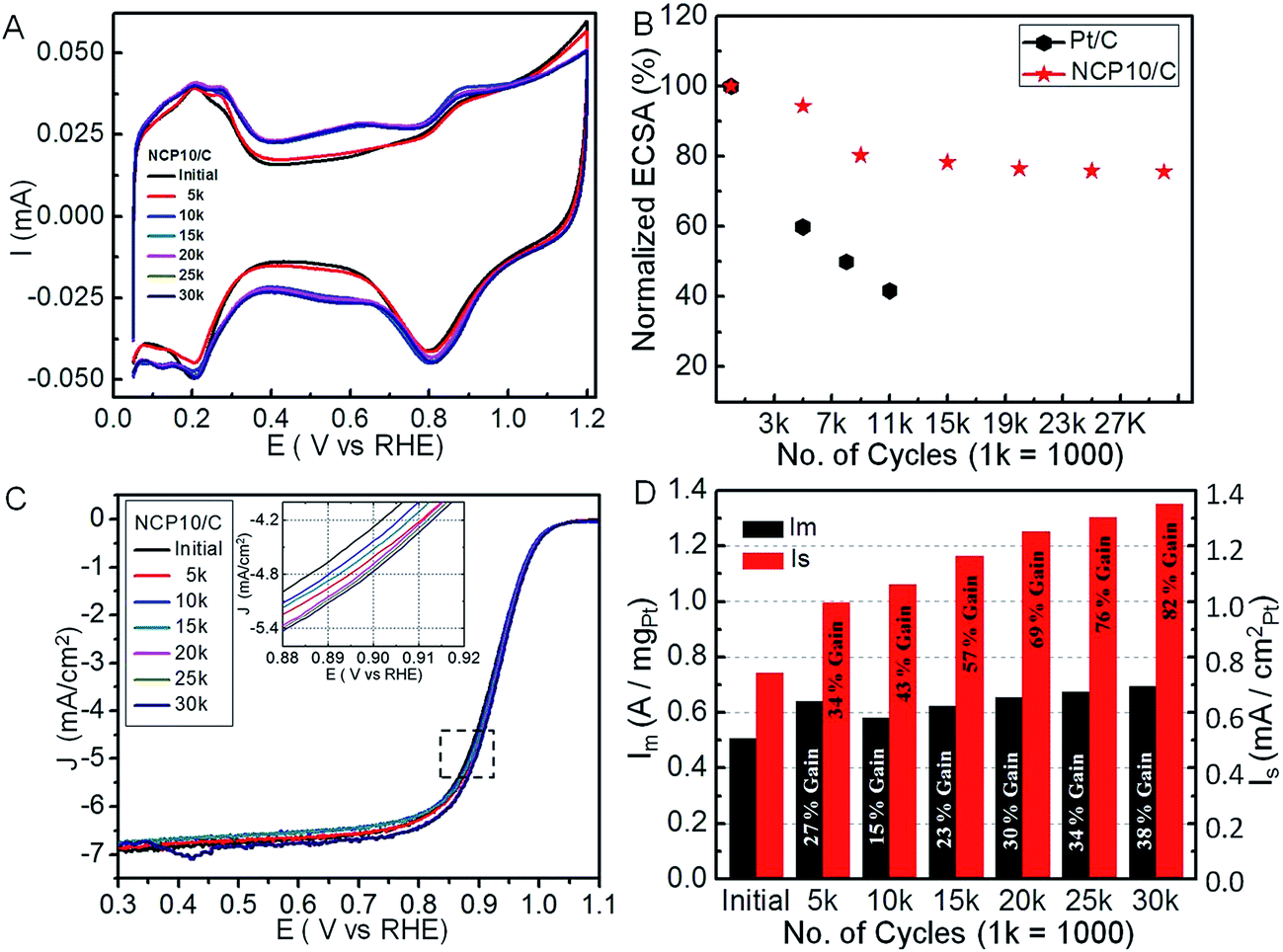 | ||
| Fig. 4 Electrochemical durability of the NCP10/C catalyst; (A) CV curves after every 5k cycles upto 30k; (B) normalized ECSA of NCP10/C and Pt/C catalyst in stability test; (C) ORR polarization curves recorded after various durability cycles up to 30k; inset of (C) clearly show the positive shift in half wave potential (E1/2) of NCP10/C at 0.9 V during the durability potential cycles; and (D) mass (Im) and specific (Is) activity comparisons of NCP10/C catalyst with different potential cycles. | ||
Furthermore, a systematic XRD study (as shown in Fig. 5A) has been performed to unravel structural changes of NCP10/C catalyst after every 5k cycles and results are correlated with the trend in ORR activities shown in Fig. 4D. Accordingly, Fig. 5A shows comparative XRD pattern recorded after every 5k cycles up to 30k cycles, where obvious changes are observed in superlattice peaks, especially in (001). Further, changes in (111) peak could be correlated with the positive shift in ORR activity of NCP10/C. The peak at 2θ = 16°, indicated by star, corresponds to carbon paper substrate. During the stability cycling in case of NCP10/C catalyst the following observations have been made;
3.1. After 5k cycle:
Inset of Fig. 5A shows the zoomed XRD pattern with (111) peaks, showing substantial shift towards higher 2θ values. Such peak shifting could be attributed to the electrochemical surface cleaning (etching) to expose maximum of (111) facets rather than (100) or (110) facets. This phenomenon furnishes highly catalytic active surface towards ORR with improvement of ∼27% and ∼34% in mass (0.64 A mgPt−1) and specific activity (0.996 mA cm−2) respectively for NCP10/C compared to that of initial activity at +0.9 V vs. RHE. Also, a positive shift of 8 mV in E1/2 has been observed after 5k cycles (shown in inset of Fig. 4C). The dynamic increment in the activity after 5k cycles indicates an electronic structure modification due to formation of large number of active sites and changes in the near-surface atomic configurations including dealloying of the surface structures that further affects the Pt–O2 binding mechanism. Similar effects of improvement in activity due to dealloying has been reported by Wang et al.32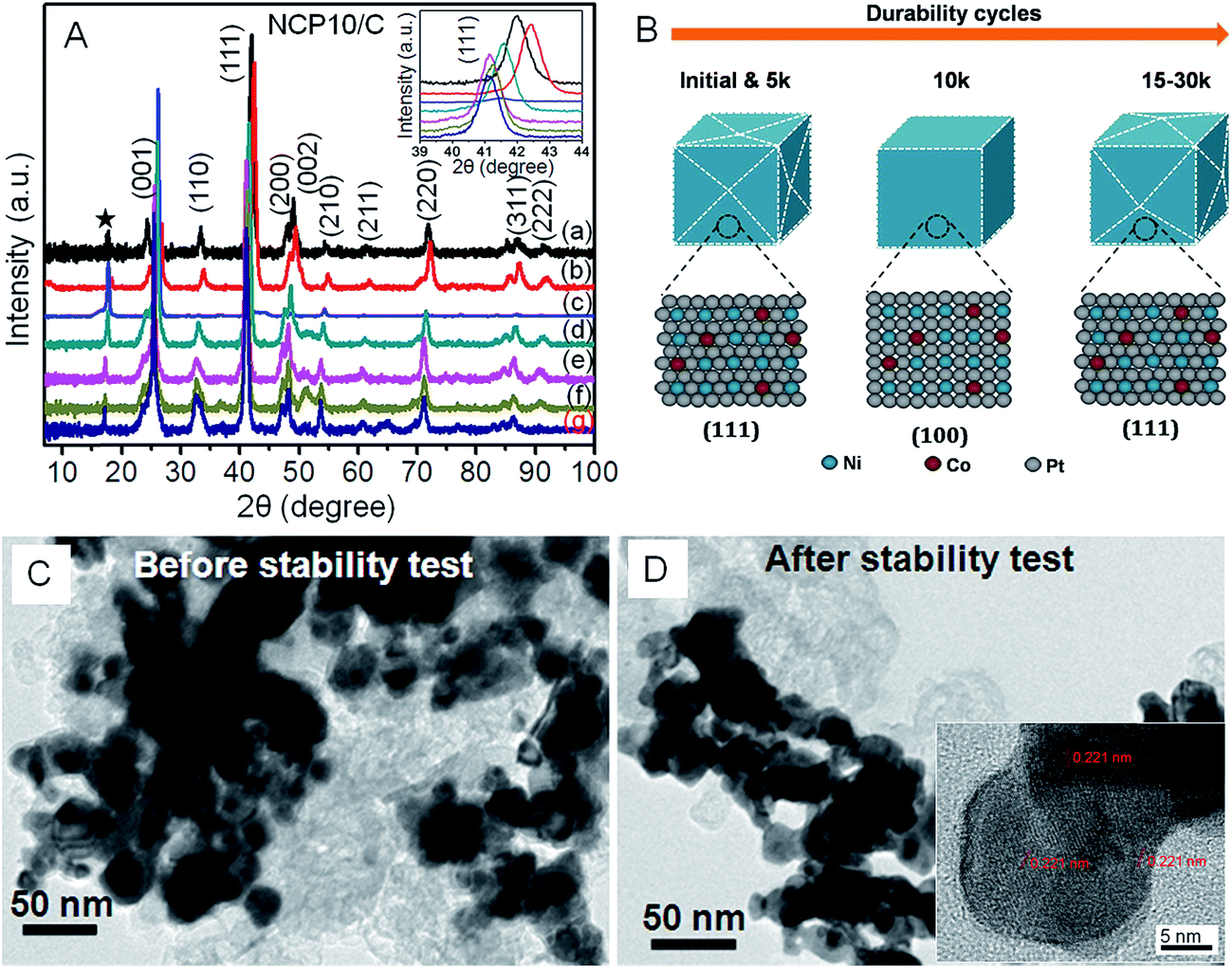 | ||
| Fig. 5 (A) XRD patterns of NCP10/C catalyst recorded after every 5k durability cycles up to 30k cycles ((a) initial, (b) @5k, (c) @10k, (d) @15k, (e) @20k, (f) @25k and (g) @30k), whereas, inset shows zoomed (111) peak for NCP10/C catalyst; (B) schematic representation of proposed mechanism for increasing ORR activity during cycling; and TEM images of NCP10/C catalyst (C) before and (D) after durability tests; inset of (D) shows HRTEM image of NCP10/C catalyst after stability test. | ||
3.2. After 10k cycle
It is interesting to note that an unusual negative shift with concomitant suppressed intensity of (111) peak after 10k cycles indicates obvious structural changes, which is evident in the XRD pattern in Fig. 5A. Consequently, a slight negative shift in half wave potential (E1/2 = −4 mV) compared to 5k is observed, which is still a +4 mV positive shift to that of initial E1/2 value. Thus, near-surface atomic rearrangement after 10k cycles leads to the disappearance of surface (111) facets and evolution of majority of (100) facets with still ∼15% and ∼43% higher mass (0.581 A mgPt−1) and specific activity (1.06 mA cm−2) respectively for NCP10/C compared to initial activity at +0.9 V vs. RHE. Similar trend in the activity with respect to surface modification of (111) to (100) has been previously observed in case of Pt based alloys.233.3. After 15–30k cycles
A substantial lower 2θ shift has been observed after 15k durability cycles, indicating further dealloying of the catalytic surface with higher extent of (111) facets than that after 10k cycles. This would lead to further enhancement in activity with a positive shift in half wave potential (E1/2 = +6 mV), and ∼23% and ∼57% increase in the mass (0.623 A mgPt−1) and specific activity (1.164 mA cm−2) respectively, for NCP10/C compared to initial activity at +0.9 V. This behavior reveals that after 5k and 15k almost similar surface modification has been observed for NiCoPt nanoparticles. Further, cycling from 20k to 30k cycles continues dealloying process without any serious shape change and again shows an increase in active sites with ∼30% and ∼69% increase in the mass (0.656 A mgPt−1) and specific activity (1.251 mA cm−2) respectively for NCP10/C compared to initial activity at +0.9 V vs. RHE. Also, a positive shift in half wave potential (E1/2 = +10 mV) has been observed after 30k. Thus, Fig. 5B presents an excerpt of overall XRD results and corresponding structural modifications that would lead to remarkable enhancement in the ORR activity of NCP10/C.Additionally, it is interesting to note that a very marginal increase (2 mV) in the E1/2 value after 30k indicates saturation process of surface modification rather than loss in the ECSA or ORR activity. Consequently, after 30k cycles, 38% gain in mass activity (0.696 A mgPt−1) and 82% gain in specific activity (1.35 mA cm−2) compared to initial activity of NCP10/C catalyst is achieved. Surprisingly after 30k cycles, the presence of (100) and (110) peaks (Fig. 5A(g)) clearly indicates the retention of superlattice structure of NCP nanostructures and that would lead to the formation of higher coordination sites. The formation of high-index facets due to presence of large number of atomic steps and dangling bonds during electrochemical durability cycles could be another reason to maintain higher coordination sites, which are in turn responsible for the enhancement in the ORR activity.47,48
Furthermore, microscopic studies of NCP10/C were also performed after stability tests to understand any obvious changes in particle sizes due corrosion or agglomeration effects owing to ripening. Accordingly, Fig. 5C and D show comparative TEM images of NCP10/C before and after durability tests, clearly exhibiting insignificant changes in the particle size, shape and particle–support interactions. Further, inset of Fig. 5D reveals the HRTEM image of NCP10/C@30k nanostructure clearly showing (111) facets with similar d-spacing of 0.22 nm, demonstrating the retention of (111) facets even after durability cycling. It is also noted that most of the defect sites including steps and twin boundaries at the surface have been removed during the potential cycling under acidic condition, resulting into enhancement in the ORR kinetics.
Moreover, Fig. 6 represents the comparative XPS studies of NCP10/C catalyst before and after durability test. Fig. 6A shows comparative survey spectra of sample before and after durability tests, showing obvious changes especially at O1s region thanks to corrosion of carbon. It is also observed that the ratio of Pt/Co and Pt/Ni has been increased after durability cycles, indicating the decrease in the Co and Ni content at the surface of the catalyst.23 This results into the modification of electronic structure of Pt at surface compared to the bulk. The quantitative analysis from Pt 4f, Co 2p and Ni 2p core level peaks after deconvolution indicates the surface composition of Pt3Co0.5Ni0.5 from initial Pt2CoNi on surface. Formation of such Pt-rich Pt3M composition would be favorable for ORR.19 However, overall composition of the NCP10/C after 30 k cycles has been found to be Pt45Ni42Co13 using EDX analysis (Fig. S7†).
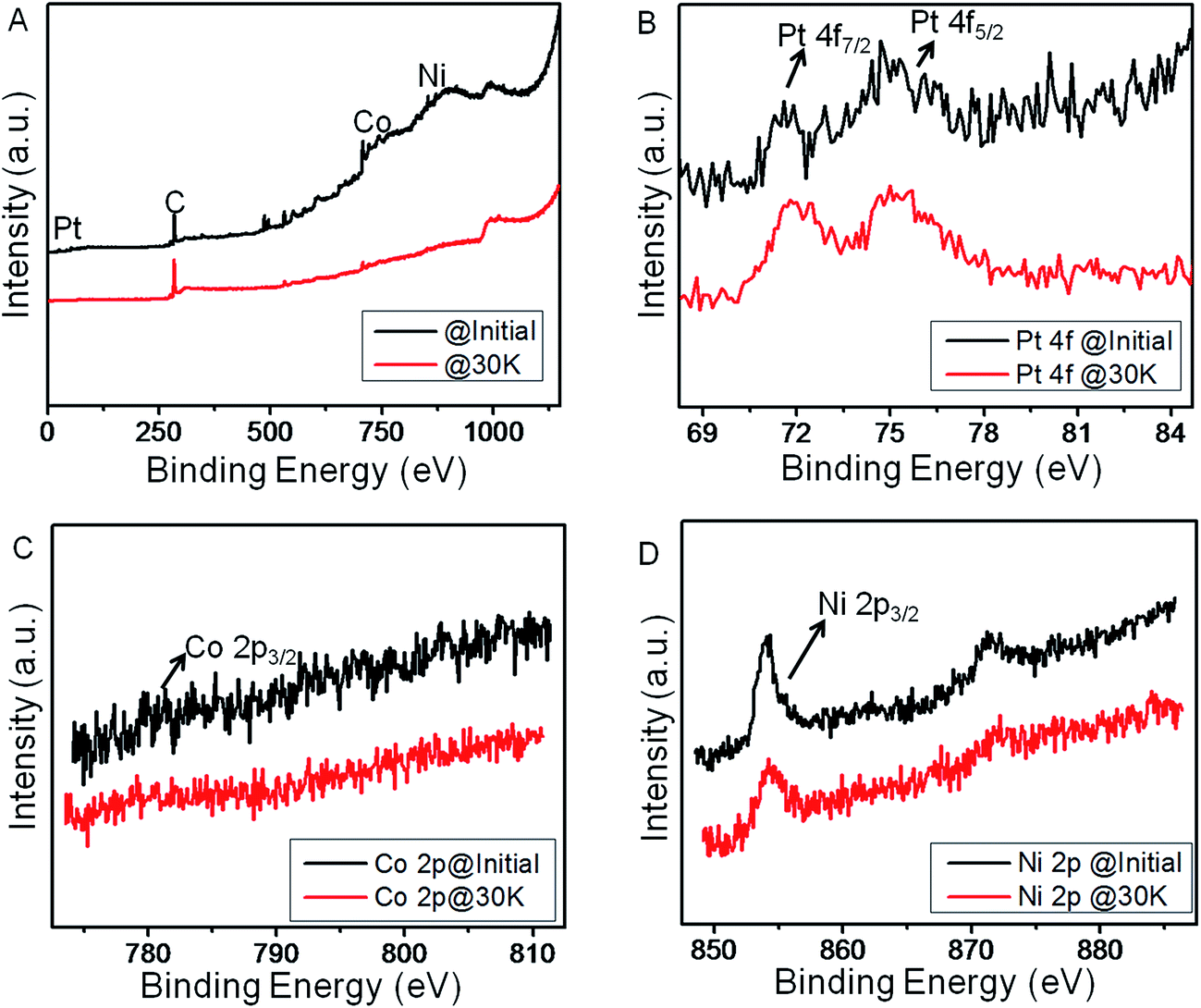 | ||
| Fig. 6 Comparative XP spectra for NCP10/C before and after 30000 cycles, showing (A) survey scan (B) core level Pt 4f, (C) core level Co 2p and (D) core level Ni 2p. | ||
The above results are in good agreement with our electrochemical studies, where just 24% loss in ECSA for NCP10/C has been observed after 30k cycles without further deterioration of the active sites of the catalyst. Also, few representative reports on trimetallic alloy nanoparticles with their ORR activities and stability results have been summarized separately in Table S2.† Further, Fig. S8† shows a very slight increase in the ‘c/a’ ratio (ratio of lattice parameters along ‘c’ axis to that of along ‘a’ axis in the fct structure) indicating a stable nature of chemically ordered structure of NiCoPt with L10 phase (Pt2ML). More significantly, Fig. S2A and B† show HAADF-STEM images of the NCP10/C with elemental mapping of Pt, Co and Ni in the nanostructure before and after stability tests respectively, where no noticeable changes have been observed validating our claim of a very stable nature of NiCoPt nanostructures with better electrocatalytic activity. Thus, a ∼7-fold higher activity of NCP10/C@30k over initial activity of commercial Pt/C would make a tremendous impact on fuel cell technology.
4. Conclusion
In summary, the chemically ordered interconnected nanostructured NiCoPt (NCP10/C) alloy catalyst was synthesized using a simple solvothermal process with post-heat treatment method. An initial mass activity of 0.744 mA cm−2 has been observed for NCP10/C, which is ∼3.7 times higher than that of commercial Pt/C. More significantly, a novel trend in the enhancement in the ORR activity during stability cycles has been observed for the first time, where a remarkable enhancement of 82% gain in the specific activity has been observed after 30k potential cycles. Thus, ∼7-fold higher activity of NCP10/C@30k over initial activity of commercial Pt/C would make a tremendous impact on fuel cell technology. A systematic XRD studies at various potential cycling suggest a changes of bulk crystal structure as well as continuous surface modification due to dealloying and formation of large number of active sites that affects the Pt–O2 binding mechanism and controls the transition metal oxidation in the ordered structure. Hence, the NCP10/C catalyst not only provides a durable and active catalyst for ORR, but also opens up various opportunities for exploring alternative energy applications.Acknowledgements
The authors thank the Department of Science and Technology-Science and Engineering Research Board (DST-SERB; No. SB/FT/CS-120/2012), India for financial support through the Young Scientist Fast track scheme. Authors also acknowledge SRM University for providing research infrastructure.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- A. Bonakdarpour, K. Stevens, G. D. Vernstrom, R. Atanasoski, A. K. Schmoeckel, M. K. Debe and J. R. Dahn, Electrochim. Acta, 2007, 53, 688–694 CrossRef CAS.
- A. Seo, J. Lee, K. Han and H. Kim, Electrochim. Acta, 2006, 52, 1603–1611 CrossRef CAS.
- S. J. Hwang, S. J. Yoo, S. Jang, T.-H. Lim, S. A. Hong and S.-K. Kim, J. Phys. Chem. C, 2011, 115, 2483–2488 CAS.
- B. Arumugam, B. A. Kakade, T. Tamaki, M. Arao, H. Imai and T. Yamaguchi, RSC Adv., 2014, 4, 27510–27517 RSC.
- B. N. Wanjala, B. Fang, S. Shan, V. Petkov, P. Zhu, R. Loukrakpam, Y. Chen, J. Luo, J. Yin and L. Yang, Chem. Mater., 2012, 24, 4283–4293 CrossRef CAS.
- R. Srivastava, P. Mani, N. Hahn and P. Strasser, Angew. Chem., Int. Ed., 2007, 46, 8988–8991 CrossRef PubMed.
- M. T. Nguyen, R. H. Wakabayashi, M. Yang, H. D. Abruña and F. J. DiSalvo, J. Power Sources, 2015, 280, 459–466 CrossRef CAS.
- S.-W. Chou, J.-J. Shyue, C.-H. Chien, C.-C. Chen, Y.-Y. Chen and P.-T. Chou, Chem. Mater., 2012, 24, 2527–2533 CrossRef CAS.
- P. Zhang, X. Dai, X. Zhang, Z. Chen, Y. Yang, H. Sun, X. Wang, H. Wang, M. Wang and H. Su, Chem. Mater., 2015, 27, 6402–6410 CrossRef CAS.
- B. N. Wanjala, R. Loukrakpam, J. Luo, P. N. Njoki, D. Mott, C.-J. Zhong, M. Shao, L. Protsailo and T. Kawamura, J. Phys. Chem. C, 2010, 114, 17580–17590 CAS.
- P. Mani, R. Srivastava and P. Strasser, J. Power Sources, 2011, 196, 666–673 CrossRef CAS.
- K. Neyerlin, R. Srivastava, C. Yu and P. Strasser, J. Power Sources, 2009, 186, 261–267 CrossRef CAS.
- A. Avekians and E. Podlaha-Murphy, ECS Trans., 2010, 28, 111–117 CAS.
- T. Tamaki, A. Minagawa, B. Arumugam, B. A. Kakade and T. Yamaguchi, J. Power Sources, 2014, 271, 346–353 CrossRef CAS.
- M. Ishida and K. Matsutani, ECS Trans., 2014, 64, 107–112 CrossRef CAS.
- C. Wang, D. Li, M. Chi, J. Pearson, R. B. Rankin, J. Greeley, Z. Duan, G. Wang, D. Van der Vliet and K. L. More, J. Phys. Chem. Lett., 2012, 3, 1668–1673 CrossRef CAS PubMed.
- M. Chi, C. Wang, Y. Lei, G. Wang, D. Li, K. L. More, A. Lupini, L. F. Allard, N. M. Markovic and V. R. Stamenkovic, Nat. Commun., 2015, 6, 8925 CrossRef CAS PubMed.
- T. Yang, G. Cao, Q. Huang, Y. Ma, S. Wan, H. Zhao, N. Li, F. Yin, X. Sun and D. Zhang, J. Power Sources, 2015, 291, 201–208 CrossRef CAS.
- B. N. Wanjala, B. Fang, R. Loukrakpam, Y. Chen, M. Engelhard, J. Luo, J. Yin, L. Yang, S. Shan and C.-J. Zhong, ACS Catal., 2012, 2, 795–806 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- J. Zhang, H. Yang, J. Fang and S. Zou, Nano Lett., 2010, 10, 638–644 CrossRef CAS PubMed.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- (a) L. Gan, C. Cui, M. Heggen, F. Dionigi, S. Rudi and P. Strasser, Science, 2014, 346, 1502–1506 CrossRef CAS PubMed; (b) V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed; (c) T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., 2011, 123, 2825–2829 CrossRef.
- S.-I. Choi, M. Shao, N. Lu, A. Ruditskiy, H.-C. Peng, J. Park, S. Guerrero, J. Wang, M. J. Kim and Y. Xia, ACS Nano, 2014, 8, 10363–10371 CrossRef CAS PubMed.
- Y. Li, F. Quan, L. Chen, W. Zhang, H. Yu and C. Chen, RSC Adv., 2014, 4, 1895–1899 RSC.
- C. Zhang, W. Sandorf and Z. Peng, ACS Catal., 2015, 5, 2296–2300 CrossRef CAS.
- B. Arumugam, T. Tamaki and T. Yamaguchi, ACS Appl. Mater. Interfaces, 2015, 7, 16311–16321 CAS.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- D. Wang, Y. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller and F. J. DiSalvo, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed.
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M.-G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719–1725 CrossRef CAS PubMed.
- B. Kakade, I. Patil, M. Lokanathan and A. Swami, J. Mater. Chem. A, 2015, 3, 17771–17779 CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659–2668 CrossRef CAS.
- K. Mayrhofer, D. Strmcnik, B. Blizanac, V. Stamenkovic, M. Arenz and N. Markovic, Electrochim. Acta, 2008, 53, 3181–3188 CrossRef CAS.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321–6328 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- P. Ferreira, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef.
- M. S. Wilson, F. H. Garzon, K. E. Sickafus and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 2872–2877 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef CAS PubMed.
- P. Yu, M. Pemberton and P. Plasse, J. Power Sources, 2005, 144, 11–20 CrossRef CAS.
- K. Sasaki, H. Naohara, Y. Choi, Y. Cai, W.-F. Chen, P. Liu and R. R. Adzic, Nat. Commun., 2012, 3, 1115 CrossRef PubMed.
- R. Loukrakpam, B. N. Wanjala, J. Yin, B. Fang, J. Luo, M. Shao, L. Protsailo, T. Kawamura, Y. Chen and V. Petkov, ACS Catal., 2011, 1, 562–572 CrossRef CAS.
- R. M. Arán-Ais, F. Dionigi, T. Merzdorf, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, M. Gliech, J. Solla-Gullón, E. Herrero and J. M. Feliu, Nano Lett., 2015, 15, 7473–7480 CrossRef PubMed.
- Y. Kang, J. Snyder, M. Chi, D. Li, K. L. More, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2014, 14, 6361–6367 CrossRef CAS PubMed.
- Y. Cai, C. Ma, Y. Zhu, J. X. Wang and R. R. Adzic, Langmuir, 2011, 27, 8540–8547 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparative electrochemical activity data of literature reports, comparative CVs and ORR polarization curve of other time dependent catalysts. SEM-EDS, RRDE and HAADF-STEM images with elemental mapping of NCP10/C are available. See DOI: 10.1039/c6ra27611c |
| This journal is © The Royal Society of Chemistry 2017 |