DOI:
10.1039/C6RA27458G
(Paper)
RSC Adv., 2017,
7, 8491-8503
Tuning the solid-state fluorescence of chalcone crystals via molecular coplanarity and J-aggregate formation†
Received
28th November 2016
, Accepted 18th January 2017
First published on 26th January 2017
Abstract
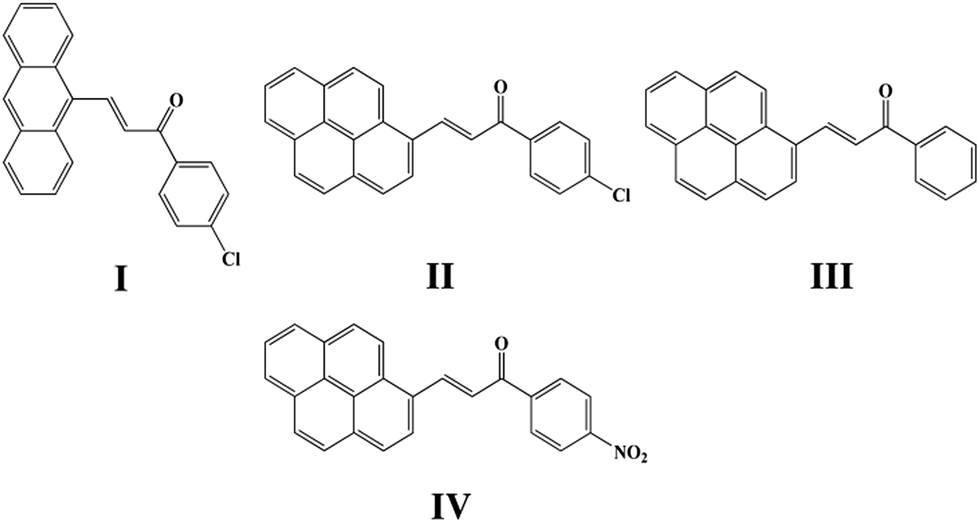
Four chalcones bearing the 1,3-diarylpropenone moiety 3-(9-anthryl)-1-(4-chloro)prop-2-en-1-one (I), 3-(1-pyrenyl)-1-(4-chlorophenyl)prop-2-en-1-one (II), 1-phenyl-3-(1-pyrenyl)prop-2-en-1-one (III) and 3-(1-pyrenyl)-1-(4-nitrophenyl)prop-2-en-1-one (IV) were synthesized and different crystal forms were obtained from different solvents by slow evaporation including I (Ia, Ib), II (IIa, IIb, IIc), III (IIIa, IIIb, IIIc, IIId) and IV (IVa, IVb). Their structures and optical properties were characterized by single crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), UV-Vis absorption spectroscopy and fluorescence spectroscopy. Hirshfeld surface calculations were also used for intermolecular interaction analysis. It has been found that polymorphism and conformational isomorphism exist in these crystals. Ib and IIIa adopt a cis configuration with respect to the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond while the other forms exhibit a trans configuration, except for those without single crystal X-ray analysis. Both Ib and IIb have two crystallographically independent molecules in their asymmetrical unit which has not been found in reported chalcone crystals. Those crystals with a cis configuration show a higher melting point and no fluorescence. Crystals with a trans configuration are fluorescent and their emission wavelengths are mainly effected by the torsion extent of the molecules and J-aggregate formation.
C bond while the other forms exhibit a trans configuration, except for those without single crystal X-ray analysis. Both Ib and IIb have two crystallographically independent molecules in their asymmetrical unit which has not been found in reported chalcone crystals. Those crystals with a cis configuration show a higher melting point and no fluorescence. Crystals with a trans configuration are fluorescent and their emission wavelengths are mainly effected by the torsion extent of the molecules and J-aggregate formation.
Introduction
Crystal polymorphism, a phenomenon having been concentrated on for years, is generally defined as the possibility of the same compound existing in more than one crystalline modification in the solid state.1,2 Polymorphs differ in a number of significant properties such as solubility, stability and bioavailability3–5 that catering for a series of potential applications in pharmaceuticals,6 pigments,7 explosives8 and so on. What's more, it is an ideal approach to compare different polymorphs for the purpose of deeply elucidating the relationships between the solid state properties and crystal structures based on the same molecules.9,10 Molecules with conformational flexibility usually show a larger extent of freedom than rigid ones so that a wide range of polymorphism might be obtained from them.11–13 Current research on the solid state fluorescence of polymorphism indicate that stacking geometries of molecules and intermolecular electronic interactions in the solid state have a significant influence on fluorescent properties of organic luminescent polymorphs.14–17 And many studies on luminescence properties suggest that J-aggregate formation is beneficial to fluorescence emission.18,19
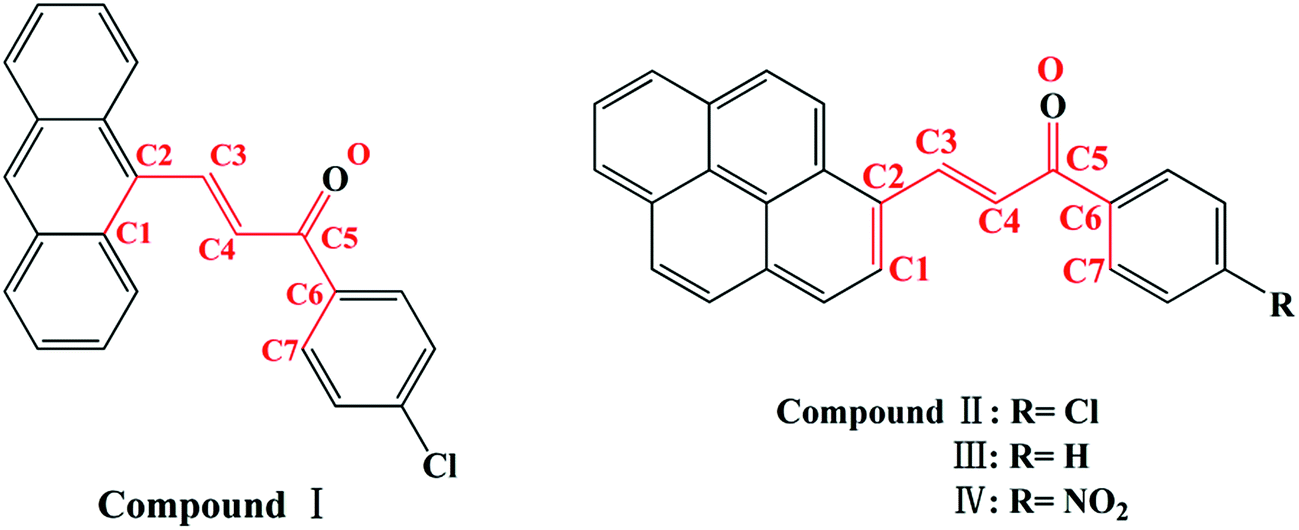
Chalcone, the important class of compound bearing α,β-unsaturated ketone moiety, increasingly attracts more attention from academic circles and pharmaceutical industry. Chalcones are found to have many applications because of their properties such as bioactivities,5 optical properties20,21 and photochemical characteristics20,22 all of which are researched and applied widespread. Furthermore, chalcones are intermediates of many heterocyclic compounds such as pyrazole, quinoline, pyrimidine and so on.23 In the phenomenon of cis–trans configuration, trans configuration exhibits greater stability than cis which is suggested by easier formation of trans configuration and the approach of quantum chemistry also gave explanation to stability difference.24,25 Thus a number of crystals of chalcone have been reported while crystals with cis configuration are hardly to find for its strikingly steric effect. In our former research done by Zhang Ruimin et al.,17 several chalcone polymorphs which show cis or trans configuration have been investigated. In this research, four new chalcones were synthesized (Scheme 1), while the polymorphism being found in this research is conformational and configurational isomorphism. Furthermore, conformational isomorphism is rarely reported for the compound of chalcone, especially when referring it to two conformers in crystal asymmetry unit.
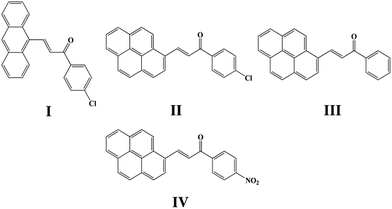 |
| | Scheme 1 Chemical structure of I, II, III, IV. | |
Fluorescent properties of chalcone polymorphism in the solid state can be rarely to found in literatures reported before. Previously, we readily prepared several high quality chalcone polymorphs and π-stacked geometries plays a key role in fluorescent properties of chalcone polymorphism.17 Therefore, we deduce that there can be some other factors related to optical properties of chalcone and we investigated in different chalcone polymorphs to find that molecular planarity and aggregation exert great influence on fluorescent properties of chalcone polymorphism in the solid state, which may offer a significant direction in application of organic light-emitting materials.26
Experimental section
Materials synthesis
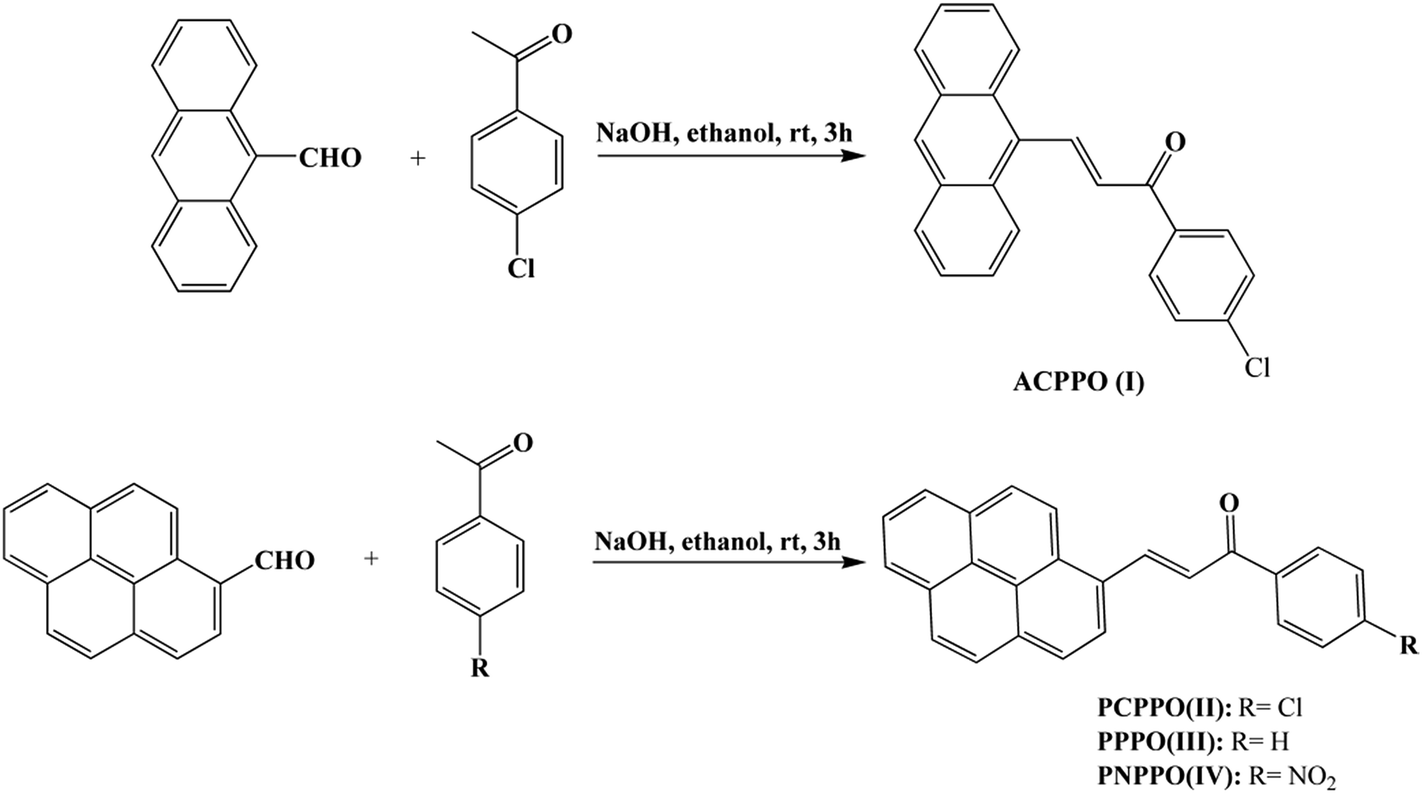
As shown in Scheme 2, a mixture of 4-chloroacetophenone (1.54 g), 9-anthrylaldehyde (2.3 g) and 3 M of aqueous sodium hydroxide (6 mL) in ethanol (20 mL) was stirred at room temperature for 3 h. The crude product was collected by filtration and recrystallized from ethyl acetate/acetic acid (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) resulting in orange crystals as 3-(9-anthryl)-1-(4-chloro)prop-2-en-1-one (I): yield: 80%, mp: 136–138 °C. 1H NMR (CDCl3, Fig. 2S, ESI†): δ (ppm) 8.81 (d, 1H), 8.49 (s, 1H), 8.36–8.19 (m, 2H), 8.04 (dd, 4H), 7.63–7.46 (m, 7H). 13C NMR (CDCl3, Fig. 3S, ESI†): δ (ppm) 188.36, 142.47, 139.59, 136.22, 131.32, 130.48, 130.12, 129.92, 129.66, 129.11, 128.98, 128.64, 126.55, 125.48, 125.20.
:1) resulting in orange crystals as 3-(9-anthryl)-1-(4-chloro)prop-2-en-1-one (I): yield: 80%, mp: 136–138 °C. 1H NMR (CDCl3, Fig. 2S, ESI†): δ (ppm) 8.81 (d, 1H), 8.49 (s, 1H), 8.36–8.19 (m, 2H), 8.04 (dd, 4H), 7.63–7.46 (m, 7H). 13C NMR (CDCl3, Fig. 3S, ESI†): δ (ppm) 188.36, 142.47, 139.59, 136.22, 131.32, 130.48, 130.12, 129.92, 129.66, 129.11, 128.98, 128.64, 126.55, 125.48, 125.20.
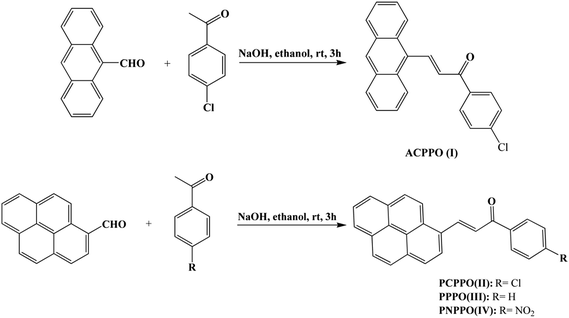 |
| | Scheme 2 Preparation of chalcone I, II, III, IV. | |
As can be seen from Scheme 2, a mixture of acetophenone (1.2 g), 1-pyrenecarboxaldehyde (2.3 g) and 3 M of aqueous sodium hydroxide (6 mL) in ethanol (20 mL) was stirred at room temperature for 3 h. The resulting solid was collected by filtration and recrystallized from ethyl acetate/acetic acid (v/v = 1:1) to get orange crystals as 1-phenyl-3-(1-pyrenyl)prop-2-en-1-one (III): yield: 70%, mp: 163–165 °C. 1H NMR (CDCl3, Fig. 4S, ESI†): δ (ppm) 8.82 (d, 1H), 8.31 (d, 1H), 8.17 (d, 1H), 8.06 (dd, 4H), 8.00–7.80 (m, 5H), 7.65 (d, 1H), 7.61–7.42 (m, 3H). 13C NMR (CDCl3, Fig. 5S, ESI†): δ (ppm) 189.67, 140.85, 137.96, 132.40, 132.36, 130.71, 130.11, 129.75, 128.27, 128.16, 128.10, 127.99, 126.78, 125.76, 125.54, 125.40, 124.46, 124.31, 123.97, 123.52, 123.08, 121.92. Similarly, 3-(1-pyrenyl)-1-(4-chlorophenyl)prop-2-en-1-one (II) and 3-(1-pyrenyl)-1-(4-nitrophenyl)prop-2-en-1-one (IV) were synthesized according to the procedure with 4-chloroacetophenone and 4-nitroacetophenone as starting material respectively. 1-3-(1-Pyrenyl)-1-(4-chlorophenyl) prop-2-en-1-one (II): 1H NMR (CDCl3, Fig. 6S, ESI†): δ (ppm) 8.88 (d, 1H), 8.40 (d, 1H), 8.26 (d, 1H), 8.14 (d, 2H), 8.09–8.01 (m, 3H), 8.01–7.93 (m, 4H), 7.63 (d, 1H), 7.45 (d, 2H). 13C NMR (CDCl3, Fig. 7S, ESI†): δ (ppm) 188.63, 141.73, 139.17, 136.60, 132.98, 131.18, 130.58, 130.32, 129.89, 128.92, 128.76, 128.70, 128.30, 127.32, 127.21, 126.26, 126.08, 125.92, 124.92, 124.83, 124.44, 124.00, 122.95, 122.33. 3-(1-Pyrenyl)-1-(4-nitrophenyl) prop-2-en-1-one (IV): 1H NMR (CDCl3, Fig. 8S, ESI†): δ (ppm) 9.05 (d, 1H), 8.54 (d, 1H), 8.44 (d, 1H), 8.41–8.33 (m, 2H), 8.33–8.19 (m, 6H), 8.17 (d, 1H), 8.14–8.01 (m, 2H), 7.77 (d, 1H). 13C NMR (CDCl3, Fig. 9S, ESI†): δ (ppm) 188.59, 143.30, 133.52, 131.31, 130.77, 130.68, 129.46, 129.23, 129.17, 127.98, 127.34, 126.52, 126.43, 126.25, 125.14, 125.05, 124.56, 124.24, 123.92, 122.80, 122.31.
Ia was recrystallized in ethyl acetate/acetic acid (v/v = 1:1) as orange rod like crystal. Ib was obtained from ethanol in which Ia was dissolved. Slow evaporation of the solvent at room temperature for 4–5 days yielded yellow rod like crystal.
IIa was recrystallized in ethyl acetate/acetic acid (v/v = 1:1) mixed solvents as orange-red plate crystal. IIb was obtained from ethyl acetate/dichloromethane (v/v = 1:1) mixed solvents in which IIa was dissolved. Slow evaporation of the solvents at room temperature for 2–3 days yielded orange rod like crystal. IIc was obtained from isopropanol in which IIa was dissolved. Slow evaporation at room temperature for 2–3 days yielded small yellow plate crystal.
IIIa was obtained from mixed solvent of ethanol/ethyl acetate (v/v = 1:1) with IIIa dissolved in it. Slow evaporation of the solvents at room temperature for 4–5 days yielded a yellow transparent crystal. IIIb was gained from acetonitrile or ethanol/ethyl acetate (v/v = 1:1) mixed solvents in which IIIa was dissolved. Slow evaporation of the solvents at room temperature for 4–5 days yielded red transparent plate crystal. IIIc was recrystallized in acetonitrile/chloroform (v/v = 1) or acetic acid/ethyl acetate (v/v = 1:1) mixed solvent as orange needle like crystal. IIId was recrystallized after 3–4 days' slow evaporation at room temperature with IIIa dissolved in isopropanol as yellow plate crystal.
IVa was obtained from acetonitrile/dichloromethane (v/v = 1:1) mixed solvents with 2–3 days slow evaporation at room temperature yielding red needle like crystal. IVb was recrystallized in acetonitrile/dichloromethane (v/v = 1:1) mixed solvents as a purple crystal after 2–3 days slow evaporation at room temperature.
Characterization methods
NMR analysis. 1H NMR and 13C NMR spectra were recorded at 303 K on a Bruker Avance 500 MHz NMR spectrometer using CDCl3 as a solvent and TMS as an internal standard.
X-ray diffraction. The PXRD patterns for the eleven crystals were recorded using a 18 kW advance X-ray diffractometer with Cu K α radiation (λ = 1.54056 Å). Single X-ray diffraction data for these crystals were collected on Nonius CAD4 diffractometer with Mo Kα radiation (λ = 0.71073 Å). The structures were solved with direct methods using the SHELXS-2014 program and refined anisotropically using full-matrix least-squares procedure.
Spectroscopic measurements. UV-Vis absorption spectra were recorded on a Shimadzu UV-2600 spectrometer. Fluorescence spectra were obtained on a Horiba FluoroMax 4 spectrofluorometer. Infrared spectra were collected on a Bruker Tensor 27 FT-IR spectrometer.
Fluorescence microscopy images. Fluorescence microscopy images were obtained on an Olympus BX51 imaging system excited at 365 nm.
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA/DSC patterns were recorded with a Mettler-Toledo TGA/DSC Thermogravimetric Analyzer with the temperature scanned from 50 to 550 °C at 10 °C min−1.
Hot stage microscopy (HSM). Hot stage microscopy was performed on a LEICA DM750P microscope using a Mettler-Toledo FP82HT hot stage. The data were visualized using the AMC Capture software.
Results and discussion
Crystal structure
Despite considerable experimental endeavor, attempts to grow single crystals of II in isopropanol (IIc), III in isopropanol (IIId) and IV in acetonitrile/dichloromethane (v/v = 1:1) (IVb) were unsuccessful. Thus, single crystal X-ray diffraction (SCXRD) analysis was performed for all other forms showing independent framework arrangements and cell parameters (Table 1). Additionally SCXRD of IIIc was reported.27
Table 1 Crystal data and structure refinement
| Crystal |
Ia |
Ib |
IIa |
IIb |
| Formula |
C23H15ClO |
C46H30Cl2O2 |
C25H15ClO |
C25H15ClO |
| Temperature/K |
293 |
293 |
293 |
293 |
| Crystal size/mm3 |
0.30 × 0.10 × 0.10 |
0.20 × 0.10 × 0.10 |
0.20 × 0.20 × 0.10 |
0.20 × 0.10 × 0.10 |
| Morphology |
Rod like |
Rod like |
Plate |
Rod like |
| Melting point/°C |
128 |
129 |
135 |
140 |
| Configuration |
trans |
cis |
trans |
trans |
| Crystal system |
Monoclinic |
Orthorhombic |
Monoclinic |
Orthorhombic |
| Space group |
P21/c |
Pca21 |
C2/c |
P212121 |
| a/Å |
5.4320(11) |
10.831(2) |
30.665(6) |
31.234(6) |
| b/Å |
19.431(4) |
10.431(2) |
5.5070(11) |
4.8180(10) |
| c/Å |
16.208(3) |
30.703(6) |
21.295(4) |
11.669(2) |
| α/° |
90.00 |
90.00 |
90.00 |
90.00 |
| β/° |
95.65(3) |
90.00 |
92.38(3) |
90.00 |
| γ/° |
90.00 |
90.00 |
90.00 |
90.00 |
| V/Å3 |
1702.4(6) |
3468.8(12) |
3593.0(12) |
1756.0(6) |
| Z |
4 |
4 |
8 |
4 |
| ρ (calcd)/Mg m−3 |
1.337 |
1.313 |
1.356 |
1.388 |
| θ range for data collection/° |
1.641–25.375 |
1.326–25.373 |
1.329–25.400 |
1.304–25.370 |
| F(000) |
712 |
1424 |
1520 |
760 |
| R1, wR2 (I > 2σ(I)) |
0.0841, 0.1314 |
0.0551, 0.0969 |
0.0986, 0.2173 |
0.0710, 0.0861 |
| R1, wR2 (all data) |
0.1980, 0.1644 |
0.1141, 0.1132 |
0.2216, 0.2764 |
0.1937, 0.1172 |
| Goodness-of-fit |
1.001 |
0.998 |
1.002 |
1.004 |
| CCDC |
1473276 |
1473277 |
1473283 |
1473284 |
| Crystal |
IIIa |
IIIb |
IIIc |
IVa |
| Formula |
C25H16O |
C50H32O2 |
C25H16O |
C25H15NO3 |
| Temperature/K |
293 |
293 |
293 |
293 |
| Crystal size/mm3 |
0.20 × 0.20 × 0.10 |
0.30 × 0.20 × 0.10 |
0.20 × 0.10 × 0.10 |
0.20 × 0.10 × 0.10 |
| Morphology |
Plate |
Plate |
Needle like |
Needle like |
| Melting point/°C |
122 |
162 |
144 |
215 |
| Configuration |
cis |
trans |
trans |
trans |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2/c |
P21 |
P21/n |
| a/Å |
5.5050(11) |
28.811(6) |
4.6720(9) |
7.3870(15) |
| b/Å |
9.6100(19) |
5.4880(11) |
22.405(5) |
7.8470(16) |
| c/Å |
16.517(3) |
21.784(4) |
8.2100(16) |
32.089(6) |
| α/° |
99.15(3) |
90.00 |
90.00 |
90.00 |
| β/° |
98.70(3) |
100.25(3) |
106.02(3) |
94.00(3) |
| γ/° |
100.80(3) |
90.00 |
90.00 |
90.00 |
| V/Å3 |
832.5(3) |
3389.4(12) |
826.0(3) |
1855.5(7) |
| Z |
2 |
4 |
2 |
4 |
| ρ (calcd)/Mg m−3 |
1.326 |
1.303 |
1.336 |
1.351 |
| θ range for data collection/° |
1.271–25.369 |
1.436–25.357 |
1.818–25.397 |
2.545–25.364 |
| F(000) |
348 |
1392 |
348 |
784 |
| R1, wR2 (I > 2σ(I)) |
0.0960, 0.1651 |
0.0693, 0.1072 |
0.0569, 0.1087 |
0.0777, 0.1285 |
| R1, wR2 (all data) |
0.2175, 0.2068 |
0.1715, 0.1313 |
0.1169, 0.1284 |
0.2004, 0.1651 |
| Goodness-of-fit |
1.002 |
1.001 |
1.002 |
1.000 |
| CCDC |
1477084 |
1473288 |
1473285 |
1473292 |
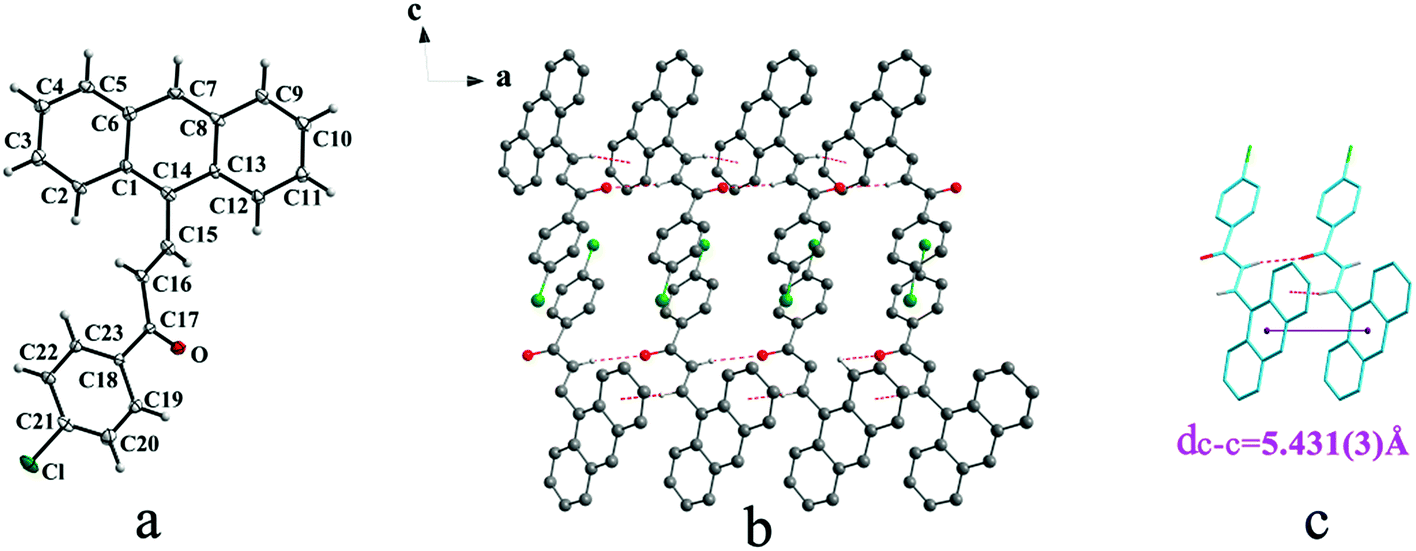
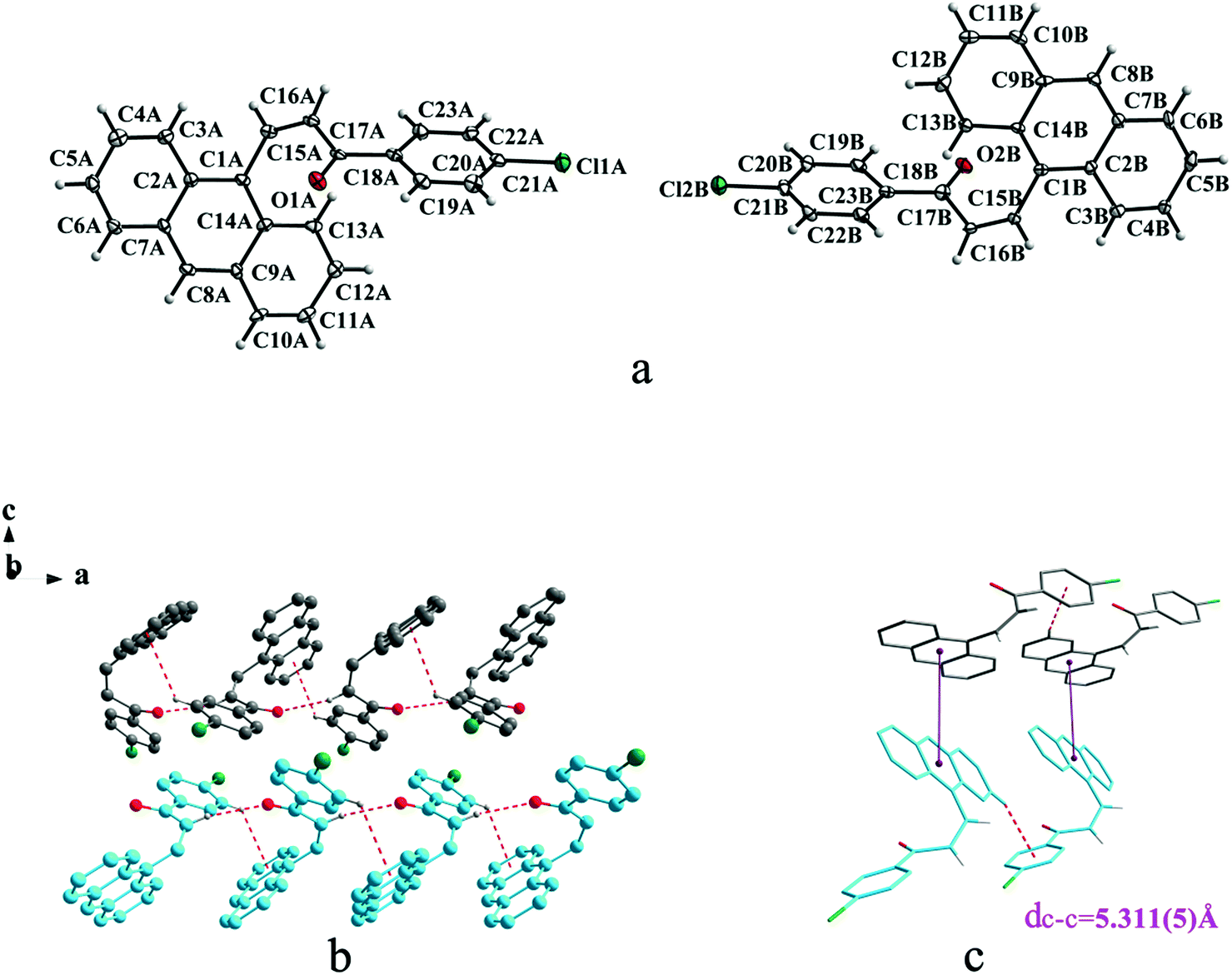
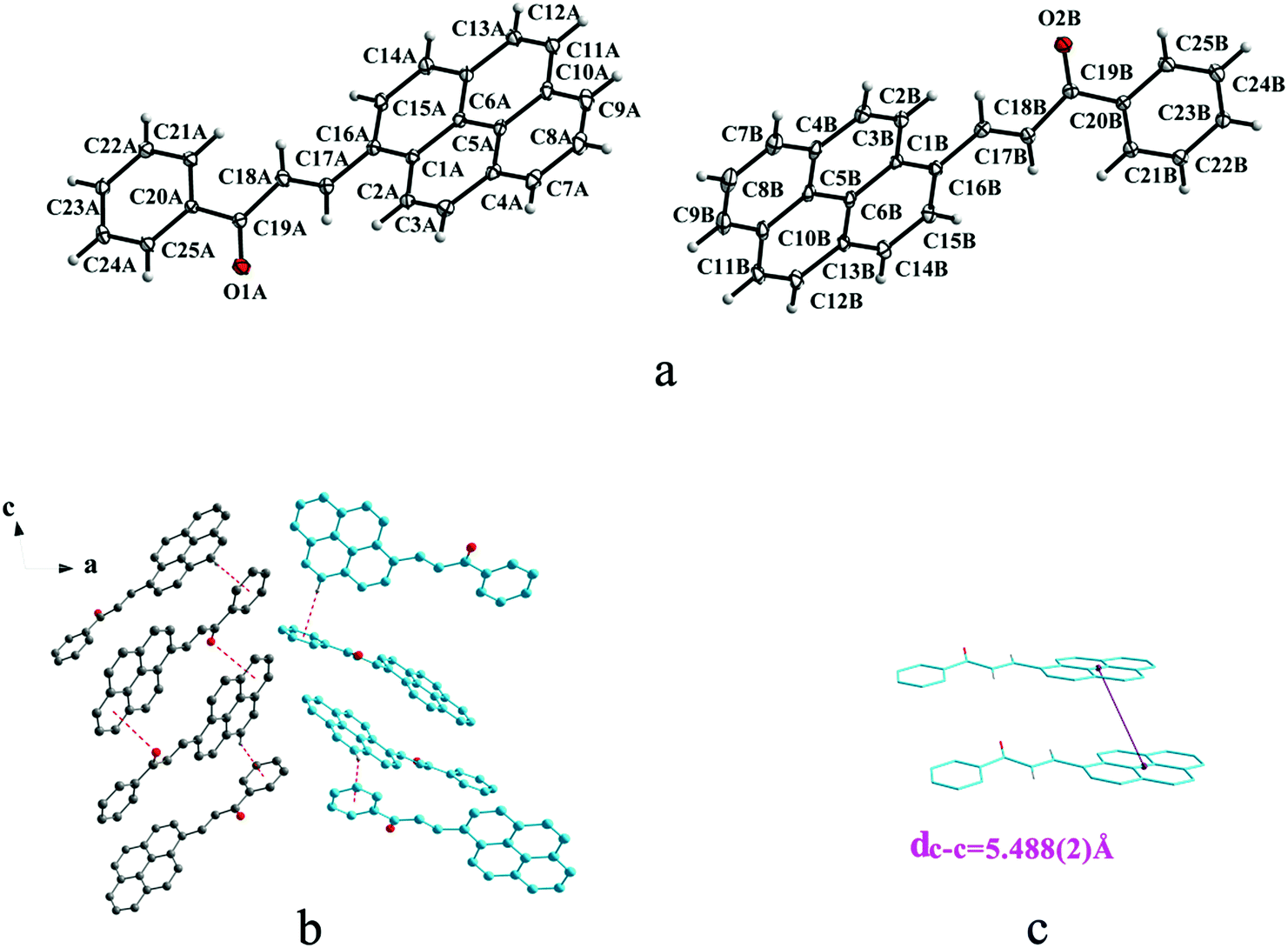
Crystal Ia crystallizes in monoclinic system with space group of P21/c while Ib crystallizes in orthorhombic system with space group of Pca21. Ia shows trans configuration while Ib exhibits cis configuration (Fig. 1a and 2a). Ia is connected by weak hydrogen bond (Table 2) (O⋯H16A distance: 2.46 Å; C–O⋯H angle: 161°) and C(alkene)–H⋯π (6 atoms of anthracene) (Table 3) interactions on ac plane (Fig. 1b). Ib possesses two types of molecules in its crystal structure (Fig. 2a). Molecules A are linked by weak hydrogen bonds (Table 2) (O1A⋯H16A distance: 2.54 Å; C–O⋯H angle: 164°) and C(alkene)–H⋯π (6 atoms of anthracene) (Table 3) interactions on ac plane (Fig. 2b). Likewise, weak hydrogen bonds (Table 2) (O2B⋯H16B distance: 2.56 Å; C–O⋯H angle: 159°) and C(alkene)–H⋯π (6 atoms of anthracene) (Table 3 and Fig. 2b) connect between molecules B on ac plane. Besides, molecules A and B are separately linked by C(anthracene)–H⋯π (benzene ring) (Table 3 and Fig. 2c) along b axis.
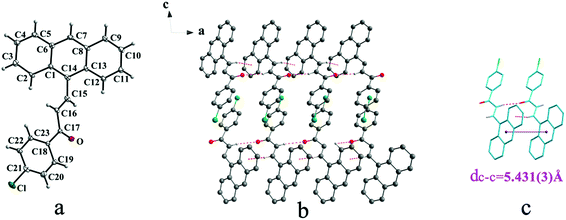 |
| | Fig. 1 Structure of form Ia: (a) trans configuration, (b) molecules are linked by hydrogen bonds and C–H⋯π interactions on ac plane, (c) molecules are stacking along a axis with hydrogen bonds and C–H⋯π interactions. The violet line represents the closest centroid distance (dc–c = 5.431(3) Å) between the adjacent anthracene rings. The dotted lines show hydrogen bonds and C–H⋯π interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
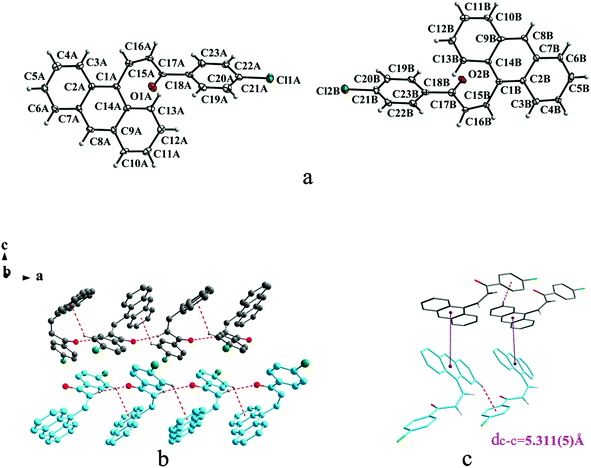 |
| | Fig. 2 Structure of form Ib: (a) cis configuration, (b) (molecule labeled A-grey B-sky blue) molecules are linked by hydrogen bonds and C–H⋯π interactions on ac plane, (c) molecules are stacking along b axis with hydrogen bonds with the closest centroid distance (dc–c = 5.311(5) Å) between the adjacent anthracene rings showed by violet lines. The dotted lines show hydrogen bonds and C–H⋯π interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
Table 2 Intermolecular hydrogen bonds parameters
| Crystal |
Interaction |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
∠D–H⋯A (°) |
| Ia |
C16–H(16A)⋯O |
0.93 |
2.46 |
3.357(6) |
161 |
| Ib |
C16A–H(16A)⋯O(1A) |
0.93 |
2.54 |
3.445(8) |
164 |
| C16B–H(16B)⋯O(2B) |
0.93 |
2.56 |
3.442(8) |
159 |
| IIIa |
C8–H(8A)⋯O |
0.93 |
2.43 |
3.182(7) |
138 |
| IVa |
C7–H(7A)⋯O(1) |
0.93 |
2.55 |
3.325(5) |
141 |
Table 3 C–X⋯π and N–O⋯π interactions in these forms
| Crystal |
Interaction |
Distancea (Å) |
Angleb (°) |
| The distances were measured from hydrogen atom or halogen atom or oxygen atom to the centre of the aromatic ring (for C–X⋯π, X = H, O, Cl). The angles were measured between C–X–c or N–O–c (for C–X⋯π or N–O⋯π). |
| Ia |
C15–H(15A)⋯6 atoms of anthracene |
2.860 |
127.00 |
| Ib |
C4–H(4A)⋯benzene ring |
2.950 |
156.00 |
| C22A–H(22A)⋯6 atoms of anthracene |
2.890 |
131.00 |
| C4B–H(4B)⋯benzene ring |
2.960 |
157.00 |
| C22B–H(22B)⋯6 atoms of anthracene |
2.920 |
130.00 |
| IIa |
C12–H(12A)⋯benzene ring |
2.710 |
147.00 |
| C22–H(22A)⋯6 atoms of pyrene ring |
2.870 |
136.00 |
| C19–O⋯6 atoms of pyrene ring |
3.900(5) |
80.1(4) |
| IIb |
C23–Cl⋯6 atoms of pyrene ring |
3.968 |
101.82 |
| C23–Cl⋯6 atoms of benzene ring |
3.678 |
90.42 |
| IIIa |
C16–H(16A)⋯benzene ring |
2.790 |
144.00 |
| IIIb |
C(12A)–H(12A)⋯benzene ring |
2.760 |
146.00 |
| C(12B)–H(12B)…benzene ring |
2.860 |
149.00 |
| C19–O(1A)⋯6 atoms of pyrene ring |
3.896 |
83.4(2) |
| IVa |
C19–O(1)⋯6 atoms of pyrene ring |
3.830(4) |
74.6(3) |
| N–O(3)⋯6 atoms of pyrene ring |
3.642(5) |
90.5(3) |
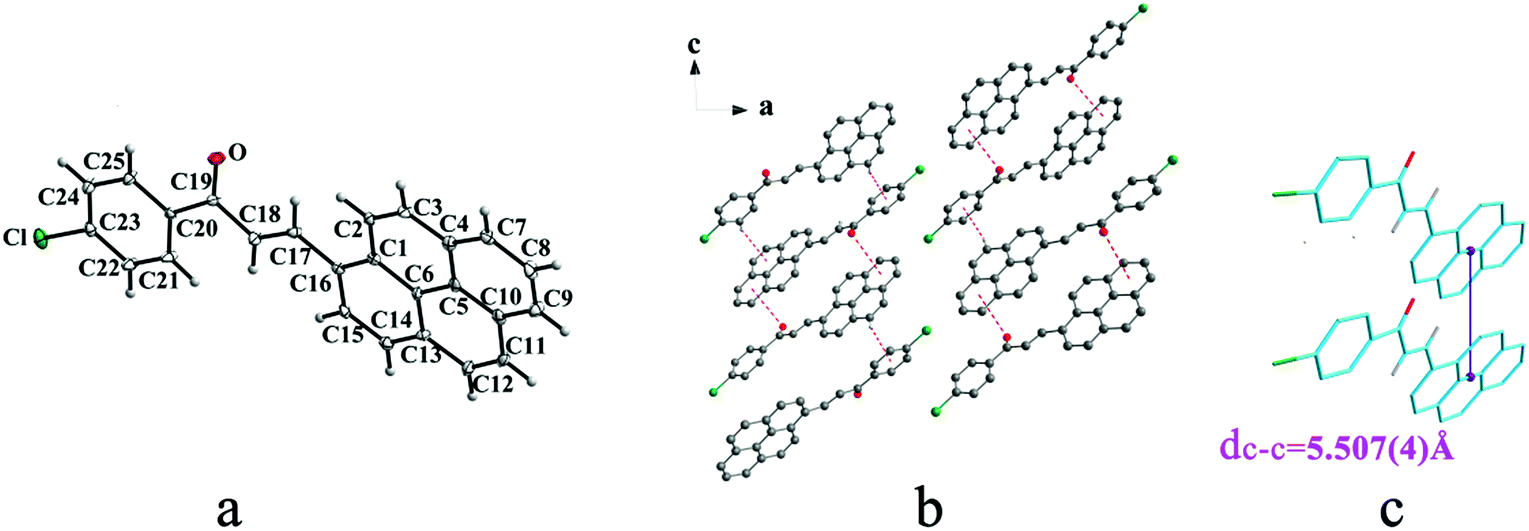
Crystal IIa crystallizes in monoclinic system with space group C2/c while IIb crystallizes in orthorhombic system with space group P212121. Both IIa and IIb show trans configuration. It is evident that molecules in the structure of IIa are connected by C–O⋯π (6 atoms of pyrene ring), C–H⋯π (6 atoms of pyrene ring) and C–H⋯π (benzene ring) interactions on ac plane (Table 3 and Fig. 3b). Molecules in IIb are linked through interactions of C–Cl⋯π (6 atoms of pyrene ring) and C–Cl⋯π (benzene ring) along c axis (Fig. 4b).
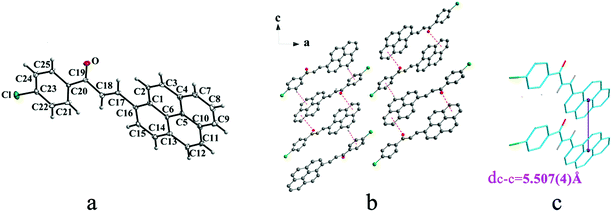 |
| | Fig. 3 Structure of form IIa: (a) trans configuration, (b) molecules are linked by C–O⋯π and C–H⋯π interactions on ac plane, (c) the violet line represents the closest centroid distance (dc–c = 5.507(4) Å) between the adjacent anthracene rings. The dotted lines show hydrogen bonds and C–H⋯π interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
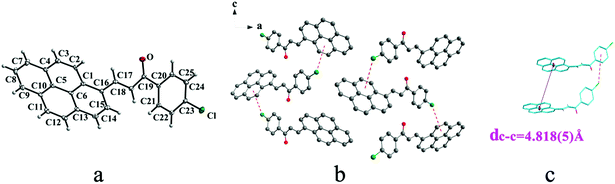 |
| | Fig. 4 Structure of form IIb: (a) trans configuration, (b) molecules are linked by C–Cl⋯π interactions on ac plane, (c) molecules are stacking along c axis with C–Cl⋯π interactions. The violet line represents the closest centroid distance (dc–c = 4.818(5) Å) between the adjacent anthracene rings. The dotted lines show C–Cl⋯π interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
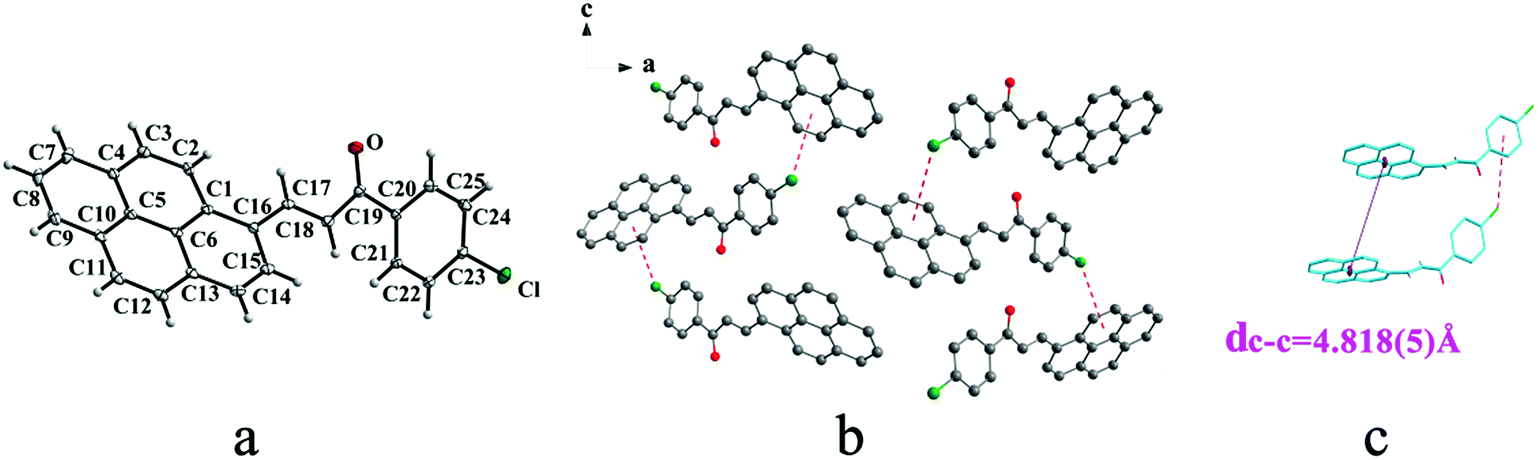
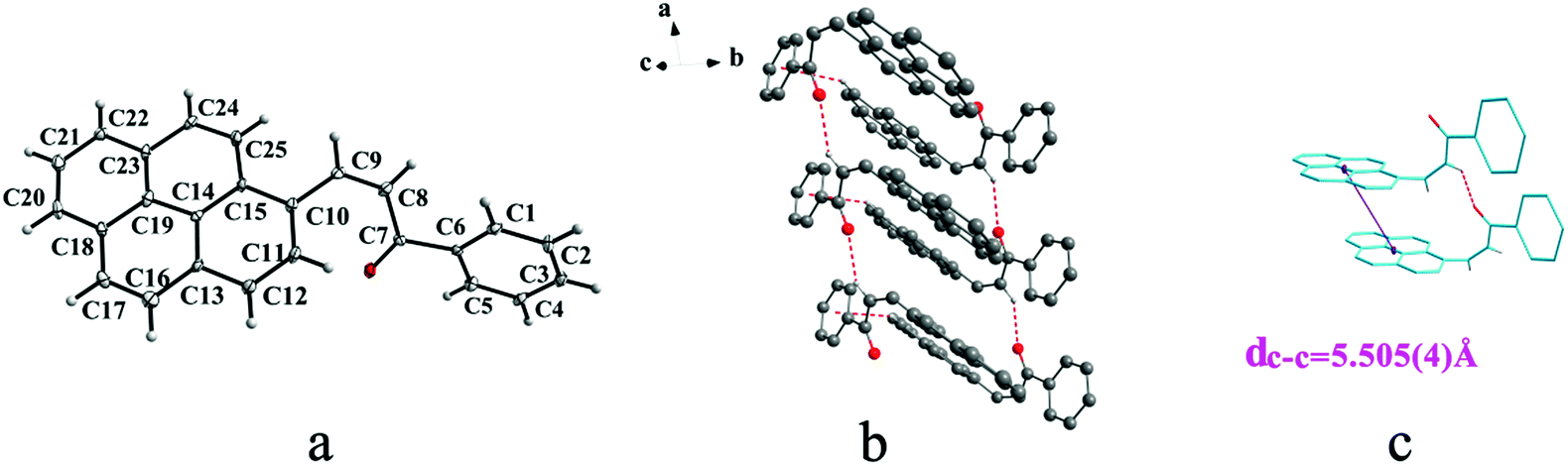
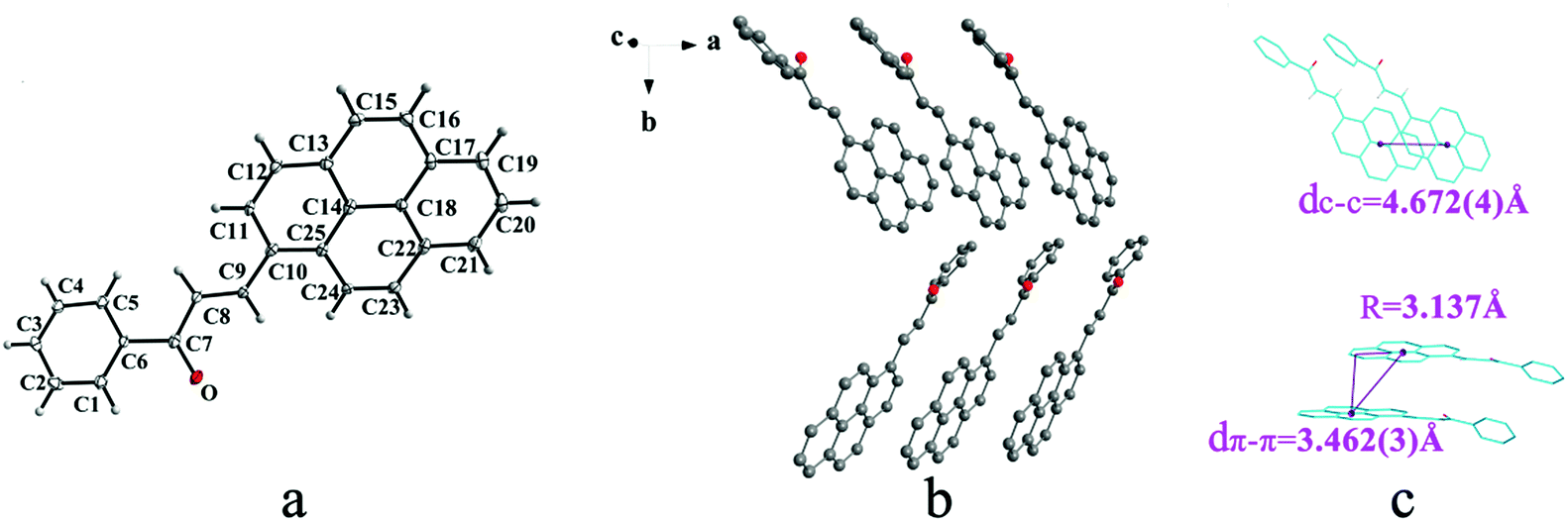
IIIa crystallizes in triclinic system with space group P and it shows cis configuration. Both IIIb and IIIc crystallizes in monoclinic system while their space groups are P2/c and P21 respectively. Besides, they all show trans configuration. As shown in Fig. 5b, molecules are packing along a axis through C–H⋯O (O⋯H8A distance: 2.43 Å; C–O⋯H angle: 138°) (Table 2) and C(pyrene)–O⋯π (benzene ring) (Table 3) interactions in IIIa. In Fig. 6a, it can be seen that IIIb owns two types of molecules in its asymmetry unit and both of them are connected by C–H⋯π and C–O⋯π interactions on ac plane. In detail, C–H⋯π (6 atoms of pyrene ring) and C–O⋯π (6 atoms of pyrene ring) (Table 3) link between neighboring type A molecules while type B molecules are only connected through C–H⋯π (6 atoms of pyrene ring) (Table 3). And in Fig. 7b, molecules in IIIc are packing on ab plane with slipped face-to-face π⋯π interaction.
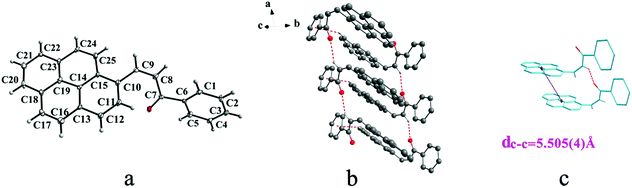 |
| | Fig. 5 Structure of form IIIa: (a) cis configuration, (b) molecules are linked by C–H⋯π and C–H⋯O interactions on ab plane, (c) the violet line represents the closest centroid distance (dc–c = 5.505(4) Å) between the adjacent anthracene rings. The dotted lines show C–H⋯π and C–H⋯O interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
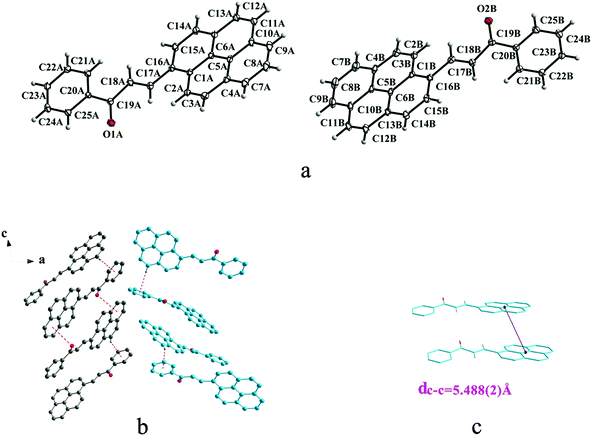 |
| | Fig. 6 Structure of form IIIb: (a) trans configuration, (b) (type A-grey type B-sky blue) molecules are linked by C–H⋯π and C–O⋯π interactions on ac plane, (c) the violet line represents the closest centroid distance (dc–c = 5.488(2) Å) between the adjacent anthracene rings. The dotted lines show C–H⋯π and C–O⋯π interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
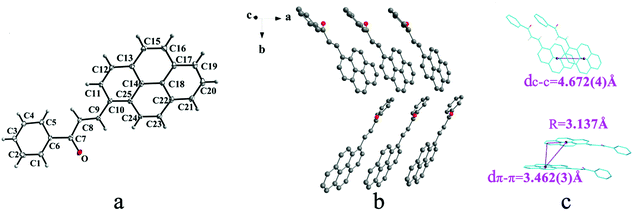 |
| | Fig. 7 Structure of form IIIc: (a) trans configuration, (b) molecules are stacking on ab plane with slipped face-to-face π⋯π interaction, (c) the dc–c, dπ–π and R are 4.672(4) Å, 3.462(3) Å and 3.137 Å, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
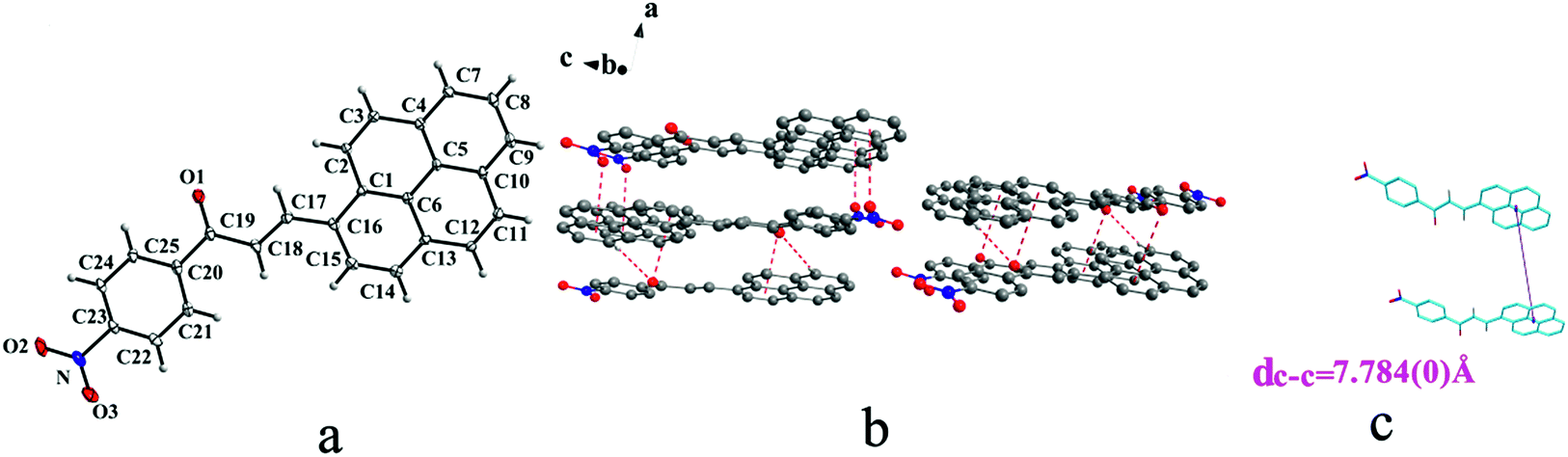
IVa crystallizes in monoclinic system with space group P21/n and it exhibits trans configuration. As shown in Fig. 8b, molecules are stacking along b axis with weak hydrogen bond C–H⋯O (O⋯H7A distance: 2.55 Å; C–O⋯H angle: 141°) (Table 2), C(alkene)–O⋯π (6 atoms of pyrene ring) and N–O⋯π (6 atoms of pyrene ring) (Table 3) interactions on ac plane.
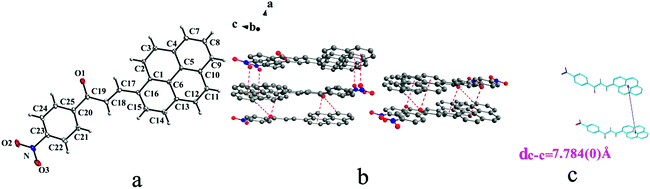 |
| | Fig. 8 Structure of form IVa: (a) trans configuration, (b) molecules are linked by C–H⋯O, C–O⋯π and N–O⋯π interactions on ac plane, (c) the violet line represents the closest centroid distance (dc–c = 7.847(0) Å) between the adjacent anthracene rings. The dotted lines show C–H⋯O, C–O⋯π and N–O⋯π interactions, respectively. Hydrogen atoms not participating in the interactions have been omitted for clarity. | |
In a word, Ia, IIa, IIb, IIIb, IIIc, IVa adopt trans configuration while Ib and IIIa adopt cis configuration with respect to the central CC bond. Thus, chalcones I and III exist configurational isomers while IIa, IIb and IIIb, IIIc are conformational isomers in the solid state. Furthermore, Ib and IIIb have two distinct molecules with different conformation in the unit cell. Besides, it can be found that all crystal forms mentioned above adopt parallel face-to-face slipped stacked arrangement for anthracene or pyrene chromophores. According to exciton coupling theory, J-aggregates (0 < α ≤ 54.7°) exhibit red shifted bands (α is the aligned angle is between transitional moments and the center-to-center axis of the two chromophores).28,29 And it can be found that J-type aggregation exists in IIa, IIIb, IVa (Fig. 3b, 6b and 8b). The closest centroid distance (dc–c) between adjacent anthracene or pyrene rings follow as below (Table 4): 5.431(3) Å, 5.311(5) Å for Ia, Ib of anthracene system and 5.507(4) Å, 4.818(5) Å, 5.505(4) Å, 5.488(2) Å, 4.672(4) Å, 7.784(0) Å for IIa, IIb, IIIa, IIIb, IIIc, IVa of pyrene system. The smallest value of closest centroid distance (dc–c) in these systems is 4.672(4) Å which corresponds to crystal IIIc. The interplanar separation (dπ–π) and lateral displacement (R) between the mean planes of the pyrene moieties for IIIc are 3.462(3) Å and 3.137 Å respectively, which indicate that weak π–π interactions existing between the neighboring pyrene chromophores. No such interactions exist in all other crystal forms.
Table 4 The closest centroid distance dc–c between anthracene or pyrene rings
| Crystal |
dc–c (Å) |
| Ia |
5.431(3) |
| Ib |
5.311(5) |
| IIa |
5.507(4) |
| IIb |
4.818(5) |
| IIIa |
5.505(4) |
| IIIb |
5.488(2) |
| IIIc |
4.672(4) |
| IVa |
7.784(0) |
As shown in Table 5, the dihedral angles between benzene ring and anthracene ring in Ib are much larger than that of Ia. The dihedral angles between benzene ring and pyrene ring in IIIa are also larger than any other crystal forms containing pyrene chromophore. This indicates that cis configuration exhibits worse molecular coplanarity between aromatic rings than trans configuration. This result can also be inferred from the torsion angles data (Table 6 and Scheme 3).
Table 5 The dihedral angles between the benzene ring and anthracene ring/pyrene ring
| Crystal |
Angles (°) |
| The first one is the dihedral angle of molecule A in asymmetry unit and the second one is the dihedral angle of molecule B. |
| Ia |
35.0(2) |
| Ib |
67.8(4)/68.1(3)a |
| IIa |
7.8(2) |
| IIb |
48.3(3) |
| IIIa |
72.0(2) |
| IIIb |
9.02(13)/15.62(13)a |
| IIIc |
39.0(3) |
| IVa |
3.32(16) |
Table 6 Torsion angles of crystal forms
| Compound |
Molecules |
C1–C2–C3–C4/° |
C3–C4–C5–O/° |
C4–C5–C6–C7/° |
| I |
Ia |
−52.3(9) |
−0.7(8) |
23.8(8) |
| A of Ib |
−58.0(1) |
−41.9(1) |
−12.4(1) |
| B of Ib |
54.6(1) |
40.0(1) |
12.2(4) |
| II |
IIa |
11.9(1) |
4.9(1) |
−5.4(9) |
| IIb |
20.0(1) |
13.6(1) |
13.4(1) |
| III |
IIIa |
43.6(8) |
18.9(8) |
29.4(7) |
| A of IIIb |
−11.1(6) |
−2.5(6) |
8.8(5) |
| B of IIIb |
−15.2(6) |
−6.5(6) |
12.0(5) |
| IIIc |
9.4(1) |
10.5(1) |
22.4(1) |
| IV |
IVa |
7.2(7) |
−0.4(7) |
−7.5(6) |
 |
| | Scheme 3 Chemical structure with torsion angles labeled. | |
Powder X-ray diffraction analysis
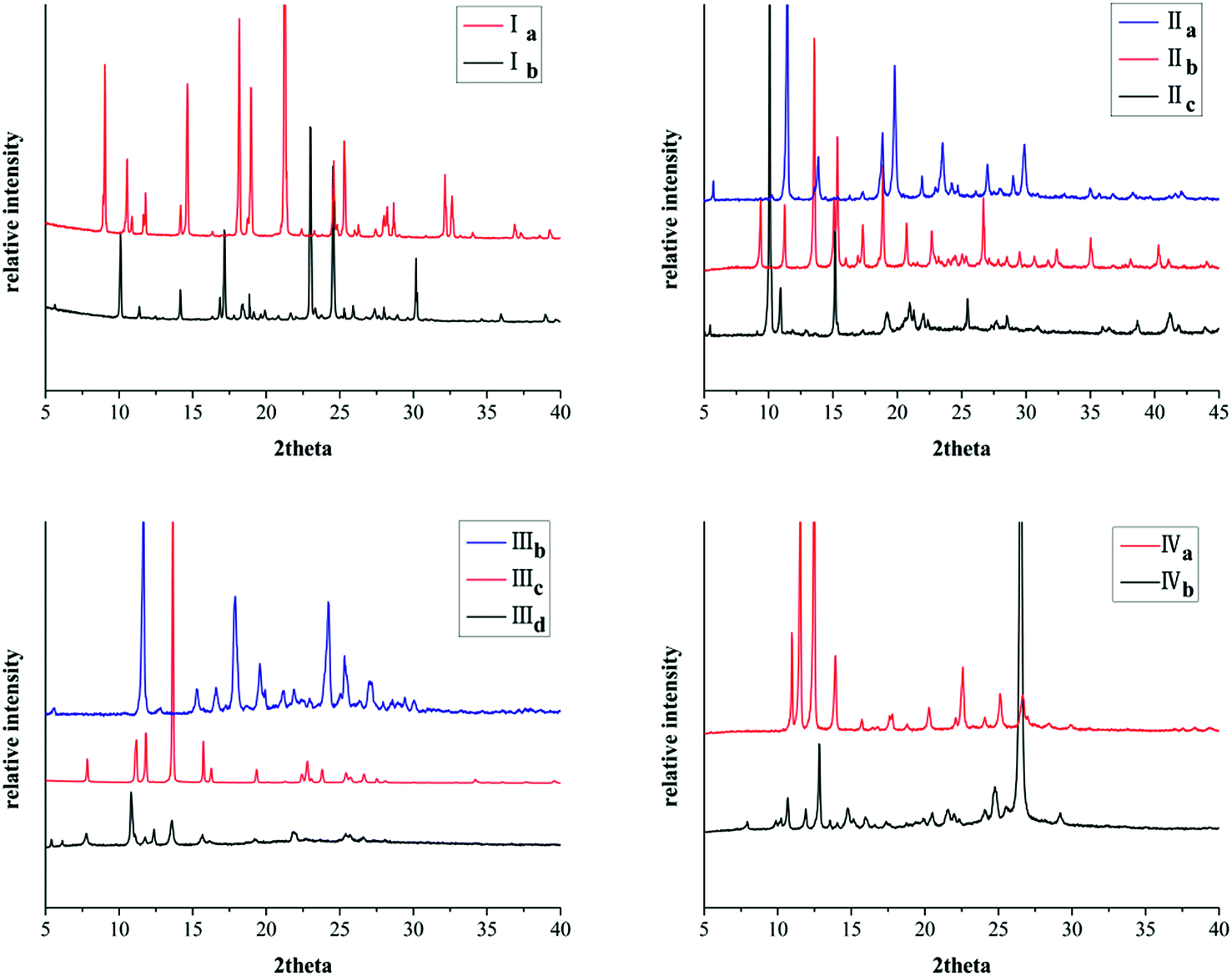
All these crystal forms were determined by powder X-ray diffraction (PXRD) analysis. Remarkably, the PXRD pattern indicates distinct difference for different crystal forms of the same compound (Fig. 9).
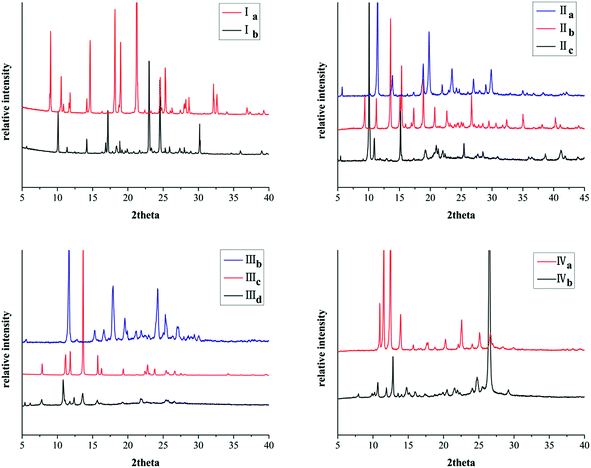 |
| | Fig. 9 PXRD patterns of crystals. | |
Furthermore, PXRD curves coincide well with simulated data calculated from the SXRD data which suggests these forms are in high purity (Fig. 10S, ESI†).
Thermal properties analysis
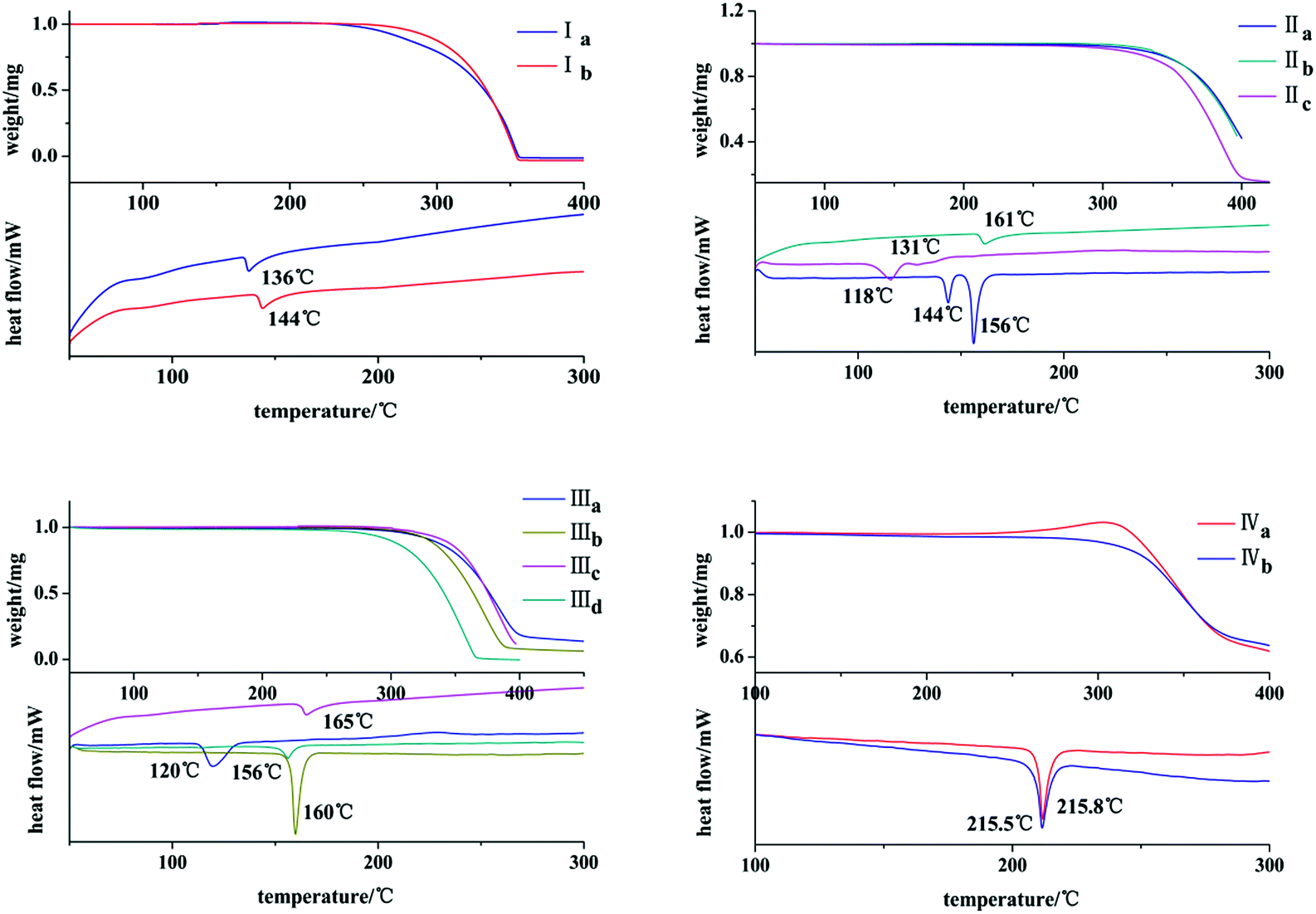
The thermal behaviors of these crystals were investigated by DSC and TGA. It can be seen from Fig. 10 that both Ia and Ib show a endothermic peak at 136 °C and 144 °C, respectively. IIb exists only one endothermic peak at 161 °C. Both IIa and IIc own two endothermic peaks at 144 °C, 156 °C and 118 °C, 131 °C. IIIa, IIIb, IIIc and IIId exhibit one endothermic peak at 120 °C, 160 °C, 165 °C and 156 °C, respectively. Both IVa and IVb show an endothermic peak with pretty similar temperatures of 215.8 °C and 215.5 °C.
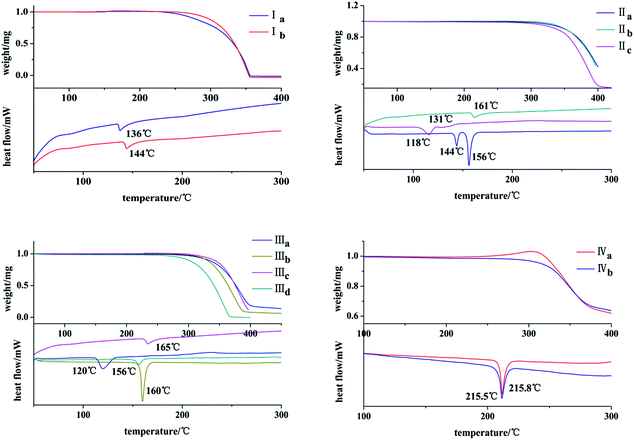 |
| | Fig. 10 TGA/DSC profiles of crystals. | |
Optical–physical properties
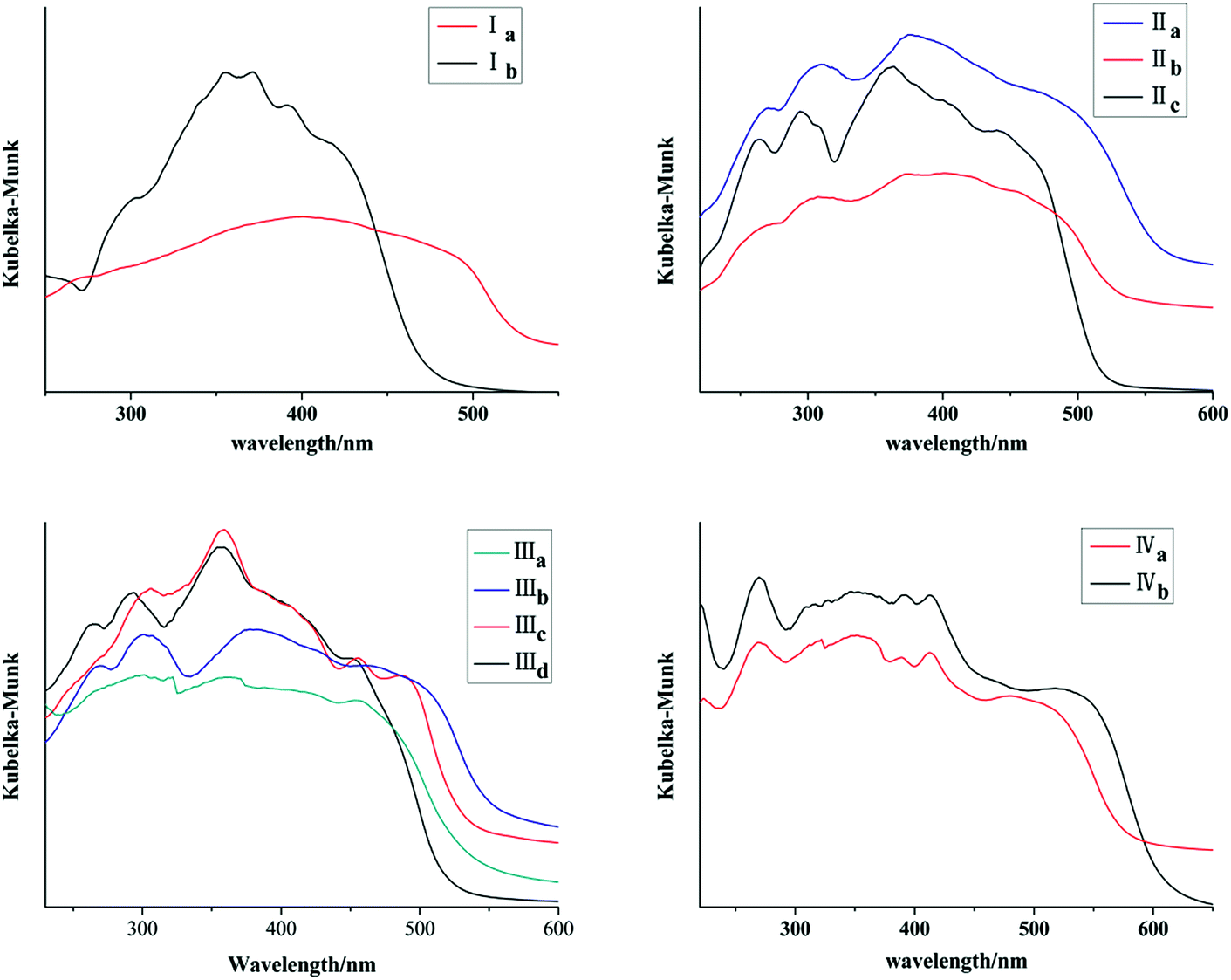
Ia and Ib show similar absorption band in 300–400 nm in acetonitrile which are attributed to anthracene chromophore. All forms with pyrene ring have similar absorption band in 300–450 nm in acetonitrile which are attributed to pyrene chromophore (Fig. 11S, ESI†). As shown in Fig. 11, absorption bands of all crystal forms have red shifted relative to their solution. Furthermore, Ia shows a red shifted absorption edge than that of Ib which can be ascribed to trans configuration of Ia. IIa has the largest absorption edge wavelength in three crystal forms of chalcone II. IIc owns the smallest absorption wavelength. Likewise, IIIb shows the largest absorption edge wavelength in four crystal forms of chalcone III. In addition, IVb shows a red shift in comparison with IVa. All these results are positively correlated with molecular coplanarity in these crystals discussed following.
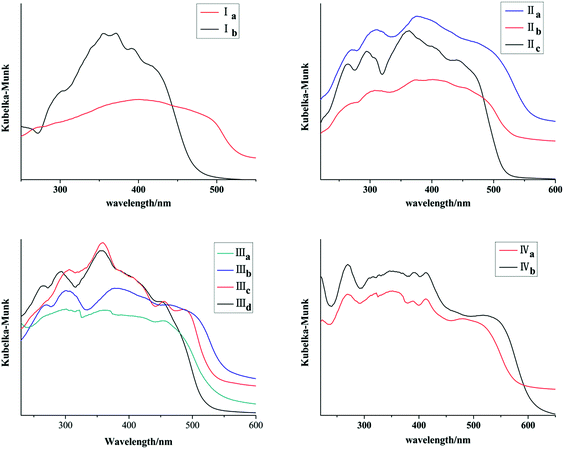 |
| | Fig. 11 Solid-state adsorption spectra of crystals. | |
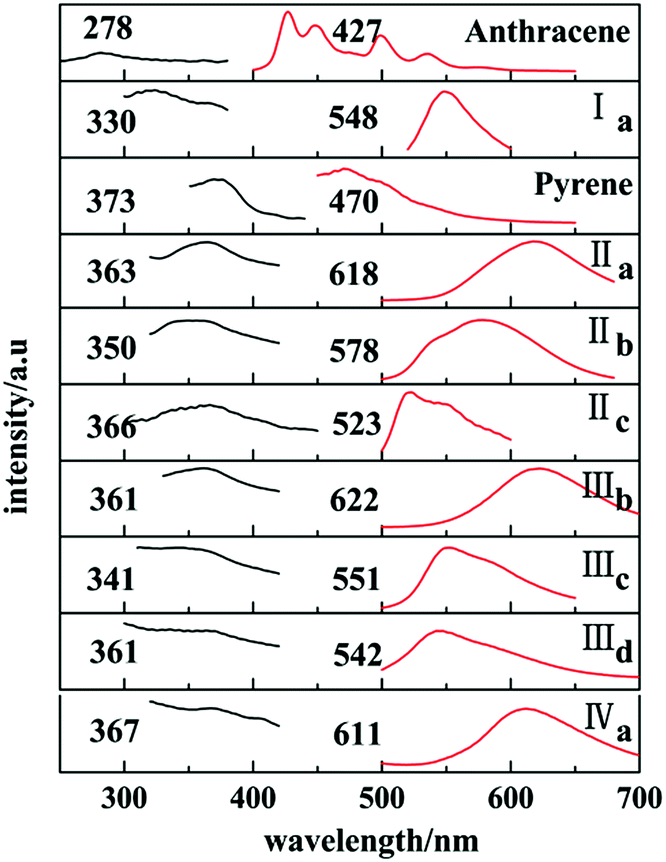
Ia and Ib show similar emission band in 450–500 nm in cyclohexane (Fig. 12S, ESI†) which are attributed to anthracene chromophore. All forms with pyrene chromophore have similar emission bands in trans configuration were found to have fluorescence and have red shifted in comparison with their solutions.
In solid states, only those with trans configuration were found to have fluorescence and have red shifted in comparison with their solutions. Ia has a emission peak at 548 nm which shows a red shifted in contrast with the emission spectra of anthracene crystal.
Similarly, emission spectra of crystals with pyrene chromophore exhibit a red shift in comparison with pyrene which shows characteristic peak at 470 nm. All these can be explained by larger conjugation system than pyrene and overwhelming intermolecular interactions in these crystals. As shown in Fig. 12, forms IIa, IIIb, IVa have longer emission peaks (600–650 nm) relative to that of forms Ia, IIb, IIIc, IIId (570–500 nm). These results can be explained by molecular coplanarity in these crystals.
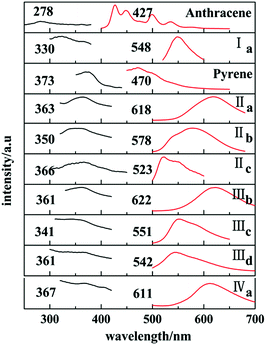 |
| | Fig. 12 Solid-state excitation spectra (black) and fluorescence emission spectra (red). | |
Crystals of forms IIa, IIIb, IVa show the least torsion extent and smallest dihedral angles among these forms so that they own the largest emission peak wavelengths which are consistent with their red fluorescence in microscopy images in Fig. 13. Besides, forms IIc and IIId may own the similar molecules planarity to those of Ia and IIIc by comparing their fluorescence even no SCXRD data.
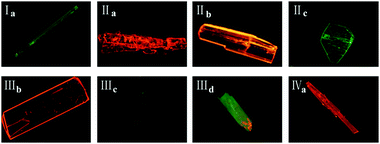 |
| | Fig. 13 Fluorescence microscopy images of eight crystals under UV light (λex = 365 nm). | |
In addition, it has been found that J-aggregates formed in the polymorphs. Researches on the luminescence properties have shown that J-aggregates are highly emissive.19 IIa show a red shifted emission band in contrast with the emission spectra in solution which suggests the J-aggregates formation in its solid state(Fig. 12S, ESI† and Fig. 12). And IIa has the larger emission wavelength than IIb for no J-aggregates form in IIb (Fig. 4 and 12). Likewise, molecules in IIIb also form J-aggregates so that IIIb shows a red shift in solid state emission spectra in comparison with its emission bands in solution. As for IIIc, π⋯π interaction exists in its crystal structure which may induce the excimer formation among pyrene rings. And excimer formation leads to the suppression of fluorescence30 so that IIIc exhibit a blue shifted emission band in contrast with IIIb. In Fig. 8 and 12, molecular aggregation of J-type exists in IVa which leads to a red shift in its solid state emission spectra.
FT-IR spectroscopy
As for FT-IR spectra (Fig. 13S, ESI†), there are two strong vibrational peaks in 1670–1520 cm−1 being magnified in the figure. These two peaks should be separately assigned to carbonyl group with high frequency and ethenyl group in all forms.
Hirshfeld surface calculation
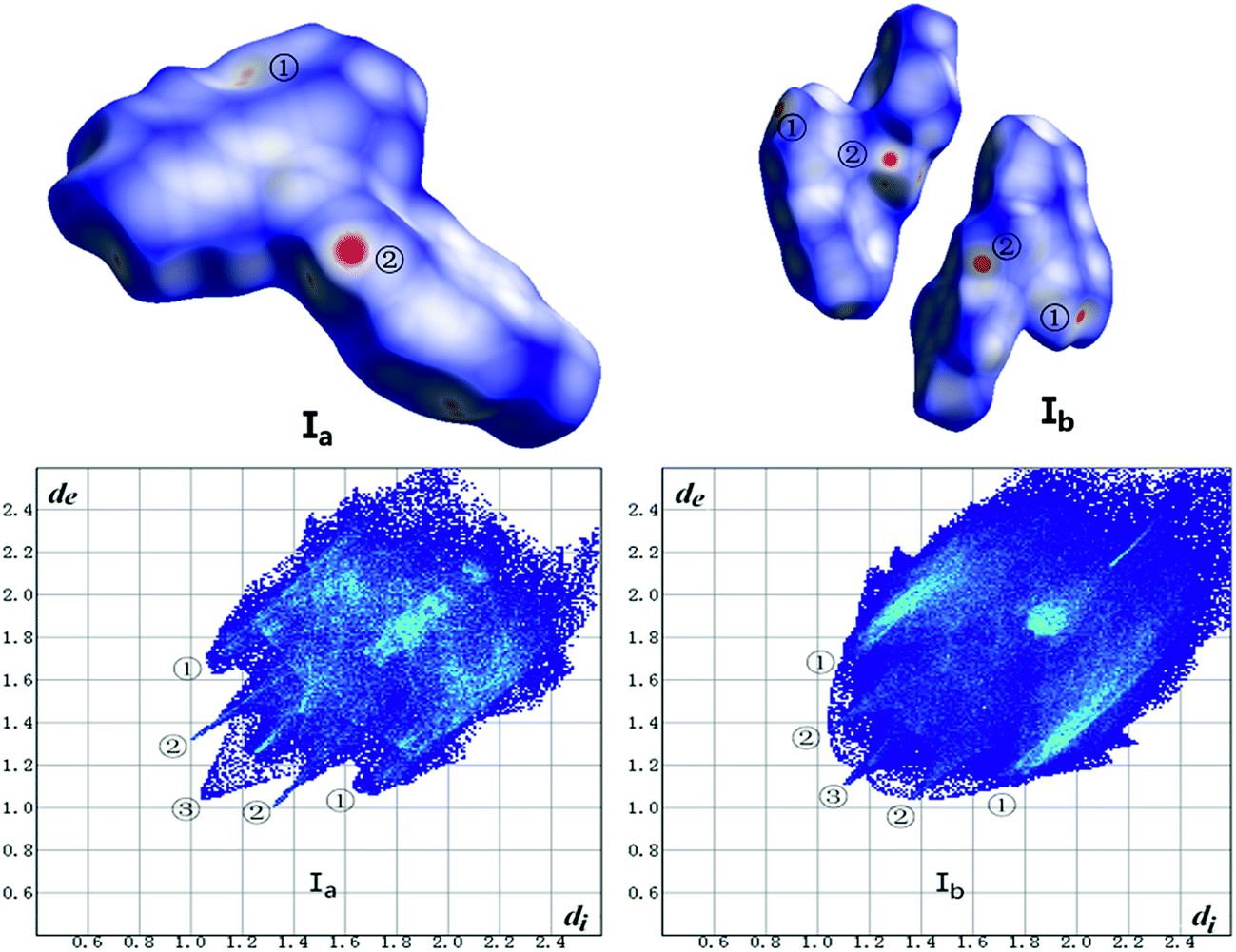
The Hirshfeld surface calculation has been used for all crystals. As shown in Fig. 14 and Table 7, the analysis of forms Ia and Ib was carried out in terms of dnorm surface and fingerprint plots. In addition, the calculation of other forms is listed in ESI (Fig. 14S, ESI†).
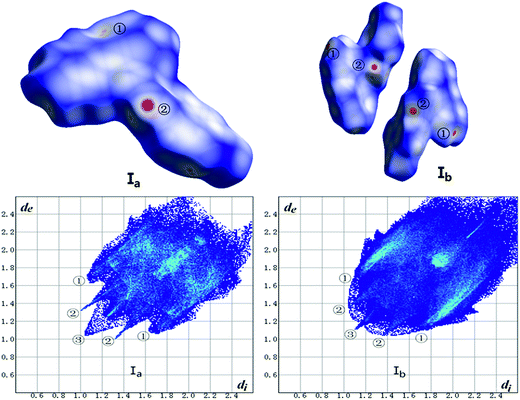 |
| | Fig. 14 Hirshfeld surface mapped with dnorm (top) and fingerprint plots (bottom) of Ib and Ib. Hirshfeld surface colored by dnorm, showing as a red-white-blue scheme as dnorm increase. | |
Table 7 Contributions of individual intermolecular interactions to the Hirshfeld surface of Ia and Ib
| Polymorph |
C–H |
O–H |
H–H |
C–C |
| Ia |
33.8% |
7.6% |
36.9% |
6.0% |
| Ib |
35.7% |
9.3% |
35.6% |
3.6% |
Hirshfeld surface mapped with dnorm takes size of atoms into consideration thus comes out normalized contact distance dnorm which is calculated from de (nearest external distance), di (nearest internal distance) and the van der Waals (vdW) radii of the two atoms to the surface.31 The fingerprint plot derives from Hirshfeld surface that represented by a coordinate (di, de). The colors indicate the number of points with a given fingerprint plot coordinate ranging from blue (relatively few points) through green (moderate fraction) to red (many points).
As shown in Fig. 14, the red hotpots marked ② on the dnorm surface of Ia and Ib correspond to strong C–H⋯O hydrogen bonds. And dnorm surface of Ib shows another two red hotpots labeled ① respectively on the surface of two molecules that can be assigned to C–H⋯π interactions while the same interactions of Ia just exhibit light red spot on the upper right of the surface which indicates weaker C–H⋯π contacts in crystal structure of Ia than Ib.
The two wings marked ① in the fingerprint plots represent C–H⋯π. The contact C–H⋯π contributes 33.8% and 35.6% of the total Hirshfeld surface respectively for Ia and Ib. The little spikes marked ② can be assigned to C–H⋯O hydrogen bonds which separately contributes 7.6% and 9.2% to the whole Hirshfeld surface of Ia and Ib. The slightly larger contribution of C–H⋯O hydrogen bonds in Ib make a contribution to its higher melting point than Ia which coincides with results of thermal analysis. Besides, the H⋯H contacts, as indicated by the blue region marked ③ diffuses around the diagonal, making a significant contribution towards the solid-state stabilization of the two polymorphs. They are 36.9% and 35.7% for Ia and Ib. The center area of the plots where de + di ≈ 3.6 Å corresponds to π⋯π (C⋯C) interactions which contributes only 6.0% and 3.6% for Ia and Ib. To some extent, the worse planarity of Ib than Ia may give a explanation to the different contribution of π⋯π (C⋯C) contacts between Ia and Ib.
Conclusion
In summary, we demonstrated eight chalcone crystal structures in detail. It is evident that the compound of chalcone can crystallize in different configurations or conformations in different solvent systems. Crystals with trans configuration are fluorescent while those with cis configuration are not. Furthermore, forms IIa, IIIb, IVa exist larger red-shifted emissions than forms Ia, IIb, IIIc which can be attributed to better molecular coplanarity and J-aggregates formation in IIa, IIIb, IVa. It implies that solid-state fluorescence of chalcone crystals can be tuned by crystal engineering. And the strategy of tuning molecular coplanarity and J-aggregates formation through polymorphism may provide useful information into organic light-emitting materials.
Acknowledgements
This project is supported by the National Basic Research Program of China (Grant 2011CB302004) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Notes and references
- R. J. Davey, Chem. Commun., 2003, 13, 1463–1467 RSC
 .
. - J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed .
- S. S. Kumar and A. Nangia, Cryst. Growth Des., 2014, 14(4), 1865–1881 CAS .
- L. Q. Yang, Q. X. Yin, B. H. Hou, Y. Bao, J. K. Wang and H. X. Hao, Ind. Eng. Chem. Res., 2013, 52(7), 2477–2485 CrossRef CAS .
- P. Singh, A. Anandb and V. Kumarc, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS PubMed .
- U. J. Griesser, R. K. R. Jetti, M. F. Haddow, T. Brehmer, D. C. Apperley, A. King and R. K. Harris, Cryst. Growth Des., 2008, 8(1), 44–56 CAS .
- G. Klebe, F. Graser and E. Hadicke, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 69–77 CrossRef .
- H. G. Gallagher and J. N. Sherwood, J. Chem. Soc., Faraday Trans., 1996, 92(12), 2107–2116 RSC .
- S. J. Yoon and S. Y. Park, J. Mater. Chem., 2011, 21, 8338–8346 RSC .
- S. H. Lapidus, A. Naik, A. Wixtrom, N. E. Massa, V. Ta Phuoc, L. del Campo, S. Lebegue, J. G. Angyan, T. Abdel-Fattah and S. Pagola, Cryst. Growth Des., 2014, 14, 91–100 CAS .
- R. A. Esteves de Castro, J. Canotilho, R. M. Barbosa, M. R. Silva, A. M. Beja, J. A. Paixao and J. S. Redinha, Cryst. Growth Des., 2007, 7(3), 496–500 CAS .
- T. M. R. Maria, R. A. E. Castro, S. S. Bebiano, M. R. Silva, A. M. Beja, J. Canotilho and M. E. S. Eusebio, Cryst. Growth Des., 2010, 10, 1194–1200 CAS .
- P. S. Pereira Silva, R. A. E. Castro, E. Melro, M. R. Silva, T. M. R. Maria, J. Canotilho and M. E. S. Eusebio, J. Therm. Anal. Calorim., 2015, 120(1), 667–677 CrossRef CAS .
- G. Q. Zhang, J. W. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed .
- S. Kervyn, O. Fenwick, F. Di Stasio, Y. S. Shin, J. Wouters, G. Accorsi, S. Osella, D. Beljonne, F. Cacialli and D. Bonifazi, Chem.–Eur. J., 2013, 19, 7771–7779 CrossRef CAS PubMed .
- S. P. Anthony, Chem.–Asian J., 2012, 7(3), 374–379 CrossRef CAS PubMed .
- R. M. Zhang, M. L. Wang, H. Sun, A. Khan, R. Usman, S. Z. Wang, X. T. Gu, J. Wang and C. X. Chun, New J. Chem., 2016, 44, 6441–6450 RSC .
- F. Wurthner, T. E. Kaiser and C. R. Saha-Moller, Angew. Chem., Int. Ed., 2011, 13, 3376–3410 CrossRef PubMed .
- Y. H. Deng, W. Yuan, Z. Jia and G. Jiu, J. Phys. Chem. B, 2014, 118, 14536–14545 CrossRef CAS PubMed .
- A. M. Asiri and S. A. Khan, Mater. Lett., 2011, 65, 1749–1752 CrossRef CAS .
- P. S. Patil, S. R. Maidur, S. V. Rao and S. M. Dharmaprakash, Opt. Laser Technol., 2016, 81, 70–76 CrossRef CAS .
- Z. K. Si, Q. Zhang, M. Z. Xue, Q. R. Sheng and Y. G. Liu, Res. Chem. Intermed., 2011, 37(6), 635–646 CrossRef CAS .
- A. M. Asiri and S. A. Khan, J. Heterocycl. Chem., 2012, 49(6), 1434–1438 CrossRef CAS .
- H. Singh, J. Sindhu and J. M. Khurana, J. Lumin., 2015, 158, 340–350 CrossRef CAS .
- R. Nithya, N. Santhanamoorthi, P. Kolandaivel and K. Senthilkumar, J. Phys. Chem. A, 2011, 115, 6594–6602 CrossRef CAS PubMed .
- Y. X. Li, H. B. Zhou, J. L. Miao, G. X. Xun, G. B. Li, Y. Nie, C. L. Chen, Z. Chen and X. T. Tao, CrystEngComm, 2012, 14, 8286–8291 RSC .
- Y. B. Wang, Z. L. Wang, T. L. Liang, H. B. Fu and J. N Yao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o630 CAS .
- E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721–722 CrossRef CAS .
- M. Kasha, R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS .
- J. N. Moorthy, P. Venkatakrishnan, D. F. Huang and T. J. Chow, Org. Lett., 2007, 9(25), 5215–5218 CrossRef CAS PubMed .
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: Microscopy graphs of all forms; NMR spectra of chalcone compounds; comparing of PXRD experimental patterns (black) of crystals with simulated patterns (red); absorption spectra of crystals in acetonitrile solvent; fluorescence spectra (λex = 365 nm) in cyclohexane solution for all forms; IR spectra of crystals; Hirshfeld surface mapped with dnorm and fingerprint plots of crystals; solvents for crystal preparation; melting point, enthalpy and decomposition temperature range of all crystals; the maximum absorption and emission peak of all solids; contributions of individual intermolecular interactions to the Hirshfeld surface of all crystals; X-ray crystallographic information files (CIF) for eight crystals. CCDC 1473276, 1473277, 1473283, 1473284, 1477084, 1473288, 1473285 and 1473292. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27458g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hao Suna,
Yaqi Shana,
Man Dua,
Arshad Khana,
Rabia Usmana,
Wei Zhanga,
Hongbin Shana and
Chunxiang Xu*b
*a,
Hao Suna,
Yaqi Shana,
Man Dua,
Arshad Khana,
Rabia Usmana,
Wei Zhanga,
Hongbin Shana and
Chunxiang Xu*b