
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iminodiacetic acid functionalized magnetic nanoparticles for speciation of Cr(III) and Cr(VI) followed by graphite furnace atomic absorption spectrometry detection†
Wenye Wei,
Bingshan Zhao,
Man He *,
Beibei Chen and
Bin Hu
*,
Beibei Chen and
Bin Hu
Key Laboratory of Analytical Chemistry for Biology and Medicine, Ministry of Education, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: heman@whu.edu.cn; Fax: +86 27 68754067; Tel: +86 27 68752162
First published on 26th January 2017
Abstract
In this work, iminodiacetic acid (IDA) functionalized magnetic nanoparticles (Fe3O4@SiO2@IDA) were prepared and the adsorption behavior of Cr(III)/(VI) on them was investigated. It was found that both Cr(III) and Cr(VI) could be quantitatively retained on Fe3O4@SiO2@IDA in the pH range of 2.5–3.5, while only Cr(III) could be adsorbed in the pH range of 4.0–9.0. Based on this, a new approach of magnetic solid phase extraction (MSPE) combined with graphite furnace atomic absorption spectrometry (GFAAS) was proposed for the speciation of trace Cr(III) and Cr(VI). Total Cr and Cr(III) were adsorbed on Fe3O4@SiO2@IDA under pH 3 and 5, respectively, followed by elution with 1.5 mol L−1 HNO3. And the concentration of Cr(VI) was calculated by subtracting Cr(III) from the total Cr. Factors affecting MSPE (pH of sample solution, concentration and volume of desorption reagent, desorption time, extraction time, sample volume) were investigated and the optimized conditions were achieved. With an enrichment factor of 100-fold, the detection limits of the proposed method were 9.1 and 12.8 ng L−1 for Cr(III) and total Cr with the relative standard deviations (RSDs) of 4.8 and 5.2% (c = 50 ng L−1, n = 7), respectively. In order to validate the accuracy of the proposed method, a certified reference material of GSBZ50009-88 environmental water was analyzed, and the determined value was in good agreement with the certified value. The proposed MSPE-GFAAS method is simple, fast, selective and sensitive for the speciation of trace Cr without any oxidation or reduction process. It has been applied in the speciation of trace Cr in natural water samples with satisfactory results.
1. Introduction
Chromium (Cr) is a heavy metal that have been widely used in industry, and it is also an essential trace element for human beings. It is worth noting that the toxicological and biological properties of Cr depend on its chemical form. Generally, Cr(III) and Cr(VI) are the primary existing forms of Cr in environmental water.1 Cr(III) is biologically essential to mammals as it maintains an effective glucose, lipid, and protein metabolism.2 In contrast, Cr(VI) is highly toxic as it can diffuse as CrO42− or HCrO4− through cell membranes and then oxidize biological species such as DNA and some proteins.3 Even at relatively low concentrations, Cr(VI) can still cause harm to the human body because of its high toxicity. Because of their wide application in electroplating, chemical engineering, textiles, oil refining, steelworks and other industries,4 Cr species are easily discharged into the environment and cause pollution especially water contamination. The United State Environmental Protection Agency (US EPA) has set the permissible limit of Cr(VI) and total Cr in drinking water at 50 and 100 μg L−1, respectively. However, World Health Organization (WHO) consider that the guideline value of 50 μg L−1 of Cr(VI) is too high, compared with its high risk of carcinogenicity.5 Consequently, developing accurate and sensitive methods for the speciation of trace Cr in environmental water samples is urgently required.The speciation of Cr in environmental waters could be performed by combining high efficient separation techniques with specific elemental detection techniques.6 Several analytical techniques including flame atomic absorption spectrometry (FAAS),7 graphite furnace atomic absorption spectrometry (GFAAS),8 inductively coupled plasma optical emission spectrometry (ICP-OES)9 and inductively coupled plasma mass spectrometry (ICP-MS)10 have been used for the determination of Cr in different matrixes. Among these elemental detection techniques, GFAAS has the advantages of high sensitivity, low cost, low sample consumption, and is suitable to single elemental analysis at trace level.11 The separation techniques used for Cr speciation include chromatographic approaches such as liquid chromatography,12 ion chromatography13 and capillary electrophoresis14 and non-chromatographic separation technologies such as coprecipitation,15 ion exchange,16 liquid phase microextraction,17 cloud point extraction18 and solid phase extraction (SPE).19 For a simple elemental speciation, especially for a different valence state of Cr, SPE is the most commonly used separation technique, featuring with high separation efficiency, high enrichment factors, low consumption of organic solvent, flexibility and plenty of sorbents available. Compared with chromatographic approaches, SPE is simple and low-cost, and it can remove the matrix effectively and preconcentrate target species simultaneously.
Various SPE materials such as functional polymer,20 resin,3 carbon materials,21 metal oxide,22 imprinted materials,23 magnetic materials24 have been used for separation and preconcentration of Cr species. Due to the magnetism, magnetic materials can be separated from aqueous samples rapidly by using a magnet instead of high-speed centrifugation or filtration. Magnetic solid phase extraction (MSPE)25 exhibits high enrichment factors, fast magnetic separation and easy batch operation, and is particularly suitable for separation and preconcentration of target analytes in large-volume liquid samples. It has been employed for the speciation of Cr in environmental water samples. Fe3O4@ZrO2 nanoparticles (NPs)24 or TritonX-114 modified alumina-coated magnetic nanoparticles (MNPs)26 have been employed for the speciation of Cr(III) and Cr(VI). While both of these two kinds of MNPs were selective towards Cr(III) rather than Cr(VI), so a reduction step was needed to obtain total Cr concentration and the amount of Cr(VI) was obtained by subtracting the amount of Cr(III) from total Cr. Diniz et al.27 achieved a selective preconcentration of Cr(VI) onto mesoporous amino-functionalized Fe3O4/SiO2 NPs, and then the supernatant containing un-adsorbed Cr(III) was subjected to cloud point extraction. Although such a combination of two extraction techniques requires no extra oxidation/reduction, the two-step operation was complex and time-consuming. Hence, it is necessary to develop a practical and efficient MSPE method for Cr species analysis.
Iminodiacetic acid (IDA) can form stable complexes with many metal ions and –COOH and –NH– are the functional groups.28–30 The IDA-chelating resin (Muromac A-1) has been applied for the selective separation of Cr(III),5,31 which is attributable to the three coordination sites of IDA. Considering IDA contains both carboxyl group and imino group, it is expected to have selectivity for Cr(III)/Cr(VI). In this work, IDA functionalized magnetic nanoparticles (Fe3O4@SiO2@IDA) were synthesized and the adsorption behavior of Cr(III)/(VI) on Fe3O4@SiO2@IDA were investigated. Based on the adsorption difference of Cr(III) and Cr(VI) on Fe3O4@SiO2@IDA, a novel method of MSPE-GFAAS was proposed for the speciation of trace Cr(III)/(VI) in the environmental water without any oxidation or reduction process. The parameters affecting the extraction and desorption of target Cr species, such as sample pH, desorption time, extraction time and sample volume were investigated and the optimized conditions were established. The developed method has been validated by the speciation of Cr(III) and Cr(VI) in a certified reference material (GSBZ50009-88, environmental water) and real water samples.
2. Experimental
2.1 Instrumentation
Constant temperature heating magnetic stirrer (DF-101S, Shanghai, China), electromotion stirrer (RW 20 digital, IKA, Germany), vacuum drying oven (DZG-6020, Shanghai, China) were used in the synthesis procedure. The self-prepared Fe3O4@SiO2@IDA was characterized by Fourier transform infrared spectroscopy (Nicolet iS10, Thermo, USA), transmission electron micrograph (JEOL, JEM-2010 HT, Tokyo, Japan) and vibrating sample magnetometer (PPMS-9T, USA). The pH values were measured by a Mettler Toledo 320-S pH meter (Shanghai, China) supplied with a combined electrode. Numerical control ultrasonic apparatus (KQ5200DE, Kunshan, Jiangsu, China) was used for ultrasonic (40 kHz and 100 watts ultrasonic power) assisted extraction and desorption of target species. An Nd-Fe-B magnet (8.0 mm × 6.0 mm × 1.6 mm) was used for magnetic separation. Determination of trace Cr was carried out by GFAAS (Z-2000, Hitachi, Japan) and the operation conditions are summarized in Table S1.†2.2 Reagents and materials
Tetraethoxysilane (TEOS) and 3-glycidoxypropyltrimethoxysilane (GLYMO) were purchased from Organic Silicon Material Company of Wuhan University (Wuhan, China). IDA, FeCl3·6H2O, FeCl2·4H2O were purchased from the First Reagent Factory (Shanghai, China). All reagents were of specpure or at least of analytical reagent grade. Stock solution of Cr(III) and Cr(VI) were prepared by dissolving corresponding amount of Cr(NO3)3·9H2O and K2CrO4 in 2% (v/v) HNO3. Working solutions were prepared by stepwise dilution with 2% (v/v) HNO3. The high purity deionized water (18.2 MΩ cm) obtained from a Milli-Q water purification system (Millipore, Bedford, MA, USA) was used throughout this work. 0.1 mol L−1 HNO3 and 0.1 mol L−1 ammonia solution were used to adjust the pH of the solution.All the laboratory wares were made of polyethylene or glass material and thoroughly cleaned by soaking in 10% (v/v) nitric acid for at least 24 h. All these acid-washed laboratory wares were rinsed three times with high purity deionized water prior to use. All stock standard solutions were stored in polyethylene bottles and maintained at 4 °C in refrigerator.
2.3 Preparation of Fe3O4@SiO2@IDA
The procedure for the preparation of Fe3O4@SiO2@IDA was referred to ref. 30 and 32 with minor modifications: 3.5 g IDA was dissolved in 1 mol L−1 sodium hydroxide aqueous solution (50 mL) and the solution was adjusted to pH 11. After magnetic stirring for 1.0 h in an ice-bath, 2.0 g GLYMO was added dropwise with magnetic stirring for about 0.5 h. The reaction system was then slowly heated to 65 °C, maintained for 6 h and cooled down to room temperature. Finally, the obtained IDA-derived silane coupling agent (GLYMO–IDA) solution was adjusted to pH 6 with concentrated HCl.Fe3O4 MNPs were synthesized by the conventional coprecipitation method.33 Briefly, 11.68 g FeCl3·6H2O and 4.30 g FeCl2·4H2O were dissolved in 200 mL high purity deionized water and heated to 85 °C under nitrogen gas. Then 25 mL ammonia solution (15 mol L−1) was added with vigorous stirring for 0.5 h. The obtained Fe3O4 MNPs were washed several times with high purity deionized water and ethanol, and stored in 100 mL ethanol at a concentration of 40 mg mL−1. Then, 50 mL magnetic fluid prepared above was homogeneously dispersed in a mixture of ethanol (70 mL) and high purity deionized water (20 mL) by sonication for 15 min, followed by the addition of 7 mL ammonia solution (15 mol L−1) and 8 mL TEOS sequentially. After being stirred at room temperature for 12 h, silica coated MNPs (Fe3O4@SiO2) were obtained and rinsed with high purity deionized water and ethanol for several times. Then the washed Fe3O4@SiO2 were stored in 200 mL ethanol at a concentration of 20 mg mL−1. 50 mL ethanol fluid containing 1 g Fe3O4@SiO2 were mixed with 25 mL water and 25 mL ethanol, then sonicated for 15 min, followed by the addition of 2 mL ammonia solution (15 mol L−1). Then, 50 mL of GLYMO–IDA solution was added dropwise to the dispersion with continuous stirring. After reaction at room temperature for 12 h, the product of Fe3O4@SiO2@IDA was collected with a magnet and washed repeatedly with ethanol and water and finally dried in vacuum at 60 °C.
2.4 Sorption experiments
The adsorption capacity of Cr(III) and Cr(VI) on Fe3O4@SiO2@IDA were investigated, respectively. 10 mL of aqueous solution containing Cr(III) or Cr(VI) with a series of concentrations of 1–50 mg L−1 were adjusted to pH 5 and 3, respectively. Then 10 mg Fe3O4@SiO2@IDA was added and dispersed by ultrasonication for 10 min. All the sorption experiments were carried out in triplicate. After adsorption and magnetic separation, the supernatant was analyzed by GFAAS. The adsorption capacity (Q, mg g−1) was calculated using the following equations: Q = (Ci − Cf)V/m; where Ci and Cf represent the initial and final concentration of Cr in the solution (mg L−1), m is the mass of Fe3O4@SiO2@IDA (g), V is the volume of sample solution (L).2.5 Sample preparation
Water samples were collected from the East Lake and Yangtze River (Wuhan, China) respectively and stored at 4 °C after acidification with HNO3. Immediately after sampling, all water samples were filtered through a 0.45 μm membrane and stored in pre-cleaned polypropylene bottles in a refrigerator at 4 °C. Certified Reference Material of GSBZ50009-88 environment water (Institute for Reference Materials of State Environmental Protection Administration, Beijing, China) was diluted by 5000-fold. All the water samples were subjected to the proposed MSPE procedure and subsequent GFAAS detection. High purity deionized water without sample addition was employed as the blank and subjected to the same procedure described above. All the tests were carried out in triplicate.2.6 Magnetic solid phase extraction procedure
50 mL of the sample solution containing Cr(III) or total Cr was transferred into a 50 mL beaker, and adjusted to pH 5 and 3 with 0.1 mol L−1 HNO3 and 0.1 mol L−1 ammonia solution, respectively. In the extraction step, 10 mg of Fe3O4@SiO2@IDA was added and dispersed by ultrasonication for 10 min at room temperature, then isolated from the suspension with an Nd-Fe-B strong magnet and washed with 1 mL high purity water. In the desorption step, 0.5 mL of 1.5 mol L−1 HNO3 was added to desorb the metal ions from the sorbent under ultrasonication for 30 min. Finally, the desorption solution was directly introduced into GFAAS for Cr determination.3. Results and discussion
3.1 Characterization
The FT-IR spectra of Fe3O4@SiO2, IDA and the as-prepared Fe3O4@SiO2@IDA are shown in Fig. 1. The peaks at 573 cm−1, 571 cm−1 and 1088 cm−1, 1083 cm−1 are the stretching vibration of Fe–O bond and Si–O bond in Fe3O4@SiO2 and Fe3O4@SiO2@IDA, respectively, indicating that silicone were coated on the Fe3O4 particles successfully. The characteristic absorption bands of C–O symmetric stretching vibration (–COOH) at 1592 cm−1, 1605 cm−1 and 1399 cm−1, 1401 cm−1 are observed in the spectra of IDA and Fe3O4@SiO2@IDA, indicating that IDA was modified on the Fe3O4@SiO2 NPs successfully. However, the peak at 3104 cm−1 corresponding to the N–H stretching vibration is observed in IDA rather than Fe3O4@SiO2@IDA, which may be due to the overlapping of broad absorption band at 3407 cm−1 that corresponds to the stretching vibration of residual Si–OH and adsorbed water. Magnetization hysteresis loop of Fe3O4@SiO2@IDA (Fig. S1†) shows that Fe3O4@SiO2@IDA was superparamagnetic and the saturation magnetization was 23.8 emu g−1, which is sufficient for the magnetic separation. Due to the strong magnetism, MNPs had the aggregative trend and we can hardly observe single nanoparticle in TEM images (Fig. S2a†). While, it can be seen that Fe3O4 nanoparticles are embedded in the microspheres and the NPs possess typical core–shell structure (Fig. S2b†). The particle size of Fe3O4@SiO2@IDA is about 20–30 nm.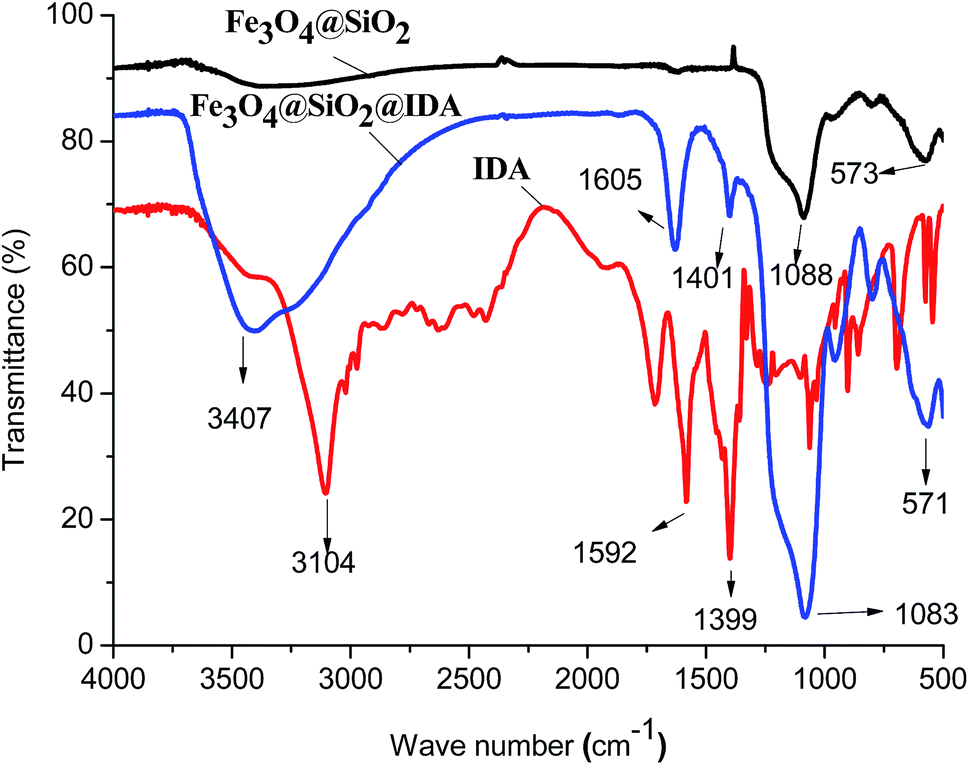 | ||
| Fig. 1 FT-IR spectra of IDA, Fe3O4@SiO2 and Fe3O4@SiO2@IDA. | ||
3.2 Optimization of MSPE conditions
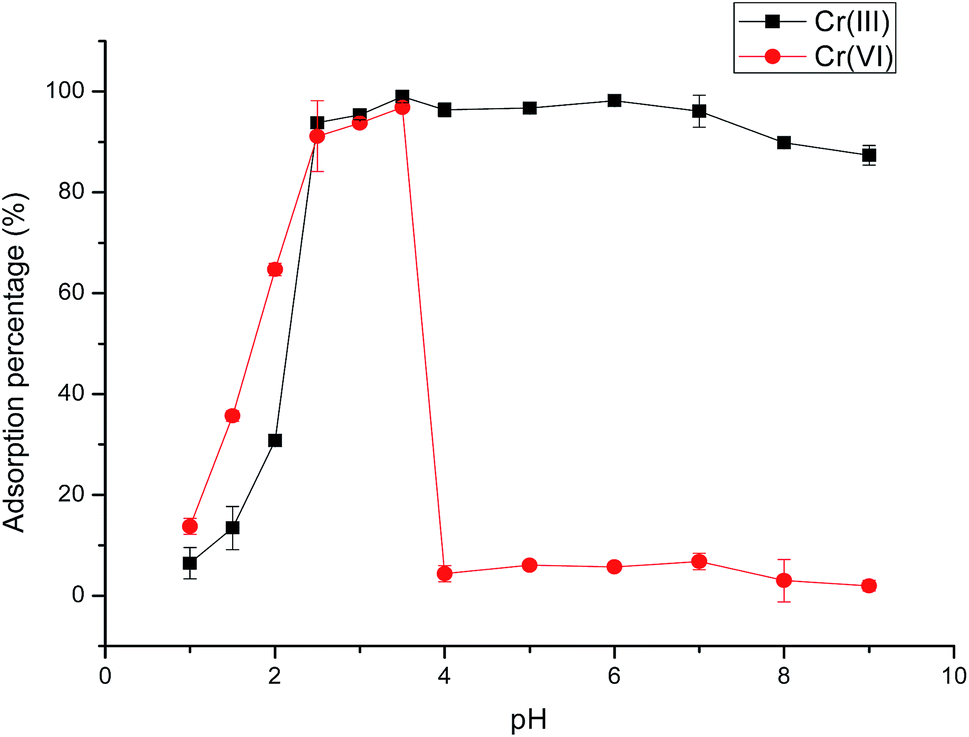 | ||
| Fig. 2 Effect of pH on the adsorption percentage of Cr(III) and Cr(VI) on Fe3O4@SiO2@IDA. Conditions: materials, 10 mg; sample volume, 2 mL; concentration, 10 μg L−1; ultrasound extraction time, 20 min. Error bars show the standard deviation (n = 3). | ||
The above results could be explained by the different adsorption mechanism of Cr(III)/(VI) on Fe3O4@SiO2@IDA. At low pH values, the protonated imino group and carboxyl group on the sorbent have an electrostatic repulsion force to the trivalent Cr cations, therefore the adsorption percentage was low. As the pH increased, the imino group and carboxyl group were deprotonated, which could chelate with Cr(III) through two nitrogen atoms and four oxygen atoms of two IDA groups31 (as shown in Scheme 1). For Cr(VI), it mainly exists in the form of chromic acid (H2CrO4) under strong acid condition (below pH 1), which cannot be retained on the sorbent. As the pH increased, chromic acid dissociates (pKa1 = 0.74, pKa2 = 6.5) and exists as anions of CrO42− or HCrO4− in the pH range of 2.5–3.5, thus can be retained on the protonated –NH+(CH2COOH)2 by electrostatic attraction. If the pH further increased, deprotonated IDA (pKa1 = 2.5, pKa2 = 9.1) has the electrical repulsion with negatively charged hexachromic anions, then the adsorption is decreased.
 | ||
| Scheme 1 Schematic illustration of the adsorption of Cr(III) and Cr(VI) on the prepared Fe3O4@SiO2@IDA NPs. | ||
It should be noted that the reusability of Fe3O4@SiO2@IDA was investigated, and the recovery of Cr on the reused MNPs was decreased along with the reuse time. It may due to the relative high concentration of nitric acid as the desorption solvent and 30 min ultrasonication process that caused damage to the MNPs coating. While, about 1.2 g of the sorbent could be obtained by each preparation process, and only 10 mg was used in each MSPE batch operation. Thus, the prepared Fe3O4@SiO2@IDA was disposable in our experiment.
In order to shorten the analytical time, the effect of extraction time in the range of 5–25 min on the adsorption was investigated, and the results are shown in Fig. S5.† It can be seen that 10 min of extraction time was adequate for the quantitative recovery of Cr(III) and total Cr. Thus, the extraction time of 10 min was selected for further studies.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:10, indicating that the ratio of Cr(III)/Cr(VI) hardly affects their recoveries in the tested range.
:1 to 1:10, indicating that the ratio of Cr(III)/Cr(VI) hardly affects their recoveries in the tested range.
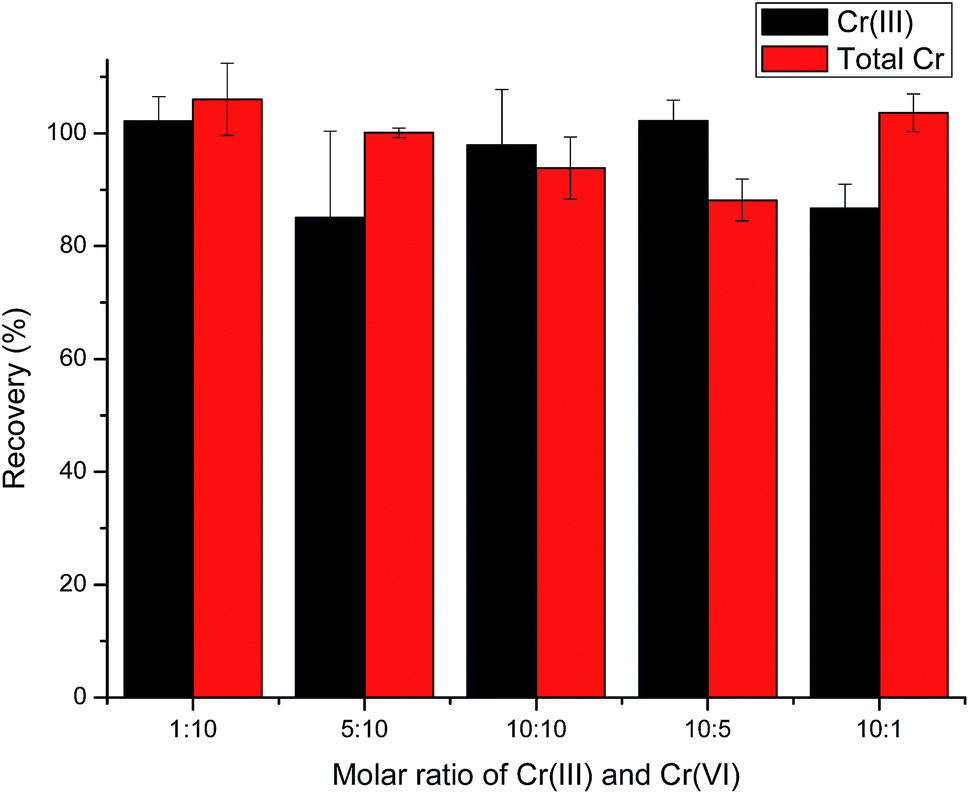 | ||
| Fig. 3 Effect of molar ratio of Cr(III) and Cr(VI) on the recovery of Cr(III) and total Cr. Conditions: materials, 10 mg; sample volume, 50 mL; pH, 5(Cr(III)), 3(total Cr); ultrasound extraction time, 10 min; desorption reagent, 1.5 mol L−1 HNO3; desorption volume, 0.5 mL; ultrasound desorption time, 30 min. | ||
3.3 Effect of coexisting ions
The effect of common coexisting ions on the extraction and determination of Cr(III) and total Cr by MSPE-GFAAS with Fe3O4@SiO2@IDA as adsorbent was investigated. In these experiments, solutions containing 100 ng L−1 of Cr(III) or total Cr and different amount of coexisting ions were subjected to the proposed procedure. The tolerance of the coexisting ions, defined as the maximum tolerance amount with which the recovery of Cr species can be maintained in the range of 85–115%, are listed in Table 1. It can be seen that the presence of major cations and anions has no obvious influence on the extraction and determination of Cr(III) and total Cr under the optimum conditions, which shows that the proposed method has a good anti-interference ability.| Coexisting ions | Tolerance limit of ions (mg L−1) | |
|---|---|---|
| Cr(III) | Total Cr | |
| K+ | 8000 | 8000 |
| Na+ | 5000 | 5000 |
| Ca2+ | 1000 | 2000 |
| Mg2+ | 1000 | 2000 |
| Cu2+ | 10 | 10 |
| Zn2+ | 10 | 10 |
| Fe3+ | 10 | 10 |
| Al3+ | 10 | 5 |
| Cl− | 8000 | 8000 |
| NO3− | 12000 |
12000 |
| SO42− | 2000 | 7000 |
3.4 Adsorption capacity and adsorption mechanism
Adsorption capacity of Fe3O4@SiO2@IDA for Cr(III) and Cr(VI) were studied and results are shown in Fig. S6.† As can be seen, the adsorption capacity was firstly increased with increasing the initial concentrations of target species in the range of 1–30 mg L−1, and then reached a plateau at the concentration range of 40–50 mg L−1. The maximum adsorption capacity of Fe3O4@SiO2@IDA for Cr(III) and Cr(VI) were calculated to be 8.2 and 2.9 mg g−1, respectively.The above data for the uptake of Cr(III) and Cr(VI) on Fe3O4@SiO2@IDA were then fitted to Langmuir and Freundlich isotherm equation in the linearized form, which were respectively expressed as
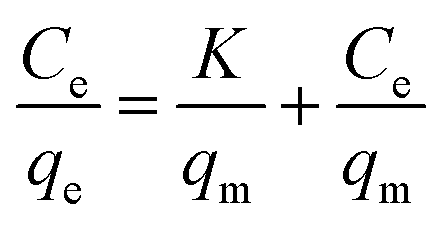
where qe was the adsorption amount (mg g−1) of Cr species under equilibrium, Ce was Cr concentration in solution (mg L−1) under equilibrium, qm represented the maximum amount of Cr species that could be adsorbed on Fe3O4@SiO2@IDA (mg g−1), and K was a constant of the Langmuir model (mg L−1). KF (mg g−1) and n were the Freundlich constants depicting the adsorption process.

The plot of Ce/qe against Ce and lnqe versus lnCe for the experimental data are shown in Fig. 4. The correlation coefficients of Freundlich isotherm model (Fig. 4a and c) (R2 = 0.8776, 0.9775) are lower than that of Langmuir isotherm model (Fig. 4b and d) (R2 = 0.9978, 0.9933). The theoretical qm values of Cr(III) and Cr(VI) calculated from the Langmuir adsorption model (6.1 and 1.9 mg g−1) are close to the experimental values (8.2 and 2.9 mg g−1). As a result, the Langmuir adsorption model, which is considered as the formation of monolayer coverage of analytes at the surface of the sorbent, can be used to explain the adsorption behavior of Cr(III) and Cr(VI) on Fe3O4@SiO2@IDA.
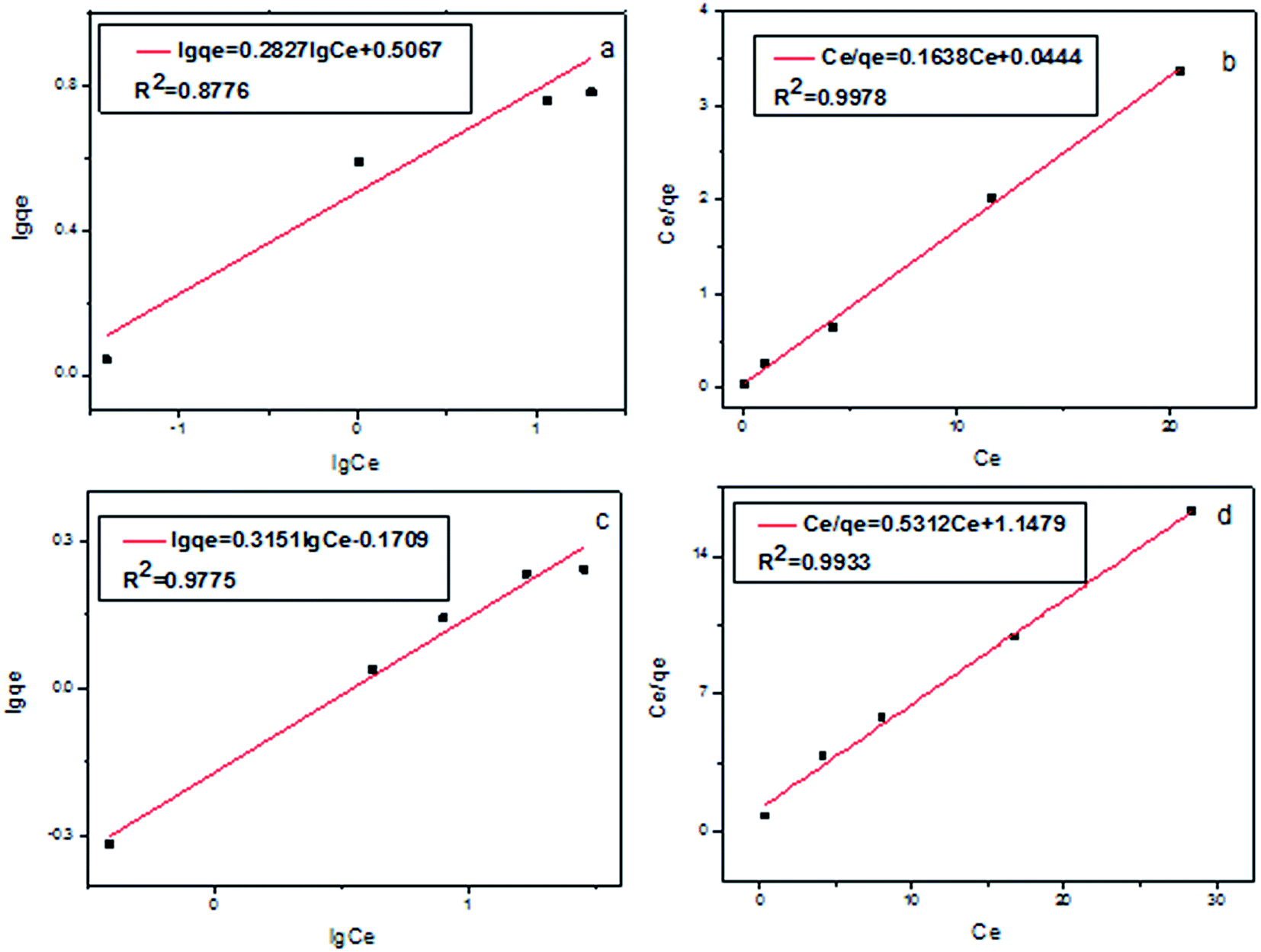 | ||
| Fig. 4 Freundlich isotherms (a, c) and Langmuir isotherms (b, d) for Cr(III) (a, b) and Cr(VI) (c, d) on Fe3O4@SiO2@IDA. | ||
3.5 Analytical performance
Under the optimized experimental conditions, the analytical performance of the proposed method was evaluated and summarized in Table 2. The developed method exhibited good linearity within the range of 20–800 ng L−1. The limits of detection (LODs, 3σ) of the method were 9.1 and 12.8 ng L−1 for Cr(III) and total Cr, respectively. The relative standard deviation (RSDs) for seven replicate determinations of 50 ng L−1 target species were 4.8 and 5.2% for Cr(III) and total Cr, respectively. The enrichment factor was 100-fold.| Target ions | Linear equation | R2 | Linear range (pg mL−1) | Enrichment factor | LODs (pg mL−1) | RSD (%, n = 7) |
|---|---|---|---|---|---|---|
| Cr(III) | y = 0.0012x + 0.0037 | 0.9946 | 20–800 | 100 | 9.1 | 4.8 |
| Total Cr | y = 0.0013x + 0.0056 | 0.9981 | 20–800 | 100 | 12.8 | 5.2 |
A comparison of the developed method with other reported methods for speciation of Cr(III)/(VI) is summarized in Table 3. As can be seen, the developed MSPE-GFAAS method offers comparable LODs to the methods involving ICP-MS detection.21–23,34 Compared with the MSPE-CPE-GFAAS method,27 the developed MSPE-GFAAS method presents a lower LOD and the operation is much easier. Furthermore, it should be noted that sorbents in ref. 21 and 35 can only adsorb one Cr species and thus reduction/oxidation reagent was needed. Compared with these methods mentioned above, the proposed method is simple, sensitive, inexpensive and easy-to-operate, it does not require any reduction/oxidation and the total time for speciation of Cr(III) and Cr(VI) is reduced.
| Analytical technique | Sorbents SPE materials | Redox | Enrichment factor | LODs (ng mL−1) | Application | Ref. |
|---|---|---|---|---|---|---|
| SPE-ICP-MS | AAPTS functionalized MWCNTs | Oxidation | 10 | 0.038, Cr(VI) | River, lake, rain water | 21 |
| CME-ICP-MS | Ordered mesoporous Al2O3 coated capillary | None | 5 | 0.074, Cr(III); 0.018, Cr(VI) | Tap, lake, mineral water | 22 |
| SPE-ICP-MS | Cr(III)-imprinted AAPTS functionalized silica gel | None | 21 | 0.0044, Cr(III); 0.0083, Cr(VI) | Tap, lake, well water | 23 |
| MSPE-ICP-MS | Amine-functionalized magnetite microspheres | None | 96, Cr(III); 47, Cr(VI) | 0.0015, Cr(III); 0.0021, Cr(VI) | Drinking water | 34 |
| MSPE-CPE-GFAAS | Amino-functionalized mesoporous magnetite Fe3O4/SiO2 nanoparticles | None | 16, Cr(III); 12, Cr(VI) | 3.2, Cr(III); 1.1, Cr(VI) | Tap, lake, mineral water | 27 |
| SPE-GFAAS | Crosslinked carboxymethyl starch | Reduction | 50 | 0.044, Cr(III) | Tap, lake water | 35 |
| MSPE-GFAAS | IDA functionalized magnetic silica | None | 100 | 0.0091, Cr(III); 0.0128, total Cr | River, lake water | This work |
3.6 Real sample analysis
In order to verify the accuracy of the method, the developed method was applied to the determination and speciation of Cr(III) and Cr(VI) in Certified Reference Materials of GSBZ50009-88 environmental water. The analytical results along with t-test values are given in Table 4. As can be seen, the determined value of total Cr is in good agreement with the certified value.The developed method was also applied to the analysis of Cr(III) and Cr(VI) in the East Lake water and Yangtze River water samples. Table 5 lists the analytical result along with the recovery of Cr species in East Lake water and Yangtze River water. As can be seen, the concentrations of Cr(VI) in the East Lake water and the Yangtze River water are below the maximum allowable concentration (MAC) (0.05 mg L−1) of Cr(VI) in surface water according to GB3838-2002 (the national environmental quality standard for surface water). The recoveries for the spiked samples were in the range of 83.8–98.7%. These results demonstrated the applicability of the MSPE-GFAAS method for interference-free determination of trace/ultra-trace Cr(III) and total Cr in environmental water samples.
| Samples | Added (pg mL−1) | Found (pg mL−1) | Recovery (%) | ||||
|---|---|---|---|---|---|---|---|
| Cr(III) | Cr(VI) | Cr(III) | Total Cr | Cr(VI) | Cr(III) | Cr(VI) | |
| East Lake water | 0 | 0 | 34.5 ± 1.7 | 59.3 ± 0.6 | 24.8 ± 2.3 | — | — |
| 100 | 100 | 129 ± 4 | 252 ± 3 | 124 ± 7 | 94.3 | 98.7 | |
| 200 | 200 | 202 ± 2 | 416 ± 7 | 214 ± 9 | 83.8 | 94.4 | |
| Yangtze River water | 0 | 0 | 108 ± 3 | 163 ± 2 | 54.7 ± 4.1 | — | — |
| 100 | 100 | 199 ± 1 | 346 ± 2 | 147 ± 3 | 90.8 | 91.8 | |
| 200 | 200 | 302 ± 1 | 535 ± 6 | 233 ± 6 | 96.6 | 89.4 | |
4. Conclusions
In this study, sensitive speciation of trace Cr(III)/(VI) in water was achieved based on selective MSPE with IDA functionalized MNPs followed by GFAAS detection without the need of extra oxidation/reduction. Compared with other methods for speciation of Cr, the developed MSPE-GFAAS method is simple, sensitive, inexpensive and selective toward Cr(III)/(VI). Besides, it exhibits good tolerance toward co-existing cations and anions and can be applied for the analysis of trace/ultra-trace Cr in environmental water samples.Acknowledgements
Financial support from the National Nature Science Foundation of China (No. 21675118, 21575108, 21375097), Science Fund for Creative Research Groups of NSFC (No. 20921062) and Large-scale Instrument and Equipment Sharing Foundation of Wuhan University are gratefully acknowledged.References
- B. Hu and Z. Jiang, The hyphenated techniques of chromatography-atomic spectrum/mass spectrometry and their application in elemental speciation analysis, 2005, vol. 8, pp. 217–221 Search PubMed.
- A. S. Krishna Kumar, S. J. Jiang and W. L. Tseng, J. Mater. Chem. A, 2015, 3, 7044–7057 CAS.
- S. Şahan, Ş. Saçmacı, Ş. Kartal, M. Saçmacı, U. Şahin and A. Ülgen, Talanta, 2014, 120, 391–397 CrossRef PubMed.
- B. Dhal, H. N. Thatoi, N. N. Das and B. D. Pandey, J. Hazard. Mater., 2013, 250–251, 272–291 CrossRef CAS PubMed.
- T. Sumida, T. Ikenoue, K. Hamada, A. Sabarudin, M. Oshima and S. Motomizu, Talanta, 2005, 68, 388–393 CrossRef CAS PubMed.
- V. Gómez and M. P. Callao, TrAC, Trends Anal. Chem., 2006, 25, 1006–1015 CrossRef.
- M. Gawin, J. Konefal, B. Trzewik, S. Walas, A. Tobiasz, H. Mrowiec and E. Witek, Talanta, 2010, 80, 1305–1310 CrossRef CAS PubMed.
- G. Y. Yang, W. B. Fen, C. Lei, W. L. Xiao and H. D. Sun, J. Hazard. Mater., 2009, 162, 44–49 CrossRef CAS PubMed.
- E. L. Silva, P. D. Roldan and M. F. Gine, J. Hazard. Mater., 2009, 171, 1133–1138 CrossRef CAS PubMed.
- C. Huang and B. Hu, Spectrochim. Acta, Part B, 2008, 63, 437–444 CrossRef.
- D. J. Butcher, Appl. Spectrosc. Rev., 2012, 48, 261–328 CrossRef.
- J. Ščančar, B. Berlinger, Y. Thomassen and R. Milačič, Talanta, 2015, 142, 164–169 CrossRef PubMed.
- H. Hagendorfer and W. Goessler, Talanta, 2008, 76, 656–661 CrossRef CAS PubMed.
- Y. Chen, J. Chen, Z. Xi, G. Yang, Z. Wu, J. Li and F. Fu, Electrophoresis, 2015, 36, 1208–1215 CrossRef CAS PubMed.
- V. N. Bulut, D. Ozdes, O. Bekircan, A. Gundogdu, C. Duran and M. Soylak, Anal. Chim. Acta, 2009, 632, 35–41 CrossRef CAS PubMed.
- M. C. Yebra and R. M. Cespon, At. Spectrosc., 2008, 29, 27–31 CAS.
- S. Sadeghi and A. Z. Moghaddam, RSC Adv., 2015, 5, 60621–60629 RSC.
- L. M. Zhang, X. L. Li, X. Y. Wang, W. T. Wang, X. S. Wang and H. Y. Han, Anal. Methods, 2014, 6, 5578–5583 RSC.
- R. Saxena, S. Tiwari and N. Sharma, RSC Adv., 2015, 5, 69196–69204 RSC.
- M. Z. Corazza, E. S. Ribeiro, M. G. Segatelli and C. R. T. Tarley, Microchem. J., 2014, 117, 18–26 CrossRef CAS.
- H. Peng, N. Zhang, M. He, B. Chen and B. Hu, Talanta, 2015, 131, 266–272 CrossRef CAS PubMed.
- W. Hu, F. Zheng and B. Hu, J. Hazard. Mater., 2008, 151, 58–64 CrossRef CAS PubMed.
- N. Zhang, J. S. Suleiman, M. He and B. Hu, Talanta, 2008, 75, 536–543 CrossRef CAS PubMed.
- Y. W. Wu, J. Zhang, J. F. Liu, L. Chen, Z. L. Deng, M. X. Han, X. S. Wei, A. M. Yu and H. L. Zhang, Appl. Surf. Sci., 2012, 258, 6772–6776 CrossRef CAS.
- M. Safarikova and I. Safarik, J. Magn. Magn. Mater., 1999, 194, 108–112 CrossRef CAS.
- H. Tavallali, G. Deilamy Rad and P. Peykarimah, Environ. Monit. Assess., 2013, 185, 7723–7738 CrossRef CAS PubMed.
- K. M. Diniz and C. R. T. Tarley, Microchem. J., 2015, 123, 185–195 CrossRef CAS.
- P. P. Ling, F. Q. Liu, L. J. Li, X. S. Jing, B. R. Yin, K. B. Chen and A. M. Li, Talanta, 2010, 81, 424–432 CrossRef CAS PubMed.
- M. Zhang, D. Cheng, X. W. He, L. X. Chen and Y. K. Zhang, Chem.–Asian J., 2010, 5, 1332–1340 CAS.
- N. Zhang, H. Y. Peng, S. Wang and B. Hu, Microchim. Acta, 2011, 175, 121–128 CrossRef.
- S. Hirata, K. Honda, O. Shikino, N. Maekawa and M. Aihara, Spectrochim. Acta, Part B, 2000, 55, 1089–1099 CrossRef.
- S. Su, B. Chen, M. He, Z. Xiao and B. Hu, J. Anal. At. Spectrom., 2014, 29, 444–453 RSC.
- C. Huang and B. Hu, J. Sep. Sci., 2008, 31, 760–767 CrossRef CAS PubMed.
- Y. F. Huang, Y. Li, Y. Jiang and X. P. Yan, J. Anal. At. Spectrom., 2010, 25, 1467–1474 RSC.
- M. W. Gao, M. Shen, A. F. Xue, S. Q. Li and H. Chen, Chin. J. Anal. Chem., 2011, 39, 690–694 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27544c |
| This journal is © The Royal Society of Chemistry 2017 |