
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-cost dual cocatalysts BiVO4 for highly efficient visible photocatalytic oxidation
Feng Lin a,
Zhiyu Shaoa,
Ping Libc,
Zhenpan Chenbc,
Xinyi Liubc,
Mingrun Libc,
Bao Zhangbc,
Jindou Huanga,
Guangqi Zhu*bc and
Bin Dong*a
a,
Zhiyu Shaoa,
Ping Libc,
Zhenpan Chenbc,
Xinyi Liubc,
Mingrun Libc,
Bao Zhangbc,
Jindou Huanga,
Guangqi Zhu*bc and
Bin Dong*a
aKey Laboratory of New Energy and Rare Earth Resource Utilization, State Ethnic Affairs Commission, School of Physics and Materials Engineering, Dalian Nationalities University, Dalian, 116600, China. E-mail: dong@dlnu.edu.cn; linfeng@dlnu.edu.cn; Fax: +86-411-87658872; Tel: +86-411-87556959
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
cDalian National Laboratory for Clean Energy, Dalian 116023, China
First published on 7th March 2017
Abstract
The cocatalysts of noble metals are reported to play a significant role in improving the photocatalytic activity in water splitting and pollutant degradation reactions. The high price of noble metals limits their further application in industry. Herein, for the first time, we report that thiophene, rhodamine B (RhB) and methyl orange (MO) can be efficiently oxidized on BiVO4 co-loaded with Ni and CuO cocatalysts (denoted as Ni–CuO/BiVO4) under visible light irradiation with molecular oxygen as the oxidant. Moreover, 0.05 wt% Ni–0.5 wt% CuO/BiVO4 possesses high photocatalytic activity (over 94% conversion of thiophene), which is close to Pt–RuO2/BiVO4 (99% conversion of thiophene). XPS and ESR measurements showed that the activation of molecular oxygen and oxidation of pollutant molecules simultaneously take place on BiVO4 co-loaded with Ni/Cu and CuO/Cu2O cocatalysts. The considerable enhancement of photocatalytic activity can be attributed to the simultaneous presence of the reduction cocatalyst Ni/Cu and oxidation cocatalyst CuO/Cu2O, which are beneficial for the efficient separation and transfer of the photo-generated electrons and holes. Such visible-light-responsive semiconductor loaded with earth-abundant dual cocatalysts has great potential in both solar energy conversion and further industrial applications.
1. Introduction
Solar driven catalysis on semiconductors is widely considered as a promising route to mitigate environmental issues caused by the combustion of fossil fuels and to meet increasing worldwide demands for energy.1,2 SOx produced from the automobile exhaust gas via the burning of sulfur-containing components present in fuels results in serious air pollution. Among the sulfur-containing components, thiophene, with the aromaticity and the low electron density of the sulfur atom, is most difficult to oxidize with conventional oxidative desulfurization processes.3 Some effective photocatalysts have been reported for the removal of sulfur-containing compounds in gasoline via photocatalytic oxidation.4–7 Polluted waste water from industry is a global environmental issue. The azo dyes rhodamine B (RhB) and methyl orange (MO) are widely used for coloring textiles. Consequently, several materials that enable the cleanup of polluted water via a far less aggressive approach have been developed.8,9The major restriction factors affecting the efficiency of photocatalysis include (i) light absorption, (ii) charge separation and transport and (iii) surface chemical reaction. In recent years, some visible-light-responsive bulk semiconductors10–15 were investigated in photocatalytic water splitting and pollutants degradation reaction, such as WO3, BiVO4, Bi2WO6, CaBi2O4, Bi2Ti2O7, and Ag3PO4. These semiconductors can expand the light absorption of photocatalysts to the visible light region, which is far beyond the light absorption of TiO2. In order to improve the efficiency of photocatalysis, a promising strategy is to load cocatalysts or secondary semiconductors that can act as either electron or hole acceptors for improved charge separation.16,17 Since the early report of CdS loaded with dual cocatalysts Pt and PdS by Can Li, which can achieve an extremely high QE (93%) in photocatalytic H2 production,18 there have been few investigations on the effect of dual cocatalysts in photocatalytic reactions. In our previous study, thiophene could be oxidized to SO3 on BiVO4 co-loaded with Pt and RuO2 cocatalysts under visible light irradiation with molecular oxygen as the oxidant.5 Domen et al. synthesized WO3 co-loaded with Pt and RuO2 cocatalysts and investigated the performance in the photocatalytic IO3− reduction and water oxidation.19 The reports show that the synergistic effect between suitable cocatalysts on the photocatalytic activity is very important to the photocatalytic reaction. However, the high price of noble metals limits further application in industry. The exploration of earth-abundant elements cocatalysts in photocatalytic reactions is highly desired.
Recently, some abundant and low-cost materials have been reported to replace noble metal catalysts for photocatalysis. Domen et al. prepared CuCrOx/GaN:ZnO composite photocatalyst20 with its photocatalytic activity being 25–30% the activity of Rh2−yCryO3/GaN:ZnO. Using earth-abundant element Cu, a cheap and efficient photocatalyst can be obtained. Ghim Wei Ho et al. loaded a core–shell structure Cu–CuO cocatalyst on TiO2 (ref. 21) to achieve a high photocatalytic activity of H2 production, which was attributed to the promoted charge separation and transport. Moreover, Co doped BiVO4 (ref. 22) and LaTiO2N23 are reported to perform well in photocatalytic degradation and water oxidation reactions. Other cocatalysts, such as Ni, NiO, Ni(OH)2 and NiS,24–27 were also studied for photocatalysis, but the activities and stability were much lower than those of the two systems mentioned above.28,29 These studies show that although the enhanced activity on noble-metal-free cocatalyst is lower than that on the noble metals, the earth-abundant elements have great potential in both theory and industry. The suitable noble-metal-free dual cocatalysts may enhance the photocatalytic activity significantly. However, little research has been reported on the effect of the dual cocatalysts on the photocatalytic activity of a BiVO4-based photocatalyst for photocatalytic waste oxidation.5 Moreover, the synergistic effect of low-cost dual cocatalysts is far less investigated in photocatalysis for environmental protection.
Herein, we report the photocatalytic oxidation of thiophene, RhB and MO by a visible-light responsive photocatalyst Ni–CuO/BiVO4. We found that BiVO4 co-loaded with Ni and CuO showed a strong synergistic effect between the two cocatalysts on the photocatalytic activity of thiophene oxidation and pollutant degradation. A high photocatalytic activity close to Pt–RuO2/BiVO4 could be achieved under visible light irradiation (λ ≥ 420 nm) using molecular oxygen as the oxidant.
2. Experimental
2.1 Catalyst preparation
All chemicals in these experiments were of analytical reagent grade and used without further treatment. BiVO4 samples were synthesized according to our previous study,5 while BiVO4 (i) corresponds to the pH value of the resulting solution adjusted to i with ammonia solution (i = 2.0, 4.0, 9.0).The loading of copper oxide on BiVO4 (2) was performed by the impregnation method. Cu(NO3)2 was used as the precursor. BiVO4 powder was impregnated in an aqueous solution containing a given amount of Cu(NO3)2. The solution was then evaporated over a water bath at 80 °C, followed by calcination in air at 350 °C for 4 h. Metal nickel was co-loaded onto CuO/BiVO4 by the photo deposition method. The photocatalyst, BiVO4 (2) co-loaded with the metal Ni and metal oxide CuO was denoted as Ni–CuO/BiVO4.
2.2 Catalyst characterization
The catalysts were characterized by X-ray powder diffraction (XRD) on a Rigaku D/Max-2500/PC powder diffractometer. Each sample was scanned using Cu-Kα radiation with an operating voltage of 40 kV and an operating current of 200 mA. A scan rate of 5° min−1 was applied to record the patterns in the range of 10°–80° at a step of 0.02°.UV-Visible diffuse reflectance spectra (UV-Vis DRS) were recorded on a UV-Vis spectrophotometer (PerkinElmer Lambda 750) equipped with an integrating sphere. The morphologies and particle sizes were examined by scanning electron microscopy (SEM) equipped with a Quanta 200 FEG scanning electron microscope with a 0.5–30 kV accelerating voltage. High-resolution transmission electron microscopy (HRTEM) images, scanning transmission electron microscopy (STEM) images and energy dispersive spectroscopy (EDS) were obtained on a Tecnai G2 F30 S-Twin (FEI Company) instrument.
X-ray photoelectron spectroscopy (XPS) was acquired on a Thermo ESCALAB 250Xi with an Al Kα X-ray (hν = 1486.6 eV). Base pressure in the analysis chamber was maintained at 10−8 Pa. Energy resolution of the spectrometer was set at 0.8 eV at a pass energy of 20 eV. Full width at half maximum (FWHM) was calibrated with respect to Ag 3d5/2 FWHM at 0.65 eV. The error in all the BE values reported was 0.22 eV.
ESR signals of radicals trapped by DMPO were recorded at ambient temperature on a Brucker ESR A200 spectrometer. After bubbling O2 for 10 min, the samples were introduced into the homemade quartz cup inside the microwave cavity and illuminated with a 300 W Xe lamp (CERAMAX LX-300). The settings for the ESR spectrometer were as follows: center field, 3486.70 G; sweep width, 100 G; microwave frequency, 9.82 GHz; modulation frequency, 200 kHz; power, and 10.00 mW. Magnetic parameters of the radicals detected were obtained from direct measurements of magnetic field and microwave frequency.
2.3 Photocatalytic reaction
The photocatalytic reactions of thiophene were carried out in a Pyrex reaction cell with O2 or air bubbled in a constant flow. Photocatalyst (1 g L−1) was dispersed in an acetonitrile solution containing given amounts of thiophene ([sulfur content]initial = 600 ppm). The suspension was irradiated by a 300 W Xe lamp (CERAMAX LX-300) equipped with an optical filter (λ ≥ 420 nm) to cut off the light in the ultraviolet region. The temperature of the reaction solution was maintained at 10 °C ± 2 °C by a flow of cooling water. The products were analyzed by GC-FPD (Agilent 7890, pona column).The photocatalytic degradation reactions of dyes rhodamine B (RhB) and methyl orange (MO) were carried out in a Pyrex reaction cell. Photocatalyst (1 g L−1) was dispersed in an aqueous solution containing given amounts of the pollutants (C0 = 5 ppm). The temperature of the reaction solution was maintained at 10 °C ± 2 °C by a flow of cooling water. The concentration of RhB and MO was monitored by colorimetry with a JASCO V-550 UV-vis spectrometer. The λmax for RB and MO are 553 and 467 nm, respectively. Calibration based on the Beer–Lambert law was used to quantify the dye concentration.
3. Results and discussion
3.1 Characterization of catalysts
Fig. 1 shows the morphology of BiVO4 samples synthesized under different experimental conditions and loaded with cocatalysts. SEM images present several morphologies for BiVO4 that are also reported in other studies.30,31 As shown in Fig. 1, large compact particles (about 2 μm in size) of decagonal shape are observed for BiVO4 (2). The particles show a smooth surface and well-defined edges. The BiVO4 (4) sample exhibited a polyhedral shape with the size of about 3 μm. | ||
| Fig. 1 SEM images of BiVO4 (2), BiVO4 (4), BiVO4 (9), CuO/BiVO4 and Ni–CuO/BiVO4. | ||
The BiVO4 (9) sample showed a trunk shape. We investigated the photocatalytic thiophene oxidation on these BiVO4 samples, and the conversion of thiophene in 3 h on BiVO4 (2), BiVO4 (4) and BiVO4 (9) was ca. 40%, 12% and 16%, respectively. As BiVO4 (2) was much more photoactive than BiVO4 (4) and BiVO4 (9), BiVO4 (2) was synthesized to load with cocatalysts for the following experiments. In the following discussion, BiVO4 represents BiVO4 (2). After loading cocatalysts CuO and Ni, the nanoparticles were highly dispersed on the smooth surface of BiVO4 for CuO/BiVO4 and Ni–CuO/BiVO4 (as shown in Fig. 1).
Fig. 2 shows the HRTEM (STEM) images and EDS analysis for the microstructure of the catalysts CuO/BiVO4 and Ni–CuO/BiVO4. The images (Fig. 2a and b) show that CuO loaded on BiVO4 was mainly in the form of semispherical particles. The typical particle size of CuO is estimated to be about 5–10 nm. For Ni–CuO/BiVO4 catalyst (Fig. 2d and e), Ni nanoparticles were dispersed on the surface of BiVO4 in the form of flat spheres besides CuO. The typical particle size of Ni is estimated to be about 2–6 nm. In addition, the EDS analysis revealed the presence of Cu on both CuO/BiVO4 and Ni–CuO/BiVO4 (Fig. 2c and f).
 | ||
| Fig. 2 HRTEM images of (a) and (b) CuO/BiVO4, (d) and (e) Ni–CuO/BiVO4. STEM images and EDS analysis of (c) CuO/BiVO4, (f) Ni–CuO/BiVO4. | ||
BiVO4 prepared by hydrothermal treatment is a monoclinic scheelite according to the standard card no. 14-0688 (Fig. 3a). After loading cocatalysts CuO and Ni, there was no evident diffraction peak of elements Cu or Ni. The XRD pattern of the Ni–CuO/BiVO4 sample demonstrated the monoclinic phase. Fig. 3b shows the UV-vis diffuse reflectance spectra of BiVO4 and Ni–CuO/BiVO4. BiVO4 showed strong absorption in the UV light region and the visible light region until 535 nm. The band gap of BiVO4 is estimated to be 2.3 eV from the absorption edge of the UV-vis DRS. After loading cocatalysts onto BiVO4, no evident shift of the absorption edge was observed. Consequently, Ni–CuO/BiVO4 can absorb light (λ ≥ 400 nm) beyond the absorption edges of gasoline and light diesel.5
 | ||
| Fig. 3 (a) XRD patterns of Ni–CuO/BiVO4, BiVO4 and the standard card of BiVO4 (no. 14-0688). (b) UV-Vis DRS of BiVO4 and of Ni–CuO/BiVO4 photocatalyst. | ||
3.2 The effect of cocatalysts on photocatalytic oxidation reaction
 | ||
| Fig. 4 Photocatalytic oxidation of RhB on BiVO4 loaded with cocatalysts (a) CuO and (b) Ni–CuO. Herein 0.05 Cu represents 0.05 wt% CuO/BiVO4, 0.03Ni–0.5 Cu represents 0.03 wt% Ni–0.5 wt% CuO/BiVO4, and so on. Reaction conditions: the concentration of photocatalyst: 1 g L−1; the initial concentration of RhB, C0 = 5 ppm; light source: 300 W Xe lamp (CERAMAX LX-300, λ ≥ 420 nm); temperature: 10 ± 2 °C. | ||
 | ||
| Fig. 5 Photocatalytic oxidation of MO on BiVO4 loaded with cocatalysts Ni and CuO. Reaction conditions: the concentration of photocatalyst: 1 g L−1; the initial concentration of MO, C0 = 5 ppm; light source: 300 W Xe lamp (CERAMAX LX-300, λ ≥ 420 nm); temperature: 10 ± 2 °C. | ||
 | ||
| Fig. 6 Photocatalytic oxidation of thiophene on BiVO4 and BiVO4 loaded with various cocatalysts under visible light irradiation (λ ≥ 420 nm). Herein 0.5 Cu represents 0.5 wt% CuO/BiVO4, 0.05 Ni–0.5 Cu represents 0.05 wt% Ni–0.5 wt% CuO/BiVO4, and so on… Reaction conditions: [sulfur content]initial = 600 ppm; the concentration of photocatalyst: 1 g L−1; O2 (bubbled into the system); temperature: 10 ± 2 °C; reaction time: 3 h. | ||
According to the abovementioned results, Ni–CuO/BiVO4 turned out to be the most effective photocatalyst for the photocatalytic oxidation of thiophene. The synergistic effect of cocatalysts on BiVO4 is very favorable for improving the photocatalytic activity of thiophene oxidation. It is also implied that the suitable energy level position of metals may be very important when choosing dual cocatalysts.
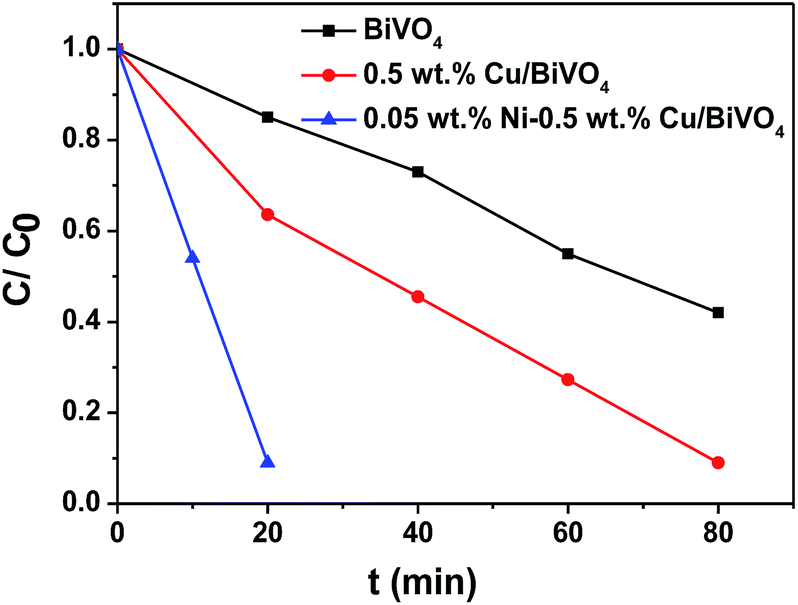
Fig. 7 shows the time course of photocatalytic oxidation of thiophene on BiVO4, CuO/BiVO4, Ni–CuO/BiVO4 and Pt–RuO2/BiVO4. After 3 h of visible light irradiation, the residual thiophene concentration (C/C0) was ca. 58% on BiVO4, ca. 56% on CuO/BiVO4, ca. 5% on Ni–CuO/BiVO4, and ca. 1% on Pt–RuO2/BiVO4. The rate of degradation of thiophene with Ni–CuO/BiVO4 catalyst was extremely fast compared with BiVO4 and CuO/BiVO4. In our previous study, thiophene (600 ppm) could be almost completely converted (ca. 99%) in 3 h for the optimized 0.03 wt% Pt–0.01 wt% RuO2/BiVO4 catalyst.5 Under the same experimental conditions, the performance of the photocatalyst 0.05 wt% Ni–0.5 wt% CuO/BiVO4 was very close to that of 0.03 wt% Pt–0.01 wt% RuO2/BiVO4. According to the abovementioned results, we can conclude that the synergistic effect of cocatalysts Ni and CuO on BiVO4 is very favorable for improving the photocatalytic activity of thiophene oxidation.
 | ||
| Fig. 7 Photocatalytic oxidation of thiophene on BiVO4 and BiVO4 loaded with various cocatalysts under visible light irradiation (λ ≥ 420 nm). Reaction conditions: [sulfur content]initial = 600 ppm; the concentration of photocatalyst: 1 g L−1; O2 (bubbled into the system); temperature: 10 ± 2 °C; reaction time: 3 h. | ||
On the other hand, the noble-metal-free catalyst possess a high photocatalytic activity close to Pt–RuO2/BiVO4, which is very important for further application requirements for ultra-low sulfur-containing fuels.
3.3 The proposed reaction mechanism
To investigate the reaction process of photocatalytic oxidation of thiophene on Ni–CuO/BiVO4, we used XPS to investigate the combined-state of the Cu element on photocatalyst after the reaction. Fig. 8 shows the asymmetrical X-ray photoelectron spectrum of Cu 2p levels of Ni–CuO/BiVO4 catalyst after the photocatalytic oxidation reaction. Cu 2p3/2 and Cu 2p1/2 XPS of the used catalyst exhibited sharp peaks at 932.4 eV and 952.2 eV, respectively, without any shift in the binding energy (BE). A strong satellite peak was observed on the higher BE side above 940 eV indicating the existence of divalent Cu.32 In addition, CuO 2p3/2 and CuO 2p1/2 XPS also exhibited small peaks at 933.6 eV and 953.6 eV respectively. The results indicate that a certain amount of CuOx (mixture of CuO and metal Cu) remained on the surface of the used catalyst. Cu2+ is thus partially reduced to metallic copper (Cu0) and/or Cu1+ in the photocatalytic oxidation reaction.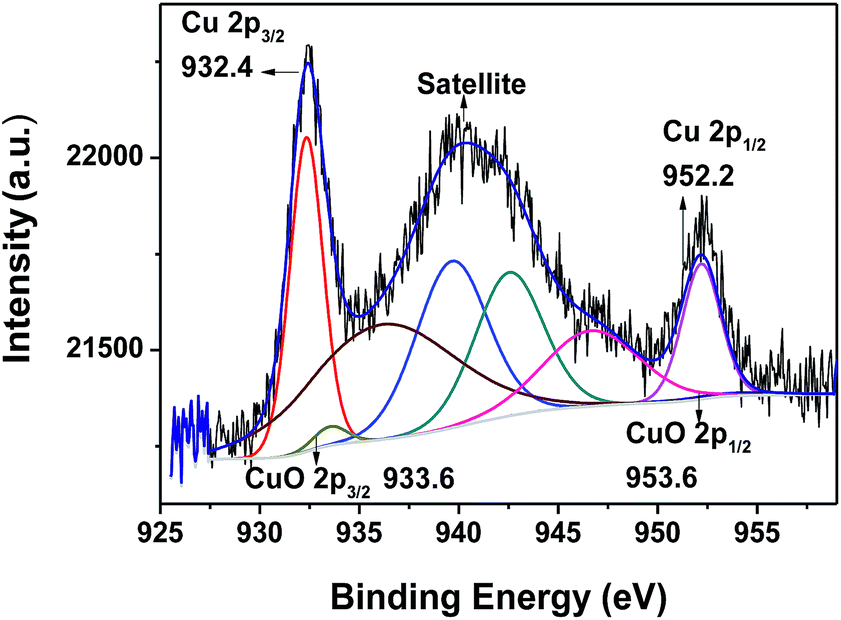 | ||
| Fig. 8 Cu 2p XPS of Ni–CuO/BiVO4 catalyst after the photocatalytic oxidation reaction. | ||
In order to convert the XPS intensity ratio into the surface atomic ratio, the following expression was applied:33
| (Cu/Cu2+) XPS = C(ICu 2p3/2/ICu2p3/22+) |
| Item | Cu 2p3/2 | Cu 2p1/2 | CuO 2p3/2 | CuO 2p1/2 | Satellite | Cu/CuO (atomic ratio) |
|---|---|---|---|---|---|---|
| BE (eV) | 932.4 | 952.2 | 933.6 | 953.6 | 941.0 | |
| Peak area | 1744.8 | 872.4 | 105.4 | 52.7 | — | 16.6 |
To clarify the reaction mechanism for the photocatalytic oxidation of thiophene on Ni–CuO/BiVO4, the ESR spin-trap technique (with DMPO) was employed to probe the active oxygen species generated under the illumination. Fig. 9 shows the ESR signals obtained from the in situ photocatalytic reaction. The ESR signals that appeared in the presence of photocatalysts were centered at g = 2.0065, which can be assigned to oxygen species.34 No ESR signals were observed either when the photocatalyst was absent or the reaction was performed with BiVO4 in the dark. After light irradiation, a sextet ESR signal was observed that is assigned to DMPO–O2˙−. The hyperfine splittings were aN = 1.27 mT, aβH = 0.99 mT and aγH = 0.14 mT, where aN, aβH and aγH are hyperfine splitting constants of nitroxyl nitrogen, one β-hydrogen and one γ-hydrogen, respectively.6,35,36 These results provide evidence of O2˙− formed in the presence of photocatalysts BiVO4 and Ni–CuO/BiVO4. May be the ˙OH, which has strong oxidizing ability, was also generated with the illumination, but the characteristic quartet peaks of the DMPO–OH adduct were submerged in the sextet signal of DMPO–O2˙− adduct. Moreover, the signals of O2˙− generated after illumination on Ni–CuO/BiVO4 for 8 min were more obvious than those for BiVO4. This might be one reason for the higher photocatalytic activity of Ni–CuO/BiVO4. During the photocatalytic oxidation reaction with Ni–CuO/BiVO4 catalyst, more active oxygen species were generated under the illumination for the oxidation of pollutants. After 16 min of illumination, the signals intensity was decreased. This is due to the consumption of the dissolved O2 and the oxidation of DMPO–O2˙− adduct by h+ generated during the illumination. In conclusion, for Ni–CuO/BiVO4, the simultaneous existence of reduction cocatalyst and oxidation cocatalyst is beneficial for efficient separation and transport of the photo-excited electrons and holes, respectively. The production of O2˙− and photo-excited holes can be enhanced simultaneously, resulting in the high photocatalytic activity of thiophene oxidation.
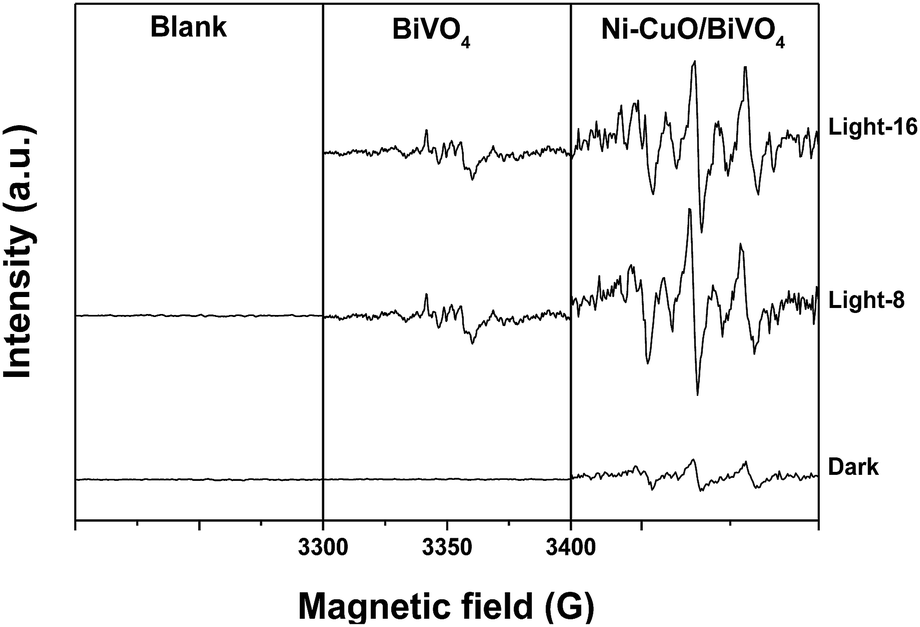 | ||
| Fig. 9 In situ ESR spectra of DMPO–O2˙− generated in the photocatalytic oxidation reaction of thiophene with different photocatalysts. The sample tested without photocatalyst is denoted as “Blank”. The signals obtained without light irradiation are denoted as “Dark”. The signals obtained after irradiating for 8 min are denoted as “Light-8”, similarly, for 16 min named “Light-16”. | ||
According to the results of XPS and ESR, we propose a reaction mechanism for the photocatalytic oxidation of pollutants on Ni–CuO/BiVO4 photocatalyst. The right part in Scheme 1 illustrates the charge transfer processes between the Ni, Cu based dual cocatalysts and semiconductor BiVO4. First, under the visible light irradiation, electron–hole pairs are photo generated in BiVO4. Due to the energy level matching of BiVO4 and CuO, the photo-generated electron transfer from the conduction band of BiVO4 to that of CuO, and photo-generated holes transfer from the valence band of BiVO4 to that of CuO. In this electron transfer process, CuO can be partially reduced to Cu2O. Second, because of the energy level matching of CuO and Cu2O, the photo-generated holes transfer from the valence band of CuO to that of Cu2O, but the photo-generated electrons cannot transfer. Herein, Cu2O/CuO acts as the oxidation cocatalyst for generated hole transfer. Third, the Cu2O acts as a barrier for the electrons of BiVO4/CuO to reach the Cu2O surface and the electrons are trapped in CuO, which may be the reason for the valence of most Cu being Cu0 for the used photocatalyst (XPS results). Consequently, most holes can reach the Cu2O/CuO and substrate interface, and thus, the charge separation and transport in photocatalysts is greatly accelerated without recombination, leading to the oxidation of pollutants. On the other side (as shown in the left part in Scheme 1), Ni0 and Cu0 with low Fermi levels on the surface of BiVO4 act as the reduction cocatalyst, and thus photo-generated electrons transfer to O2 via Ni (0) and Cu (0) cocatalysts, where superoxide species O2˙− is formed when O2 reacts with the photo-generated electrons. Namely, the adsorbed oxygen acts as an electron trap that efficiently inhibits electron–hole recombination.37 Then, ˙OH, which has a strong oxidation ability, might be generated via reaction of OH− with holes in the reaction system under the illumination. The active oxygen species can react with pollutant molecules in the presence of Ni–CuO/BiVO4 catalyst. Thus, the series of photocatalytic oxidation reactions are accelerated on Ni/Cu and CuO/Cu2O co-loaded on BiVO4 catalyst. The synergistic effect of CuO acting as an oxidation cocatalyst and Ni acting as a reduction cocatalyst is beneficial for the efficient separation and transfer of the photo-excited electrons and holes, being responsible for the high photocatalytic oxidation activity of pollutants.
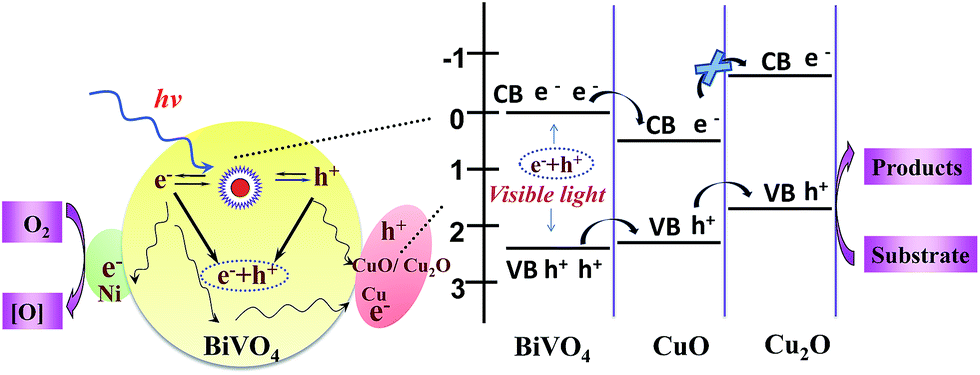 | ||
| Scheme 1 Schematic of the mechanism for photocatalytic oxidation of pollutants on Ni–CuO/BiVO4 photocatalyst. | ||
4. Conclusions
The visible light responsive photocatalyst BiVO4 co-loaded with noble-metal-free Ni and CuO can achieve over 94% conversion of thiophene oxidation under visible light irradiation using molecular oxygen as the oxidant. This high activity of Ni–CuO/BiVO4 was also successfully presented in photocatalytic oxidation of organic dyes. Dual cocatalysts Ni and CuO showed a strong synergistic effect on the enhanced photocatalytic activity. The noble-metal-free catalyst possessed high photocatalytic activity close to Pt–RuO2/BiVO4 (in the same condition), which is very important for further application requirements in industry. XPS and ESR measurements showed that the activation of molecular oxygen and oxidation of pollutants molecule simultaneously take place on Ni/Cu and CuO/Cu2O co-loaded on BiVO4 catalyst. Herein, Ni/Cu acted as the reduction cocatalyst for photo-generated electron transfer, while CuO/Cu2O acted as the oxidation cocatalyst for photo-generated hole transfer. The co-existing low-cost dual cocatalysts thus play the significant role in greatly enhancing the photocatalytic oxidation activity. The visible light responsive semiconductor loaded with earth-abundant dual cocatalysts has great potential in both solar energy conversion and further industrial applications.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 21603025, 11274057, 11474046, 21506203) and Open Project of State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences (Grant No. N-15-06). The authors thank Prof. Can Li of Dalian Institute of Chemical Physics (Chinese Academy of Sciences) very much for his advice in experiment and discussion.Notes and references
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z. X. Guo and J. W. Tang, Energy Environ. Sci., 2015, 8, 731 CAS.
- Z. J. Sun, H. F. Zheng, J. S. Li and P. W. Du, Energy Environ. Sci., 2015, 8, 2668 CAS.
- B. Y. Zhang, Z. X. Jiang, J. Li, Y. N. Zhang, F. Lin, Y. Liu and C. Li, J. Catal., 2012, 287, 5 CrossRef CAS.
- Y. Z. Zhen, J. Li, D. J. Wang, F. Fu and G. L. Xue, J. Inorg. Mater., 2015, 30, 408 CrossRef CAS.
- F. Lin, D. E. Wang, Z. X. Jiang, Y. Ma, J. Li, R. G. Li and C. Li, Energy Environ. Sci., 2012, 5, 6400 CAS.
- F. Lin, Y. N. Zhang, L. Wang, Y. L. Zhang, D. E. Wang, M. Yang, J. H. Yang, B. Y. Zhang, Z. X. Jiang and C. Li, Appl. Catal., B, 2012, 127, 363 CrossRef CAS.
- F. Lin, Z. X. Jiang, N. F. Tang, C. Zhang, Z. P. Chen, T. F. Liu and B. Dong, Appl. Catal., B, 2016, 188, 253 CrossRef CAS.
- F. Chen, J. C. Zhao and H. Hidaka, Res. Chem. Intermed., 2003, 29, 733 CrossRef CAS.
- X. Lin, X. Y. Guo, W. L. Shi, F. Guo, G. B. Che, H. J. Zhai, Y. S. Yan and Q. W. Wang, Catal. Commun., 2015, 71, 21 CrossRef CAS.
- X. Lin, Y. S. Wang, J. Zheng, C. Liu, Y. Yang and G. B. Che, Dalton Trans., 2015, 44, 19185 RSC.
- J. W. Tang, Z. G. Zou and J. H. Ye, Angew. Chem., Int. Ed., 2004, 43, 4463 CrossRef CAS PubMed.
- J. F. Ma, J. Zou and L. Y. Li, Appl. Catal., B, 2014, 144, 36 CrossRef CAS.
- F. Guo, W. L. Shi, X. Lin, X. Yan, Y. Guo and G. B. Che, Sep. Purif. Technol., 2015, 141, 246 CrossRef CAS.
- X. Lin, X. Y. Guo, W. L. Shi, L. N. Zhao, Y. S. Yan and Q. W. Wang, J. Alloys Compd., 2015, 635, 256 CrossRef CAS.
- X. Lin, D. Xu, J. Zheng, M. S. Song, G. B. Che, Y. S. Wang, Y. Yang, C. Liu, L. N. Zhao and L. M. Chang, J. Alloys Compd., 2016, 688, 891 CrossRef CAS.
- M. M. Liu, F. Y. Li, Z. X. Sun, L. Xu, Y. F. Song and A. Munventwali, RSC Adv., 2015, 5, 47314 RSC.
- A. Y. Meng, J. Zhang, D. F. Xu, B. Cheng and J. G. Yu, Appl. Catal., B, 2016, 198, 286 CrossRef CAS.
- H. J. Yan, J. H. Yang, G. J. Ma, G. P. Wu, X. Zong, Z. B. Lei, J. Y. Shi and C. Li, J. Catal., 2009, 266, 165 CrossRef CAS.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390 CAS.
- K. Maeda, T. Ohnoa and K. Domen, Chem. Sci., 2011, 2, 1362 RSC.
- W. J. Foo, C. Zhang and G. W. Ho, Nanoscale, 2013, 5, 759 RSC.
- B. Zhou and J. H. Qu, Appl. Catal., B, 2010, 99, 214 CrossRef CAS.
- F. X. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348 CrossRef CAS PubMed.
- G. C. Bi, J. Q. Wen, X. Li, W. Liu, J. Xie, Y. P. Fang and W. W. Zhang, RSC Adv., 2016, 6, 31497 RSC.
- Y. Xu and R. Xu, Appl. Surf. Sci., 2015, 351, 779 CrossRef CAS.
- T. Pham, C. Nguyen-Huy and E. W. Shin, Appl. Surf. Sci., 2016, 377, 301 CrossRef CAS; J. Q. Wen, X. Li, H. Q. Li, S. Ma, K. L. He, Y. H. Xu, Y. P. Fang, W. Liu and Q. Z. Gao, Appl. Surf. Sci., 2015, 358, 204 CrossRef.
- L. L. Li, B. Cheng, Y. X. Wang and J. G. Yu, J. Colloid Interface Sci., 2015, 449, 115 CrossRef CAS PubMed.
- Q. Z. Wang, G. X. Yun, Y. Bai, N. An, Y. T. Chen, R. F. Wang, Z. Q. Lei and W. F. Shangguan, Int. J. Hydrogen Energy, 2014, 39, 13421 CrossRef CAS.
- Q. Q. Hu, J. Q. Huang, G. J. Li, J. Chen, Z. J. Zhang, Z. H. Deng, Y. B. Jiang, W. Guo and Y. G. Cao, Appl. Surf. Sci., 2016, 369, 201 CrossRef CAS.
- J. Q. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163 CrossRef CAS.
- W. Z. Yin, W. Z. Wang, L. Zhou, S. M. Sun and L. Zhang, J. Hazard. Mater., 2010, 173, 194 CrossRef CAS PubMed.
- S. Velu, K. Suzuki, M. Vijayaraj, S. Barman and C. S. Gopinath, Appl. Catal., B, 2005, 55, 287 CrossRef CAS.
- G. Fierro, M. L. Jacono, M. Inversi, R. Dragone and P. Porta, Top. Catal., 2000, 10, 39 CrossRef CAS.
- S. Leonard, P. M. Gannett, Y. Rojanasakul, D. Schwegler-Berry, V. Castranova, V. Vallyathan and X. L. Shi, J. Inorg. Biochem., 1998, 70, 239 CrossRef CAS PubMed.
- J. R. Harbour and M. L. Hair, J. Phys. Chem., 1978, 82, 1397 CrossRef CAS.
- Y. Huang, J. Li, W. Ma, M. Cheng, J. Zhao and J. C. Yu, J. Phys. Chem. B, 2004, 108, 7263 CrossRef CAS.
- J. Robertson and T. J. Bandosz, J. Colloid Interface Sci., 2006, 299, 125 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |