
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel polymeric micelle used for in vivo MR imaging tracking of neural stem cells in acute ischemic stroke†
Liejing Lu‡
a,
Yong Wang‡b,
Minghui Caoa,
Meiwei Chena,
Bingling Lina,
Xiaohui Duana,
Fang Zhanga,
Jiaji Maoa,
Xintao Shuaibc and
Jun Shen *a
*a
aDepartment of Radiology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, No. 107 Yanjiang Rd. West, Guangzhou 510120, Guangdong, China. E-mail: shenjun@mail.sysu.edu.cn; Fax: +86 20 8133 2702; Tel: +86 20 8133 2702
bPCFM Lab of Ministry of Education, School of Materials Science and Engineering, Sun Yat-Sen University, Guangzhou 510275, China
cBME Center, Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou 510080, China
First published on 7th March 2017
Abstract
Transplantation of neural stem cells (NSCs) is a promising treatment strategy for acute ischemic stroke. In vivo tracking of the therapeutic stem cells in the host brain after transplantation is essential not only to ensure the safety and efficacy of the treatment, but also to better understand their migrational dynamics and regeneration potential. Many polymeric nanoparticles have been developed to label stem cells for in vivo tracking by magnetic resonance imaging (MRI), optical imaging (OI) or other imaging modalities. However, the non-degradability and presence of cellular toxicity of the nanoparticles restrict their clinical applications. In this study, we developed a novel cationic polymeric micelles based on amphipathic polymer of biodegradable hydrophilic poly(aspartic acid-dimethylethanediamine) (PAsp(DMA)) that were conjugated with two molecules of hydrophobic cholic acid (CA) by lysine. Image labels, superparamagnetic iron oxide nanoparticles (SPIONs) and fluorescent nile red were simultaneously loaded into the micelles to label NSCs. The labeling capacity, efficiency and cytotoxicity of the cationic micelles were determined. The in vivo MRI tracking of the therapeutic NSCs in acute ischemic stroke was also explored. Our results showed that this type of cationic polymeric micelles achieved a high efficient and safe labeling of NSCs and resulted in reliable in vivo MRI tracking of therapeutic stem cells in acute ischemic stroke, but without detrimental effect. The cationic, biodegradable polymeric micelles are highly translatable for clinical application and can be used as a versatile nanoplatform for stem cell labeling and subsequently in vivo tracking in regenerative medicine.
Introduction
Acute ischemic stroke inevitably involves the loss of neurons, astroglia and oligodendroglia, and disruption of synaptic architecture.1 The natural capacity of the brain to recover from the ischemic injury is obviously limited, since cell replacement to damaged tissues from endogenous neural stem cells is restricted by their finite numbers, limited renewal ability and slow turnover.2 Thus, cellular therapy using exogenous stem cells has emerged as a promising candidate approach for the therapy of stroke. Neural stem cell (NSCs) are the main therapeutic candidate cells, as they can repair the central nerve system (CNS) insults probably by neuroprotection via secreting various neural trophic factors or by differentiating into a broad range of neuronal and glial lineages to replace damaged neural tissues.1,3–5Despite the great therapeutic potential of stem cells, many issues, such as in vivo monitoring of cell fate, remain to be resolved. Tracking the status of cells after transplantation using noninvasive imaging techniques is essential for future clinical applications and can help to maximize the therapeutic effect.6 Magnetic resonance imaging (MRI) is one of the most powerful imaging tools to track therapeutic cells in vivo, due to its high spatial resolution, without exposure to ionizing radiation, and wide clinical applicability.6,7 For cellular MRI, superparamagnetic iron oxide nanoparticles (SPIONs) are the most commonly used imaging label owing to its high sensitivity, low toxicity and biocompatibility.8 However, because of the poor uptake of native SPIONs by stem cells, a high concentration of SPIONs, a potentially toxic transfection agent or complicate surface modification of SPIONs are usually required to achieve sufficient cellular labeling for sensitive tracking of grafted cells in vivo. However, these labeling strategies may interfere with the viability of labeled cells and restrict their clinical applications.9 Therefore, developing a novel type of SPIONs that can be efficiently internalized by stem cells featured by convenient labeling procedure, robust biocompatibility and high imaging sensitivity, is of great interest.10
Polymeric nanoparticles as versatile carriers have attracted tremendous attention due to their colloidal stability, tunable membrane properties and ability to encapsulate or integrate a broad range of drugs and molecules.11 Previously, we had developed a new class of nano-sized polymersomes to label stem cells, which showed a high efficiency in delivering optical imaging (OI) agent and SPIONs into stem cells while in a rapid way.12 Nevertheless, this class of polymersomes was dependent on the cationic polyetherimide (PEI) to deliver image labels into stem cells. Concerning the non-degradability and cellular toxicity of PEI, in this study, we developed a novel cationic polymeric micelles based on amphipathic polymer of biodegradable hydrophilic poly(aspartic acid-dimethylethanediamine) (PAsp(DMA)), conjugated with two molecules of hydrophobic cholic acid (CA, approved for clinical application by Food and Drug Administration) by lysine. The images labels, SPIONs and fluorescent nile red were simultaneously loaded to label NSCs to achieve in vivo MRI tracking of transplanted stem cells in stroke. A neutral micelles based on polyethylene glycol (PEG) instead of PAsp(DMA) was also prepared as a control (Fig. 1).
 | ||
| Fig. 1 Schematic diagram of the synthesis of cationic and neutral polymeric micelles and the labeling of neutral stem cells. | ||
Experimental
Materials
The following reagents were purchased from Sigma-Aldrich and used as received: 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium (HBTU), N-hydroxybenzotriazole (HOBt), N,N-diisopropylethylamine (DIPEA), di-tert-butoxycarbonyl-L-lysine (Boc-Lys(Boc)-OH), α-methoxy-ε-hydroxy-poly(ethylene glycol) (mPEG-OH, Mn = 2 kDa), anhydrous dimethyl sulfoxide (DMSO) and anhydrous dimethylformamide (DMF). N,N-Dimethyl-1,2-ethanediamine (DMA), trifluoroacetic acid (TFA), cholic acid (CA) and n-butylamine (BA) were purchased from Alfa Aesar China Co., Ltd. (Tianjin, China). Dialysis bag (MWCO: 3.5 kDa) was purchased from Shanghai Green Bird Technology Development Co., Ltd., China. Chloroform (CHCl3), acetic ether, petroleum ether, and dichloromethane (DCM, CH2Cl2) were dried over CaH2 and then distilled under ambient pressure. Diethyl ether was of analytical grade and purchased from Guangzhou Chemical Reagent Factory, China. mPEG-NH2 and N-carboxyanhydride of β-benzyl-L-aspartate (BLA-NCA) were synthesized as previously reported.13 The cell counting kit-8 reagent (CCK-8) was purchased from Dojindo Molecular Technologies Inc. (Kumamoto, Japan). Mouse anti-rat nestin, anti-rat β-III-tubulin and rabbit anti-rat GFP antibodies were purchased from Millipore Co., Ltd (Temecula, CA, USA). Alexa Fluor 594-conjugated goat anti-mouse IgG antibody was purchased from Life Technologies (Carlsbad, CA, USA). 4′,6-Diamidino-2-phenylindole (DAPI) were purchased from Invitrogen. HRP-labeled goat anti-rabbit secondary antibody and 3,3-diaminobenzidine (DAB) were purchased from Zsbio (Beijing, China). Monofilament nylon sutures with the polysiloxane-coated tip were purchased from Jialing Biotechnology Co., LTD (3600-AA; Guangzhou, China).Synthesis of polymers
The cationic amphipathic polymer of PAsp(DMA), lysine and hydrophobic CA, i.e. PAsp(DMA)–Lys–CA2 was synthesized via multi-step reactions (ESI Fig. 1†). Firstly, N-butylamine-termined poly(β-benzyl-L-aspartate) (BA-PBLA) was synthesized by ring-opening polymerization of BLA-NCA using n-butylamine as an initiator.13 In brief, 73.14 mg of n-butylamine (1.0 mmol, 0.74 g mL−1) was charged into a 50 mL reaction flask and dissolved with 30 mL anhydrous CHCl2. 2.49 g of BLA-NCA (10 mmol) dissolved in 3 mL of anhydrous DMF was then added into the flask under the protection of N2 gas. The solution was kept stirring for 72 h at 35 °C, followed by precipitated into excessive cool diethyl ether. The precipitate was filtered, washed with diethyl ether, and vacuum-dried until a constant weight was attained (BA-PBLA: Mn = 2.0 kDa, calculated from 1H NMR spectrum). Secondly, Boc-Lys(Boc)-OH (1.5 equiv.) was coupled onto the N-terminal of PBLA using HBTU (1.5 equiv.) and HOBt (1.5 equiv.) as coupling reagents in DMF overnight.14 The targeted molecules were precipitated and washed three times by cold diethyl ether. Subsequently, the Boc groups were removed by treating with TFA for 30 min, the mixture solution was precipitated into cold diethyl ether, and the precipitate was filtered, washed with diethyl ether, and vacuum-dried to get PBLA–Lys. Thirdly, CA molecules (3.0 equiv.) were coupled onto the N-terminal of PBLA–Lys via amidation reaction with HBTU (3.0 equiv.) and HOBt (3.0 equiv.) as coupling reagents. The reaction was proceeded in DMF overnight and then precipitated and washed by cold methanol, followed by filtering and vacuum-drying to get PBLA–Lys–CA2. Finally, PAsp(DMA)–Lys–CA2 was synthesized by aminolysis of DMA with PBLA–Lys–CA2.13 In brief, 0.90 g of PBLA–Lys–CA2 (0.3 mmol) and 2.6 g of DMA (30 mmol, about 10 equiv. to benzyl group) were dissolved in 20 mL of anhydrous DMSO, and then the reaction was stirred for 24 h at 35 °C. The mixture was added dropwise to 100 mL of water under sonication then dialyzed (MWCO: 14 kDa) against deionized water for 3 days and freeze-dried to obtain final polymer PAsp(DMA)–Lys–CA2 (Mn = 2.6 kDa, calculated from 1H NMR spectrum, yield: 74%). mPEG–Lys–CA2 was analogously synthesized by coupling Boc-Lys(Boc)-OH and CA orderly onto the N-terminal of PEG.13Preparation of SPIONs and nile red co-loaded micelles
The hydrophobic SPIONs measuring 6 nm were synthesized according to a reported method.12 To prepare the SPIONs and nile red co-loaded micelles, 2 mg of SPIONs, 0.2 mg of nile red and 20 mg of polymers (PAsp(DMA)–Lys–CA2 or PEG–Lys–CA2) were co-dissolved into 2 mL of DMSO and chloroform (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Under sonication (VCX130, Sonics, USA, 20 kHz, 40% power level), the above solution was added dropwise to 20 mL of phosphate-buffered saline (PBS, PH = 7.4, 0.05 mol L−1). After the organic solvent chloroform was removed by rotary evaporation, the solution was filtered through a syringe filter (pore size: 450 nm) to eliminate free SPIONs, nile red and large aggregates, followed by ultrafiltration using a Millipore centrifugal filter device (MW cut off: 100 kDa) to remove DMSO and other hydrophilic impurities. Finally, SPIONs and nile red co-loaded cationic nanoparticles of PAsp(DMA)–Lys–CA2 (C-NPs) or neutral PEG–Lys–CA2 (N-NPs) were obtained.
:3). Under sonication (VCX130, Sonics, USA, 20 kHz, 40% power level), the above solution was added dropwise to 20 mL of phosphate-buffered saline (PBS, PH = 7.4, 0.05 mol L−1). After the organic solvent chloroform was removed by rotary evaporation, the solution was filtered through a syringe filter (pore size: 450 nm) to eliminate free SPIONs, nile red and large aggregates, followed by ultrafiltration using a Millipore centrifugal filter device (MW cut off: 100 kDa) to remove DMSO and other hydrophilic impurities. Finally, SPIONs and nile red co-loaded cationic nanoparticles of PAsp(DMA)–Lys–CA2 (C-NPs) or neutral PEG–Lys–CA2 (N-NPs) were obtained.
Characterization of polymeric micelles
1H NMR spectra were carried out on a Varian Unity 400 MHz spectrometer using DMSO-d6 as a solvent. Fourier transform infrared (FTIR) spectral measurements were recorded using a Nicolet/Nexus 670 FTIR spectrometer with a resolution of 2 cm−1 and the powder samples were compressed into KBr pellets. Gel permeation chromatography (GPC) was employed to determine the molecular weight and molecular weight distribution. GPC analysis was carried out using a SHODEX 7.8 × 300 mm column with chloroform as an eluent (1 mL min−1) and polystyrene was used as a standard for column calibration. Twenty micro liter samples were injected with a microsyringe, and the eluent was analyzed with a Waters 2414 differential refractive index (RI) detector (Waters Corporation, USA). The size and zeta potential were determined using dynamic light scattering (DLS). Measurements were carried out at 25 °C on a 90° Plus/BI-MAS equipment (Brookhaven Instruments Corporation, USA). For the zeta potential measurement, a standard electrophoresis mini-cell from Brookhaven was used. The data for particle size and zeta potential were collected on an auto-correlator with a detection angle of scattered light at 90° and 15°, respectively. For each sample, the data from five measurements were averaged to obtain the mean ± standard deviation (SD). For transmission electron microscopy (TEM), the samples were prepared by drying a drop (5 μL, 0.5 mg mL−1) of the sample solution on a copper grid coated with amorphous carbon and then blotted with a filter paper after 1 h. To stain the samples, 10 μL of uranyl acetate solution (2 wt% in water) was added to the copper grid, and then blotted with a piece of filter paper after 1 min. The grid was finally dried overnight at room temperature inside a desiccator and observed using a transmission electron microscope (Hitachi H-7650, Tokyo, Japan) operated at 80 kV. The iron content was detected by atomic absorption spectrometry (AAS). Samples were dissolved in 1 M HCl solution for thorough release and dissolution of SPIONs, then determined at the specific Fe absorption wavelength (248.3 nm) based on a pre-established calibration curve using a polarized Zeman atomic absorption spectrometer (Hitachi Z-200; Tokyo, Japan). To detect the content of nile red, the freeze-dried micelles were redissolved in DMSO at a concentration of 0.5 mg mL−1, and then the nile red fluorescence intensities were measured on a Fluorescence Spectroscopy PE-LS55 (PerkinElmer Ltd., United Kingdom) based on a pre-established calibration curve, using excitation wavelength of 550 nm, excitation slit width of 5 nm, emission slit width of 5 nm, and scanning speed of 500 nm min−1. The r2 relaxivity of C-NPs and N-NPs was detected by using 3.0 T MR scanner (Intera; Philips Medical Systems, Best, The Netherlands) with a synergy head coil. The measurements were performed by using single-slice multi-echo spin echo (SE) imaging for transverse relaxation time (T2) calculations to generate T2-maps. The acquisition parameters were: repetition time (TR)/echo time (TE) = 2000/20–80 ms, 4 stepped echoes, and number of acquisitions (NSA) = 1, acquisition matrix = 160 × 266, field of view (FOV) = 80 × 80 mm2, and slice thickness = 2 mm.Cell culture and characterization
Transgenic green fluorescence protein (GFP)-C17.2 neural stem cells (NSCs) were purchased from Sainer Biotechnology Inc. (Tianjing, China). The NSCs were cultured with Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 5% horse serum (Sigma Aldrich, St. Louis, MO, USA) and 2 mM glutamine in a humidified incubator (5% CO2, 95% air) at 37 °C. When the cell monolayers were grown to approximately 90% confluence, the cells were passaged by trypsinization. For cellular characterization, the obtained cells were planted on poly-D-lysine-coated coverslips and immunostained for nestin (ESI Fig. 2†), as previously described.15Effect of surface charges on cell labeling
The effect of surcharge charges on cellular uptake of micelles was assessed by Prussian blue staining. The GFP-C17.2 NSCs were seeded on coverslips planted in 12-well plate (Corning Inc., Corning, NY) at a density of 1 × 105 cells per well. When cells grew to 60% confluence, the culture medium was replenished, C-NPs and N-NPs were then directly added to the culture medium with final iron concentrations of 10 μg mL−1 and 20 μg mL−1 in each well. After incubating for 2, 4 and 6 h, the cells were washed with PBS three times and fixed with 4% paraformaldehyde. Then, 1 mL of solution containing 5 wt% potassium ferrocyanide and 10 vol% HCl was added to each well and incubated at room temperature for 20 min. After washing with PBS three times, cells were counterstained with nuclear fast red solution for 5 min, followed by washing with PBS three times. Finally, the coverslips were dehydrated and mounted on the slide glass. Imaging data were obtained by an Olympus AX80 microscope (Olympus, Tokyo, Japan) with objective lenses of 100× through an AxioCam HRc digital camera (Carl Zeiss, Oberkochen, Germany). All experiments were done in triplicate.Optimal cell labeling conditions
The GFP-C17.2 NSCs were cultured in 12-well plates at a density of 1 × 105 cells per well. When cells grew to 90% confluence, the culture medium was replenished. To determine the optimal labeling concentration, N-NPs and C-NPs were directly added to the culture medium with final iron concentrations of 1.25, 2.5, 5, 10, 15 and 20 μg mL−1 and incubated for 4 h. To determine the optimal incubation time, N-NPs or C-NPs were added to cells with final iron concentration of 10 μg mL−1 and incubated with cells under standard culture conditions for 1, 2, 3, 4, 5 and 6 h, respectively. After incubation, the cells were collected and washed three times with PBS, then prepared for in vitro MRI. For MRI, cells were resuspended in 200 μL 1% agarose solution and imaged after gelation. The MRI were performed on the clinical 3.0-T system with parameters described above. All experiments were done in triplicate.Cellular uptake and intracellular distribution of micelles
Cellular uptake and intracellular distribution of micelles were further analyzed using fluorescence microscope and TEM, respectively. The GFP-C17.2 NSCs were seeded on coverslips at a density of 1 × 105 cells per well. When grew to 60% confluence, the cells were treated with different nanoparticle formulations at final iron concentration of 10 μg mL−1 and incubated for different incubation times (1, 2, 3, 4, 5 and 6 h). Afterwards, the cells were washed twice with PBS and fixe with 4% PFA, the nucleus were stained with DAPI (1 μg mL−1) for 2 minutes. The coverslips were mounted on the slide glass and observed using a fluorescence microscope (BX63; Olympus, Tokyo, Japan). For TEM, 1 × 106 labeled cells (at iron concentration of 10 μg mL−1 for 6 h of incubation) were centrifuged and fixed in 2.5% glutaraldehyde cacodylate buffer at 4 °C overnight. After incubation in 1% OsO4 for 1 h, the cells were dehydrated in a graded ethanol series and embedded in artificial resin (Epon; Merck, Darmstadt, Germany). Ultrathin sections were cut, followed by stained with uranyl acetate and lead citrate, then imaged with a H-7650 transmission electron microscope (Hitachi, Tokyo, Japan) at 80–120 kV.Labeling efficiency assay
The GFP-C17.2 NSCs were seeded in 12-well plates at a density of 1 × 105 cells per well 24 h before labeling. After treated with N-NPs and C-NPs at iron concentration of 10 μg mL−1 for 4 h, the cells were collected and washed twice with PBS. The labeling efficiency was assessed by flow cytometry (FACScalibur, BD, Mountain View, CA, USA) via calculating the percentage of cells with red fluorescence. The experiments were done in triplicate.Cell viability assay
The apoptosis and CCK-8 assay were employed to assess the cytotoxicity of different micelles. Cell apoptosis was assessed using the Annexin V/Propidium Iodide (PI) double staining method 1 d after cell labeling under the pre-determined optimal conditions. Briefly, after labeling, 1 × 106 of GFP-C17.2 NSCs were labeled with Annexin V-FITC and PI according to the manufacturer's protocols, and then analyzed on a flow cytometer. For CCK-8 assay, GFP-C17.2 NSCs after labeling were planted in 96-well plates at a density of 5000 cells per well with the total culture media of 200 μL and incubated for another 1, 2, and 3 d. Then 10 μL CCK-8 reagent was added into each well. After incubation for 2 h, the absorbance was recorded at 450 nm on a microplate reader (SpectraMaxM5; Molecular Devices, CA, USA). All experiments were conducted in triplicate. To investigate the effect of micelles on the differentiation capability of stem cells, GFP-C17.2 NSCs after labeling were tested for their neuronal differentiation potential in vitro. Cells were planted on coverslips and cultured with DMEM/F12 containing 2% B27 and 1% penicillin–streptomycin up to 2 weeks. Then cells were fixed with 4% paraformaldehyde and immunostained with cell-specific markers for neurons (β-III-tubulin). After cells were counterstained with DAPI, the coverslips were mounted on the slide glass and directly observed using a fluorescence microscope.Animal surgery and cell transplantation
12 Sprague-Dawley (SD) rats were obtained from the Animal Experiment Centre of our University. All animal experiments were conducted according to the guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care Committee. Focal cerebral ischemia was established by using transient middle cerebral artery occlusion (MCAO) model16 with a minor modification.17 Two days after surgery, animals were randomly selected to receive stereotactic injection of 5 × 105 GFP-C17.2 NSCs pre-labeled with N-NPs or with C-NPs (n = 6 each). The cells were injected into the striatum ipsilateral to the ischemic hemisphere (stereotaxic coordinates: 3.0 mm lateral to bregma, 0.5 mm rostral to bregma and 6.0 mm deep from the pial surface) using a 28 s gauge needle. The cells were suspended in 2.5 μL PBS and was injected at a constant rate of 0.2 μL min−1. After injection, the needle was kept in place for another 15 min, and then slowly withdrawn.In vivo MR imaging
At 1, 3, 7 and 14 d after transplantation, MRI was performed on a clinical 3.0-T system (Achieva; Philips Medical Systems) with a 50 mm × 50 mm 4-channel phased array rat coil (Suzhou Zhongzhi Medical Technologies, China) to detect the distribution of implanted cells. For in vivo MRI, axial brain images were obtained by using fast spin echo (FSE) T2-weighted imaging (TR/TE = 200/31 ms; NSA = 3), fast field echo (FFE) -weighted images (TR/TE = 500/18 ms; flip angle = 20°; NSA = 3). Other parameters for these sequences were FOV = 60 × 60 mm, acquisition matrix = 267 × 268, reconstruction matrix = 512 × 512, section thickness = 1.0 mm and no intersection gap.
-weighted images (TR/TE = 500/18 ms; flip angle = 20°; NSA = 3). Other parameters for these sequences were FOV = 60 × 60 mm, acquisition matrix = 267 × 268, reconstruction matrix = 512 × 512, section thickness = 1.0 mm and no intersection gap.
Immunocytochemistry
At 14 d after transplantation, animals were sacrificed after MRI by means of anaesthetic overdose, followed by transcardially perfusion with saline and 4% PFA in PBS. The brain was removed, post-fixed for 1–2 h and dehydrated in 20 and 30% sucrose solution sequentially. After embedding in O.C.T. (cryo-embedding medium) at −80 °C, the corresponding 10 μm slices were prepared with a Zeiss Microm HM500 microtome cryostat (Zeiss, Walldorf, Germany). Prussian blue staining alone, and Prussian blue staining and immunohistochemical double-staining were performed. For immunohistochemical staining, the slides were incubated with 0.3% H2O2 for 10 min at room temperature to eliminate endogenous peroxidase activity, and then incubated with rabbit anti-rat GFP antibody (1:400), or rabbit anti-rat nestin antibody (1:400) for 40 min. After washing with PBS, the sections were incubated with HRP-conjugated goat anti-rabbit antibody (1:200) for 30 min and were subsequently stained with DAB. After that, the sections were washed with PBS and stained with Prussian blue staining and nuclear fast red.
Statistical analysis
All results were expressed as the mean ± standard error (SD), unless otherwise noted. Statistical analysis was performed by Student's t-test for comparison between two groups, and one way ANOVA for comparison among multiple groups. Statistical analysis was performed by using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). A value of P < 0.05 was considered statistically significant.Results and discussion
Preparation and characterization of micelles
The synthesis of polymers via multi-step reactions was confirmed by 1H NMR, FTIR and GPC analyses (ESI Fig. 3–5†). PBLA was synthesized by ring-opening polymerization of BLA-NCA with BA used as initiator. The polymerization degree of PBLA was 12 according to the integral area ratio of proton from methyl and benzyl group. The characteristic shift of t-butyloxycarbonyl (Boc) proton appeared at 1.35 ppm after Boc-Lys(Boc)-OH was conjugated onto PBLA, and then disappeared when Boc was removed by trifluoroacetic acid (TFA), indicating a successful obtaining of PBLA–Lys. After cholic acid (CA) reacted onto PBLA–Lys, the major CA proton shifts appeared at 0.56, 0.80 and 0.91 ppm. The disappeared resonance peaks of benzyl group (4.96–5.14 ppm and 7.22–7.42 ppm) in 1H spectrum of PAsp(DMA)–Lys–CA2 showing a complete ammonolysis reaction take place. For the FTIR spectra, differences appeared when the ammonolysis reaction was carried out. As shown in ESI Fig. 4,† after ammonolysis reaction, compared with PBLA–Lys–CA2, the prominent ester absorption around 1740 cm−1 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) disappeared, which was attributed to the deprotection of benzyloxycarbonyl groups and resulted in the formation of amide bonds in the side chains. We successfully synthesized a new type of cationic polymeric micelles based on PAsp(DMA), conjugated with two molecules of hydrophobic CA by lysine. The molecular weight and distribution of final polymers of PAsp(DMA)–Lys–CA2 and mPEG–Lys–CA2 were detected by GPC analyses. Two polymers both showed a unimodal molecular weight distribution in their GPC chromatograms (ESI Fig. 5†), and the molecular weight was in line with the result calculated by 1H NMR (ESI Table 1†).
O) disappeared, which was attributed to the deprotection of benzyloxycarbonyl groups and resulted in the formation of amide bonds in the side chains. We successfully synthesized a new type of cationic polymeric micelles based on PAsp(DMA), conjugated with two molecules of hydrophobic CA by lysine. The molecular weight and distribution of final polymers of PAsp(DMA)–Lys–CA2 and mPEG–Lys–CA2 were detected by GPC analyses. Two polymers both showed a unimodal molecular weight distribution in their GPC chromatograms (ESI Fig. 5†), and the molecular weight was in line with the result calculated by 1H NMR (ESI Table 1†).
The morphology, particle size and surface charge of each nanoparticles preparation were subsequently characterized. TEM demonstrated that both types of micelles were spherically shaped with high-density SPIONs loaded in the nanoparticles (Fig. 2A). The C-NPs and N-NPs were all spherically shaped with a narrow distribution of sizes. The cationic and neutral micelles had a size of 64.1 ± 1.7 nm and 69.4 ± 5.8 nm, respectively. The zeta potential of C-NPs was 15.32 ± 2.74 mV, while N-NPs showed an almost neutral zeta potential of 0.1 ± 0.46 mV (Fig. 2B). Their iron content was the same (180 μg Fe per mL) and their r2 relaxivity was almost similar (0.4605 × 106 M−1 s−1 vs. 0.4629 × 106 M−1 s−1) (Fig. 2C). Fluorescence spectroscopy showed the presence of nile red and the loading content of nile red was 1.1 ± 0.1% for both types of micelles (ESI Fig. 6†). Notably, both SPIONs and nile red were successfully encapsulated in the N-NPs and C-NPs to establish a co-delivery system. Such system would enable potential tracking of grafted cells with bimodal MRI and optical imaging simultaneously.
 | ||
| Fig. 2 Characterization of polymeric micelles. (A) Transmission electronic microscopy (TEM) images demonstrated that both cationic PAsp(DMA)–Lys–CA2 (C-NPs) and neutral PEG–Lys–CA2 (N-NPs) were all spherically shaped with SPIONs (high density) loaded in the nanoparticles, scale bar = 50 nm. (B) Graphs show the size and zeta potential of C-NPs and N-NPs. (C) Graphs show the r2 relaxivity of C-NPs and N-NPs. | ||
Large iron oxide particles, over 200 nm in diameter, possess much greater amounts of iron per particle, thus theoretically few, even single particle is required to label per cell. However, large particles suffer from gravitational sedimentation that could impact cell labeling efficiency, especially for non-phagocytic suspending cells.18 Moreover, commercially available SPIONs used as MRI contrast agents usually have a size of 120–180 nm, but tend to be degraded by intracellular enzymes and acids, and diluted with rapid cell division.9 Therefore, nanoparticles used for MRI tracking of stem cells need to be engineered with a “larger” number of iron oxide but a “smaller” size, as such the iron oxide nanoparticles will be distributed enough in the off-spring cells after cell proliferation.9,18 It was also shown that nonphagocytic cells take up spherical nanoparticles with a size between 20 and 50 nm at the highest rate.19,20 Collectively, the C-NPs with relatively small size and high payload of SPIONs in our study are well suitable for high efficient labeling of stem cells.
The cellular uptake of micelles
To determine the effect of surface charge on the cellular uptake, Prussian blue staining was performed after GFP-C17.2 NSCs incubated with N-NPs or C-NPs at different concentration (10 μg mL−1 and 20 μg mL−1) for various incubation times (2, 4 and 6 h). As shown in Fig. 3, the cells labeled with C-NPs demonstrated obvious intracellular accumulation of SPIONs, as many blue staining particles were observed in the cytoplasm (Fig. 3A and B). The intracellular accumulation of iron became more obvious with the increased micelles concentration and incubation time. However, no blue staining particles could be detected in the cells labeled with N-NPs (Fig. 3C and D). No obvious morphological changes were found between cells labeled with C-NPs and with N-NPs. | ||
| Fig. 3 The effect of surface charges on cellular uptake of polymeric micelles. (A and B) Prussian blue staining micrographs show that GFP-C17.2 NSCs incubated with cationic PAsp(DMA)–Lys–CA2 (C-NPs) have numerous blue-stained particles inside the cytoplasm compared with cells labelled with neutral PEG–Lys–CA2 (N-NPs). For C-NPs, no more blue staining nanoparticles could be observed when the labeling concentration reached 10 μg mL−1 or the incubation time extended to 4 h. (C and D) No blue staining particles could be detected in the cells labeled with N-NPs. Scale bar = 20 μm. | ||
The optimal labeling conditions with the micelles were assessed by in vitro MRI. As shown in Fig. 4, the signal intensity of GFP-C17.2 NSCs labeled with C-NPs exhibited a remarkable decline on MRI. Furthermore, exposure time longer than 4 h or micelles iron concentration greater than 10 μg mL−1 did not improve the cellular uptake. However, no significant signal change was observed between cells treated with N-NPs and unlabeled cells. The labeling efficiency of neutral and cationic micelles was analyzed at pre-determined optimal cell labeling conditions (4 h incubation and iron concentration of 10 μg mL−1). Flow cytometry analysis revealed the labeling efficiency of C-NPs reached up to 99.27 ± 0.65%. However, the N-NPs had a labeling efficiency merely of 8.68 ± 4.56% (Fig. 5).
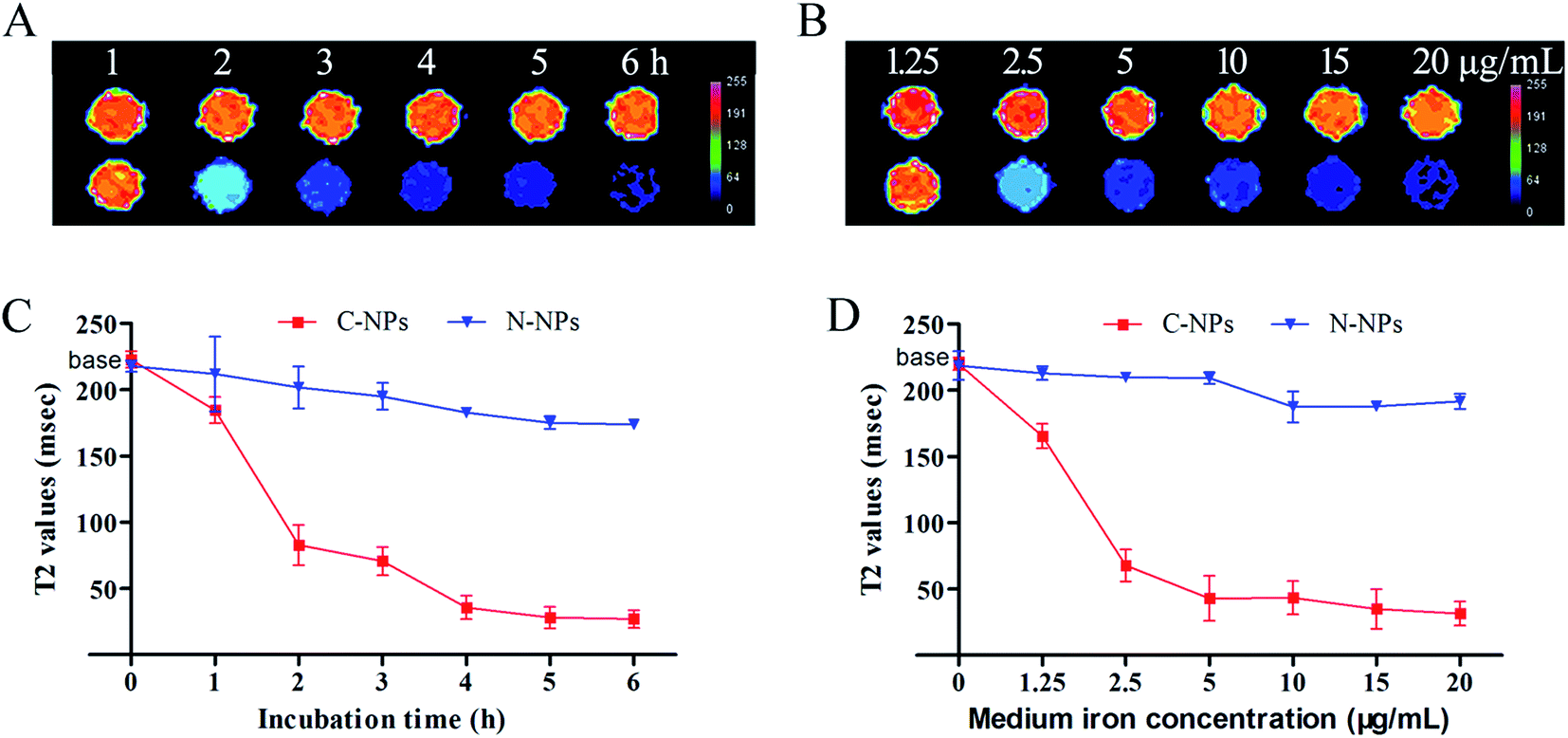 | ||
| Fig. 4 The optimal labeling conditions. (A) T2-Maps of GFP-C17.2 NSCs labeled with cationic PAsp(DMA)–Lys–CA2 (C-NPs) and untreated cells. (B) T2-Maps of GFP-C17.2 NSCs labeled with neutral PEG–Lys–CA2 (N-NPs) and untreated cells. (C) Graphs show the T2 values of GFP-C17.2 NSCs labeled with C-NPs and N-NPs in different incubation times. (D) Graphs show the T2 values of GFP-C17.2 NSCs labeled with C-NPs and N-NPs in different micelle concentration. | ||
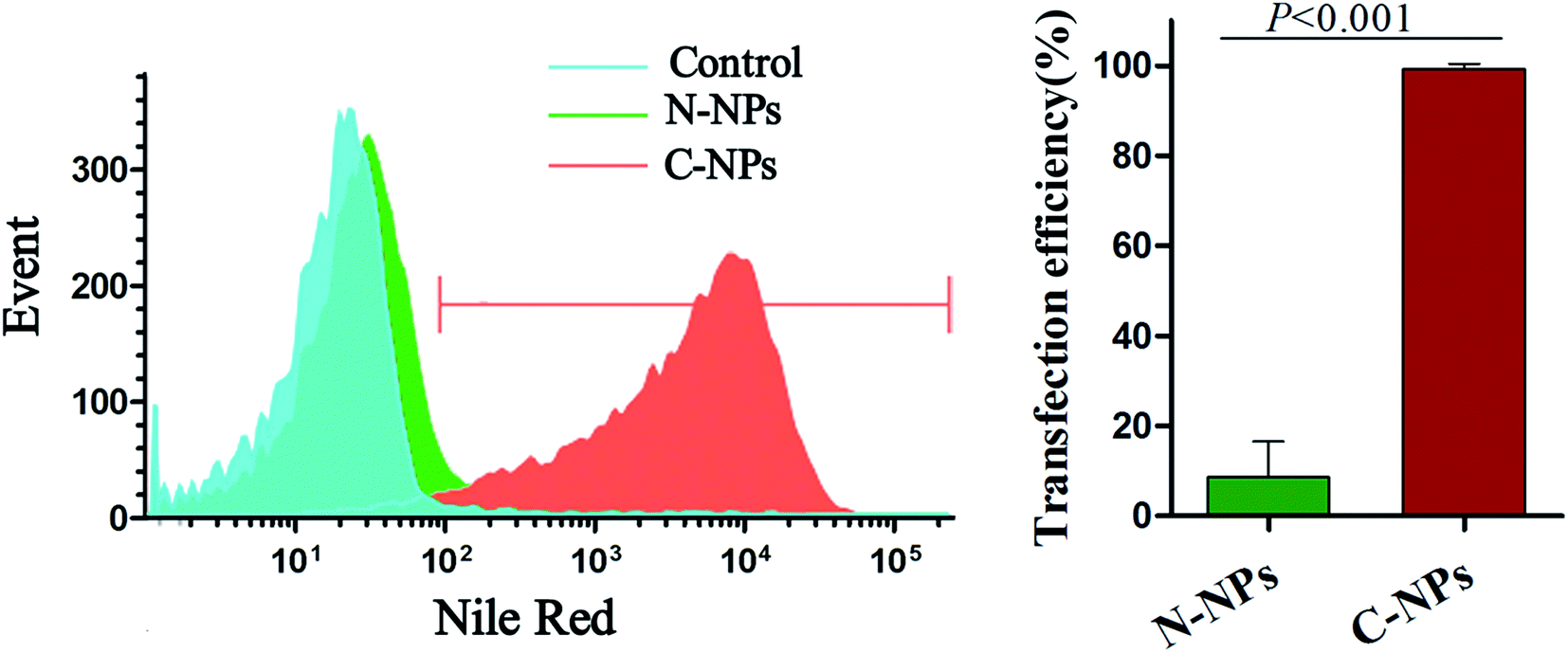 | ||
| Fig. 5 Flow cytometry of cell labeling efficiency. Flow cytometry shows a significant difference in the labeling efficiency between cationic PAsp(DMA)–Lys–CA2 (C-NPs) and neutral PEG–Lys–CA2 (N-NPs). | ||
To further detect the kinetics of cellular uptake, GFP-C17.2 NSCs were incubated with N-NPs and C-NPs for 1 h up to 6 h at iron concentration of 10 μg mL−1 and then observed directly using fluorescence microscope (Fig. 6). In the cells incubated with C-NPs, red particles (nile red) initially accumulated around the cell membrane, then they gradually aggregated in the cytoplasm within the first 4 h. After 4 h, the number of red particles no longer increased. In contrast, only a few red particles were found in the membrane and to enter the cells labeled with N-NPs during the whole labeling procedure (ESI Fig. 7†). TEM showed that many iron oxide particles were present in the lysosomes in the cells labeled by C-NPs (Fig. 7A and B). However, no iron oxide particles could be observed in cells labeled by N-NPs (Fig. 7C).
 | ||
| Fig. 6 The dynamics of cellular uptake of cationic polymeric micelles. Fluorescence micrographs show the red particles (nile red) in the membrane of GFP-C17.2 NSCs 1 h after incubation with PAsp(DMA)–Lys–CA2 (C-NPs), which are increasingly accumulated in the cytoplasm within the first 4 h. After 4 h, the number of red particles no longer increased. Scale bar = 50 μm. | ||
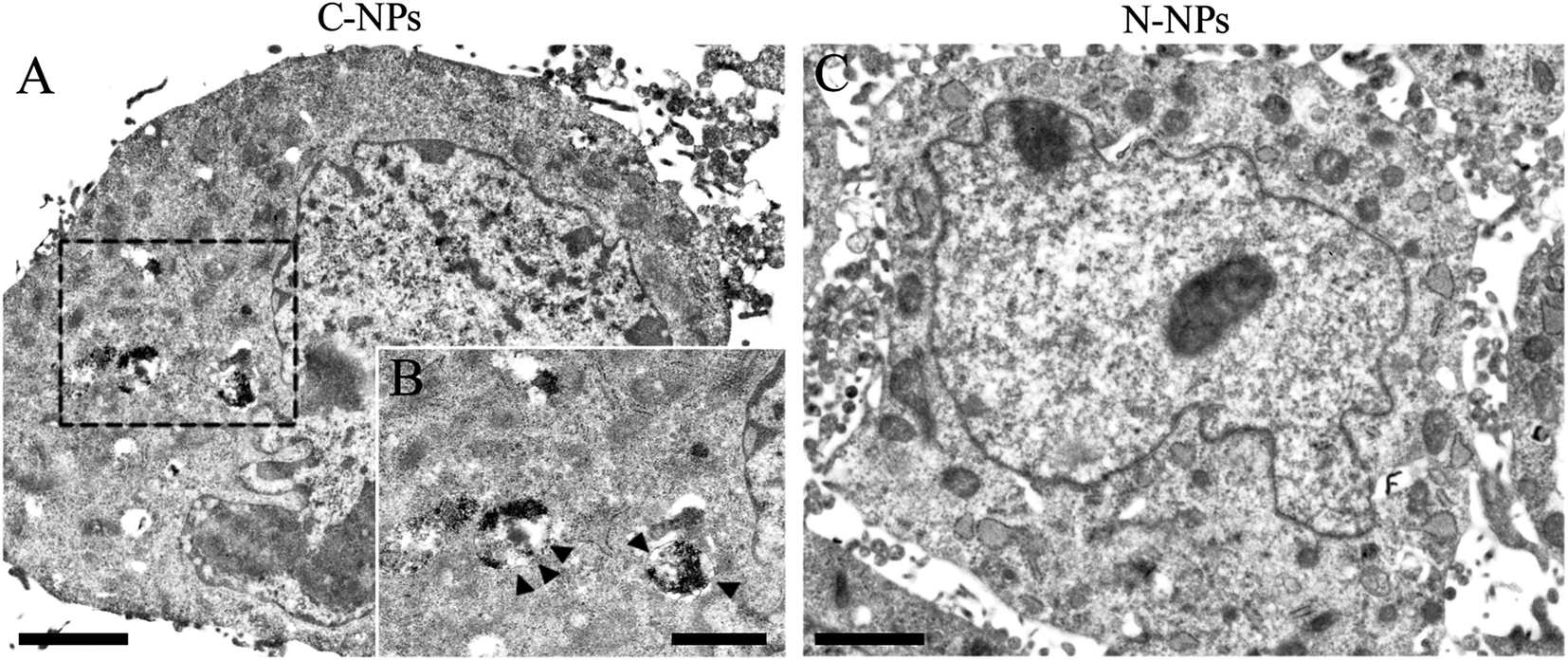 | ||
| Fig. 7 Intracellular distribution of polymeric micelles. (A) Transmission electron microscopy images show many iron oxide particles in the cytoplasm of cells labeled with cationic PAsp(DMA)–Lys–CA2 (C-NPs). (B) The inserted, magnified view shows iron oxide particles in the lysosomes (black arrow head). (C) No iron oxide particles were observed in cells labeled with neutral PEG–Lys–CA2 (N-NPs). Scale bar = 2 μm in (A) and (C); scale bar = 1 μm in (B). | ||
Surface charge is a very important factor not only to influence the stability and dispersion of nanoparticles, and the efficiency and mechanism of cellular uptake, but also to significantly determine the cytotoxicity of nanoparticles.19,21 Studies on the effects of charge density and the type of charge (positive, negative) of nanoparticles in non-phagocytic cell labeling showed that cationic surface charge correlates with higher cellular uptake than negatively charged or neutral ones.19 A possible explanation for this phenomenon is the electrostatic interaction between the positive charges on the surface of nanoparticles and the negative charges on the cell membrane from sialic acids and phospholipid head groups, which facilitates the subsequent endosomolytic uptake of nanoparticles.19,22 Further studies showed that the cellular uptake of nanoparticles was accordingly increased with the increasing of positive charge density.23,24 However, high positive charge also means high cytotoxicity, because positively charged nanoparticles could cause plasma-membrane disruption to a greater extent either directly or by detachment of adsorbed polymers (e.g., polyethylenimine).19 In our study, the positively charged C-NPs exhibited a high labeling efficiently for C17.2 NSCs as compared with neutral N-NPs, which was consistent with previous results.21,23 However, C-NPs in our study had a weak positive charge that could ensure a low cytotoxicity. In addition, compared with modulating the particle surface properties through post-synthesis modification, the development of cationic micelles in our study by the self-assembling of pre-functionalized subunits would eliminate the need of post-particle modification, and may have less batch-to-batch variation.25
Cell viability
To assess cytotoxicity, CCK-8 and apoptosis assay were performed after GFP-C17.2 NSCs were labeled. No significant difference of apoptosis was found among cells labeled with C-NPs, N-NPs and untreated control cells 24 h after incubation (Fig. 8A). Moreover, no significant decrease of viability was observed in cells labeled with C-NPs compared with untreated cells and cell labeled with N-NPs, even until 3 d after labeling (Fig. 8B). In vitro differentiation analysis showed that neuronal differentiation capability was not affected in cells labeled with C-NPs and N-NPs (Fig. 8C). | ||
| Fig. 8 The apoptosis, viability and differentiation capability of neural stem cells labeled with polymeric micelles. (A) Graphs show no significant difference in apoptosis rate among cells labeled with cationic PAsp(DMA)–Lys–CA2 (C-NPs), neutral PEG–Lys–CA2 (N-NPs) and untreated cells (control) at 24 h after incubation. (B) Graphs show no significant difference in viability between among cells labeled with C-NPs, N-NPs and untreated cells up to 3 d after labeling (P > 0.05). (C) Immunofluorescence micrographs show neuronal differentiation of cells labeled with C-NPs, N-NPs and untreated cells (control). Scale bar = 50 μm. | ||
SPIONs are the most widely used imaging labels in MRI tracking of stem cells. The major shortcoming of SPIONs for stem cell labeling is that cationic transfection reagents, such as Superfect, DOTAP, Lipofectamin and poly-L-lysine (PLL) are needed to help cell uptake of SPIONs. These transfection reagents are toxic at high concentration and have not been approved for clinical use.9 Polymeric nanoparticles are promising alternative candidates.11 After delivering the encapsulated medication to target tissues, polymeric nanoparticles should be degraded into biologically acceptable compounds by hydrolysis. As such, no toxicity would be induced by the carriers themselves. This is especially important in stem cell-based regenerative medicine, because stem cells are always vulnerable for labeling procedures and should retain a state of stemness for future therapeutic application. Ideally, the polymers can be degraded into lactic or glycolic acids, which are eventually reduced to carbon dioxide and water by the Krebs cycle.9 However, among the existing nanoparticles investigated for delivery of SPIONs, only a few have been approved by the Food and Drug Administration (FDA). This may be attributed to the potential toxicity, poor biocompatibility, low drug loading capacity, or batch-to-batch inconsistencies in the bio-physicochemical properties.26 Biodegradable polymers with improved characteristics to maximize delivery efficiency while minimize the side effects, are in tremendous demand.
As building blocks for the micelles, lysine, PAsp(DMA) and cholic acid, are normal compounds found in human body. They are biocompatible and biodegradable, thus have favorable characteristics for drug entrapment and delivery.27 In addition, it is generally considered that iron oxide is non-toxic to stem cells, since it can be degraded and eventually utilized by cells via the physical iron metabolism pathway.28,29 The high biocompatible formulation of C-NPs would offer benefit to cell viability. Indeed, the proliferation and apoptosis of the stem cells were not affected after labeled by the cationic SPIONs and nile red co-loaded PAsp(DMA)–CA2. Moreover, the neuronal differentiation ability of C17.2, which was very important for the stem cell therapy, was also not affected. These results revealed the biocompatibility and safety of the cationic polymeric micelles in our study.
In vivo MRI tracking and histological assessment of grafted, labeled NSCs in stroke
After GFP-C17.2 NSCs that were pre-labeled with C-NPs or N-NPs were implanted to the striatum ipsilateral to the ischemic hemisphere, in vivo distribution of these cells was assessed by using a 3.0-T clinical MRI. The cells labeled with C-NPs appeared as marked hypointense signal area on -weighted images (Fig. 9) and less pronounced hypointense signal area on T2-weighted images (ESI Fig. 8†). These hypointense signal area slowly reduced over time. However, only a black needle tract could be observed in the striatum of animals treated with N-NPs-labeled cells. The infarction showed hyperintense signal on T2-weighted images, which gradually decline in the size in animals treated with C-NPs- or N-NPs-labeled NSCs.
-weighted images (Fig. 9) and less pronounced hypointense signal area on T2-weighted images (ESI Fig. 8†). These hypointense signal area slowly reduced over time. However, only a black needle tract could be observed in the striatum of animals treated with N-NPs-labeled cells. The infarction showed hyperintense signal on T2-weighted images, which gradually decline in the size in animals treated with C-NPs- or N-NPs-labeled NSCs.
 | ||
Fig. 9 In vivo MRI tracking of grafted, labeled NSCs. (A)  -Weighted images show the hypointense signal of GFP-C17.2 NSCs labeled with cationic PAsp(DMA)–Lys–CA2 (C-NPs) in the striatum ipsilateral to the infarcted hemisphere. (B) Only a black, thin signal representing needle track appears in the striatum in animals treated with cells pre-labeled with neutral PEG–Lys–CA2 (N-NPs). -Weighted images show the hypointense signal of GFP-C17.2 NSCs labeled with cationic PAsp(DMA)–Lys–CA2 (C-NPs) in the striatum ipsilateral to the infarcted hemisphere. (B) Only a black, thin signal representing needle track appears in the striatum in animals treated with cells pre-labeled with neutral PEG–Lys–CA2 (N-NPs). | ||
Prussian blue staining showed numerous blue iron deposits in the striatum of animals treated with C-NPs-labeled NSCs (Fig. 10A). No such blue particles were observed at the same region in the animals treated with N-NPs-labeled NSCs (Fig. 10D). Double Prussian blue staining and GFP immunostaining confirmed the co-localization between SPIONs and transplanted GFP-NSCs. In animals treated with C-NPs-labeled NSCs, almost all SPIONs were located inside living cells (Fig. 10B and E). Immunostaining for nestin demonstrated the survival of GFP-C17.2 NSCs in the transplantation site (Fig. 10C and F). No malignant transformation of grafted NSCs was found.
 | ||
| Fig. 10 Histopathology assessment of grafted, labeled cells. (A) Prussian blue staining micrographs show that blue iron particles deposited in the striatum in an animal treated with cationic PAsp(DMA)–Lys–CA2 (C-NPs)-labeled GFP-C17.2 NSCs. (B) Double Prussian blue staining and GFP immunostaining micrographs reveal the co-localization of C-NPs with GFP-positive cells. (C) Nestin immunostaining micrographs show positive staining of GFP-C17.2 NSCs in the striatum in an animal treated neutral C-NPs-labeled GFP-C17.2 NSCs. (D) Prussian blue staining micrographs show no blue iron particles in the striatum in an animal treated with neutral PEG–Lys–CA2 (N-NPs)-labeled GFP-C17.2 NSCs. (E) Double Prussian blue staining and GFP immunostaining micrographs reveal the presence of GFP-positive cells but absence of iron particles in the striatum in an animal treated with N-NPs-labeled GFP-C17.2 NSCs. (F) Nestin immunostaining micrographs show positive staining of GFP-C17.2 NSCs in the striatum in an animal treated with N-NPs-labeled GFP-C17.2 NSCs. Scale bar = 500 μm in (A) and (D); scale bar = 50 μm in (B–E). | ||
Upon the encapsulation of SPIONs, the cationic polymeric micelles showed a high imaging sensitivity of cellular MRI for detecting grafted cells. The distribution of labeled NSCs was clearly tracked by MRI after their transplantation in the infarcted hemisphere. The characteristic hypointense signals from the cell mass on MRI were well-matched with histologically detected distribution and surviving of implanted cells. Notably, the polymeric micelles we designed encapsulated SPIONs inside the hydrophobic core, where SPIONs formed a closed packing structure, i.e. clustering, which would result in stronger T2 effects than single particle-containing micelles at the same iron concentration.30,31 This unique structure provides opportunities for the design and development of ultrasensitive MR probes for in vivo tracking of the dynamics of therapeutic cells.
Conclusion
In conclusion, we successfully synthesized a novel class of cationic polymeric nanoparticles consisted of PAsp(DMA) conjugated with two molecules of hydrophobic CA by lysine. This biocompatible polymeric micelle showed a variety of favorable properties for stem cell labeling, such as non-toxicity composition, spherical shape, weak positive charge, relatively small size while high payload of image labels. With a high efficient and safe cell labeling, these cationic polymeric micelles can achieve a reliable MRI tracking of stem cells in acute ischemic stroke in vivo, while without any detrimental effect on cell viability, proliferation, apoptosis and differentiation. This class of cationic polymeric micelles are highly translatable and can be used as a versatile nanoplatform for in vivo tracking of therapeutic stem cells in regenerative medicine.Acknowledgements
This work is supported by National Natural Science Foundation (Grant No. 81371607, 81571739, and 51225305) and National Basic Research Program of China (Grant No. 2015CB755500) and Natural Science Foundation of Guangdong Province, China (Grant No. 2014A030312018) and Elite Young Scholars Program of Sun Yat-Sen Memorial Hospital (J201403).References
- L. Hao, Z. Zou, H. Tian, Y. Zhang, H. Zhou and L. Liu, BioMed Res. Int., 2014, 2014, 468748 Search PubMed.
- A. Arvidsson, T. Collin, D. Kirik, Z. Kokaia and O. Lindvall, Nat. Med., 2002, 8, 963–970 CrossRef CAS PubMed.
- A. Björklund and O. Lindvall, Nat. Neurosci., 2000, 3, 537–544 CrossRef PubMed.
- S. Haas, N. Weidner and J. Winkler, Curr. Opin. Neurol., 2005, 18, 59–64 CrossRef PubMed.
- S. U. Kim, J. Neurosci. Res., 2009, 87, 2183–2200 CrossRef CAS PubMed.
- C. M. Long and J. W. Bulte, Expert Opin. Biol. Ther., 2009, 9, 293–306 CrossRef CAS PubMed.
- J. Trekker, C. Leten, T. Struys, V. V. Lazenka, B. Argibay, L. Micholt, I. Lambrichts, R. W. Van, L. Lagae and U. Himmelreich, Biomaterials, 2014, 35, 1627–1635 CrossRef CAS PubMed.
- J. W. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS PubMed.
- M. Mahmoudi, H. Hosseinkhani, M. Hosseinkhani, S. Boutry, A. Simchi, W. S. Journeay, K. Subramani and S. Laurent, Chem. Rev., 2011, 111, 253–280 CrossRef CAS PubMed.
- K. Andreas, R. Georgieva, M. Ladwig, S. Mueller, M. Notter, M. Sittinger and J. Ringe, Biomaterials, 2012, 33, 4515–4525 CrossRef CAS PubMed.
- L. Zhao, N. Li, K. Wang, C. Shi, L. Zhang and Y. Luan, Biomaterials, 2014, 35, 1284–1301 CrossRef CAS PubMed.
- X. Wen, Y. Wang, F. Zhang, X. Zhang, L. Lu, X. Shuai and J. Shen, Biomaterials, 2014, 35, 4627–4635 CrossRef CAS PubMed.
- Y. Wang, H. Xiao, J. Fang, X. Yu, Z. Su, D. Cheng and X. Shuai, Chem. Commun., 2015, 52, 1194–1197 RSC.
- J. Luo, K. Xiao, Y. Li, J. S. Lee, L. Shi, Y. H. Tan, L. Xing, R. H. Cheng, G. Y. Liu and K. S. Lam, Bioconjugate Chem., 2010, 21, 1216–1224 CrossRef CAS PubMed.
- R. L. Rietze and B. A. Reynolds, Methods Enzymol., 2006, 419, 3–23 CAS.
- E. Z. Longa, P. R. Weinstein, S. Carlson and R. Cummins, Stroke, 1989, 20, 84–91 CrossRef CAS PubMed.
- M. Kameda, T. Shingo, K. Takahashi, K. Muraoka, K. Kurozumi, T. Yasuhara, T. Maruo, T. Tsuboi, T. Uozumi and T. Matsui, Eur. J. Neurosci., 2007, 26, 1462–1478 CrossRef CAS PubMed.
- D. L. Thorek and A. Tsourkas, Biomaterials, 2008, 29, 3583–3590 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- X. Duan, Y. Wang, F. Zhang, L. Lu, M. Cao, B. Lin, X. Zhang, J. Mao, X. Shuai and J. Shen, J. Biomed. Nanotechnol., 2016, 12, 2112–2124 CrossRef CAS.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32, 3435–3446 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- F. Gu, L. Zhang, B. A. Teply, N. Mann, A. Wang, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2586–2591 CrossRef CAS PubMed.
- K. Xiao, J. Luo, W. L. Fowler, Y. Li, J. S. Lee, L. Xing, R. H. Cheng, L. Wang and K. S. Lam, Biomaterials, 2009, 30, 6006–6016 CrossRef CAS PubMed.
- J. E. Gautrot and X. X. Zhu, J. Biomater. Sci., Polym. Ed., 2006, 17, 1123–1139 CrossRef CAS.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lan, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- C. M. Long and J. W. Bulte, Expert Opin. Biol. Ther., 2009, 9, 293–306 CrossRef CAS PubMed.
- Q. Guo, L. Kuang, H. Cao, W. Li and J. Wei, Colloids Surf., B, 2015, 136, 687–693 CrossRef CAS PubMed.
- H. Ai, C. Flask, B. Weinberg, X. T. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. Gao, Adv. Mater., 2005, 17, 1949–1952 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00345e |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |