
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-neuroinflammatory asarone derivatives from the rhizomes of Acorus tatarinowii†
Da-Peng Qin‡
a,
Xiao-Lin Feng‡a,
Wei-Yang Zhangd,
Hao Gaoa,
Xiao-Rui Chengc,
Wen-Xia Zhouc,
Yang Yu*ab and
Xin-Sheng Yao *a
*a
aInstitute of Traditional Chinese Medicine & Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632, P. R. China. E-mail: 1018yuyang@163.com; tyaoxs@jnu.edu.cn; Fax: +86-20-85221559; Tel: +86-20-85221559
bState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, P. R. China
cBeijing Institute of Pharmacology and Toxicology, Beijing 100850, P. R. China
dState Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Macau, P. R. China
First published on 26th January 2017
Abstract
A novel 7-O-7′ type mesomeric neolignan, meso-asarolignan A (1), six pairs of new neolignan enantiomers, (±)-asarolignan B–G (2a/2b, 3a/3b, 4–6, 7a/7b), along with 16 known analogues (8–23) were isolated from the rhizomes of Acorus tatarinowii. The structures of the new compounds were elucidated by extensive analysis of spectroscopic, X-ray diffraction, and computational data. Compounds 1–3 represent rare examples of naturally occurring lignans with a 7-O-7′ linkage pattern. All isolated compounds were assayed for their anti-neuroinflammatory effects on tumor necrosis factor α (TNF-α) production in an activated murine microglia cell line. Compounds 7(a/b), 14(a/b), 16, 20, and 22 reduced TNF-α levels without high cell toxicity in LPS-activated BV-2 cells.
Introduction
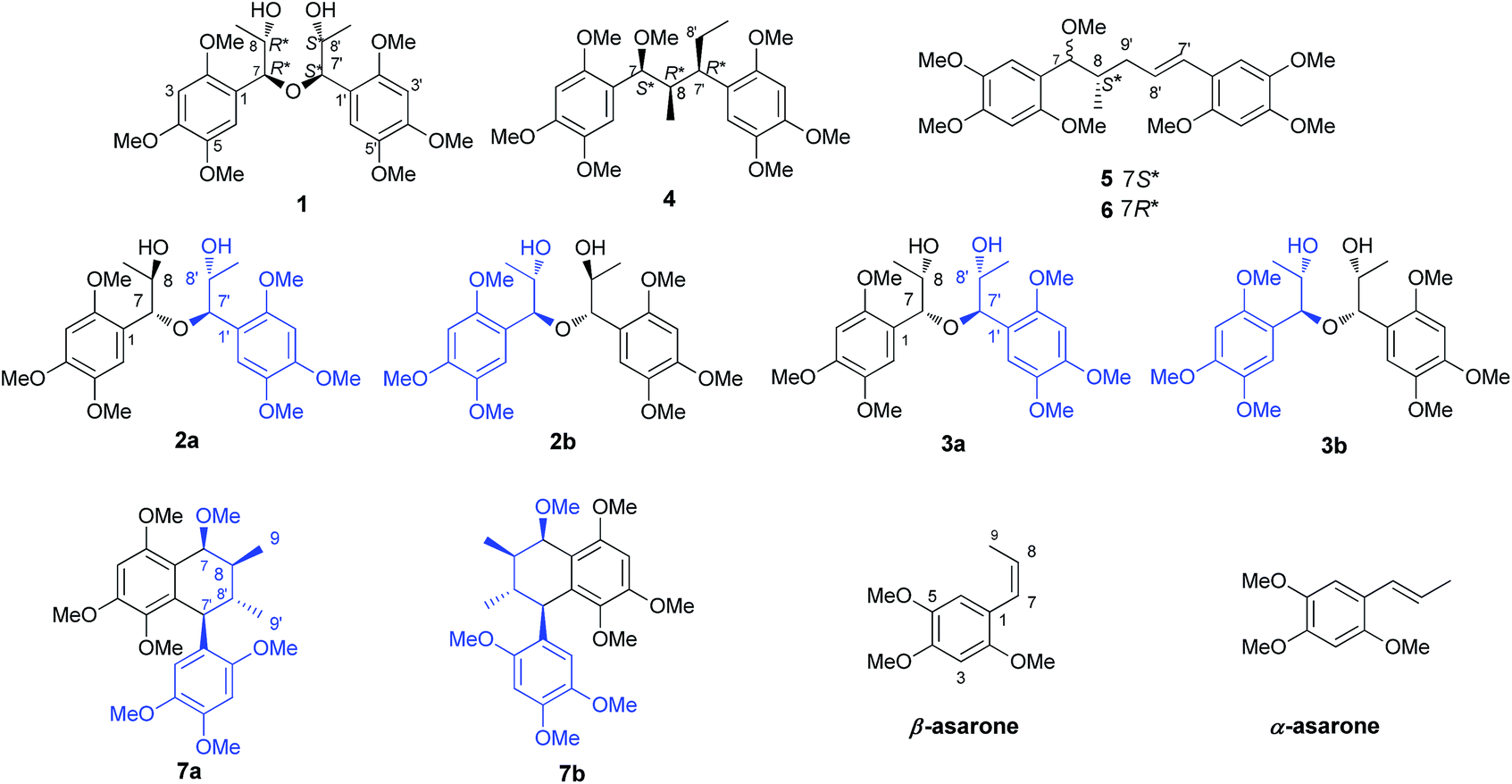
Acorus tatarinowii has been historically used as a principal medicine in traditional Chinese formulas for the treatment of learning and memory disorders, dementia and dysmnesia.1,2 Previous studies on Acorus tatarinowii have revealed that the most notable constituents are α- and β-asarones, which exhibit a wide spectrum of biological activities, such as cerebrovascular protection, anti-cancer, antioxidation, neuroprotection, anti-neuroinflammation, improving cognitive function, antidepressant-like effects and antibacterial activity.3–8Over the course of our continuing investigations on bioactive asarone analogues from the rhizomes of Acorus tatarinowii, 14 neolignans (1–7, 9–15), one quinone (8), six asarone analogues (16–21), along with α- and β-asarones (22 and 23) were isolated. Interestingly, compounds 1–3 were obtained as stereoisomers, represented the first examples of naturally occurring 7-O-7′ type neolignans. meso-Asarolignan A (1) was defined as a mesomer, while (±)-asarolignan B (2) and C (3) were two pairs of 7-O-7′ neolignan enantiomers. The resolution of enantiomers 2 and 3 by chiral HPLC led to the isolation of 2a/2b and 3a/3b. Considering the racemic nature of the compounds from this plant, compounds 3–8 were also determined as racemic mixtures due to their negligible optical activities. In addition, all isolated compounds were assayed for their anti-neuroinflammatory effects on tumor necrosis factor α (TNF-α) production in the murine microglia BV-2 cells.
Results and discussion
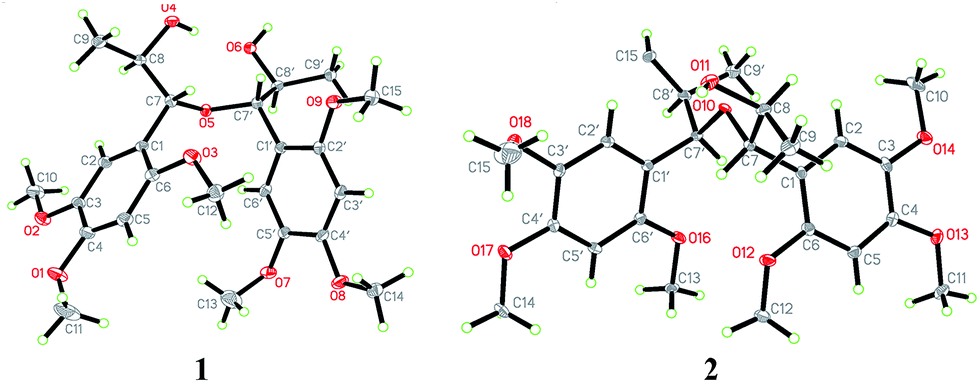
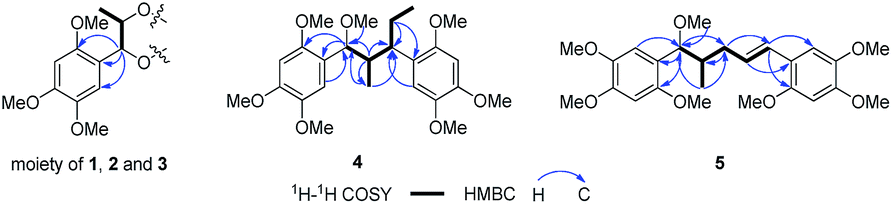
Compound 1 was obtained as colourless block crystal. Its molecular formula was determined to be C24H34O9 by HRESIMS at m/z 489.2094 [M + Na]+ (calcd 489.2101). The UV spectrum exhibited absorption maxima at 205, 231 (sh) and 291 nm. Its IR spectrum disclosed absorption bands assignable to hydroxyl (3378 cm−1) and benzene ring moiety (1614, 1512, and 1465 cm−1). The 1H NMR spectrum of 1 (Table 1) showed the signals for a 1,2,4,5-tetrasubstituted benzene ring [δH 6.67 (s, H-6/6′), 6.41 (s, H-3/3′)], two oxygenated methines [δH 4.57 (d, J = 7.9 Hz, H-7/7′), 3.88 (m, H-8/8′)], one methyl [δH 0.92 (d, J = 6.4 Hz, Me-9/9′)] and three methoxy groups [δH 3.76, 3.62, 3.56 (each 3H, s)]. The 13C NMR in combination with DEPT135 experiments resolved 12 carbon signals, corresponding to six aromatic carbons, two oxygenated methine carbons, one methyl, and three methoxy groups. These data together with the molecular formula could make us assume that 1 might be a highly symmetrical structure. Further analysis of the 1H–1H COSY, HSQC and HMBC spectra allowed the establishment of the partial C6–C3 moiety (1′,2′-dihydroxyasarone moiety, half of the structure) in compound 1 (Fig. 4). The large coupling constant between H-7 (H-7′) and H-8 (H-8′) (J = 7.9 Hz) indicated a threo relative configuration. Connection of the two C6–C3 units in 1 was mainly carried out by using the results of HMBC and X-ray analyses. The key HMBC correlations from H-7 to C-7′ or H-7′ to C-7 indicated the linkage of the two C6–C3 units through C-7-O-C-7′ bonds. The fact that compound 1 bears four chiral centres and symmetric nature, was undoubtedly confirmed by X-ray (Fig. 2) using anomalous scattering of Mo Kα radiation. The crystals of 1 belong to monoclinic space group P21/c, showing a helical conformation with an intramolecular mirror symmetry. These findings, combined with the lack of optical activity supported that compound 1 was obtained as a mesomer. Therefore, the structure of 1 was established as a novel 7-O-7′ neolignan mesomer, and named meso-asarolignan A.| 1 | 2 | 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | δC | δH (J in Hz) | δC | δH (J in Hz) | No. | δC | δH (J in Hz) | No. | δC | δH (J in Hz) |
| a Recorded at 400 (1H) and 100 MHz (13C) in CD3OD. Multiplets or overlapped signals are reported without designating multiplicity. | ||||||||||
| 1,1′ | 121.9 | 120.4 | 1 | 120.5 | 1′ | 119.1 | ||||
| 2,2′ | 153.1 | 154.1 | 2 | 154.4 | 2′ | 154.3 | ||||
| 3,3′ | 98.7 | 6.41 (1H, s) | 99.2 | 7.09 (1H, s) | 3 | 99.0 | 6.62 (1H, s) | 3′ | 99.3 | 6.62 (1H, s) |
| 4,4′ | 150.5 | 150.7 | 4 | 150.9 | 4′ | 150.6 | ||||
| 5,5′ | 144.2 | 144.7 | 5 | 144.8 | 5′ | 144.5 | ||||
| 6,6′ | 114.4 | 6.67 (1H, s) | 114.3 | 6.62 (1H, s) | 6 | 113.8 | 7.01 (1H, s) | 6′ | 115.3 | 7.19 (1H, s) |
| 7,7′ | 83.0 | 4.57 (1H, d, 7.9) | 77.0 | 4.35 (1H, d, 6.5) | 7 | 77.3 | 4.39 (1H, d, 7.4) | 7′ | 75.9 | 4.45 (1H, d, 3.2) |
| 8,8′ | 73.1 | 3.88 (1H, m) | 72.1 | 3.85 (1H, m) | 8 | 72.4 | 3.85 (1H, m) | 8′ | 71.3 | 3.98 (1H, m) |
| 9,9′ | 18.8 | 0.92 (3H, d, 6.4) | 19.1 | 0.94 (3H, d, 6.4) | 9 | 18.9 | 0.91 (3H, d, 6.3) | 9′ | 18.6 | 1.00 (3H, d, 6.6) |
| 2,2′-OMe | 56.8 | 3.76 (3H, s) | 56.8 | 3.62 (3H, s) | 2-OMe | 56.8 | 3.61 (3H, s) | 2′-OMe | 56.8 | 3.61 (3H, s) |
| 4,4′-OMe | 57.2 | 3.56 (3H, s) | 56.7 | 3.85 (3H, s) | 4-OMe | 56.9 | 3.85 (3H, s) | 4′-OMe | 55.7 | 3.85 (3H, s) |
| 5,5′-OMe | 56.8 | 3.62 (3H, s) | 57.2 | 3.81 (3H, s) | 5-OMe | 57.4 | 3.80 (3H, s) | 5′-OMe | 57.2 | 3.83 (3H, s) |
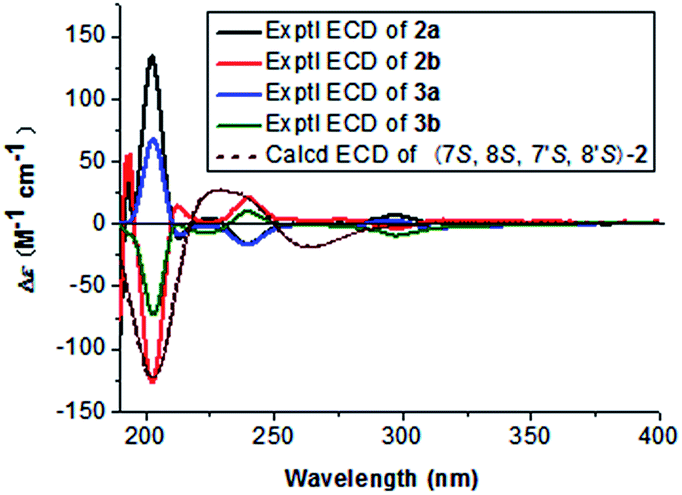
Compound 2 (2a/2b), obtained as a colourless block crystal, possesses the same molecular formula (C24H34O9) as compound 1, as determined by HREIMS [M + Na]+ ion at m/z 489.2101 (calcd 489.2101). The NMR data of 2 (Table 1) were similar to those of 1, except that broad signal of C-6/6′ was obviously observed at δC 114.3 ppm and the broad signal of oxygenated methine considerably upfield shifted to δC 77.0 ppm [δC 80.8 (C-7/7′) in 1]. Similarly, the same C6–C3 moiety (1′,2′-dihydroxyasarone, Fig. 4) with a threo relative configuration was assigned in 2. Crystals of 2 were obtained from CH3OH–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which allowed the establishment of the structure of 2 by X-ray analysis (Fig. 2). Thus, 2 was unambiguously defined as a stereoisomer of 1. The chiral HPLC and optical rotation data indicated that 2 was racemic mixture. Subsequent chiral resolution of 2 by HPLC yielded the enantiomers 2a and 2b in a 1:1 ratio. The enantiomers displayed typical antipodal ECD curves (Fig. 3) and specific rotations of opposite sign (2a: [α]23D +173; 2b: [α]23D −174). Furthermore, the configuration of 2b was determined as 7S,8S,7′S,8′S on the basis of the good agreement with calculated ECD spectra of (7S,8S,7′S,8′S)-2, and the configuration of 2a was determined to be 7R,8R,7′R,8′R. Finally, the enantiomers 2a and 2b were named as (+)-asarolignan B and (−)-asarolignan B, respectively.
:1), which allowed the establishment of the structure of 2 by X-ray analysis (Fig. 2). Thus, 2 was unambiguously defined as a stereoisomer of 1. The chiral HPLC and optical rotation data indicated that 2 was racemic mixture. Subsequent chiral resolution of 2 by HPLC yielded the enantiomers 2a and 2b in a 1:1 ratio. The enantiomers displayed typical antipodal ECD curves (Fig. 3) and specific rotations of opposite sign (2a: [α]23D +173; 2b: [α]23D −174). Furthermore, the configuration of 2b was determined as 7S,8S,7′S,8′S on the basis of the good agreement with calculated ECD spectra of (7S,8S,7′S,8′S)-2, and the configuration of 2a was determined to be 7R,8R,7′R,8′R. Finally, the enantiomers 2a and 2b were named as (+)-asarolignan B and (−)-asarolignan B, respectively.
The molecular formula of compound 3 (3a/3b), C24H34O9, was identical to those of 2, as indicated by HRESIMS at m/z 489.2103 [M + Na]+ (calcd 489.2101). The NMR data of 3 (Table 1) were similar to those of 2, except that two sets of signals for C6–C3 units were distinctly observed in 3. A detailed comparison and analyses of the NMR data of 3 revealed the establishment of two same C6–C3 moieties (1′,2′-dihydroxyasarone, Fig. 4). The relative configurations were assigned as 7,8-threo and 7′,8′-erythro due to the observed coupling constants of H-7/H-8 (J7,8 = 7.4 Hz) and H-7′/H-8′ (J7′,8′ = 3.2 Hz), respectively. Thus, the structure of 3 was defined as a stereoisomer of 2 with different relative configurations as shown in Fig. 1. Similarly, the enantiomers (+)-3a and (−)-3b were obtained and showed opposite ECD curves and optical rotations of opposite sign. Comparing the CD spectra of 2 (a/b) and 3 (a/b) in Fig. 3, the configurations of 3a and 3b were determined to be 7R,8S,7′R,8′R and 7S,8R,7′S,8′S, respectively. Thus, two enantiomers of 3 were assigned as (+)-asarolignan C (3a) and (−)-asarolignan C (3b).
 | ||
| Fig. 1 Chemical structures from the rhizomes of A. tatarinowii. | ||
 | ||
| Fig. 2 X-ray crystallographic structures of 1 and 2. | ||
 | ||
| Fig. 3 Experimental ECD spectra of 2a, 2b and 3a, 3b, and calculated ECD spectrum of (7S,8S,7′S,8′S)-2. | ||
(±)-Asarolignan D (4), white solid, was assigned molecular formula C25H36O7 by HRESIMS (m/z 471.2363, [M + Na]+) with eight degrees of unsaturation. The IR spectrum showed the presence of aromatic ring (1509, and 1458 cm−1). The NMR data of 4 (Table 2) showed four aromatic proton signals at [δH 6.86 (1H, s, H-6), 6.84 (1H, s, H-6′), 6.68 (1H, s, H-3′), and 6.64 (1H, s, H-3)], which were ascribed to the protons of two tetrasubstituted aromatic rings. Moreover, three methines groups [δH 4.22 (1H, d, J = 3.5 Hz, H-7), 3.04 (1H, br s, H-7′), 1.94 (1H, m, H-8)], one methylene [δH 1.78 (1H, m, H-8′a), 1.52 (1H, m, H-8′b)], two methyls [δH 0.84 (3H, d, J = 7.0 Hz, Me-9), and 0.65 (3H, t, J = 7.4 Hz, Me-9′)], and seven methoxy groups [δH 3.83, 3.81, 3.80, 3.78, 3.75, 3.70, 2.99 (each 3H, s)] were also observed. The 1H–1H COSY spectrum showed two spin coupling systems arising from two C3 substructures (C-7–C-8–C-9 and C-7′–C-8′–C-9′) shown in bold lines in Fig. 4. In the HMBC spectrum, correlations from H-6 to C-7, H-7 to C-1/C-2/C-6, H-6′ to C-7′, and H-8′a to C-1′, allowed the establishment of two C6–C3 units. Furthermore, the 1H–1H COSY correlations from H-8 to H-7′, together with the key HMBC correlations from Me-9 to C-7/C-7′, and H-8 to C-1′/C-8′, indicated the two C6–C3 units were connected via C-8–C-7′ bond form a neolignan (Fig. 4). A methoxy group at δH 2.99 was located at C-7 as confirmed by the HMBC correlation from MeO-7 to C-7. The remaining six methoxy resonances were unambiguously located on the aromatic rings by using the HMBC and NOE experiments. Finally, an X-ray diffraction analysis was performed, from which the relative configuration of 4 (7S*,8′R*,7′R*) was determined (Fig. 5). The lack of optical activity suggested that 4 was obtained as a racemate. Attempts to separate the enantiomers of 4 using different chiral phase columns were not successful.
| No. | 4a | 5b | 6c | 7b | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a Recorded at 600 (1H) and 125 MHz (13C) in acetone-d6.b Recorded at 400 (1H) and 100 MHz (13C) in CD3OD.c Recorded at 300 (1H) and 75 MHz (13C) in CD3OD. Multiplets or overlapped signals are reported without designating multiplicity. | ||||||||
| 1 | 122.5 | 122.0 | 122.0 | 120.5 | ||||
| 2 | 152.7 | 154.1 | 153.8 | 155.1 | ||||
| 3 | 99.2 | 6.64 (1H, s) | 99.7 | 6.58 (1H, s) | 99.4 | 6.58 (1H, s) | 96.0 | 6.55 (1H, s) |
| 4 | 150.0 | 150.6 | 150.4 | 154.3 | ||||
| 5 | 144.2 | 144.7 | 144.5 | 141.8 | ||||
| 6 | 113.7 | 6.86 (1H, s) | 113.2 | 6.87 (1H, s) | 113.1 | 6.87 (1H, s) | 137.9 | |
| 7 | 79.7 | 4.22 (1H, d, 3.5) | 82.0 | 4.39 (1H, d, 7.6) | 81.6 | 4.44 (1H, d, 6.1) | 76.2 | 4.55 (1H, d, 1.9) |
| 8 | 43.3 | 1.94 (1H, m) | 40.9 | 1.90 (1H, m) | 40.6 | 1.88 (1H, m) | 42.0 | 1.37 (1H, m) |
| 9 | 11.8 | 0.84 (3H, d, 7.0) | 16.2 | 0.75 (3H, d, 6.9) | 15.9 | 0.94 (3H, d, 6.6) | 17.2 | 1.10 (1H, d, 6.8) |
| 1′ | 125.2 | 120.6 | 120.3 | 131.0 | ||||
| 2′ | 153.9 | 152.6 | 152.5 | 152.9 | ||||
| 3′ | 99.2 | 6.68 (1H, s) | 99.1 | 6.64 (1H, s) | 98.9 | 6.62 (1H, s) | 99.1 | 6.65 (1H, s) |
| 4′ | 149.3 | 150.6 | 150.5 | 144.6 | ||||
| 5′ | 144.2 | 144.8 | 144.6 | 148.8 | ||||
| 6′ | 115.9 | 6.84 (1H, s) | 112.1 | 6.96 (1H, s) | 111.7 | 6.92 (1H, s) | 115.5 | 6.58 (1H, s) |
| 7′ | 43.3 | 3.04 (1H, br s) | 126.5 | 6.57 (1H, d, 14.7) | 126.1 | 6.50 (1H, d, 15.9) | 41.8 | 4.16 (1H, d, 8.7) |
| 8′ | 25.3 | 1.78 (1H, m), 1.52 (1H, m) | 128.7 | 6.03 (1H, dt, 14.7, 6.5) | 128.7 | 5.90 (1H, dt, 15.9, 7.1) | 39.5 | 1.86 (1H, m) |
| 9′ | 12.7 | 0.65 (3H, t, 7.4) | 37.8 | 2.53 (1H, m), 2.06 (1H, m) | 38.7 | 2.18 (1H, m), 2.00 (1H, m) | 18.1 | 0.93 (3H, d, 6.7) |
| 7-OMe | 57.3 | 2.99 (3H, s) | 57.0 | 3.15 (3H, s) | 57.1 | 3.16 (3H, s) | 57.7 | 3.45 (3H, s) |
| 2-OMe | 56.4 | 3.70 (3H, s) | 57.1 | 3.77 (3H, s) | 57.0 | 3.78 (3H, s) | 56.7 | 3.85 (3H, s) |
| 4-OMe | 56.5 | 3.81 (3H, s) | 56.7 | 3.83 (3H, s) | 56.7 | 3.81 (3H, s) | 56.7 | 3.79 (3H, s) |
| 5-OMe | 57.6 | 3.75 (3H, s) | 57.5 | 3.76 (3H, s) | 57.4 | 3.77 (3H, s) | 57.0 | 3.06 (3H, s) |
| 2′-OMe | 56.5 | 3.78 (3H, s) | 56.8 | 3.77 (3H, s) | 56.7 | 3.77 (3H, s) | 56.7 | 3.88 (3H, s) |
| 4′-OMe | 57.2 | 3.83 (3H, s) | 56.7 | 3.81 (3H, s) | 56.6 | 3.81 (3H, s) | 57.0 | 3.58 (3H, s) |
| 5′-OMe | 56.5 | 3.80 (3H, s) | 57.4 | 3.76 (3H, s) | 57.4 | 3.71 (3H, s) | 56.7 | 3.81 (3H, s) |
 | ||
| Fig. 4 Key 1H–1H COSY and HMBC correlations of 1–5. | ||
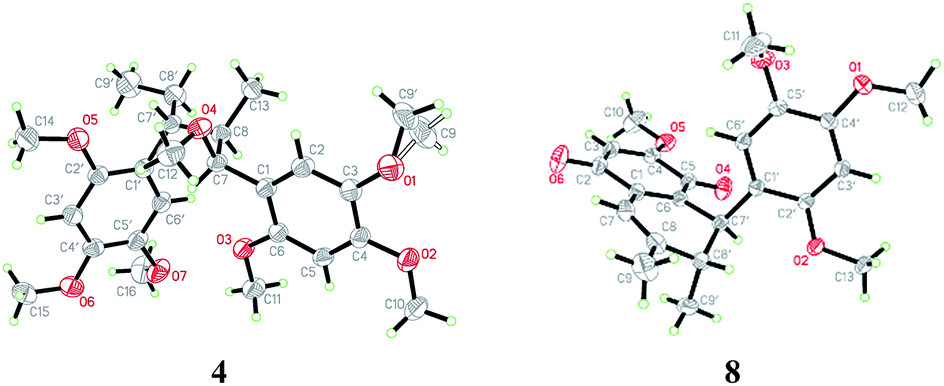 | ||
| Fig. 5 X-ray crystallographic structure of 4 and 8. | ||
(±)-Asarolignan E (5), colourless oil, was assigned the molecular formula C25H34O7 by HRESIMS (m/z 469.2203, [M + Na]+) with nine degrees of unsaturation. The 1H and 13C NMR data (Table 2) indicated its structural similarity to 4, except for the replacement of a methine and a methylene groups by two olefinic carbons. By 1H–1H COSY, HSQC, and HMBC spectra, two C6–C3 units were assigned as shown in Fig. 4. Similarly, the connection between C-8 and C-9′ was determined as the correlations from Me-9 to C-7/C-9′, H-7 to C-9′, and H-9′ to C-8/C-9/C-7′. The large coupling constant of H-7/H-8 (J7,8 = 7.6 Hz), indicated a threo relative configuration. On the basis of the negligible optical activity, compound 5 was also deduced as a racemic mixture and named as (±)-asarolignan E.
(±)-Asarolignan F (6), colourless oil, had the same molecular formula as 5, established by HRESIMS. The 1H and 13C NMR spectroscopic data of 6 (Table 2) indicated them to be very similar to 5, differing only in the relative configurations at C-7/8. The coupling constant of H-7/H-8 was 6.1 Hz, which suggested that the configuration was erythro in comparison with a higher value in threo (J7,8 = 7.6 Hz) configuration of 5. Thus, 6 was identified as (±)-asarolignan F.
(±)-Asarolignan G (7) was obtained as yellow oil. Its molecular formula was assigned C25H34O7 by HRESIMS (m/z 469.2196, [M + Na]+) with nine degrees of unsaturation. The 13C NMR and DEPT-135 spectra exhibited 25 carbon signals, corresponding to two aromatic rings, four methines, two methyls and seven methoxy groups. Based on these data, the skeleton of 7 was speculated to be a lignan with seven methoxy substitutes. The 1H–1H COSY spectrum suggested the presence of H-7/H-8/H-9, H-7′/H-8′/H-9′ and H-8/H-8′ units (bold lines in Fig. 6), and the HMBC correlations of H-7 to C-1/C-2/C-6/C-8′, and H-7′ to C-1′/C-2′/C-6′/C-1/C-5/C-6, were indicative of a tetrahydronaphthalene lignan structure. The other six methoxy groups linked to two substituted aromatic rings by HMBC and NOE experiments, respectively (Fig. 6). Furthermore, the coupling constant of H-7/H-8 (J7,8 = 1.9 Hz) and H-7′/H-8′ (J7′,8′ = 8.7 Hz), demonstrated the cis and trans relative configurations of H-7/H-8 and H-7′/H-8′, respectively. In the NOESY spectrum, the correlations of Me-9 and MeO-7, H-8 and H-7′, as well as Me-9′ and H-7′ suggested that H-7, H-8 and H-7′ were β-oriented, whereas H-8′ was situated on the opposite side. Considering the racemic nature, chiral HPLC analysis was performed and the resolved enantiomers (7a and 7b) possessed the opposite CD curves (Fig. 7). The absolute configuration of 7a was determined as 7S,8S,7′S,8′R by the CD data showing a negative Cotton effect at 285 nm. Similarly, the absolute configuration of 7b was elucidated as 7R,8R,7′R,8′S. Thus, two enantiomers were established and named as (+)-asarolignan G and (−)-asarolignan G, respectively.
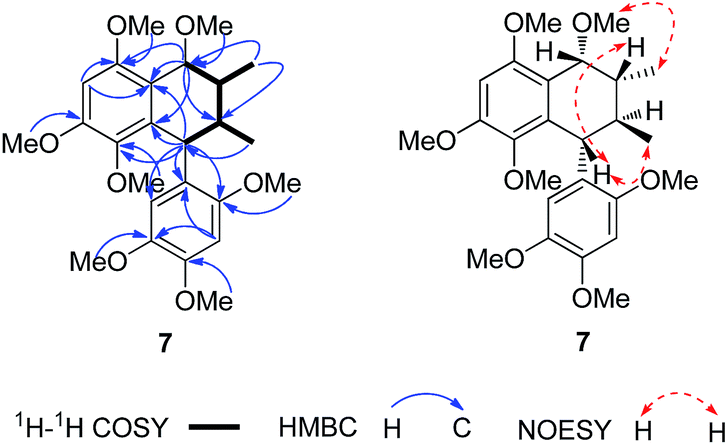 | ||
| Fig. 6 Selected 1H–1H COSY, HMBC and NOE correlations of 7. | ||
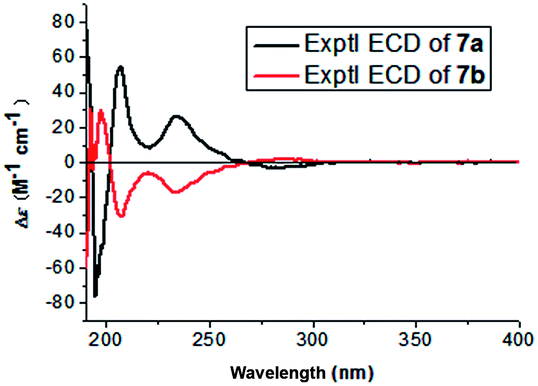 | ||
| Fig. 7 Experimental ECD spectra of 7a, 7b. | ||
The remaining known compounds, including acortatarinowin H (8),9 tatarinan A (9),10 magnosalin (10),11 andamanicin (11),11 5,5′-((1R*,2R*,3R*,4S*)-3,4-dimethylcyclobutane-1,2-diyl)bis(1,2,4-trimethoxybenzene) (12),12 magnoshinin (13),13 tatarinoid C (14),14 and tatanan C (15),15 (7R*,8S*)-7,8-dihydroxyasarone (16),16,17 (7S*,8S*)-7,8-dihydroxyasarone (17),16,17 (7R*,8S*)-(7-methoxy-8-hydroxy)-asarone (18),16,17 (7S*,8S*)-(7-methoxy-8-hydroxy)-asarone (19),16,17 isoacoramone (20),18 asaraldehyde (21),18 β-asarone (22)19 and α-asarone (23)19 were identified by comparison of their physiochemical properties and spectral data with those reported in the literature. Among them, the known compound (±)-acortatarinowin H (8) was obtained as red-brown block crystals, the X-ray diffraction experiment showed an unambiguous assignment of the configurations as 7′S*,8′R* (Fig. 5). It was worth noting that the crystals of 8 had a P21/n space group, indicating a racemic nature, which was in accordance with its small optical value {[α]23D 0 (c 0.5, MeOH)}. Subsequent chiral resolution was performed on a chiral column to yield 8a and 8b, which were opposite in terms of ECD curves (see ESI†). The absolute configurations of 8a and 8b were determined as (7′S,8′R) and (7′R,8′S), respectively, by comparing their cotton effects at 280–300 nm in CD spectra with those reported in the literatures.20,21 Tatarinoid C (14) was firstly separated by chiral HPLC analysis. Compared with calculated ECD spectra of (8R)-14, the two enantiomers named as (+)-tatarinoid C (14a) and (−)-tatarinoid C (14b), respectively (see ESI†).
Pharmacological studies have indicated that neuroinflammation plays a major role such as dysregulation of genes in the pathophysiology of Alzheimer's disease (AD) and Parkinson's disease (PD).22 TNF-α is one of the critical proinflammatory cytokines.23,24 To a certain extent, reducing TNF-α levels has the potential to medicate anti-neuroinflammation action. In our study, all the isolated compounds were evaluated for anti-neuroinflammatory activities on TNF-α production in LPS-activated BV-2 cells. As shown in Fig. 8, compounds 7a, 7b, 14a, 14b, 16, 20, and 22 could reduce TNF-α levels in the murine microglia BV-2 cells. A comparison of the inhibition of the enantiomers 7 (7a/7b), and 14 (14a/14b) in this assay, suggested the configuration was not critical to the anti-neuroinflammation activity. In addition, all the compounds were assayed for cytotoxicity against BV-2 cells by CCK-8 method, and α-asarone (23) displayed cytotoxicity at 20 and 50 μM.
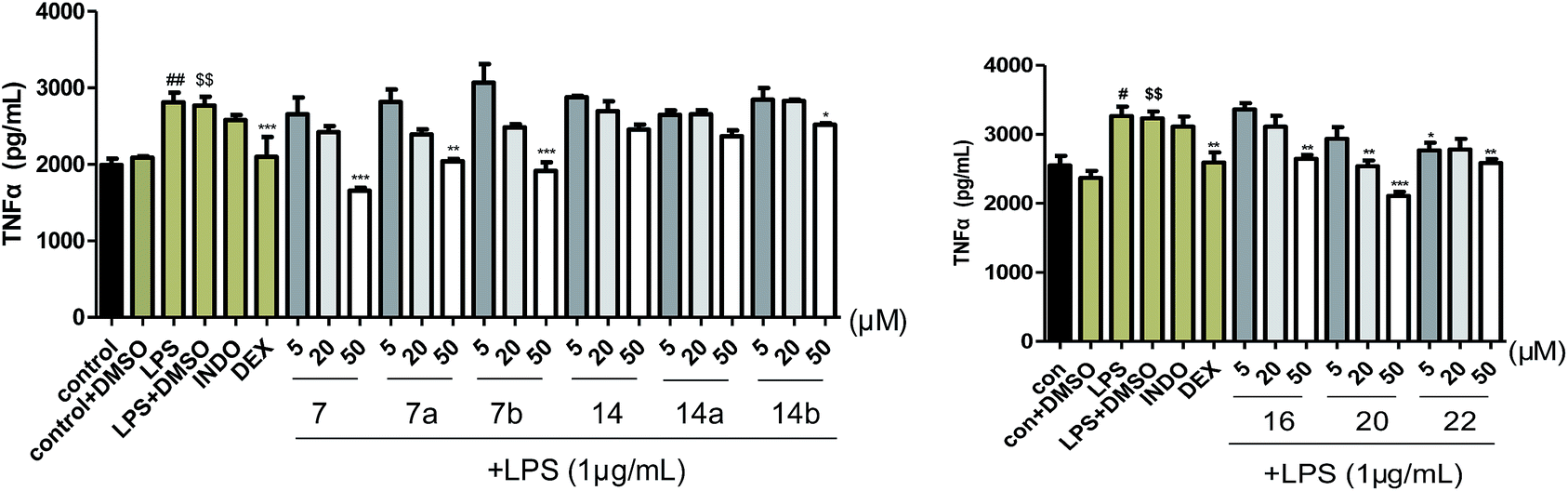 | ||
| Fig. 8 Compounds 7, 7a, 7b, 14, 14a, 14b, 16, 20, 22 inhibit TNF-α levels in the murine microglia BV-2 cells. Results are expressed as mean ± SEM and include data from three independent experiments performed in triplicate. #p < 0.05, ##p < 0.01, when compared to the control group, $$p < 0.01, when compared to the control + DMSO group, and *P < 0.05, **P < 0.01, ***P < 0.001, when compared to the LPS + DMSO group. | ||
Conclusions
In summary, twenty-three compounds derived from asarone biogenetic pathway were isolated and identified from the rhizomes of Acorus tatarinowii, including seven new neolignans (1–7). Their structures including absolute configurations were determined by comprehensive spectroscopic data together with X-ray crystallography, as well as experimental and calculated ECD spectra. Besides, all the compounds were evaluated for anti-neuroinflammatory activities on TNF-α production in LPS-activated BV-2 cells. INDO (indomethacin, 10 μM) and DEX (dexamethasone, 10 μM) were used as positive control. As results, compounds 7, 7a, 7b, 14b and 16 exhibited obvious inhibitory effects on the release of TNF-α at a concentration of 50 μM, their TNF-α level were 1.7, 2.0, 1.9, 2.5 and 2.6 μg mL−1, respectively; compound 20 could inhibit the release of TNF-α at a concentration of 20 μM (TNF-α level was 2.5 μg mL−1) and 50 μM (TNF-α level was 2.1 μg mL−1); compound 22 could dramatically reduce TNF-α level at a concentration of 5 μM (TNF-α level was 2.8 μg mL−1) and 50 μM (TNF-α level was 2.6 μg mL−1). These findings will provide some scientific foundation for the utilization of Acorus tatarinowii as CNS disorders treatment.Experimental
General experimental procedures
Optical rotations were measured on a JASCO P-1020 polarimeter with a 1 cm cell at room temperature. UV spectra were recorded on a JASCO V-550 UV/Vis spectrometer. IR spectra were obtained using a JASCO FT/IR-480 plus spectrometer. CD spectra were obtained on a JASCO J-810 spectropolarimeter at room temperature. HRESIMS spectra were acquired using a Waters Synapt G2 mass spectrometer. 1D and 2D NMR data were acquired with Bruker AV 600, and AV 400 using solvent signals (CDCl3: δH 7.26/δC 77.0; DMSO-d6: δH 2.50/δC 39.5) as the internal standard. Silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., China), octadecyl silanized (ODS) silica gel (YMC Ltd., Japan), and Sephadex LH-20 (Amersham Pharmacia Biotech, Sweden) were used for column chromatography (CC). TLC was performed on precoated silica gel plate (SGF254, 0.2 mm, Yantai Chemical Industry Research Institute, China). The analytical HPLC was performed on a Waters 2695 separations module equipped with a 2998 photodiode array detector using an RP-18 column (5 μm, 4.6 × 250 mm; COSMOSIL). Chiral semi-preparative HPLC was performed using a Lux Amylose-2 column (5 μm, 4.6 × 250 mm, Phenomenex). The semi-preparative and preparative HPLC were carried on a Waters 1515 isocratic HPLC pump equipped with a 2489 UV/Vis detector and RP-18 columns (5 μm, 10 × 250 mm; 5 μm, 20 × 250 mm; COSMOSIL).Plant material
The dried rhizomes of Acorus tatarinowii were purchased from PuraPharm Corporation in 2011 and identified by Jia-Fu Wei, the pharmacist of Guangxi Institute for Food and Drug Control, China. A voucher specimen (20110301) was deposited in the Institute of Traditional Chinese Medicine & Natural Products, Jinan University, Guangzhou, P. R. China.Extraction and isolation
The air-dried and powdered rhizomes of Acorus tatarinowii (19.5 kg) were refluxed with 60% EtOH for 2 h (×2) to give an crude extract (2.2 kg), which was suspended in water (10 L) followed by an exhaustive partition with EtOAc and n-BuOH. The EtOAc soluble extract (312.0 g) was separated by silica gel CC (ϕ 12.0 × 36.0 cm) eluted with gradient cyclohexane–EtOAc (100:0 → 0:100) to afford seven fractions (Fr. 1–7). Fr. 2 (144.9 g) was fractionated further by silica gel CC (ϕ 2.5 × 54.0 cm) eluted gradiently with ether–acetone (100:1, 98:2, 95:5 and 0:100, v/v) to yield 8 fractions (Fr. 2.1–2.8). Fr. 2.3 (5.5 g) were purified by recrystallization from ether to afford compound 22 (5.4 g). Fr. 2.6 (4.2 g) was subjected to semipreparative RP-HPLC (65% MeOH–H2O) to afford 23 (2.0 mg, tR = 28.9 min) and 21 (100.0 mg, tR = 35.0 min). Fr. 3 (22.9 g) was further purified by using ODS CC (ϕ 3.0 × 40.0 cm) eluted gradiently with MeOH–H2O followed by semipreparative RP-HPLC (65% MeOH–H2O) to afford compound 7 (10.9 mg, tR = 42.7 min). Compound 7 was isolated using a chiral analytical column (60% MeCN–H2O, 0.7 mL min−1) to yield compounds 7a (2.2 mg) and 7b (2.2 mg). Fr. 4 (8.8 g) was further subjected to ODS CC (ϕ 3.3 × 25.0 cm) with a gradient system of MeOH–H2O gradient (3:7, 1:1, 7:3, 8:2, 9:1 and 1:0, v/v) to afford 14 subfractions (Fr. 4.1–4.14). Fr. 4.2 (1.1 g) was separated by silica gel CC (ϕ 2.5 × 54.0 cm) eluted gradiently with cyclohexane–acetone (98:2 → 7:3) to yield 11 fractions (Fr. 4.2.1–4.2.11). Fr. 4.2.6 (78.3 mg) was subjected to RP-HPLC (55% MeOH–H2O) to afford compound 20 (24.0 mg, tR = 50.0 min). Fr. 4.3 (3.4 g) was separated by the same manner as Fr. 4.2 to give 16 fractions (Fr. 4.3.1–4.3.16). Compound 10 (40.4 mg, tR = 25.2 min) was obtained by RP-HPLC eluted with 65% MeOH–H2O from Fr. 4.3.9 (961.3 mg). Fr. 4.4 (840.4 mg) was separated by the same manner as Fr. 4.2 to yield 9 fractions (Fr. 4.4.1–4.4.9). Fr. 4.4.5 (238.1 mg) was separated by Sephadex LH-20 eluted with CHCl3–MeOH (1:1, v/v) to yield 11 (10.0 mg). Fr. 5 (43.1 g) was further subjected to ODS CC (ϕ 4.4 × 35.0 cm) with a gradient system of MeOH–H2O (3:7, 1:1, 6:4, 7:3 and 1:0, v/v) to afford 11 subfractions (Fr. 5.1–5.11). Fr. 5.2 (5.0 g) was separated by silica gel CC (ϕ 3.0 × 40.0 cm) eluted gradiently with cyclohexane–acetone (100:0 → 0:100) to yield 13 fractions (Fr. 5.2.1–5.2.13). Fr. 5.2.3 (524.3 mg) was separated by Sephadex LH-20 CC eluted with MeOH to afford 2 subfractions (Fr. 5.2.3.1–5.2.3.2). Fr. 5.2.3.1 (961.3 mg) was further purified by semipreparative RP-HPLC eluted with 25% MeCN–H2O to yield compounds 1 (1.9 mg, tR = 29.2 min), 2 (4.2 mg, tR = 35.2 min) and 3 (10.2 mg, tR = 37.0 min). Compound 2 was isolated using a chiral analytical column (35% MeCN–H2O, 0.7 mL min−1) to yield compounds 2a (1.6 mg) and 2b (1.6 mg). Compound 3 was isolated using a chiral analytical column (30% MeCN–H2O, 0.7 mL min−1) to yield compounds 3a (2.3 mg) and 3b (2.3 mg). Fr. 5.3 (1.4 g) was separated by the same manner as Fr. 5.2 to yield 10 fractions (Fr. 5.3.1–5.2.10). Fr. 5.3.6 (247.9 mg) was separated by Sephadex LH-20 CC eluted with MeOH followed by RP-HPLC eluted with 30% MeCN–H2O to yield 14 (7.3 mg, tR = 20.2 min). Compound 14 was isolated using a chiral analytical column (40% MeCN–H2O, 0.7 mL min−1) to yield compounds 14a (1.5 mg) and 14b (1.6 mg). Fr. 5.6 (8.8 g) was separated by the same manner as Fr. 5.2 to yield 10 fractions (Fr. 5.6.1–5.6.16). Fr. 5.6.10 (323.9 mg) was separated by Sephadex LH-20 CC eluted with MeOH followed by semipreparative RP-HPLC eluted with 60% MeOH–H2O to yield compound 8 (32.0 mg, tR = 16.2 min). Compound 8 was separated using a chiral analytical column (40% MeCN–H2O, 0.7 mL min−1) to yield compounds 8a (0.4 mg) and 8b (0.4 mg). Fr. 5.8 (1.2 g) was separated by silica gel CC (ϕ 2.5 × 40.0 cm) eluted gradiently with cyclohexane–acetone (100:0 → 0:100) to yield 11 fractions (Fr. 5.8.1–5.8.11). Compounds 4 (12.1 mg, tR = 36.2 min) and 9 (13.6 mg, tR = 60.0 min) were obtained by using semipreparative RP-HPLC eluted with 65% MeOH–H2O from Fr. 5.8.3. Compounds 5 (31.0 mg, tR = 35.3 min), 6 (27.9 mg, tR = 38.3 min), 12 (19.6 mg, tR = 43.5 min) and 13 (4.6 mg, tR = 50.0 min) were obtained by semipreparative RP-HPLC eluted with 65% MeOH–H2O from Fr. 5.8.4 (168.6 mg). Fr. 6 (20.6 g) was further subjected to ODS CC (ϕ 3.3 × 25.0 cm) with a gradient system of MeOH–H2O (1:9, 3:7, 1:1, 7:3, 9:1 and 1:0, v/v) to afford 11 subfractions (Fr. 6.1–6.11). Fr. 6.4 (3.0 g) was separated by silica gel CC (ϕ 3.5 × 48.0 cm) eluted gradiently with CHCl3–MeOH to yield 12 fractions (Fr. 6.4.1–6.4.12). Compounds 18 (26.1 mg, tR = 28.5 min) and 19 (15.8 mg, tR = 34.6 min) were obtained by using RP-HPLC eluted with 35% MeOH–H2O from Fr. 6.4.2. (92.6 mg). Compounds 16 (20.5 mg, tR = 35.5 min) and 17 (15.0 mg, tR = 50.2 min) were obtained by semipreparative RP-HPLC eluted with 30% MeOH–H2O from Fr. 6.4.4 (208.0 mg). Fr. 6.8 (1.8 g) was separated by silica gel CC (ϕ 3.5 × 57.0 cm) eluted gradiently with CHCl3–acetone followed by RP-HPLC (53% MeOH–H2O) to yield 15 (16.7 mg, tR = 35.5 min).
ε) 205 (4.92), 231 (sh, 4.44), 291 nm; IR (KBr) νmax 3378, 1614, 1512, 1465 cm−1; 1H and 13C NMR data (Table 1); ESIMS m/z 489 [M + Na]+; HRESIMS m/z 489.2094 [M + Na]+ (calcd for C24H34O9Na, 489.2101).ε) 208 (4.46), 230 (4.14), 291 (3.92) nm; ECD (MeCN) λmax (Δε) 202 (+134.17), 213 (−11.41), 226 (+4.03), 240 (−16.81), 297 (+6.05) nm; IR (KBr) νmax 3420, 1610, 1509, 1458 cm−1; 1H and 13C NMR data (Table 1); ESIMS m/z 489 [M + Na]+; HRESIMS m/z 489.2101 [M + Na]+ (calcd for C24H34O9Na, 489.2101).ε) 218 (3.99) nm; ECD (MeCN) λmax (Δε) 203 (+81.57), 212 (−14.91), 225 (−2.12), 240 (−18.38), 299 (+2.13) nm; IR (KBr) νmax 3421, 2931, 1735, 1653, 1073, 1029 cm−1; 1H and 13C NMR data (Table 1); HRESIMS m/z 489.2103 [M + Na]+ (calcd for C24H34O9Na, 489.2101).ε) 205 (5.08), 230 (4.45), 291 (4.22) nm; IR (KBr) νmax 1509, 1458 cm−1; 1H and 13C NMR data (Table 2); ESIMS m/z 471 [M + Na]+; HRESIMS m/z 471.2363 [M + Na]+ (calcd for C25H36O7Na, 471.2359).ε) 206 (5.00), 262 (4.58), 310 (4.30) nm; IR (KBr) νmax 1609, 1509, 1465 cm−1; 1H and 13C NMR data (Table 2); HRESIMS m/z 469.2203 [M + Na]+ (calcd for C25H34O7Na, 469.2202).ε) 209 (4.97), 261 (4.59), 309 (4.33) nm; IR (KBr) νmax 1609, 1508, 1458 cm−1; 1H and 13C NMR data (Table 2); HRESIMS m/z 469.2203 [M + Na]+ (calcd for C25H34O7Na, 469.2202).) 207 (5.44), 235 (4.90), 289 (4.52) nm; ECD (MeCN) λmax (Δε) 207 (+54.06), 221 (+8.25), 234 (+26.09), 285 (−3.04) nm; IR (KBr) νmax 1598, 1512, 1463 cm−1; 1H and 13C NMR data (Table 2); HRESIMS m/z 469.2196 [M + Na]+ (calcd for C25H34O7Na, 469.2202).X-ray crystallographic analysis
765 unique reflections were collected until θmax = 25.00°, in which 4347 reflections were observed [F2 > 4σ(F2)]. The structure was solved by direct methods using the SHELXS-97 program, and refined by the program SHELXL-97 and full-matrix least-squares calculations. In the structure refinements, non-hydrogen atoms were placed on the geometrically ideal positions by the “ride on” method. Hydrogen atoms bonded to oxygen were located by the structure factors with isotropic temperature factors. The final refinement gave R = 0.0554(3359), Rw = 0.1473(4347), S = 1.035. Crystallographic data for structure 1 has been deposited at the Cambridge Crystallographic Data Centre (CCDC 1518425).†662 reflections were collected until θmax = 29.64°, in which independent unique 12501 reflections were observed [F2 > 4σ(F2)]. The structure was solved by direct methods using the SHELXS-97 program, and refined by the program SHELXL-97 and full-matrix least-squares calculations. In the structure refinements, non-hydrogen atoms were placed on the geometrically ideal positions by the “ride on” method. Hydrogen atoms bonded to oxygen were located by the structure factors with isotropic temperature factors. The final refinement gave R = 0.0697(9471), Rw = 0.1815(12501), S = 1.069, and Flack = 0.0(5). Crystallographic data for structure 2 has been deposited at the Cambridge Crystallographic Data Centre (CCDC 1518426).†344 reflections were collected until θmax = 62.72°, in which independent unique 3954 reflections were observed [F2 > 4σ(F2)]. The structure was solved by direct methods using the SHELXS-97 program, and refined by the program SHELXL-97 and full-matrix least squares calculations. In the structure refinements, non-hydrogen atoms were placed on the geometrically ideal positions by the “ride on” method. Hydrogen atoms bonded to oxygen were located by the structure factors with isotropic temperature factors. The final refinement gave R = 0.0478(3012), Rw = 0.1338(3954), S = 1.040. Crystallographic data for structure 4 has been deposited at the Cambridge Crystallographic Data Centre (CCDC 1518427).†ECD calculation method
In theoretical calculations, the geometry of the molecules was optimized with Gaussian 09 package 33 at B3LYP/6-31G(d) computational level. The minimum nature of the structure was confirmed by frequency calculations at the same computational level. Then ECD calculations were carried out in the methyl cyanide solvent medium using time-dependent density functional theory (TDDFT) with B3LYP functional and DGDZVP basis set.Anti-neuroinflammatory activity assays
BV-2 cells were obtained from Peking Union Medical College (PUMC) Cell Bank (Beijing, People's Republic of China) and were maintained at 5 × 105 cells per mL in DMEM medium supplemented with 10% heat-inactivated FBS, penicillin G (100 U mL−1), streptomycin (100 mg L−1), and L-glutamine (2 mM), and they were incubated at 37 °C in a humidified atmosphere containing 5% CO2. The cell viability was determined by adding 0.8 mL of CCK-8 to each well and incubated at 37 °C for 4 h. Then, the OD value was read at 450 nm using a Bio-Rad ELISA microplate reader.To measure TNF-α production, cells were seeded in a 96-well plate at a density of 1.5 × 106 cells per well in DMEM and incubated for 24 h. The cultures were prepared and stimulated with LPS (1 μg mL−1) in the presence or absence of LNE. Thereafter, the cells were treated with 5, 20 and 50 μM concentrations of 1–23 respectively. The purity of compound 10 is 94.7%, the purity of compound 13 is 90.1%, the purity of other compounds range from 95.0% to 99.0% (see ESI†). After 24 h incubation, supernatant from the culture medium was harvested and used for measuring the level of TNF-α. TNF-α were measured by using an ELISA kit.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81422054, 81172945), and the support from State Key Laboratory of Bioactive Substance and Function of Natural Medicines (GTZK201505), the Guangdong Natural Science Funds for Distinguished Young Scholar (S2013050014287).Notes and references
- X. L. Feng, Y. Yu, D. P. Qin, H. Gao and X. S. Yao, RSC Adv., 2015, 5, 5173–5182 RSC
.
- J. Mao, S. Huang, S. Liu, X. L. Feng, M. Yu, J. Liu, Y. E. Sun, G. L. Chen, Y. Yu, J. Zhao and G. Pei, Aging Cell, 2015, 14, 784–796 CrossRef CAS PubMed
- Z. Q. Li, G. P. Zhao, S. Q. Qian, Z. J. Yang, X. Y. Chen, J. Chen and C. Cai, J. Ethnopharmacol., 2012, 144, 305–312 CrossRef CAS PubMed
- H. Y. Qi, L. Chen, L. Ning, H. Ma, Z. Y. Jiang, Y. Fu and L. Li, J. Pharm. Biomed. Anal., 2015, 115, 292–299 CrossRef CAS PubMed
- M. Sundaramahalingam and S. D. Rathinasamy, Pharmacol. Res., 2005, 52, 467–474 CrossRef PubMed
- G. Wei, Y. B. Chen, D. F. Chen, X. P. Lai, D. H. Liu and R. D. Deng, J. Alzheimer's Dis., 2013, 33, 863–880 CAS
- B. W. Kim, S. Koppula, H. Kumar, J. Y. Park, I.-W. Kim, S. V. More, I.-S. Kim, S. D. Han and S. K. Kim, Neuropharmacology, 2015, 97, 46–57 CrossRef CAS PubMed
- C. Ranjithkumar, P. Vijayapandi and M. Zahurin, Front. Pharmacol., 2016, 7, 72/1–72/8 Search PubMed
- Y. Y. Lu, Y. B. Xue, S. J. Chen, H. C. Zhu, J. W. Zhang, X. N. Li, J. P. Wang, J. J. Liu, C. X. Qi, G. Du and Y. H. Zhang, Sci. Rep., 2016, 6, 22909/1–22909/10 Search PubMed
- G. Ni, Study on chemical constituents and bioactivities and of Acorus tatarinowii and Morus cathayana, Chinese Academy of Medical Sciences and Peking Union Medical College, People Rep China, 2010 Search PubMed
- J. H. Ryu, H. J. Son, S. H. Lee and D. H. Sohn, Bioorg. Med. Chem. Lett., 2002, 12, 649–651 CrossRef CAS PubMed
- S. Kadota, K. Tsubono, K. Makino, M. Takeshita and T. Kikuchi, Tetrahedron Lett., 1987, 28, 2857–2860 CrossRef CAS
- T. Kikuchi, S. Kadota, K. Yanada, K. Tanaka, K. Watanabe, M. Yoshizaki, T. Yokoi and T. Shingu, Chem. Pharm. Bull., 1983, 31, 1112–1114 CrossRef CAS
- X. G. Tong, G. S. Wu, C. G. Huang, Q. Lu, Y. H. Wang, C. L. Long, H. R. Luo, H. J. Zhu and Y. X. Cheng, J. Nat. Prod., 2010, 73, 1160–1163 CrossRef CAS PubMed
- G. Ni, Z. F. Shen, Y. Lu, Y. H. Wang, Y. B. Tang, R. Y. Chen, Z. Y. Hao and D. Q. Yu, J. Org. Chem., 2011, 76, 2056–2061 CrossRef CAS PubMed
- N. Matsuda and M. Kikuchi, Chem. Pharm. Bull., 1996, 44, 1676–1679 CrossRef CAS
- Z. Yuan and X. Li, Chin. J. Magn. Reson., 2003, 20, 307–314 CAS
- C. H. Park, K. H. Kim, I. K. Lee, S. Y. Lee, S. U. Choi, J. H. Lee and K. R. Lee, Arch. Pharmacal Res., 2011, 34, 1289–1296 CrossRef CAS PubMed
- A. Patra and A. K. Mitra, J. Nat. Prod., 1981, 44, 668–669 CrossRef CAS
- T. D. Silva and L. M. X. Lopes, Phytochemistry, 2006, 67, 929–937 CrossRef PubMed
- L. M. X. Lopes, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1982, 21, 751–755 CrossRef CAS
- C. K. Johnathan, K. Janine, F. Laura, R. H. Paul, R. Magnus and J. S. Pamela, Nat. Rev. Neurol., 2012, 8, 518–530 CrossRef PubMed
- X. Jiang, L. Chen, L. L. Shen, Z. W. Chen, L. X. Xu, J. J Zhang and X. F Yu, Brain Res., 2016, 1649, 30–37 CrossRef CAS PubMed
- V. Barbara, B. Mariaserena, M. Natalia and M. Marina, Neurotoxicology, 2014, 43, 10–20 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of all new compounds. CCDC 1518425–1518428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27786a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |