
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new fluorescent PET sensor probe for Co2+ ion detection: computational, logic device and living cell imaging applications†
Sonaimuthu Mohandoss and
Thambusamy Stalin *
*
Department of Industrial Chemistry, School of Chemical Sciences, Alagappa University, Karaikudi-630 003, Tamilnadu, India. E-mail: tstalinphd@rediffmail.com; drstalin76@gmail.com; Tel: +91 9944266475
First published on 15th March 2017
Abstract
The hydrophobic nature of β-cyclodextrin (β-CD) increases the solubility and stability of 1,2-dihydroxyanthraquinone (1,2-DHAQ; 1) and hence permits the coordination of 1 with cations in aqueous solution; this has been investigated by means of UV-visible and fluorescence spectroscopy. β-CD:1,2-DHAQ (2) demonstrates colorimetric recognition behavior toward Co2+ ion by changing the color of the solution, which can be easily detected with the naked eye. The chemosensor 2 showed good fluorescence behavior upon interaction with various cations; it displayed strong fluorescence quenching (Co2+; ∼80% switch-off) as a fluorescent chemosensor based on photoinduced electron transfer (PET). Moreover, when the 2·Co2+ complex was tested with various anions, only nitrate (NO3−) enabled cobalt binding (as CoNO3) and led to fluorescence enhancement (∼82%; switch-on). The detection limits of sensor 2 with Co2+ and NO3− were found to be 22.7 nM and 2.4 nM. Theoretical molecular docking studies and density functional theory (DFT) calculations were performed to study the binding of Co2+ and NO3− ions with 2. The changes in the fluorescence of 2 upon addition of Co2+ followed by NO3− can be utilised as an XNOR logic gate. Furthermore, 2 has potential for use in bio-imaging as a fluorescent probe to detect Co2+ ion, followed by sequential detection of NO3− ion by 2·Co2+, in living cells (human cervical cancer HeLa cell line) using confocal laser scanning microscopy.
Introduction
Cobalt is an essential trace element found in rocks, minerals, soils and seawater; it plays an important role in various biological systems.1 It is well known that Co2+ plays essential parts in the metabolism of iron and the synthesis of hemoglobin; it is also a main component of vitamin B12 and other biological compounds.2 However, exposure to high levels of cobalt can cause toxicological effects, including heart disease, thyroid enlargement, asthma, decreased cardiac output, lung disease, dermatitis and vasodilation.3 In addition, cobalt is a significant environmental pollutant. On the other hand, cobalt deficiency in the human body may also lead to pathological conditions.4 Therefore, a highly sensitive and selective analytical method to detect Co2+ ions is of great importance to prevent these toxic effects. Nitrate anion is an important intermediate in the biological nitrogen cycle and is plentiful in the environment.5 Nitrates are often used as food preservative agents6 for meat and fish and are also essential nutrients for the growth of plants.7 Water is another widely accessible source through which the safety of human health is threatened by dangerous levels of nitrite (or nitrate).8 Furthermore, increased nitrate levels have been implicated in the formation of carcinogenic nitrosamines and can cause methemoglobinemia (blue-baby syndrome) in infants.9,10 Therefore, it is of significant importance to detect nitrate anion in water, food and agricultural products. The World Health Organization (WHO) recommends maximum limits of cobalt and nitrate ions in drinking water of 1.7 and 65 μM, respectively.11Therefore, it is essential to monitor the concentration of these species in biological and environmental samples. A variety of analytical methods have been developed for the determination of cobalt and nitrate, such as atomic absorption spectroscopy,12 inductively coupled plasma atomic emission spectrometry,13 fabricated nanomaterials,14 ion chromatography,15 flow-injection analysis,16 capillary electrophoresis,17 and electrochemical methods.18 However, many of these methods are complicated and are not suitable for rapid, on-site analysis. In contrast, colorimetric methods can enable convenient and facile monitoring of target ions without the use of sophisticated equipment. Therefore, chemosensors that allow “naked eye” detection have advantages over other methods in that they are easy to use, are portable, and do not require sophisticated instrumentation. Fluorimetry is one of the most powerful trace analysis techniques; efforts have been made to determine Co2+ and NO3− based on fluorescence quenching and enhancement.19 These studies suggest that the use of fluorescent chemosensors for the detection of certain cations and anions can avoid the use of expensive classical methods and can allow in situ, real-time detection, although their accuracy may be less than desired. However, most of these chemosensors are limited by interfering background fluorescence, non-specific quenching from competing ions, and incompatibility with water.
1,2-Dihydroxyanthraquinone (1,2-DHAQ; probe 1, Scheme 1) is a well-known colorimetric pH indicator that can be used with cations and anions. A highly pH-sensitive colorimetric chemosensor with significant color changes would be very useful in the study of physiological fluids. In basic pH media, these substances exist in anionic forms (catocholate), which is attributed to intramolecular charge transfer (ICT) transitions from the donating phenolic groups to the π* orbitals of the quinone.20 1,2-DHAQ is practically insoluble in water; however, it forms supramolecular complexes with a class of cyclic oligosaccharides, cyclodextrins (CDs), which are water-soluble host molecules which can include various kinds of guest molecules inside their cavities. For some substances with low water solubility, inclusion in CDs may improve their physicochemical proprieties, such as solubility, stability and spectroscopic properties.21 In fact, CDs have been used in a number of systems to enhance the fluorescence of hydrophobic fluorophores; this can be used to improve the detection limits for analytes through signal enhancement.22 We recently published an investigation of the absorption and fluorescence emission spectral properties of 1,2-DHAQ in aqueous solutions of β-cyclodextrin (β-CD).23 We showed that the β-CD:1,2-DHAQ complex exhibits substantial color changes and fluorescence quenching upon complexation with Co2+ and NO3−, which could be utilized for the selective colorimetric and fluorescent determination of Co2+ and NO3− in aqueous solutions.
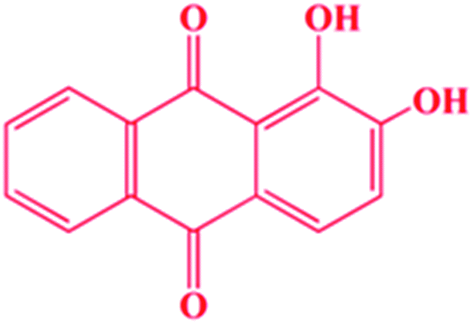 | ||
| Scheme 1 Chemical structure of 1,2-DHAQ. | ||
In this work, we report a highly selective and sensitive 1-based chemosensor probe for the rapid detection of Co2+ and the sequential recognition of NO3− by the inclusion complex (as β-CD). Towards the application of this sensor, computational studies were performed, and the two chemical inputs were found to mimic the performance of an exclusive-NOR (XNOR) logic gate; human cervical cancer HeLa cell imaging studies were also conducted. To the best of our knowledge, there are no experimental or theoretical studies of the complexation probe 1 with β-CD or its use as a chemosensor.
Experimental section
Materials and instruments
β-Cyclodextrin (β-CD) was obtained from Hi-Media chemical company and was used without further purification. 1,2-Dihydroxyanthraquinone (1,2-DHAQ) (Sigma Aldrich) and other chemical reagents were of analytical reagent grade and were used as received. Human cervical cancer HeLa cell lines were collected from the National Center for Cell Science, Pune, India. All the metal salts and anions were purchased from Sigma Aldrich and were used as received. The stock solutions of cations and anions (1 mM) were prepared in triply distilled water from the chloride salts of Ca2+, Ni2+, Co2+, Cu2+, Hg2+, and Fe3+, the nitrate salts of Pb2+, Mg2+, and Zn2+, and the potassium salts of Br−, Cl−, NO3−, I−, AcO−, Cr2O72−, H2PO4− and SO42−. Solutions with various pH levels were prepared by adding appropriate amounts of NaOH and H3PO4; the solutions were prepared immediately prior to taking measurements. The stock solution of 1,2-DHAQ (1 × 10−4 M) was prepared using dimethylformamide, and β-CD solutions were prepared with the following concentrations: 2, 4, 6, 8, 10 and 12 × 10−3 M.The UV-Vis spectra (absorption spectral measurements) were acquired with a Shimadzu UV-2401PC double-beam spectrophotometer, and the fluorescence spectra (emission spectral measurement) were acquired with a JASCO FP-8200 spectrofluorometer. The pH values were measured on an ELICO model LI-10T pH meter. 1H NMR spectra were acquired on a BRUKER-NMR 500 MHz spectrometer. Bio-imaging studies were performed with a CLSM-LSM 710 confocal laser scanning microscope (objective 40; numerical aperture, 0.6); confocal images were obtained using Zen 2009 image software (model 710; Carl Zeiss Imaging, Germany) in deconvolution mode. Computational studies were performed using a single 1.0 GHz PC processor under the Linux operating system.
Computational studies
The most probable structures of the complexes of 1 and 2 (Co2+ and NO3−) were determined by molecular docking studies using the PatchDock server (http://bioinfo3d.cs.tau.ac.il/PatchDock/). A docking protocol consisting of a global search by PatchDock and a refinement by FireDock was extensively tested (http://bioinfo3d.cs.tau.ac.il/FireDock/). The 3D structures were taken from Cambridge Structural Database (CSD) Chemdraw Ultra and Chem3D Ultra (version 8.0, http://Cambridge.soft.com, USA). The 3D structures of the Co2+ and NO3− complexes of 1 and 2 were generated by the Chimera 1.8.1 server (https://www.cgl.ucsf.edu/chimera). All geometries for the complexes of 1 and 2 with ions (Co2+ and NO3−) were optimized by density functional theory (DFT) calculations using the B3LYP function with a 6-31G basis set of the Gaussian 09W program, except for Co2+ and NO3−, for which LANL2DZ was used.Confocal laser scanning microscopy
The effective biofilm inhibitions of the complexes were studied on the surfaces of glass slides. The test cultures were grown for 24 h and dispensed in 24-well polystyrene plates supplemented with the complexes, then incubated for 24 h at 28 °C. The biofilms were monitored by confocal laser scanning microscopy (CLSM) (model: LSM 710) (Carl Zeiss, Jena, Germany) after washing with PBS buffer (pH 7.4) and staining with Cu2+ and Cr2O72−. The 485 nm argon laser and a 500 to 750 nm band pass emission filter were used to excite and detect the stained cells (human lung cancer A549). CLSM images were obtained from the 24 h old control treated biofilms and were processed using Zen 2009 image software (Zen, Frickenhausen, Germany).Results and discussion
Colorimetric and fluorescent sensing of Co2+
The response of chemosensors is useful to detect silent substances, such as various cations, by spectroscopic methods (UV-visible and fluorescence spectroscopy). Selectivity is an important parameter for the evaluation of the performance of a chemosensor. The selectivities of 1,2-DHAQ and β-CD:1,2-DHAQ were measured by observing the variations in the absorption and fluorescence spectra in pH 11.0 solution caused by Co2+ and other selected metal ions, including Ca2+, Ni2+, Cu2+, Pb2+, Mg2+, Hg2+, Zn2+ and Fe3+; the results are shown in Fig. S1 and S2.† The binding of 1,2-DHAQ (1) and β-CD:1,2-DHAQ (2) with Co2+ ion resulted in large blue-shifts in the absorption spectra (1; 234, 266.5 and 530.5 nm, 2; 230.5, 259.5 and 528 nm), while other metal ions produced insignificant changes, except Cu2+ (1; 236, 262.5 and 524.5 nm, 2; 223, 262.0 and 519.5 nm), which showed slight interference. Then, the selectivities of 1 and 2 for Co2+ were further evaluated in complicated systems. As shown in Fig. S1 and S2,† in the presence of miscellaneous metal ions, 1 and 2 still exhibited absorption peak shifts as well as color changes from violet to brown and yellow; these changes are visible to the naked eye (inset image in Fig. S1†), indicating that the blue shifted absorption resulting from the addition of Co2+ was not influenced by any added metal ions.24 No noticeable color changes were observed upon addition of Ca2+, Ni2+, Pb2+, Mg2+, Hg2+, Zn2+ and Fe3+ cations; however, Cu2+ showed dark brown and light yellow coloration. 1,2-DHAQ is practically insoluble in water; however, when it forms a supramolecular complex with β-CD in water, the entire system is soluble and stable. Both the absorption and fluorescence intensity of 1 were enhanced by β-CD, e.g., the fluorescence intensity of 1 was at least 2-fold higher in the presence of β-CD. Namely, β-CD has a sensitizing effect on the determination of cations. Moreover, β-CD has a stabilizing effect on the entire system. An aqueous solution of the 2 complex remained clear and transparent upon the addition of cations. Thus, cation sensing experiments were performed in aqueous solutions of β-CD. The use of β-CD at a concentration of 12 × 10−3 M gave satisfactory results. By comparison, 1 and 2 show high selectivity towards Co2+ ion (increasing absorption intensity and quenching of fluorescence intensity). It is interesting to note that chemosensor 2 showed more drastic changes in absorption and fluorescence intensity than 1 upon addition of Co2+ ion; this is represented in Fig. S3.†The sensitivity effect of Co2+ ion on the absorption spectrum of 1 (Fig. S4†) indicates that the free 1 exhibited four maxima at 266, 327.5, 562.5 nm and a shoulder peak at 608 nm. The maxima at 266 nm and 327.5 nm are assigned to the π–π* electronic transitions, whereas the maxima at 562.5 and 608 nm appear due to the n–π* transition. Upon incremental addition of Co2+ ion to 1, sharp peaks of the π–π* transition appeared at 234 and 266.5, sharp peaks of the n–π* transition appeared at 530.5 nm (a blue-shift of 32 nm from 562.5 nm), and the peak ascribed to the π–π* transition at 327.5 nm disappeared (Fig. S4†). The appearance of the large blue-shift from 530.5 to 522 nm strongly suggests an interaction of Co2+ with the deprotonated phenolic oxygen of 1 to form the complex 1·Co2+.24 This suggests an interaction between the lone pairs of the complex, either at the quinone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or at the coordinated (catechol-type) oxygen atoms. This blue-shift is consistent with hydrogen bonding, which withdraws electron density from the coordinating oxygen atoms and stabilizes the Co2+ orbitals, causing a blue-shift in the charge transfer transitions from Co2+ (d) to 1 (assuming that the d orbitals are stabilized more than 1-π*). The spectral shifts accompanied by the change of color from violet to brown clearly delineated the formation of a complex species between 1 and Co2+; this could be clearly seen under visible light by the naked eye (inset image in Fig. S4†). The color change occurs as a consequence of the coordination of Co2+ with phenolate in 1. This is an interesting new feature that can be used to detect Co2+ without using any other instrumental techniques. 1 shows a pronounced selectivity for Co2+ ion over other metal ions with the addition of increasing amounts of Co2+ (1.2 to 3.6 μM). This indicates the formation of 1·Co2+ complex due to the metal to ligand charge transfer (MLCT) process in the formation of the complex at 530.5 nm.25 In order to determine the stoichiometry of the 1·Co2+ complex, the method of continuous variation (Job's plot) was used.26 The total concentrations of 1 and Co2+ were constant, with a continuous variable of the molar fraction of the guest ([Co2+]/[1] + [Co2+]). In the Job's plot of the concentration of 1 with Co2+ at 530.5 nm, the 1·Co2+ complex concentration approaches a maximum when the molar fraction of Co2+ is 0.5; this indicates that 1 and Co2+ form a 1
O or at the coordinated (catechol-type) oxygen atoms. This blue-shift is consistent with hydrogen bonding, which withdraws electron density from the coordinating oxygen atoms and stabilizes the Co2+ orbitals, causing a blue-shift in the charge transfer transitions from Co2+ (d) to 1 (assuming that the d orbitals are stabilized more than 1-π*). The spectral shifts accompanied by the change of color from violet to brown clearly delineated the formation of a complex species between 1 and Co2+; this could be clearly seen under visible light by the naked eye (inset image in Fig. S4†). The color change occurs as a consequence of the coordination of Co2+ with phenolate in 1. This is an interesting new feature that can be used to detect Co2+ without using any other instrumental techniques. 1 shows a pronounced selectivity for Co2+ ion over other metal ions with the addition of increasing amounts of Co2+ (1.2 to 3.6 μM). This indicates the formation of 1·Co2+ complex due to the metal to ligand charge transfer (MLCT) process in the formation of the complex at 530.5 nm.25 In order to determine the stoichiometry of the 1·Co2+ complex, the method of continuous variation (Job's plot) was used.26 The total concentrations of 1 and Co2+ were constant, with a continuous variable of the molar fraction of the guest ([Co2+]/[1] + [Co2+]). In the Job's plot of the concentration of 1 with Co2+ at 530.5 nm, the 1·Co2+ complex concentration approaches a maximum when the molar fraction of Co2+ is 0.5; this indicates that 1 and Co2+ form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex (inset in Fig. S4a†). The association constant (Ka) values of the 1·Co2+ complex were calculated from the UV-visible titration experiments by a Benesi–Hildebrand plot; they show a good linear correlation coefficient (R2 = 0.9916, inset in Fig. S4b†).
:1 complex (inset in Fig. S4a†). The association constant (Ka) values of the 1·Co2+ complex were calculated from the UV-visible titration experiments by a Benesi–Hildebrand plot; they show a good linear correlation coefficient (R2 = 0.9916, inset in Fig. S4b†).
It is noteworthy that the absorbance of the system was enhanced by β-CD, reflecting the sensitizing effect of β-CD on the determination of Co2+ with 1 (Fig. 1).27 The change in the color of 2 from violet to yellow upon addition of Co2+ was clearly visible under visible light by the naked eye (inset image in Fig. 1). This shows that the intensity of 1 is slightly influenced by the presence of the cation. However, in the inclusion complex, the intensity of the 2·Co2+ system incrementally increases more remarkably than 1·Co2+, which can be attributed to the formation of the supramolecular complex after binding with metal ions in the β-CD solution.22,28 It is expected that the novel functions produced are different from those found in free molecules. Under the same experimental conditions, the blue shift of the absorption maxima from 528 to 524.5 nm due to the simultaneous increase of Co2+ ion concentration can be attributed to the increasing intensity of 2·Co2+ (0.3 to 0.85 μM). The Job's plot analysis revealed that a stoichiometry system between 2 and Co2+ in the complex can be determined at 528 nm (inset in Fig. 1a). As expected, when the molar fraction of the complex sensor was 0.5, the absorbance reached a maximum, which demonstrates that the interaction between 2 and Co2+ forms a 1:1 complex. The association constant (Ka) value of the 2·Co2+ complex was calculated from the UV-visible titration experiments by the Benesi–Hildebrand method; this plot shows a good linear correlation coefficient (R2 = 0.9937, inset in Fig. 1b).
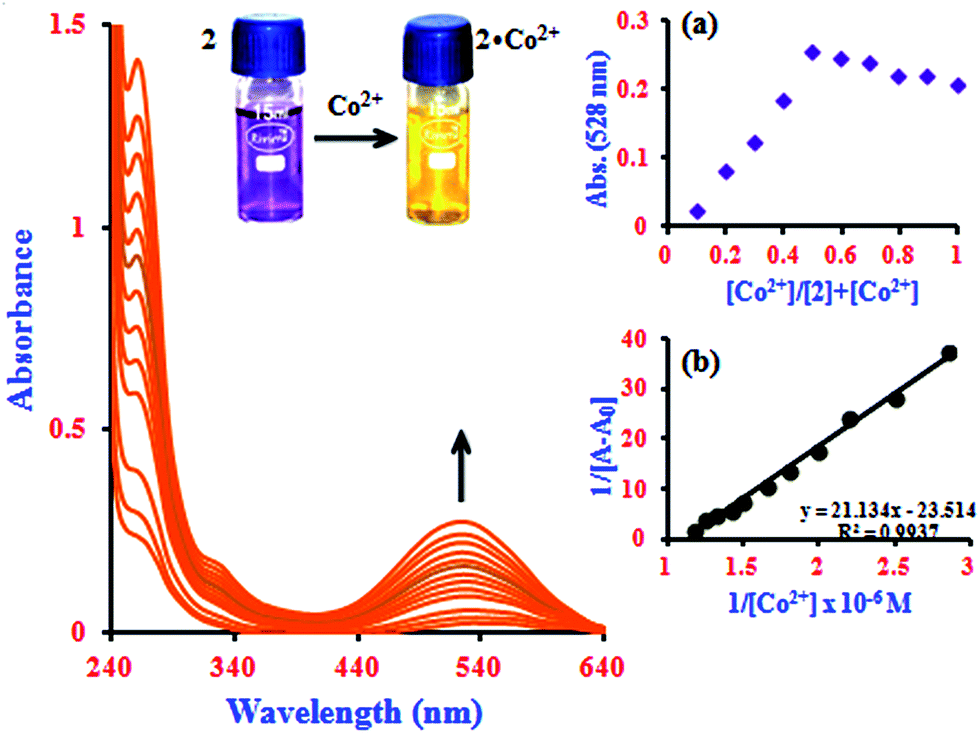 | ||
| Fig. 1 Absorption spectra of 2 with the addition of Co2+ ion concentrations from 0.3 to 0.85 μM. Inserts: images of the naked eye colorimetric detection of the complex formation by a color change from violet to yellow (2·Co2+), (a) Job's plot analysis for the complexation between 2 and Co2+ at 528 nm and (b) Benesi–Hildebrand plot of 1/[A − A0] vs. 1/[Co2+]. | ||
Sensitivity is a very important parameter to evaluate the performance of a fluorescent chemosensor. In good agreement with the findings from the absorption studies, 1 and 2 also exhibited specific emission responses towards Co2+ under similar conditions. The quenching effects of 1 by Co2+ have been studied, and the quenching process was attributed to the formation of a complex between 1 and Co2+. At high pH, the base-induced deprotonation of phenolic –OH groups may lead to photoinduced electron transfer (PET) from the electron-rich catechol moiety to the anthraquinone fluorophore,29 which could account for the fluorescence quenching of 1. The experiments were repeated five times, and the phenomenon was consistent. Fig. S5† displays the fluorescence spectra of 1 in the presence of Co2+ with varying concentrations. As shown, with increasing concentration of Co2+, the fluorescence intensity at 629 nm (λex = 530.5 nm) decreased gradually; it was found that 1·Co2+ presented a quenching effect (switch-off ∼ 80%). This sharp decrease of fluorescence intensity is caused by Co2+ with high sensitivity based on the 1·Co2+ sensing system. It can be seen that the characteristic emission intensity of 1 at 629 nm decreased in the presence of Co2+ ions (1·Co2+). It is expected that the formation of a metal complex would suppress both PET and the conformational transition and thereby provoke fluorescence emission. Thus, PET-type fluorescence quenching is less probable, and the native fluorescence of the fluorophore is restored (Scheme 2). In this regard, it is known that Co2+ is a paramagnetic ion with an unfilled d shell and can strongly quench the emission of a nearby fluorophore via electron or energy transfer from the metal d-orbital.30 The Job's plot analysis revealed that a stoichiometric system of the complex between 1 and Co2+ can be determined at 629 nm. As expected, when the molar fraction of the complex sensor was 0.5, the emission reached a maximum, which demonstrated that 1 and Co2+ form a 1:1 complex (inset in Fig. S5a†). The fluorescence quenching of 1 can be attributed to the formation of the 1·Co2+ complex. The quenching was analyzed using the Stern–Volmer equation31 and the fluorescence intensity at 629 nm in the presence and absence of Co2+ ion, respectively. The quenching of 1 by cobalt ions showed a good linear coefficient of R2 = 0.9972 for Co2+ ion concentrations in the range from 1.2 to 3.6 μM in the Stern–Volmer plot (F0/F) versus [Co2+], with a KSV value of 8.27 × 103 M−1 (inset in Fig. S5b†). The KSV value suggests that Co2+ exhibits a strong quenching ability toward the fluorescence of 1. This high quenching efficiency endows the assay with high sensitivity.
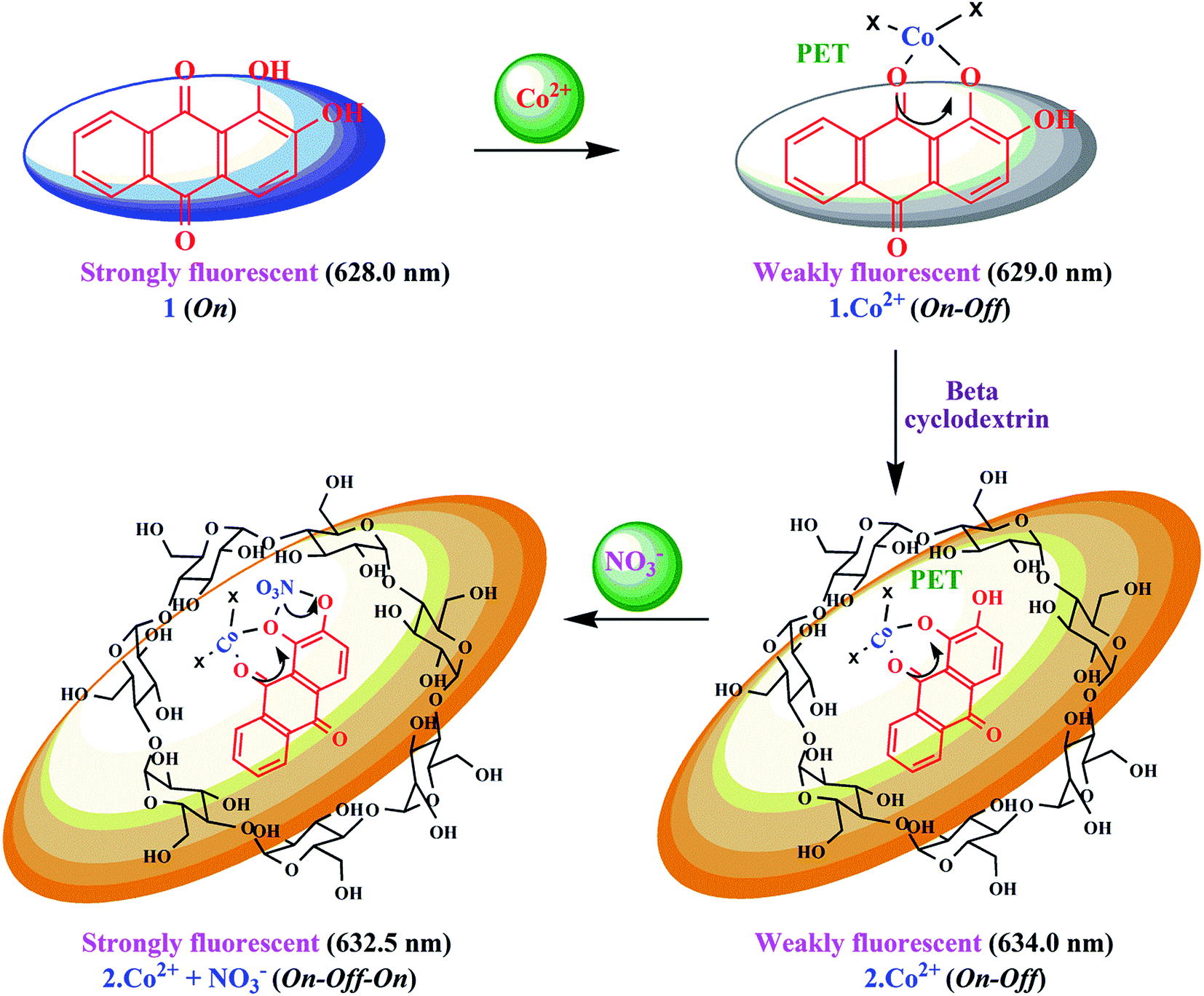 | ||
| Scheme 2 Proposed mechanistic pathway for 1 and 2 for the sensing of Co2+ and NO3− ions based on complexation. | ||
It is an experimentally demonstrated phenomenon that upon entering a free β-CD cavity, 1 can also bind guest molecules. In a preliminary report, the effects of this association of the fluorescence of 1 led to improved fluorescence sensing ability for Co2+. Herein, a similar phenomenon was also observed; that is, the fluorescence sensing ability and selectivity towards Co2+ of 1 was significantly enhanced by β-CD. The fluorescence of 1 in the presence of β-CD (2) showed a 2.3-fold enhancement with Co2+, which is three times higher than the corresponding value of 1·Co2+. The complexation induces PET-retarded fluorescence, which is responsible for the fluorescence enhancement in the presence of Co2+. That is, the emission enhancement in the 2·Co2+ complex is probably due to retardation of PET by a combination of the chelation of both oxygen atoms and phenolic hydroxyl groups with metal ions. The emission peaks of 2·Co2+ were observed at 634 nm (excitation at 528 nm). The quenching of the fluorescence intensity of the 2·Co2+ complex system and the large blue-shift from 634 to 624.5 nm of the fluorescence maxima can be attributed to supramolecular formation followed by binding with 1·Co2+ in the presence of β-CD, as shown in Fig. 2 and Scheme 2.
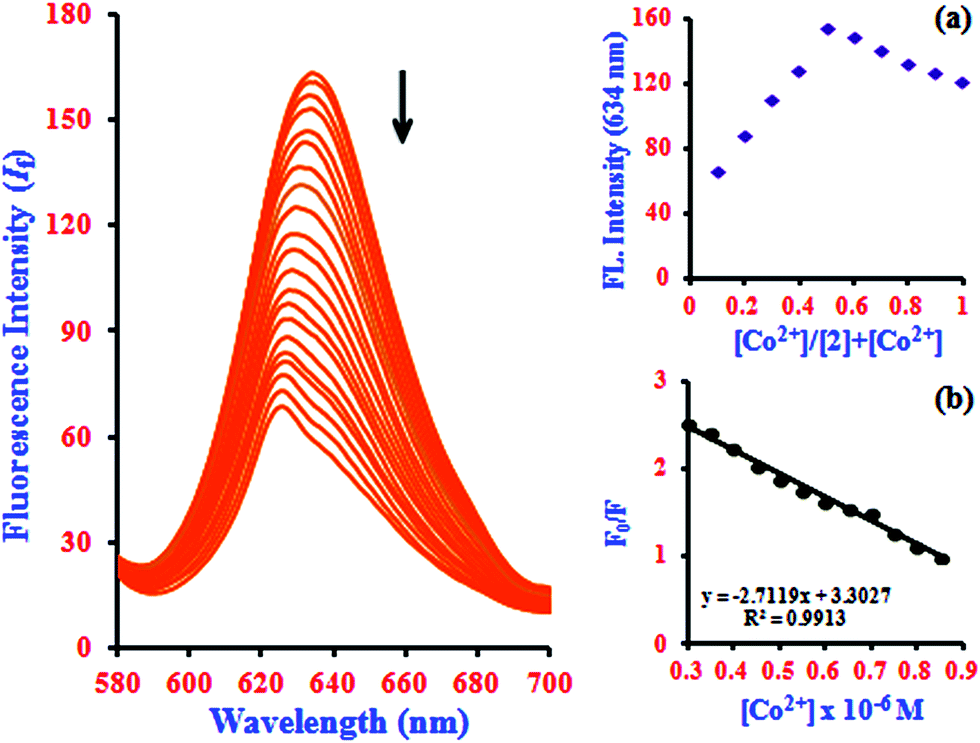 | ||
| Fig. 2 Fluorescence spectra of 2 with the addition of Co2+ ion concentrations of 0.3 to 0.85 μM. Insets: (a) Job's plot analysis for the complexation between 2 and Co2+ at 634 nm and (b) Stern–Volmer quenching plot of 2 by Co2+ presented in the form of (F0/F) vs. [Co2+]. | ||
The Job's plot analysis revealed a stoichiometry system between 2 and Co2+ in the complex, which was investigated systematically at 634 nm (inset in Fig. 2a). Additionally, the Stern–Volmer plot of (F0/F) versus [Co2+] gives a good linear correlation coefficient of R2 = 0.9913 for Co2+ ion concentration in the range from 0.3 to 0.85 μM (inset in Fig. 2b). The Stern–Volmer quenching constant (KSV) of Co2+ ion binding in the system was evaluated graphically from the slope value and was found to be 3.26 × 103 M−1. According to the IUPAC definition, the LOD can be calculated using the relationship LOD = (3 × standard deviation)/slope.32 To calculate the relative standard deviation, the absorption measurements of ten blank samples were taken. The calibration values between the minimum and the maximum intensity were obtained, and then a linear regression curve was fitted to these normalized data to obtain the slope. With this approach, the LOD was found to be 22.7 nM.
To gain a better understanding of the binding modes of Co2+ to 1 and 2, we also performed 1H NMR titration experiments (Fig. S6† and 3). The results showed that the proton signals of the phenolic hydroxyl and catecholic hydroxyl groups were observable throughout the titration. The proton peaks of OH (Ha and Hb) shifted from 5.213 ppm to 5.457 ppm (1) and from 5.402 ppm to 5.721 ppm (2), and the peak intensity weakened gradually. At the same time, the peaks from the CH protons broadened, with a small, gradual downfield shift. These results suggest that the two hydroxyl groups may provide good binding sites for Co2+ and the cavities of the β-CD skeleton. The spectra and 1H NMR titration experiments showed 1:1 stoichiometry for the binding between 1 and 2 with Co2+, demonstrating its moderate binding affinity for Co2+. The 1H NMR spectra provide evidence that the hydroxyl groups of sensors 1 and 2 provide suitable complex sites for Co2+, leading to dramatic fluorescence quenching, a red-shift of the absorption spectrum, and a unique color change.
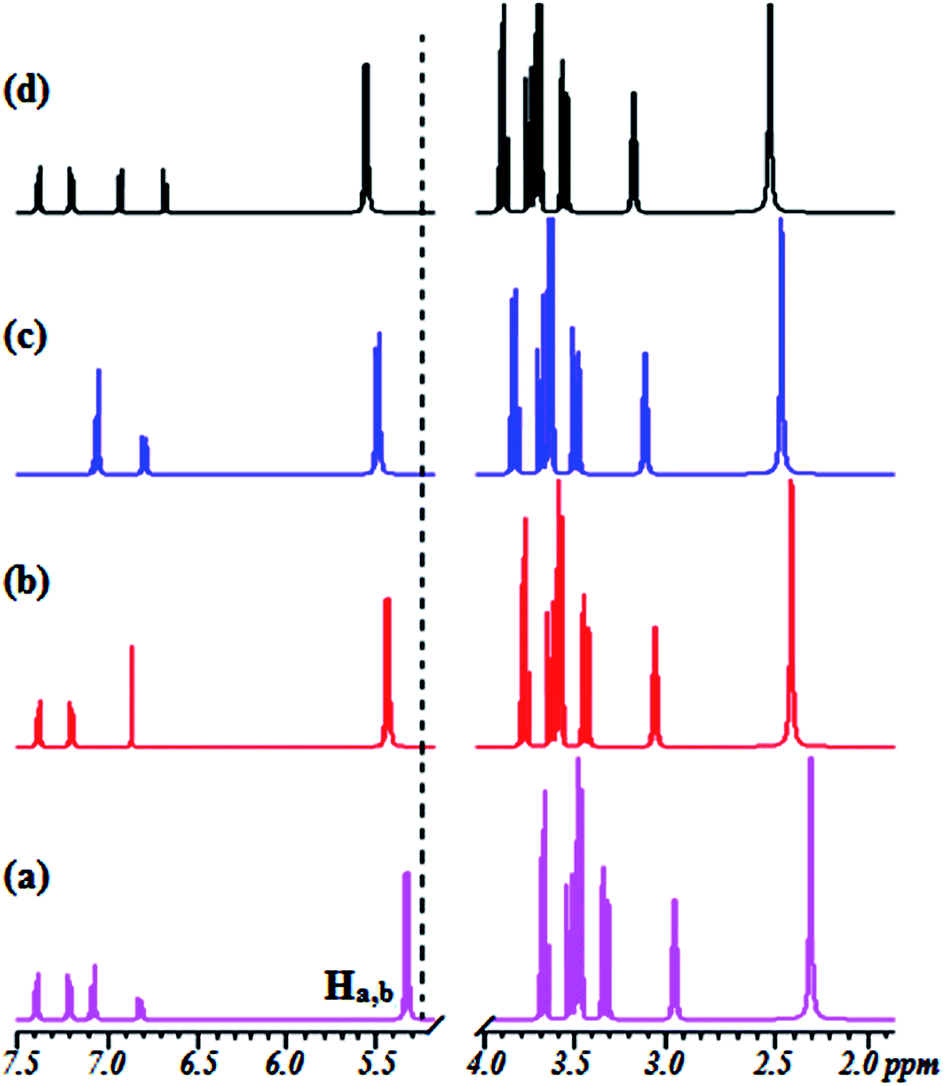 | ||
| Fig. 3 Changes in the partial 1H NMR spectrum of 2 upon titration with Co2+: (a) (no Co2+) and (b–d) in the presence of 0.25, 0.50, and 1.0 equiv. of Co2+ at 500 MHz in CDCl3/CD3CN, 4/1 (v/v). | ||
Colorimetric and fluorescence sensing of Co2+ toward NO3−
In addition to the metal ion sensing properties of β-CD:1,2-DHAQ (2), we also investigated the binding behavior of the metal-based chemosensor β-CD:1,2-DHAQ·Co2+ towards various anions using absorption and fluorescence spectra, as shown in Fig. S7.† The addition of excess selected anions, including Br−, Cl−, I−, AcO−, H2PO4−, Cr2O72− and SO42−, to solution of 2·Co2+ did not show any significant change in its absorption and fluorescence spectra. However, addition of NO3− led to significant changes in both the absorption and fluorescence spectra of 2·Co2+. Spectrophotometric monitoring of the changes revealed that a new peak appeared instantaneously at 367.5 nm, attributed to the n–π* transition, with a concomitant change of color from yellow to pink that is visible to the naked eye (inset image in Fig. S7a†). The instant appearance of 2·Co2+ in the solution indicates the possible formation of a complex of 2·Co2+ with NO3− 33 due to stabilization of the lower oxidation state by NO3−, which is a strong π-acceptor. The probe also provides oxygen bridges to minimize the distance between the Co2+ binding site and the fluorophore 1, which ensures strong fluorescence quenching of Co2+ in the off state with 2. Initially, the fluorescence of 2 is quenched by coordinating to Co2+ ion, due to the paramagnetic character of Co2+. The 2·Co2+-based NO3− probe is formed in situ by treating 2 with one equivalent of cobalt chloride. After addition of NO3− solution, the non-emissive 2·Co2+ “ensemble” formed a metal–NO3− adduct, releasing the fluorophore that was initially quenched by coordination to the paramagnetic transition-metal center by electron or energy transfer.33 The fluorophores can bind to the metal center as axial ligands. The introduction of NO3− causes displacement of the fluorophores, with concomitant fluorescence turn-on changes in NO3− under physiological conditions; this fluorogenic behaviour (∼82%; switch-on) can detected with good sensitivity and selectivity by measuring the ratio of fluorescence intensity. The histogram of absorption and the fluorescence intensity changes are listed in Fig. S8.† As shown in the histogram, significant variations in the absorption and fluorescence intensities of 2·Co2+ upon addition of NO3− were observed in comparison with other added anions. All these results indicate the high selectivity of 2·Co2+ towards NO3− over coexisting anions in aqueous solution.In the absorption spectra of 2·Co2+ in the presence of NO3− anion, the absorption intensity drastically increases and a new peak appears at 367.5 nm, as shown in Fig. 4. The increase in intensity and the absorption peak at 367.5 nm occur simultaneously with increasing addition of NO3− (1.0 equiv.) concentration; this can be attributed to the formation of a stable complex of 2·Co2+ with NO3−. The spectral shifts accompanied by the change of color from yellow to pink clearly delineate the formation of a complex species between 2·Co2+ and NO3− that can be clearly seen under visible light by the naked eye (inset image in Fig. 4). This suggests that there is a change in the absorption intensity of the complex formed between NO3− and 2·Co2+ upon further addition of NO3− anion. In the UV-visible titration analysis of different ratios of 2·Co2+ and NO3−, the Job's plot analysis revealed a 2:1 stoichiometry between 2·Co2+ and NO3− in the complex. As expected, when the molar fraction of the complex was 0.7, the intensities reached a maximum, which demonstrates that the complex formation between 2·Co2+ and NO3− has 2:1 stoichiometry (inset in Fig. 4a).34 The complex can determined according to the B–H equation from the absorption titration analysis. At 367.5 nm, a plot of 1/[A − A0] versus 1/[NO3−] gives a straight line and a linear correlation coefficient of R2 = 0.9580, confirming the formation of a complex with NO3− in the range of 0.14 to 0.32 μM; the association constant was found to be 7.6 × 104 M−1 (inset in Fig. 4b). This result clearly demonstrates the complexation of 2·Co2+ with NO3−, leading to free 2.
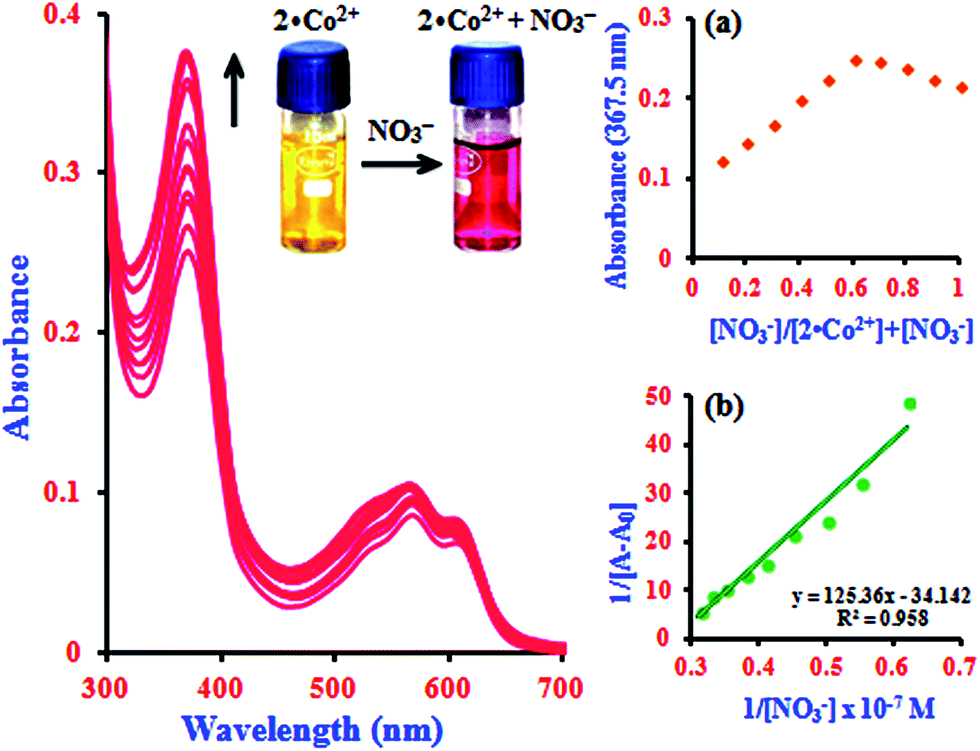 | ||
| Fig. 4 Absorption spectra of 2·Co2+ with the addition of NO3− concentrations of 0.14 to 0.32 μM. Inserts; images of naked eye colorimetric detection of complex formation by a color change from yellow to pink (2·Co2+ to NO3−), (a) Job's plot analysis for the complexation between 2·Co2+ and NO3− at 367.5 nm and (b) Benesi–Hildebrand plot of 1/[A − A0] vs. 1/[NO3−]. | ||
In fluorescence titration experiments, addition of NO3− to a 2·Co2+ solution led to a significant increase in the fluorescence intensity at 632.5 nm; the emission reached saturation when 2 equiv. of NO3− were added (relative to 2). The emission intensities were found to increase gradually, and a large red shift was observed in the emission maximum from 632.5 to 637 nm, as shown in Fig. 5. This can be ascribed to NO3−-induced fluorescence enhancement, which can be accounted for by the Co2+ ion.33 In the absence of NO3−, the 2·Co2+ complex shows a weak fluorescence emission, while in the presence of NO3−, 2·Co2+ undergoes complexation to form free 2 and Co2+ with NO3− in the medium, leading to a highly fluorescent ‘switch-On’ response with only 1 equiv. of NO3− due to the restoration of 2. Hence, a significant enhancement was observed on addition of NO3− to the 2·Co2+ complex. In the fluorescence titration analysis of different ratios of 2·Co2+ to NO3−, the Job's plot revealed a 2:1 stoichiometry between 2·Co2+ and NO3− in the complex (inset in Fig. 5a). The association constant (Ka) of 2·Co2+ for NO3− was calculated on the basis of the B–H plot, which gives a straight line and a good linear correlation coefficient of R2 = 0.9478; it was found to be 3.77 × 104 M−1 (inset in Fig. 5b). The detection limit of the 2·Co2+ complex as a fluorescent sensor for the analysis of NO3− was determined from a plot of the fluorescence intensity as a function of the concentration of NO3−. It was found that 2·Co2+ has a detection limit of 2.4 nM for NO3− in concentrations of 0.14 to 0.32 μM. The addition of NO3− to the 2·Co2+ complex led to the appearance of a new excitation peak at 367.5 nm and enhanced the emission maxima, with a red-shift from 632.5 to 637 nm. 2 selectively binds with Co2+ to form a 2·Co2+ complex, which undergoes considerable changes in its spectral properties upon addition of NO3−.33 NO3− is known to react with Co2+ to form the very stable CoNO3 species. Our preliminary report showed that 2·Co2+ and 2·NO3− are the fluorescence turn on–off mechanisms. However, here, it was noted that the interference of anions (NO3−) with the fluorescence of 2·Co2+ forms the off–on mechanism. This off–on phenomenon induced by NO3− anion enhances the fluorescence intensity; this is due to the changes in 2·Co2+ upon complexation with NO3− anion (turn on–off–on), as shown in Scheme 2.33 The inclusion complex (2) has high selectivity and sensitivity for Co2+, and Co2+ has high selectivity and sensitivity for NO3−; this supramolecular sensing process is acceptable for recognition of Co2+ and NO3− in aqueous solution. The detection limit is comparable to that of cobalt and nitrite ions (1.7 μM and 65 μM) recommended by the World Health Organization (WHO) guidelines.11 Therefore, 2 could be a good indicator for detection of cobalt and nitrite ions in drinking water. These results show that the 2·Co2+ is sensitive enough to monitor NO3− in water.
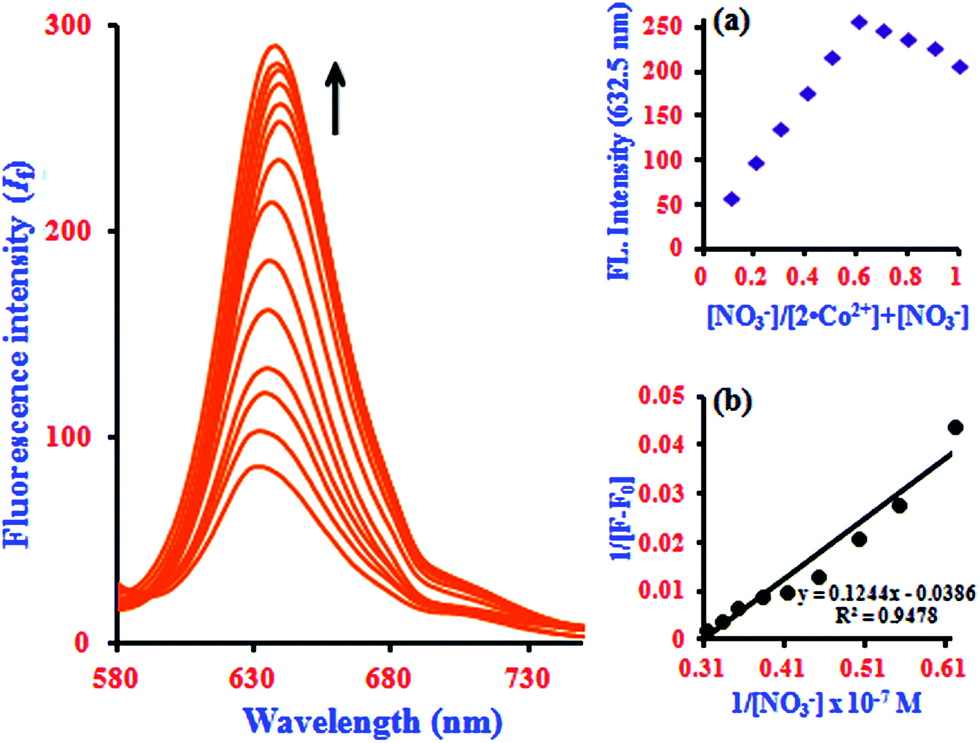 | ||
| Fig. 5 Fluorescence spectra of 2·Co2+ with the addition of NO3− concentrations of 0.14 to 0.32 μM. Insert: (a) Job's plot analysis for the complexation between 2·Co2+ and NO3− at 632.5 nm and (b) Benesi–Hildebrand plot of 1/[F − F0] vs. 1/[NO3−]. | ||
According to a survey of the literature, many relevant works concerning colorimetric and fluorescent chemosensors of Co2+ and NO3− have been reported.1–3,19,24,30,33,34 We have noticed that most of the reported Co2+ and NO3− selective fluorescent chemosensors have several drawbacks (i.e., linear range (LR), poor detection limit (LOD) and pH); these are also listed in Table 1. The main analytical characteristics of the proposed chemosensor for the determination of Co2+ and NO3− and that of Co2+ for other anions were compared with those of some previously reported fluorescent chemosensors, including benzothiazole,1 ruthenium(II) tris (bipyridine),2a resorufin,2b triazole-carboxyl AgNps,3a dicarbonyl pyridine,3b PYET,3c CPOH AuNPs,19a HPHPA,19b tripodal amide,19c phthalazine,19d coumarin,19e bis-pyridinium,19f DIPZON,24a coumarin,24b quinolone, dipodal-triazole33 and bibenzimidazole;34a these are given in Table 1. The proposed fluorescent chemosensor (β-CD:1,2-DHAQ; 2) is more sensitive for the detection of Co2+ and NO3− than the previously reported chemosensors. Thus, exploration of superior fluorescent chemosensors for the detection of Co2+ and NO3− in aqueous solution is still in strong demand.
| Reagent | Sample | Ion | LR (M) | LOD (M) | pH | Ref. |
|---|---|---|---|---|---|---|
| Benzothiazole | HEPES buffer | Co2+ | 2.0 × 10−6 to 1.0 × 10−4 | 1.0 × 10−6 | 7.2 | 1 |
| Ruthenium(II) tris(bipyridine) | NaOAc | Co2+ | 1.0 × 10−7 to 5.0 × 10−5 | 5.0 × 10−8 | 6.0 | 2a |
| HOAc | ||||||
| Resorufin | HEPES buffer | Co2+ | 1.0 × 10−6 to 2.0 × 10−5 | 2.0 × 10−5 | 7.2 | 2b |
| Triazole-carboxyl Ag Nps | — | Co2+ | 5.0 × 10−6 to 5.0 × 10−4 | 7.5 × 10−6 | 3.0–12.0 | 3a |
| Dicarbonyl pyridine | THF | Co2+ | 0.5 × 10−6 to 3.0 × 10−6 | 3.5 × 10−6 | — | 3b |
| PYET | HEPES buffer | Co2+ | 1.0–5.0 × 10−6 | 6.4 × 10−7 | 7.4 | 3c |
| F− | 0.1–1.0 × 10−5 | 7.7 × 10−8 | ||||
| CPOH Au NPs | — | Co2+ | 0.01–1.0 × 10−6 | 1.0 × 10−6 | 7.0 | 19a |
| HPHPA | DMSO: bis–tris buffer | Co2+ | 0.2–1.6 × 10−6 | 1.8 × 10−6 | 7.0 | 19b |
| Tripodal amide | DMSO: water | Co2+ | 0.8–8.0 × 10−5 | 4.9 × 10−5 | — | 19c |
| Phthalazine | CH3CN: water | Co2+ | 0.1–0.01 × 10−5 | 2.8 × 10−8 | — | 19d |
| Coumarin | HEPES buffer | Co2+ | 0–0.1 × 10−5 | 0.3 × 10−6 | 7.4 | 19e |
| Bis-pyridinium | Phosphate buffer | NO3− | 0.1–0.02 × 10−5 | 9.2 × 10−7 | 6.0 | 19f |
| DIPZON | CH3OH: water | Co2+ | 0–3.5 × 10−4 | 7.0 × 10−7 | 7.4 | 24a |
| Coumarin | HEPES buffer | Co2+ | 0–3.0 × 10−6 | 1.0 × 10−6 | 7.2 | 24b |
| Quinolone | Bis–tris buffer | Co2+ | 0.2–0.02 × 10−6 | 1.2 × 10−6 | 7.0 | 30 |
| Dipodal-triazole | HEPES buffer | Co2+ | 1.0–0.1 × 10−6 | 0.3 × 10−6 | 7.4 | 33 |
| NO2− | 1.0–0.1 × 10−6 | 1.2 × 10−6 | ||||
| Bibenzimidazole | MeCN | Co2+ to I− | 0.6–0.02 × 10−6 | 0.4 × 10−6 | 9.0 | 34a |
| β-CD:1,2-DHAQ | Water | Co2+ | 0.3–0.85 × 10−6 | 2.2 × 10−8 | 11.0 | This work |
| Co2+ to NO3− | 0.14–0.32 × 10−6 | 0.24 × 10−8 |
Molecular modeling studies of Co2+ and NO3−
The ion docking scores of the molecular docking models of 1,2-DHAQ (1) and β-CD:1,2-DHAQ (2) with Co2+ and NO3− were calculated using the PatchDock and FireDock servers; the most probable structures were based on the energetic parameters given in Table S1† and Fig. 6. The docking studies revealed the highest geometric shape complementarity scores, approximate interface area sizes and atomic contact energies of the complexes according to the PatchDock server. The refinement by FireDock was extensively tested; the highest global energy, attractive and repulsive van der Waals interaction energies and atomic contact energy were the most probable and energetically favorable models.35 The metal center closely fits into the hydroxyl and carbonyl group of 1 and reveals an excellent interface with the hydrophobic cavity of β-CD. These interactions suggest a process in which water molecules in the cavity are replaced by guest molecules via van der Waals forces between molecules.22b,36 The molecular docking results are in good correlation with the results obtained through experimental methods.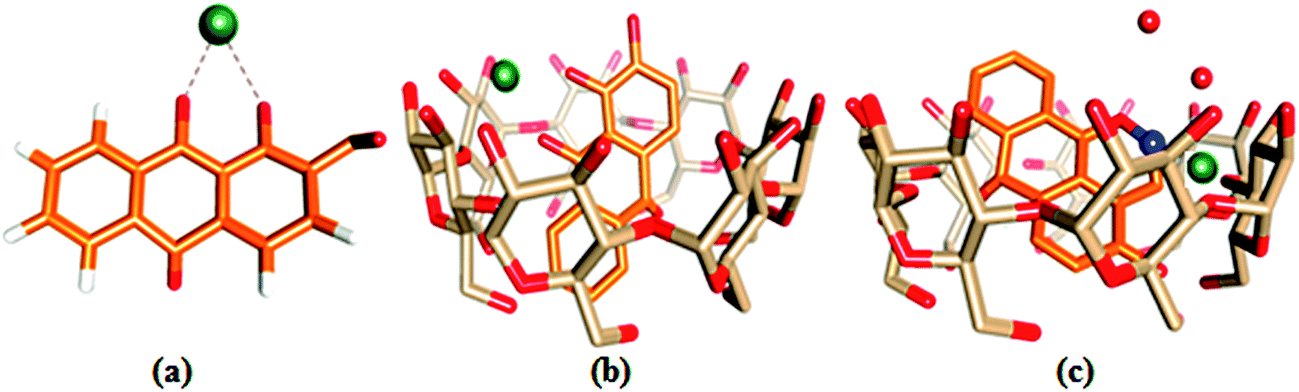 | ||
| Fig. 6 Ball and stick representations of the structures of (a) 1·Co2+, (b) 2·Co2+ and (c) 2·Co2+ with NO3−; the O, C, H, Co and N atoms are shown in red, orange, white, green and blue colors. | ||
The optimized geometries of the ion complexes of 1 and 2 with Co2+ and NO3− are shown in Fig. 7; the images show that Co2+ and NO3− ions bind readily to 1 through the coordination sites, and the entire molecular system forms a nearly square planar structure. The ligand structures tend to optimize with rather long CO distances; constraining these bond lengths to typical CO distances for quinones improves the fit between the calculated and observed electronic spectra. The C–O distances of ligand 1 vary for the different molecules in the unit cell and appear to be strongly influenced by intermolecular hydrogen bonding. The geometry optimization demonstrated conformational changes of the anthraquinone ring, i.e., the quinone and catecholate moieties, to accommodate the Co2+ and NO3− ions. In the process, the carbonyl oxygen of the quinone moiety and the phenolate oxygen of the phenol moiety rotated toward the same direction to form a binding core. In the optimized structures of 2·Co2+ and 2·NO3−, the Co2+ and NO3− ions are bound by two oxygen atoms (carbonyl oxygen and catecholate oxygens) in the binding core, leading to highly distorted geometries about the Co2+ and NO3− ions. The distances and angles (Co–O, O–Co–O, N–O–H and O–N–O; phenolate and quinone oxygen atoms) associated with the central ions of the optimized complexes of Co2+ and NO3− with 1 and 2 are given in Table S2.†10b,37
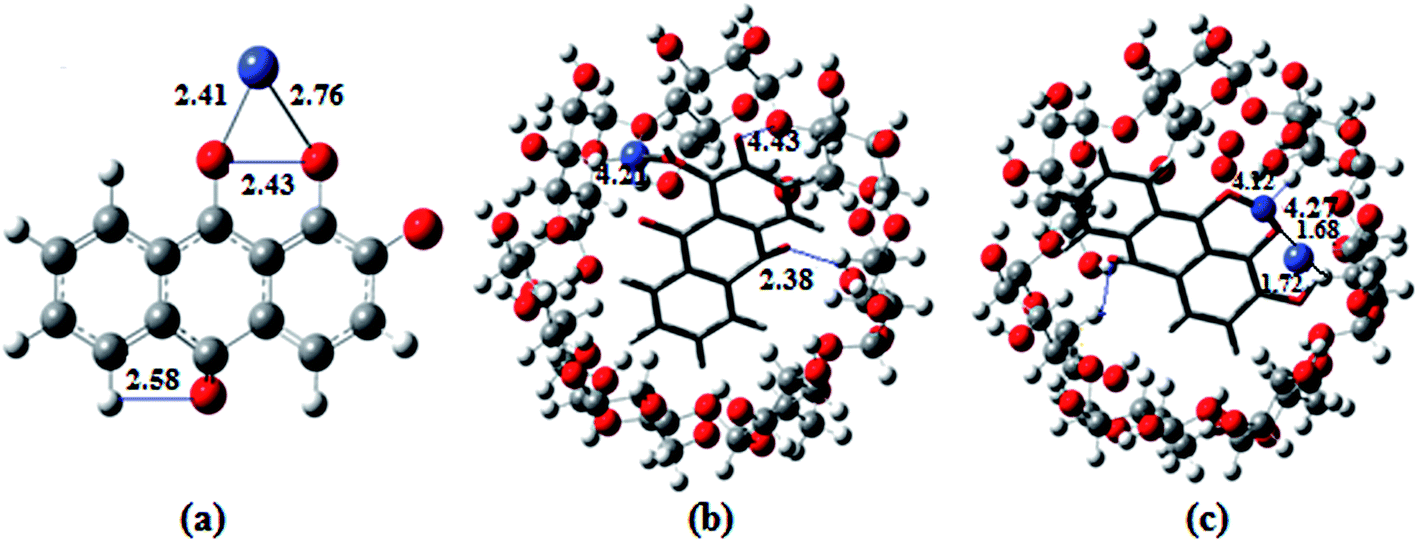 | ||
| Fig. 7 The DFT optimized structures of (a) 1·Co2+, (b) 2·Co2+ and (c) 2·Co2+ with NO3−, calculated at the B3LYP/6-31G/LANL2DZ level. The red, gray, white, cornflower blue and blue balls refer to O, C, H, Co and N, respectively. All the distances are given in Å. | ||
The optimized geometries, highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) of 1 and 2 and their complexes with Co2+ and NO3− were calculated; the results are listed in Table S3† and presented in Fig. 8. In contrast to the HOMO of 1, the amplitude of the carbonyl groups increases significantly; this indicates that the HOMO–LUMO transition of 1 has less hydroxyl-to-carbonyl charge transfer character. The same level of theory was used to calculate the electronic absorption spectra of the hypothetical 1:1 complexes. A comparison between the UV-Vis spectrum 1:1 complex, obtained from chemosensor methods, and theoretical transitions calculated for the possible structures is presented in Fig. 8. Better agreement is found when Co2+ and NO3− are coordinated with the catecholate group. The theoretical spectra are quite similar in the longer wavelength range (500 to 600 nm); the maxima recorded at 562.5 nm were calculated at 530.5 nm for the structures with chelation at the catechol site and hydroxy-keto site, respectively. The results show that Co2+ is unambiguously coordinated to the hydroxy-keto function. Thus, it is clearly established by ab initio calculations that the catechol function of 1 is the chelating site involved the fixation of other cations.38
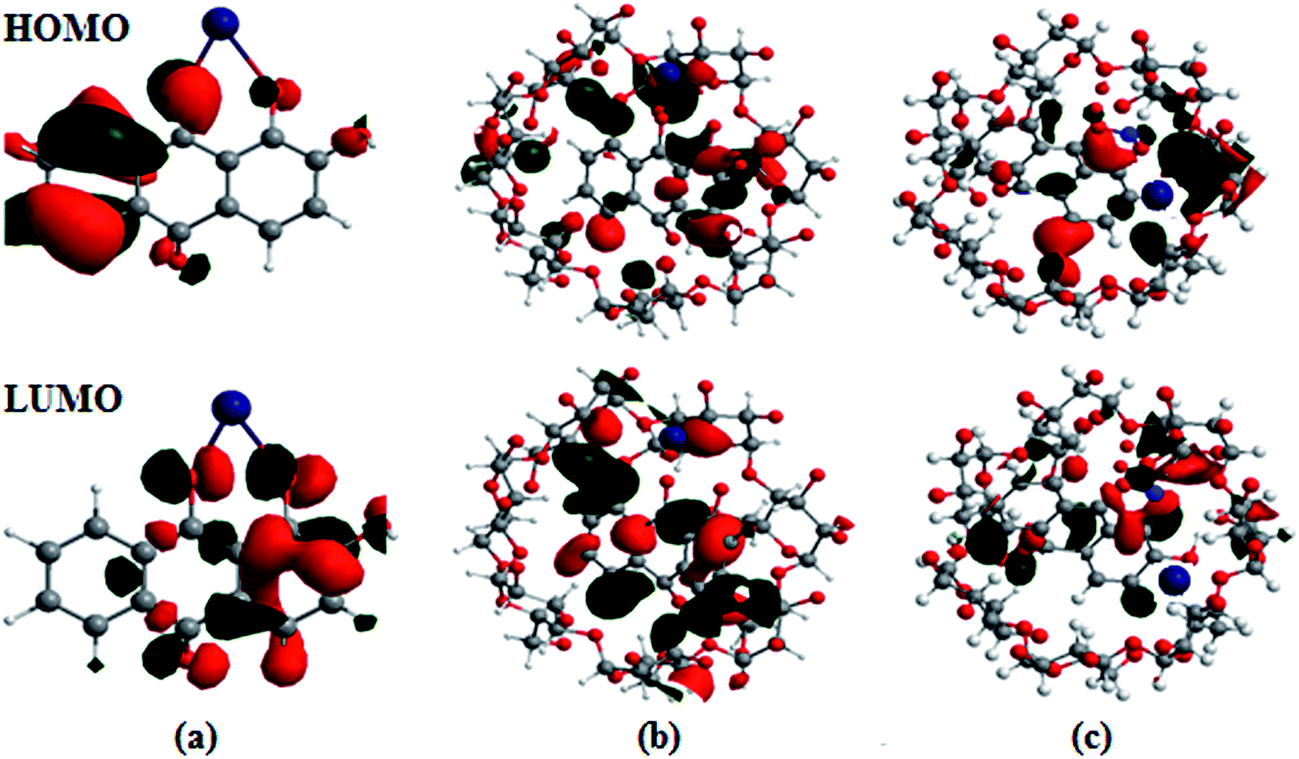 | ||
| Fig. 8 Frontier molecular orbitals (HOMO and LUMO) of the B3LYP/6-31G/LANL2DZ optimized complexes of (a) 1·Co2+, (b) 2·Co2+ and (c) 2·Co2+ with NO3−. | ||
The HOMO–LUMO excitation was found to be related to MLCT and its contribution to the low-energy excitation. This excitation corresponds to charge transfer from the excited Co2+ center to the catecholate and quinone moieties, which provides the excited state. For the fluorescence quenching by Co2+, different calculation results are obtained, as shown in Fig. 8. The removal of Co2+ from the 1·Co2+ complex cannot restore the emission of 1. The energy gap (ΔE) between EHOMO − ELUMO in the 1·Co2+ complex was 1.16 eV. In the inclusion complex of 1 with β-CD, the HOMO and LUMO energies differed from that in free 1, indicating that the inclusion complexation exhibits negligible influence on the distribution of the frontier orbitals and the active site of 1. However, the inclusion complexation shows the remarkable influence of the HOMO and LUMO energies. Compared with free 1, inclusion in β-CD induces slight decreases in the HOMO and LUMO energies. This indicates that the electron donating capacity of 1 is enhanced by β-CD.22b Moreover, the cavity size of the host molecules and the inclusion mode shows the remarkable influence of the HOMO energies.
Furthermore, the inclusion complex of 2 with Co2+ and NO3− shows a remarkable influence on the HOMO and LUMO energies. Binding energies obtained from theoretical studies have proved that the energy values of 2 decrease with the addition of Co2+ and NO3−. The ΔE between EHOMO and ELUMO in the complexes of 2·Co2+ and 2·Co2+ with NO3− are 1.04 and 0.91 eV, respectively. The energy gap between the HOMO and LUMO in the complex decreases and shows favorable complexation according to the proposed coordination mode, leading to the formation of stable complexes.39 The energy decrease in the case of 2·Co2+ to NO3− is significantly more stable than that of 1·Co2+ and 2·Co2+. The negative HOMO value of this stabilization energy showed that a stable inclusion complex is formed. Thus, the complex formed by 1 with ions is preferentially encapsulated in the β-CD cavity.40–42 The calculations carried out by the DFT method show that the inclusion complexes of 1 with coordination of ions in the presence of β-CD are stable; this confirms the experimental observations.
Logic gate behaviors of Co2+ toward NO3−
The fascinating ‘on–off’ emission properties of β-CD:1,2-DHAQ (2) in the presence of Co2+ and NO3− encouraged us to constructed a molecular logic gate. The development of molecular logic gates has gained considerable interest in the design of molecular-scale computers for applications in the fields of biochemical systems, biosensing, functional materials, diagnostics, etc.43 The development of optical receptors which mimic the functions of an XNOR logic gate is very important in supramolecular chemistry.44 Thus, the molecular switching behavior of 2 with two chemical inputs (Co2+ and NO3−) can be demonstrated with the help of binary logic. Depending on the two chemical inputs, Co2+ and NO3−, 2 can switch between different fluorescence emission states, i.e. “on or off”, as given in Table S4† and Fig. 9. The presence and absence of the two chemical inputs, Co2+ and NO3−, was defined as the ‘1’ and ‘0’ states, and the fluorescence intensity of 2 at 628 nm was defined as the output of the logic gate.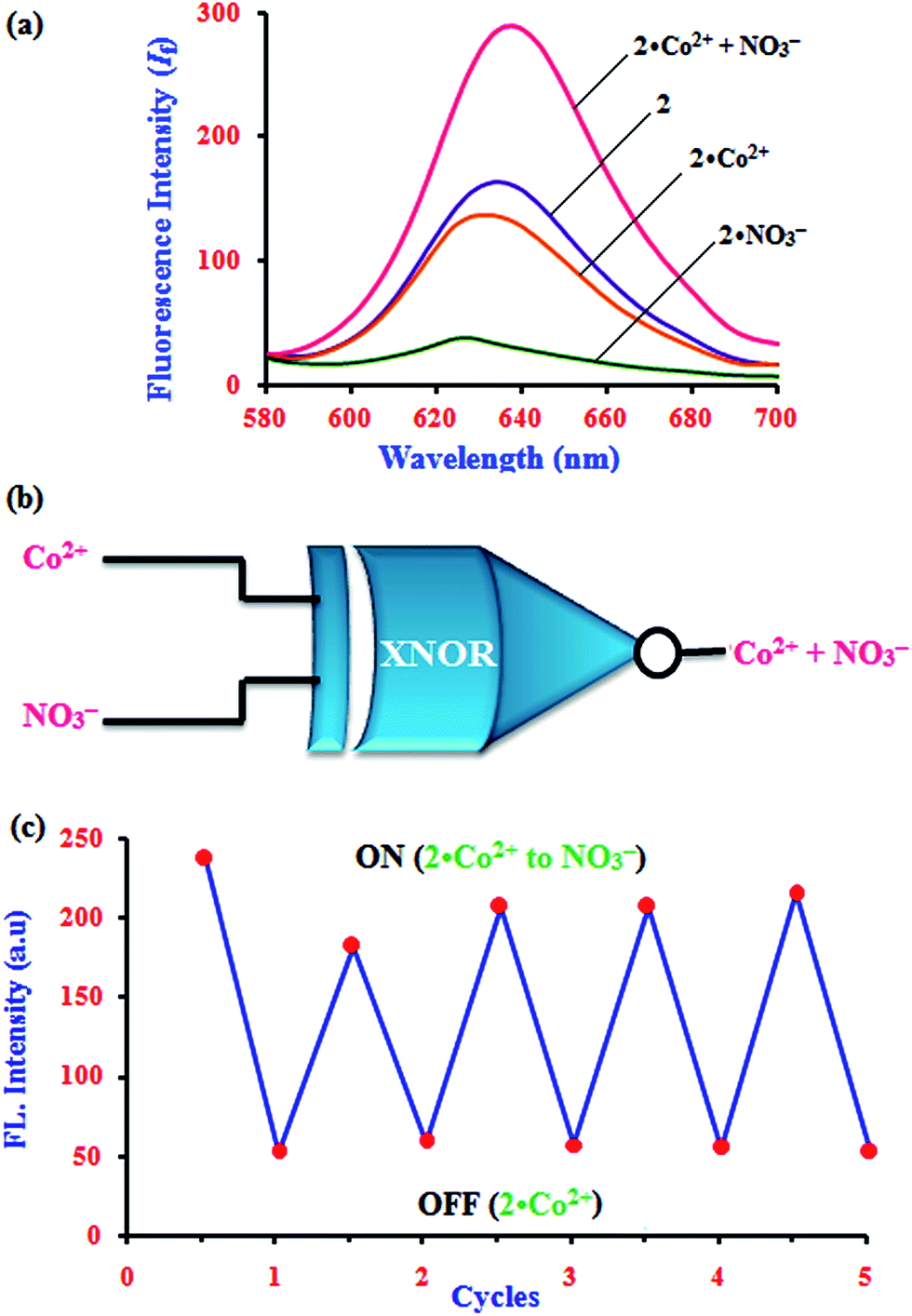 | ||
| Fig. 9 (a) Fluorescence emission spectra for an XNOR logic gate with two chemical inputs, Co2+ and NO3−, for 2 and (b) schematic of an exclusive-NOR (XNOR) gate formed with two chemical inputs, Co2+ and NO3−, for 2. (c) Fluorescence intensity of 2 in aqueous solution at pH 11 upon alternate additions of Co2+ and NO3−. | ||
The fluorescence of receptor 2 is quenched in the presence of Co2+ and NO3− (0, 0), and that of 2·Co2+ is enhanced in the presence of and NO3− (1) due to the energy transfer process. However, enhanced fluorescence of 2 above the threshold level is observed in the absence (0, 0) and presence of both inputs (1, 1). Therefore, monitoring the fluorescence at 634, 630.5 and 632.5 nm of 2 and with the two inputs (Co2+ and NO3−), an XNOR-type logic gate can be constructed at the molecular level, as shown in Fig. 9b. An XNOR logic function is a two-input device; its output is “true” if the inputs are the same and “false” if the outputs are different. Due to this property, the 2·Co2+ ensemble is a practical sensor for NO3− detection in water. The reversible sensing nature of the chemosensor is an important aspect of sensing phenomena. As shown in Fig. 9, after the addition of Co2+, the emission intensity decreased to a lower level for chemosensor 2. Similarly, alternate additions of NO3− and Co2+ to a solution of 2 exhibited a switchable change in the enhanced fluorescence intensity. Moreover, 2 displays an “On–Off–On” mode of fluorescence change (Fig. 9c) with alternate addition of Co2+ and NO3−, along with reversible formation–separation of the complex; thus, this complex is attractive for sensing applications. This reversible fluorescence behavior of 2 can be repeated several times.
Living cell imaging of Co2+ toward NO3−
The biological applications of the excellent selectivity and high sensitivity of 1,2-DHAQ (1) and β-CD:1,2-DHAQ (2) towards the Co2+ and NO3− complexes were demonstrated in the human cervical cancer HeLa cell line. Human cervical HeLa cancer cells were first incubated separately with CoCl2 and NH4(NO3)2 for 4 h and then treated with 10 μM 1 and 2 for 30 min; the cells showed fluorescence (625 to 635 nm) when observed under a fluorescence microscope using an excitation filter of 500 to 550 nm (Fig. 10). The cells were able to take up both Co2+ and NO3−, and receptors 1 and 2 produced fluorescence when the (1·Co2+, 2·Co2+ and 2·Co2+ to NO3−) complexes formed. This indicates that 1 and 2 have some autofluorogenic properties when applied to biological systems. Weak fluorescence was observed for complex 1 with increasing concentration of Co2+. The intensities of the fluorescence produced by 1 with the Co2+ complex remained the same even when the incubation time of Co2+ was increased from 3 h to 48 h. Incubating the cells with Co2+ alone failed to produce any fluorescence, confirming that the fluorescence was uniquely produced by 1. This shows that the complex of 1 with Co2+ has potential applications in live cell imaging.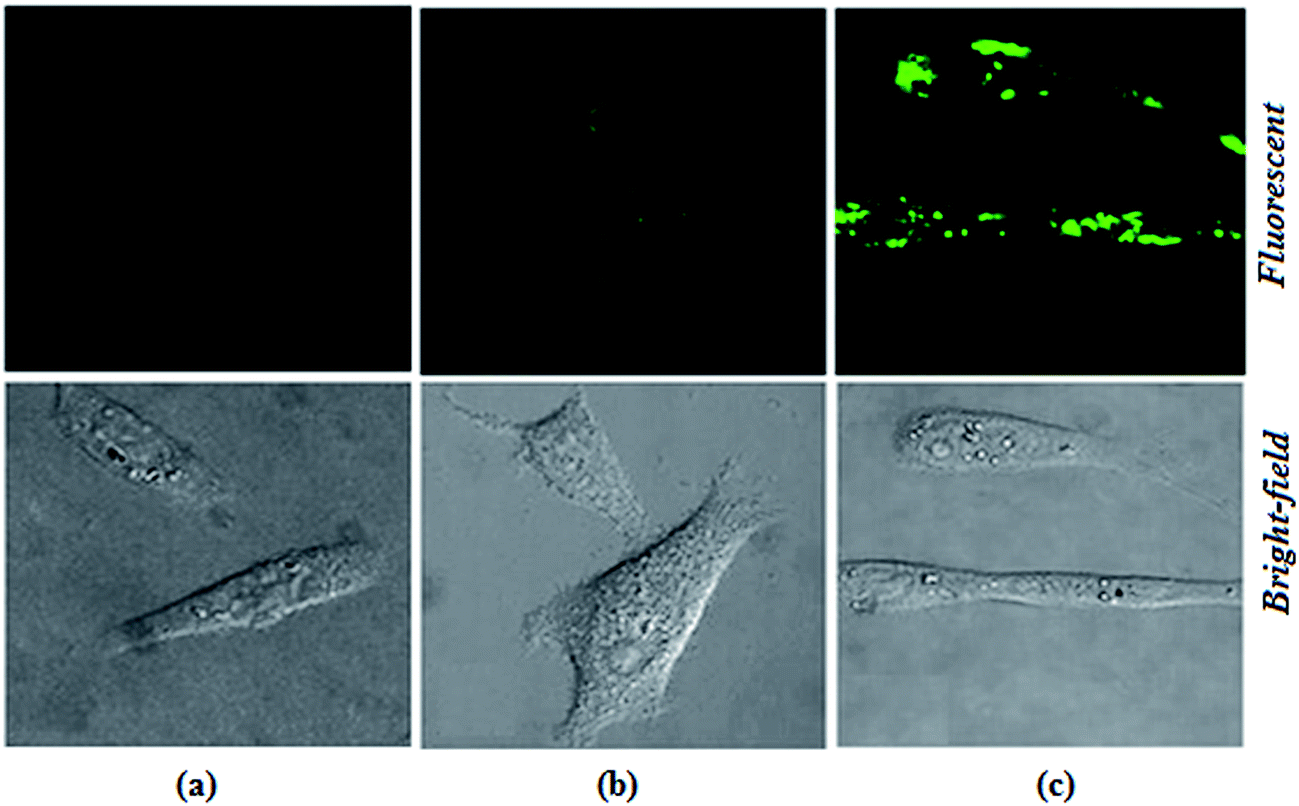 | ||
| Fig. 10 Intracellular Co2+ and NO3− imaged in living cells (HeLa cells) at 28 °C by confocal laser scanning microscopy. (a) Cells and Co2+ incubated with 1 (1·Co2+), with excitation at 530.5 nm and emission at 629 nm, (b) after incubation of cells with Co2+ and 2 (2·Co2+), with excitation at 528 nm and emission at 634 nm and (c) after incubating cells with Co2+, 2 and NO3− (2·Co2+ to NO3−), with excitation at 367.5 nm and emission at 632.5 nm. (a–c) Indicate the fluorescence and bright-field images of the cells. | ||
The more potent biological activities of β-CD can be explained by the more efficient cellular uptake of 1 with Co2+ and Co2+ to NO3− from β-CD compared to corresponding concentrations of free 1 with Co2+.24b,45 Interestingly, the complex of 1 with Co2+ and Co2+ to NO3− taken up from β-CD also remained longer in the cells, leading to prolonged exposure of the cells to intracellular Co2+ and NO3−. It has been shown for several other hydrophobic molecules that β-CD enhances their cellular uptake.24b,46 The interesting photophysical properties of 2, including high selectivity, sensitivity and rapid response towards Co2+ and subsequent recognition of NO3−, further prompted us to extend our study to the detection of Co2+ and NO3− ions in biological systems by living cell imaging experiments. The results suggested that the host–guest system of β-CD and 1 can permeate cells and provide specific fluorescence signals in the presences of Co2+ and NO3−. The obtained bioimaging results are in good agreement with the fluorescence spectra for the recognition of both Co2+ and NO3− ions. The fluorescence microscopic analysis strongly suggested that chemosensor 2 could permeate the cell membrane and rapidly cascade recognition of intracellular Co2+ and NO3− at micromolar concentrations. Therefore, this chemosensor could be used as a sensor for Co2+ and NO3− in biological systems. These results demonstrated that chemosensors 1 and 2 can be applied in the imaging of intracellular Co2+ and NO3− ions.
Conclusion
In summary, to the best of our knowledge, this is the first simple and effective study concerning the inclusion complex (2) of a new fluorescent chemosensor for selective and sensitive detection of Co2+ ion and sequential recognition of NO3− ion by the derived 2·Co2+ complex. The changes in color of 2 upon the addition of Co2+ and NO3− ions were clearly visible under visible light by the naked eye. The excellent detection limits of 2 for Co2+ (22.7 nM) and 2·Co2+ for NO3− (2.4 nM) can be useful for the detection of trace amounts of Co2+ and NO3− ions. Molecular docking studies and DFT calculations were used to corroborate the experimental observations. Moreover, the fluorescence changes of 2 upon the addition of Co2+ and NO3− can be utilized as an XNOR logic gate. In addition, we demonstrated that this new fluorescent chemosensor 2 can be utilized in live cell imaging of Co2+ and NO3− ions. These results provide a new opportunity to detect NO3− through the use of a cation chemosensor (2·Co2+).Acknowledgements
One of the authors, S. Mohandoss (SRF), wishes to thank UGC for providing financial assistance in the scheme of RFSMS-BSR to carry out this work. The fluorescence spectra were carried out with a JASCO spectrofluorometer FP-8200 facility under the scheme of DST, Government of India (Ref: DST Order No. SR/FT/CS-101/2010 (G)) is gratefully acknowledged.References
- D. Maity, V. Kumar and T. Govindaraju, Org. Lett., 2012, 14, 6008–6011 CrossRef CAS PubMed.
- (a) C. Y. Li, X. B. Znang, Z. Jin, R. Hari, G. L. Shen and R. Q. Yu, Anal. Chim. Acta, 2006, 580, 143–148 CrossRef CAS PubMed; (b) D. Maity, A. Raj, D. Karthigeyan, T. K. Kundu and T. Govindaraju, RSC Adv., 2013, 3, 16788–16794 RSC.
- (a) Y. Yao, D. Tian and H. Li, ACS Appl. Mater. Interfaces, 2010, 2, 684–690 CrossRef CAS PubMed; (b) Y. Tan, J. Yu, Y. Cui, Y. Yang, Z. Wang, X. Hao and G. Qian, Analyst, 2011, 136, 5283–5286 RSC; (c) M. Inya, D. Jenyanthi, K. Krishnaveni and D. Chellappa, RSC Adv., 2014, 4, 25393–25397 RSC.
- C. Reimann, F. Koller, G. Kashulina and H. P. Englmaier, Environ. Pollut., 2001, 115, 239–252 CrossRef CAS PubMed.
- J. Davis, K. J. McKeegan, M. F. Cardosi and D. H. Vaughan, Talanta, 1999, 50, 103–112 CrossRef CAS PubMed.
- I. A. Wolf and A. E. Wasserman, Science, 1972, 177, 15–19 Search PubMed.
- M. J. Hill, Nitrates and Nitrites in Food and Water, Woodhead Publishing Limited, England, 1st edn, 1996 Search PubMed.
- C. J. Johnson and C. K. Burton, Am. J. Ind. Med., 1990, 18, 449–456 CrossRef CAS PubMed.
- R. F. Follett, and J. L. Hatfield, Nitrogen in the environment sources, problems, and management, Elsevier, Amsterdam, NewYork, 2001 Search PubMed.
- (a) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, UK, 2nd edn, 2009, ch. 4, p. 224 Search PubMed; (b) O. A. Okunola, P. V. Santacroce and J. T. Davis, Supramol. Chem., 2008, 20, 169–190 CrossRef CAS.
- (a) J. H. Kim, H. J. Gibb and P. D. Howe, Cobalt and inorganic cobalt compounds, WHO Press, Geneva, Switzerland, 2006 Search PubMed; (b) WHO, Guidelines for Drinking Water Quality: incorporating 1st and 2nd addenda, WHO Press, Geneva, Switzerland, Recommendations. – 3rd edn, 2008, vol. 1 Search PubMed.
- (a) M. Ghaedi, F. Ahmadi and A. Shokrollahi, J. Hazard. Mater., 2007, 142, 272–278 CrossRef CAS PubMed; (b) M. Zhag, D. X. Yuan, G. H. Chen, Q. L. Li, Z. Zhang and Y. Liang, Microchim. Acta, 2008, 160, 461–469 CrossRef.
- A. R. Khorrami, T. Hashempur, A. Mahmoudi and A. R. Karimi, Microchem. J., 2006, 84, 75–79 CrossRef CAS.
- (a) S. Mittal, R. Sharma, M. Sharma, N. Singh, J. Singh, N. Kaur and M. Chhibber, J. Appl. Electrochem., 2014, 44, 1239–1251 CrossRef CAS; (b) K. Ngamdee, T. Tuntulani and W. Ngeontae, Sens. Actuators, B, 2015, 216, 150–158 CrossRef CAS.
- (a) C. Abha, K. B. Anil and V. K. Gupta, Talanta, 2001, 55, 789–797 CrossRef; (b) K. Tirumalesh, Talanta, 2008, 74, 1428–1434 CrossRef CAS PubMed.
- (a) J. C. M. Gamboa, R. C. Pena, T. R. L. C. Paixao and M. Bertotti, Talanta, 2009, 80, 581–585 CrossRef CAS PubMed; (b) C. E. Lopez Pasquali, A. Gallego-Pico, P. Fernandez Hernando, M. Velasco and J. S. Durand Alegrıa, Microchem. J., 2010, 94, 79–82 CrossRef.
- N. Bord, G. Cretier, J. L. Rocca, C. Bailly and J. P. Souchez, J. Chromatogr., A, 2005, 1100, 223–229 CrossRef CAS PubMed.
- (a) M. Morfobos, A. Economou and A. Voulgaropoulos, Anal. Chim. Acta, 2004, 519, 57–64 CrossRef CAS; (b) Y. Tian, J. X. Wang, Z. Wang and S. C. Wang, Synth. Met., 2004, 143, 309–313 CrossRef CAS.
- (a) K. D. Bhatt, D. J. Vyas, B. A. Makwana, S. M. Darjee and V. K. Jain, Spectrochim. Acta, Part A, 2014, 121, 94–100 CrossRef CAS PubMed; (b) Y. J. Na, Y. W. Choi, G. R. You and C. Kim, Sens. Actuators, B, 2016, 223, 234–240 CrossRef CAS; (c) J. R. Zhou, D. P. Liu, Y. He, X. J. Kong, Z. M. Zhang, Y. P. Ren, L. S. Long, R. B. Huang and L. S. Zheng, Dalton Trans., 2014, 43, 11579–11586 RSC; (d) S. Patil, R. Patil, U. Fegade, B. Bondhopadhyay, S. K. Sahoo, N. Singh, A. Basu, R. Bendre and A. Kuwar, Photochem. Photobiol. Sci., 2015, 14, 439–443 RSC; (e) Z. Liu, W. Wang, H. Xu, L. Sheng, S. Chen, D. Huang and F. Sun, Inorg. Chem. Commun., 2015, 62, 19–23 CrossRef CAS; (f) Y. Yang, S. Chen and X. L. Ni, 2015, 87(14), 7461–7466.
- (a) Y. Wang, H. Sun, L. Hou, Z. Shang, Z. Donga and W. Jin, Anal. Methods, 2013, 5, 5493–5500 RSC; (b) Y. Wang, L. Wang, L. L. Shi, Z. B. Shang, Z. Zhang and W. J. Jin, Talanta, 2012, 94, 172–177 CrossRef CAS PubMed; (c) J. Zhou, H. Liu, B. Jin, X. Liu, H. Fu and D. Shangguan, J. Mater. Chem. C, 2013, 1, 4427–4436 RSC.
- (a) S. Mohandoss, M. Maniyazagan and T. Stalin, Mater. Sci. Eng., C, 2015, 48, 94–102 CrossRef CAS PubMed; (b) S. Mohandoss, J. Sivakamavalli, B. Vaseeharan and T. Stalin, RSC Adv., 2015, 5, 101802–101818 RSC; (c) S. Mohandoss, J. Sivakamavalli, B. Vaseeharan and T. Stalin, Sens. Actuators, B, 2016, 234, 300–315 CrossRef CAS.
- (a) J. Sun, X. S. Zhu and M. Wu, J. Fluoresc., 2007, 17, 265–270 CrossRef CAS PubMed; (b) M. Y. Xu, S. Z. Wu, F. Zeng and C. M. Yu, Langmuir, 2010, 26, 4529–4534 CrossRef CAS PubMed.
- S. Mohandoss and T. Stalin, Photochem. Photobiol. Sci., 2016 10.1039/C6PP00285D.
- (a) S. H. Mashraqui, M. Chandiramani, R. Betkar and K. Poonia, Tetrahedron Lett., 2010, 51, 1306–1308 CrossRef CAS; (b) D. Maity and T. Govindaraju, Inorg. Chem., 2011, 50, 11282–11284 CrossRef CAS PubMed.
- P. Alreja and N. Kaur, RSC Adv., 2016, 6, 23169–23217 RSC.
- P. Job, Ann. Chim. Appl., 1928, 9, 113–203 CAS.
- (a) M. Yu, L. Pan, L. Sun, J. Li, J. Shang, S. Zhang, D. Liu and W. Li, J. Mater. Chem. B, 2015, 3, 9043–9052 RSC; (b) I. Abulkalam Azath and K. Pitchumani, Sens. Actuators, B, 2013, 188, 59–64 CrossRef; (c) P. Suresh, I. Abulkalam Azath and K. Pitchumani, Sens. Actuators, B, 2010, 146, 273–277 CrossRef CAS.
- C. Jullian, S. F. Sandoval, M. R. Aranguiz, H. G. Machuca, P. S. Figueroa, C. C. Barros, G. Z. Torres, R. Adam and B. Abarca, Carbohydr. Polym., 2014, 107, 124–131 CrossRef CAS PubMed.
- (a) T. Tang, Y. Zhou, Y. Chen, M. Li, Y. Feng, C. Wang, S. Wang and X. Zhou, Anal. Methods, 2015, 7, 2386–2390 RSC; (b) S. Dalapati, S. Jana and N. Guchhait, Spectrochim. Acta, Part A, 2014, 129, 499–508 CrossRef CAS PubMed.
- G. J. Park, Y. J. Na, H. Y. Jo, S. A. Lee and C. Kim, Dalton Trans., 2014, 43, 6618–6622 RSC.
- O. Stern and M. Volmor, Z. Phys, 1919, 20, 183–188 CAS.
- L. A. Currie, Anal. Chim. Acta, 1999, 391, 127–134 CrossRef CAS.
- A. K. Mahapatra, G. Hazra, S. K. Mukhopadhyay and A. R. Mukhopadhyay, Tetrahedron Lett., 2013, 54, 1164–1168 CrossRef CAS.
- (a) R. Indumathy, P. S. Parameswaran, C. V. Aiswarya and B. U. Nair, Polyhedron, 2014, 75, 22–29 CrossRef CAS; (b) V. G. Marini, E. Torri, L. M. Zimmermann and V. G. Machado, ARKIVOK, 2010, 11, 146–162 Search PubMed.
- (a) D. S. Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363–W367 CrossRef PubMed; (b) N. Andrusier, R. Nussinov and H. J. Wolfson, Proteins, 2007, 69, 139–159 CrossRef CAS PubMed; (c) E. Mashiach, D. S. Duhovny, N. Andrusier, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2008, 36, W229–W232 CrossRef CAS PubMed; (d) R. Chen and Z. Weng, Proteins: Struct., Funct., Genet., 2003, 51, 397–408 CrossRef CAS PubMed; (e) R. Chen and Z. Weng, Proteins: Struct., Funct., Genet., 2002, 47, 287–294 Search PubMed; (f) S. Volkov, L. Kumprecht, M. Budesinsky, M. Lepsik, M. Dusek and T. Kraus, Org. Biomol. Chem., 2015, 13, 2980–2985 RSC.
- (a) X. Cheng, Z. Lu, Y. Li, Q. Wang, C. Lu and Q. Meng, Dalton Trans., 2011, 40, 11788–11794 RSC; (b) C. Przybylski, V. Bonnetb and C. Cezard, Phys. Chem. Chem. Phys., 2015, 17, 19288–19305 RSC.
- (a) N. Venkatramaiah, D. M. G. C. Rocha, P. Srikanth, F. A. Almeida Paz and J. P. C. Tome, J. Mater. Chem. C, 2015, 3, 1056–1067 RSC; (b) S. S. Lee, S. Park, J. Y. Kim, H. R. Kim, S. Lee and H. B. Oh, Phys. Chem. Chem. Phys., 2014, 16, 8376–8383 RSC.
- S. S. L. Fat and J. P. Cornard, Polyhedron, 2011, 30, 2326–2332 CrossRef.
- P. Wang, J. Wu, P. Su, C. Shan, P. Zhou, Y. Ge, D. Liu, W. Liu and Y. Tang, J. Mater. Chem. B, 2016, 4, 4526–4533 RSC; P. Wang, J. Wu, P. Zhou, W. Liu and Y. Tang, J. Mater. Chem. B, 2015, 3, 3617–3624 RSC; G. Sivaraman and D. Chellappa, J. Mater. Chem. B, 2013, 1, 5768–5772 RSC.
- M. Fatiha, L. Leila, K. D. Eddine and N. Leila, J. Inclusion Phenom. Macrocyclic Chem., 2012, 77, 421–427 CrossRef.
- A. Fifere, M. Spulber, N. Marangoci, N. Fifere, M. Pinteala, V. Harabagiu and B. C. Simionescu, Cellul. Chem. Technol., 2011, 45, 149–156 CAS.
- M. Fatiha, K. Djameleddine and L. Leila, Orbital, 2009, 1(1), 26–37 CAS.
- S. M. Ali and S. Shamim, J. Encapsulation Adsorpt. Sci., 2014, 4, 63–70 CrossRef.
- (a) V. Bojinov and N. Georgiev, J. Univ. Chem. Technol. Metall., 2011, 46, 3–26 CAS; (b) A. Kuwar, R. D. Patil, A. Singh, S. K. Sahoo, J. Marek and N. Singh, J. Mater. Chem. C, 2015, 3, 453–460 RSC; (c) M. K. Chahal and M. Sankar, Anal. Methods, 2015, 7, 4552–4559 RSC.
- (a) J. Ru, X. Mi, L. Guan, X. Tang, Z. Ju, G. Zhang, C. Wang and W. Liu, J. Mater. Chem. B, 2015, 3, 6205–6212 RSC; (b) S. Youming, X. Zhang, Y. Zhang, C. Zhang, J. Jin, H. Li and S. Yao, Anal. Methods, 2016, 8, 2420–2426 RSC.
- (a) S. M. Sharker, S. M. Kim, S. H. Kim, I. In, H. Lee and S. Y. Park, J. Mater. Chem. B, 2015, 3, 5833–5841 RSC; (b) S. Yu, J. Yuan, J. Shi, X. Ruan, Y. Wang, S. Gao and Y. Du, J. Mater. Chem. B, 2015, 3, 5277–5283 RSC; (c) A. Mahammad, H. A. Molla, R. Bhowmick, A. Katarkar, K. Chaudhuri and S. Gangopadhyay, Anal. Methods, 2015, 7, 5149–5156 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27497h |
| This journal is © The Royal Society of Chemistry 2017 |