
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modification of benzoxazine with aryl-ether-ether-ketone diphenol: preparation and characterization†
Yiqing Xia ,
Yifeng Lin,
Qichao Ran*,
Rongqi Zhu and
Yi Gu*
,
Yifeng Lin,
Qichao Ran*,
Rongqi Zhu and
Yi Gu*
State Key Laboratory of Polymeric Materials Engineering, College of Polymer Science and Engineering, Sichuan University, Chengdu, China. E-mail: guyi@scu.edu.cn; ranqichao@scu.edu.cn; Fax: +86 28 85405138; Tel: +86 28 85405138
First published on 5th January 2017
Abstract
A highly active aryl-ether-ether-ketone diphenol intermediate (m-DHPBP) was synthesized by molecular design. Then it was introduced into benzoxazine (BA-a) as a modifier and a new modified benzoxazine resin system (BA-a/m-DHPBP-X) with varying contents of m-DHPBP was prepared. Differential scanning calorimetry (DSC) and Fourier transform infrared spectroscopy (FT-IR) were used to characterize the curing behaviour and to explore the polymerization mechanism of the BA-a/m-DHPBP-X mixtures by following major changes in the functional groups during the curing process. A significant reduction in the peak reaction temperature in the DSC curves and in the gel-time of BA-a/m-DHPBP-X indicated that m-DHPBP was an effective catalyst for the ring-opening polymerization (ROP) of benzoxazine. As the amount of m-DHPBP increased, the variation in benzene ring substituents of poly(BA-a/m-DHPBP-X) implied that m-DHPBP reacted with BA-a and finally embedded in the three dimensional networks of poly(BA-a). The thermal properties of the BA-a/m-DHPBP-X mixtures were evaluated by dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA); they exhibited a higher initial storage modulus, maintained a high glass transition temperature (Tg) and displayed a higher char yield compared with the neat poly(BA-a). Furthermore, the tensile properties of poly(BA-a/m-DHPBP-X) were good. Thus a modified benzoxazine resin system with “high strength, high modulus and high toughness” was prepared, and a more than 60% increase in tensile strength and 78% increase in elongation at break were observed by incorporating 20 wt% m-DHPBP into poly(BA-a). We believe that this modified benzoxazine system can be applied as a matrix for high-performance composites due to its excellent processibility and superior comprehensive mechanical properties.
Introduction
Benzoxazines are newly developed heat-resistant resins and possess a series of intriguing properties, such as molecular design flexibility,1–3 near-zero shrinkage upon curing,4,5 high glass transition temperature (usually much higher than the curing temperature), low water absorption,6,7 low surface energy,8–11 good mechanical properties and good thermal stability.12–17 However, their high curing temperature and the brittleness of polybenzoxazine restrict their application. A number of studies have been done with the aim of increasing the toughness of polybenzoxazines and they can be summarized into three broad categories: one method is to introduce flexible chains into the structure of benzoxazines,18–20 or prepare main-chain type benzoxazines by using diphenol and diamine.21–24 The toughening effect of this method is good but it is complex and difficult to get the desired benzoxazine precursors. The second method is to blend polybenzoxazine directly with elastomers or plastics with flexible molecular chains,25–32 which is believed to be convenient and effective. However, the increase of the melt viscosity from the high molecular weight gives rise to deterioration in the processability of the benzoxazines, and synchronously this method hampers the thermal properties and strength. The third method is to form multiphase structures by reaction-induced phase separation (RIPS) in benzoxazine/thermoplastic33–35 or benzoxazine/thermosetting36–39 systems. The toughening effect of this method is obvious, but it is difficult and complicated to control the formation of different phase structures, especially the prospective multiphase structure during actual production. Hence, choosing high-performance engineering plastic oligomers as modifiers might be an efficient way to toughen polybenzoxazine without causing deterioration in processability.The high curing temperature was another shortcoming for benzoxazine resins. Various catalytic systems, such as organic acids,40,41 metal halides,42,43 thiol compounds,44–46 imidazoles and primary amine compounds47,48 have been employed to decrease the curing temperature and promote the curing reaction of benzoxazine. Nevertheless, the thermostability of polybenzoxazines deteriorates because these catalysts keep polybenzoxazine in the form of micro-molecules or form weak bonds in the crosslinked structures of polybenzoxazine. However, phenols can not only catalyze the ring-opening reaction of benzoxazine monomers and lower their curing temperature, but will also react with benzoxazines through electrophilic substitution reactions to form similar chemical structures to polybenzoxazine. Hamerton et al.49 added 15 wt% of bisphenol-A into bisphenol-A/aniline benzoxazine (BA-a), and found that the exothermic peak temperature of BA-a was decreased from 245 °C to 215 °C because of the catalytic action of phenol. Kolanadiyil et al.50 studied the catalytic effect of various phenolic nucleophiles on a trifunctional benzoxazine (T-BZ). The results suggested that hydroquinone, 2-methyl resorcinol and pyrogallol showed high activity in the ring-opening reaction between phenols and T-Bz. Additionally, Arnebold et al.51 and Endo et al.52 demonstrated that 2-methyl resorcinol and resorcinol displayed high reactivity whether acting as nucleophiles or phenol components of benzoxazine. These studies mainly focused on the influence of phenolic compounds on the curing reaction of benzoxazine and the thermal properties of polybenzoxazine, whereas they did not investigate the effects on mechanical properties, especially toughness.
Therefore, some strategies can be used to improve the toughness of polybenzoxazine and decrease the curing temperature. In this study, an aryl-ether-ether-ketone diphenol, with a highly reactive terminal phenol hydroxyl and ductile ether linkage (m-DHPBP), was firstly synthesized and was then used to catalyze the polymerization of BA-a. The polymerization mechanism of the blends of BA-a and the diphenol was explored. Furthermore, the effects of m-DHPBP on the thermal properties and toughness of poly(BA-a) were evaluated.
Experimental
Reagents
Chemicals, with purities greater than 99%, were purchased from ChengDu KeLong Chemical Reagent Co. Ltd. (China), including formaldehyde (37 wt% in water), bisphenol-A, aniline, 4,4′-difluorobenzophenone (DFBP), m-dihydroxybenzene (m-DHB), potassium carbonate, hydrochloric acid (37 wt% in water), N-methyl-2-pyrrolidone (NMP), toluene, tetrahydrofuran (THF) and ethyl alcohol. All the reagents were used without further purification.Bisphenol A-aniline benzoxazine monomer (BA-a) was synthesized from formaldehyde (37 wt% in water), aniline and bisphenol-A. The detailed procedure is provided in the ESI;† The 1H NMR spectrum and FTIR spectrum of m-DHPBP are illustrated in Fig. SIII and SIV,† respectively.
Synthesis of aryl-ether-ether-ketone diphenol (m-DHPBP)
The synthesis of m-DHPBP was conducted in a 500 ml round bottom flask equipped with a Dean–Stark collector, condenser and magnetic stirrer. A synthetic method was designed to prepare m-DHPBP, as shown in Scheme 1. | ||
| Scheme 1 Synthesis of m-DHPBP. | ||
A mixture of m-DHB (39.67 g, 0.36 mol), DFBP (15.72 g, 0.07 mol) and potassium carbonate (49.78 g, 0.36 mol) in 252 ml NMP and 72 ml toluene was slowly heated to 140 °C and retained at that temperature for 4 h. Then the temperature was increased to 180 °C and kept at that temperature for another 2 h. After that, the solution was cooled to 160 °C and concentrated under vacuum. Next the mixture was cooled to room temperature, abundant distilled water was added and the pH was adjusted to 3 through adding HCl. The new precipitate was then filtered, washed with deionized water, and dried in a vacuum oven at 60 °C overnight to give 28.7 g of m-DHPBP as a white powder (81% yield). The 1H NMR spectrum and FT-IR spectrum of m-DHPBP are illustrated in Fig. SI and SII,† respectively. 1H NMR (400 MHz, DMSO-d6, (CH3)4Si, δH, ppm): 9.78 (4H, Ar-OH, s), 7.79, 7.77 (12H, Ha, d), 7.55, 7.53, 7.51 (1H, Hb, t). FT-IR (KBr, cm−1): 3500–3200 cm−1 (Ar-OH symmetrical stretching), 1644 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetrical stretching), 1230 and 1160 cm−1 (symmetrical stretching and asymmetrical stretching of ether), 770 and 677 cm−1 (1,3-substitution). The number average molecular weight (Mn) of m-DHPBP was characterized by size exclusion chromatography (GPC) as shown in Fig. SV.† The results of GPC and 1H NMR illustrated that m-DHPBP was a blend of oligomers of two different molecular weights.
O symmetrical stretching), 1230 and 1160 cm−1 (symmetrical stretching and asymmetrical stretching of ether), 770 and 677 cm−1 (1,3-substitution). The number average molecular weight (Mn) of m-DHPBP was characterized by size exclusion chromatography (GPC) as shown in Fig. SV.† The results of GPC and 1H NMR illustrated that m-DHPBP was a blend of oligomers of two different molecular weights.
Blending and curing
BA-a and m-DHPBP were blended in various weight ratios in THF at 60 °C. The weight ratios of BA-a to m-DHPBP were 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 95:5, 90:10, 85:15 and 80:20 (named as BA-a/m-DHPBP-0, BA-a/m-DHPBP-5, BA-a/m-DHPBP-10, BA-a/m-DHPBP-15 and BA-a/m-DHPBP-20, respectively). The solutions were cast onto glass plates and then dried at 80 °C for 2 h under vacuum. Then, these blends were treated according to the following curing procedure: vacuum at 160 °C maintained for 7 h with samples removed at 3 h and 5 h, and then at 170, 180 and 190 °C for 2 h at each temperature. The obtained films were transparent and yellow, and are named as poly(BA-a/m-DHPBP-0), poly(BA-a/m-DHPBP-5), poly(BA-a/m-DHPBP-10), poly(BA-a/m-DHPBP-15) and poly(BA-a/m-DHPBP-20).
:0, 95:5, 90:10, 85:15 and 80:20 (named as BA-a/m-DHPBP-0, BA-a/m-DHPBP-5, BA-a/m-DHPBP-10, BA-a/m-DHPBP-15 and BA-a/m-DHPBP-20, respectively). The solutions were cast onto glass plates and then dried at 80 °C for 2 h under vacuum. Then, these blends were treated according to the following curing procedure: vacuum at 160 °C maintained for 7 h with samples removed at 3 h and 5 h, and then at 170, 180 and 190 °C for 2 h at each temperature. The obtained films were transparent and yellow, and are named as poly(BA-a/m-DHPBP-0), poly(BA-a/m-DHPBP-5), poly(BA-a/m-DHPBP-10), poly(BA-a/m-DHPBP-15) and poly(BA-a/m-DHPBP-20).
Measurements
FT-IR spectra (KBr, cm−1) were obtained with a Nicolette 560 FT-IR spectrometer using a spectral width ranging from 500 to 4000 cm−1, a resolution of 4 cm−1 and an accumulation of 32 scans. The samples were prepared on KBr pellets by the tableting method. 1H NMR measurements were carried out using a Bruker TD-65536 NMR (400 MHz), with deuterium dimethyl sulfoxide (DMSO-d6) as the solvent and tetramethylsilane (TMS) as the internal reference. Differential scanning calorimetry (DSC) was recorded with a DSC-Q20 (TA Instruments) at a heating rate of 10 °C min−1 from 40 °C to 350 °C under nitrogen. The gel-times were obtained by using a gel-time testing instrument. The sample cell of the instrument was first heated to the required temperature. Then, about 1 g resin was placed in the cell and spread on a disk with a stir-stick. The sample was kneaded by pressing it uniformly every second until it was no longer stuck to the spatula, and this time was defined as the gel-time (tgel). Number average molecular weights were estimated by gel permeation chromatography (GPC) on a Waters 2414 system, equipped with two consecutive polystyrene gel columns (Waters Styrage HR 2 DMF 7.8 × 300 mm and Waters Styrage HR 4 DMF 7.8 × 300 mm) and refractive index (RI) detectors, using DMF as an eluent at a flow rate of 1.0 ml min−1, calibrated with polystyrene standards. Thermogravimetric analysis (TGA) was determined with a TGA Q600 at a heating rate of 10 °C min−1 under nitrogen. Dynamic mechanical analyses (DMA) were conducted on a Q800 analyzer (TA Instruments) at 1.0 Hz at a heating rate of 5 °C min−1. Tensile properties were recorded with a KQL KD-5 testing machine according to ASTM D882 at a crosshead speed of 2 mm min−1 with samples 5 cm long. The tensile properties of each sample were determined from an average of five tests at least.Results and discussion
Effects of m-DHPBP content on polymerization of BA-a/m-DHPBP-X blends
The DSC curves of BA-a/m-DHPBP-X systems and the corresponding initial reaction temperatures (Tonset), peak reaction temperatures (Tpeak) and gel-times at 180 °C (tgel 180 °C) are represented in Fig. 1. According to Fig. 1a, after addition of m-DHPBP, the Tonset and Tpeak exhibited obvious shifts toward lower temperature. When the content of m-DHPBP reached 20 wt% (BA-a/m-DHPBP-20), the Tonset and Tpeak were 184.1 °C and 212.3 °C, respectively, which are 69 °C and 47 °C lower than those of BA-a (Tonset = 253.1 °C and Tpeak = 259.6 °C). Moreover, as seen in Fig. 1b, tgel 180 °C of BA-a/m-DHPBP-X decreased exponentially with increasing m-DHPBP. When the content of m-DHPBP was 15 wt%, the gel-time was reduced to 9.8 min. The above results suggest that the addition of m-DHPBP can effectively lower the ROP temperature of BA-a and promote the curing reaction. | ||
| Fig. 1 DSC curves of BA-a/m-DHPBP-X (X = 0, 5, 10, 15, 20) (a), and the variation of Tonset, Tpeak and tgel 180 °C with m-DHPBP content (b). | ||
FT-IR spectra of BA-a/m-DHPBP-X and poly(BA-a/m-DHPBP-X) are presented in Fig. 2. For BA-a/m-DHPBP-X, a weak and broad band appeared at 3600–3200 cm−1 due to the OH group in m-DHPBP and this increased with increasing m-DHPBP. The absorption bands around 1494 cm−1 (characteristic band of 1,2,4-trisubstituted benzene), 1230 and 1030 cm−1 (characteristic bands of asymmetric and symmetric stretching of C–O–C), and 947 cm−1 (characteristic band of the oxazine ring) were wholly retained in BA-a/m-DHPBP-X, demonstrating that there had been no ROP of the oxazine rings during the sample preparation process. Compared with the spectra of BA-a/m-DHPBP-X, the spectra of poly(BA-a/m-DHPBP-X) changed significantly (Fig. 2b). In particular, the absorption in the region of 3600–3200 cm−1 had obviously intensified, especially the band near 3385 cm−1 corresponding to the intermolecular –OH⋯O hydrogen bonding stretching frequency. These features implied that the hydrogen bonding networks of poly(BA-a) were enriched with the introduction of extra phenolic hydroxyl groups (m-DHPBP). In contrast, the absorption bands around 1494, 1230, 1030 and 947 cm−1 were significantly weakened or had even disappeared, and a new and intense band appeared at approximately 1486 cm−1 (characteristic band of 1,2,3,5-tetrasubstituted benzene).
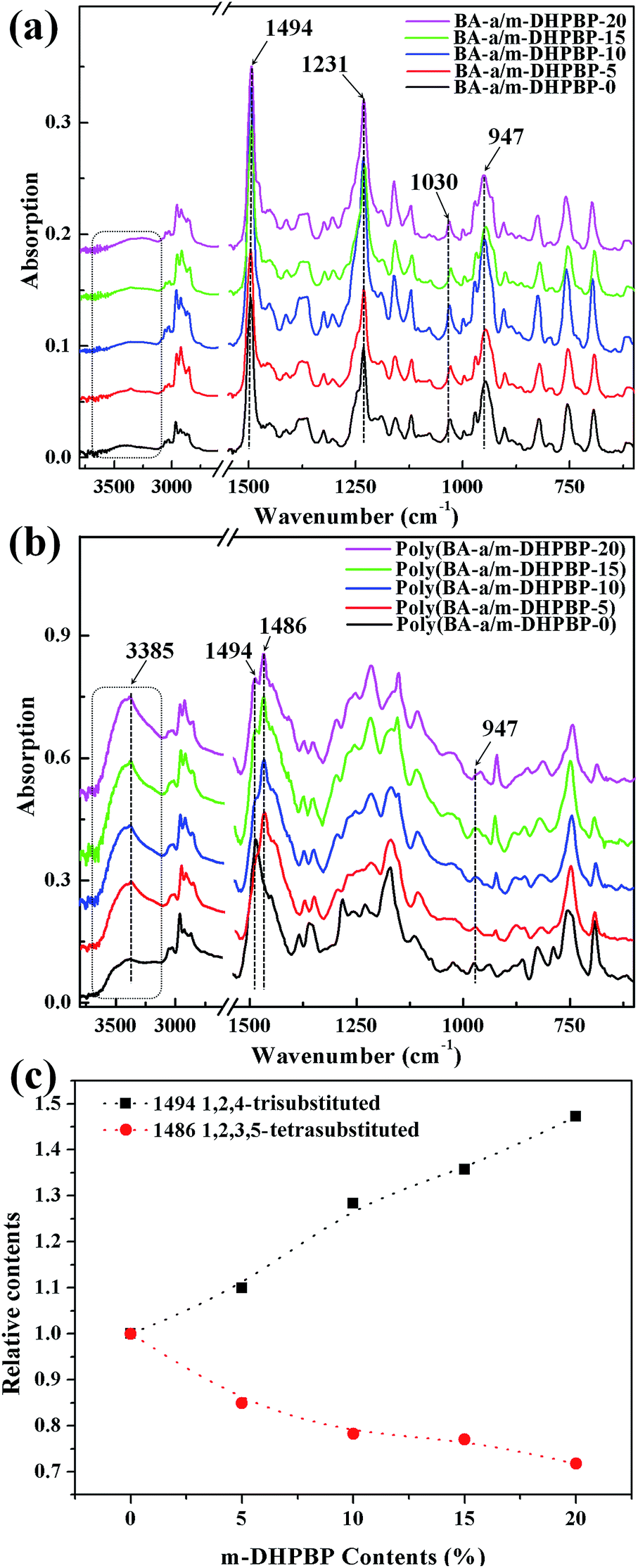 | ||
| Fig. 2 FT-IR spectra of BA-a/m-DHPBP-X (a) and poly(BA-a/m-DHPBP-X) (b) at each curing stage, and the relative contents of 1,2,4-trisubstituted benzene and 1,2,3,4-tetrasubstituted benzene in poly(BA-a/m-DHPBP-X) (c). | ||
To clarify the effect of m-DHPBP on the polymer structure, the bands in the range of 1530–1340 cm−1 (from Fig. 2b) were resolved using IR-peak-resolving analysis, and the peak heights of the peaks at 1494 and 1486 cm−1 were calculated using the band at 2964 cm−1 as an internal standard. Then, the relative peak heights of 1494 and 1486 cm−1 in poly(BA-a/m-DHPBP-X) were normalized with respect to those in poly(BA-a/m-DHPBP-0). As illustrated in Fig. 2c, it was found that the relative content of the 1,2,4-trisubstituted benzene structure increased gradually and that of the 1,2,3,5-tetrasubstituted benzene structure decreased in poly(BA-a/m-DHPBP-X) as the amount of m-DHPBP increased. The results above indicate that the introduction of m-DHPBP could change both the polymerization mechanism of benzoxazine and the chemical structure of poly(BA-a); these findings will be discussed in detail in the following section.
The polymerization mechanism of poly(BA-a/m-DHPBP-X)
To explore the influence of m-DHPBP on the curing reaction and polymer structure, the samples of BA-a/m-DHPBP-0 and BA-a/m-DHPBP-20 at each curing stage were characterized by DSC and FT-IR.Fig. 3 presents the DSC curves of BA-a/m-DHPBP-0 and BA-a/m-DHPBP-20 at each curing stage, and the corresponding reaction extents were calculated by the residual reaction enthalpy (the integrated area of the exothermic peak) employing eqn (1):
 | (1) |
 | ||
| Fig. 3 DSC curves of BA-a/m-DHPBP-0 (a) and BA-a/m-DHPBP-20 (b) at each curing stage. | ||
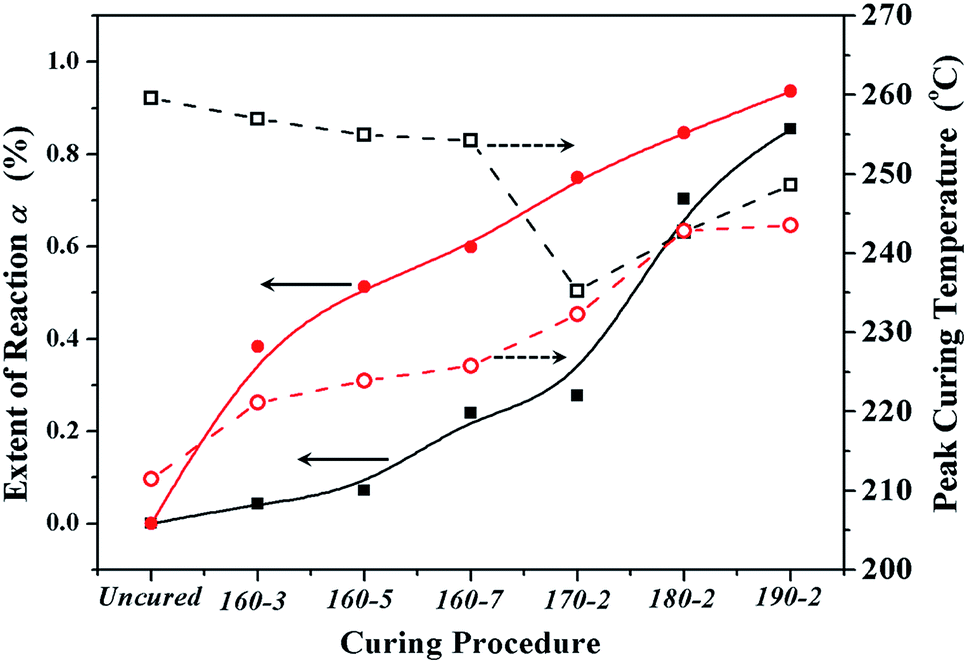 | ||
| Fig. 4 The reaction extents (solid shapes) and peak curing temperatures (open shapes) of BA-a/m-DHPBP-0 (squares) and BA-a/m-DHPBP-20 (circles) at each curing stage. | ||
As displayed in Fig. 3 and 4, all DSC curves of BA-a/m-DHPBP-0 and BA-a/m-DHPBP-20 at each curing stage showed a single curing exothermic peak. However, the curing reactions of the two resin systems exhibited different trends. For BA-a/m-DHPBP-0, the Tpeak was about 260 °C. When the sample was heated at 160 °C, the Tpeak slowly shifted to lower temperature as the curing time increased but when the curing temperature was increased to 170 °C and was held for 2 h, the Tpeak of the sample rapidly reduced to 235 °C. Meanwhile, the extent of the reaction was only 28%. It is worth noting that there was a sharp increase in the extent of the reaction (from 28% to 70%) in the sample cured at 180 °C for 2 h, and the reaction extent finally reached 90% after curing at 190 °C for 2 h. However, the Tpeak of the corresponding sample then shifted toward a higher temperature. Tpeak of the uncured blend for BA-a/m-DHPBP-20 was significantly reduced to 211.5 °C when compared to that of the BA-a/m-DHPBP-0 sample. When BA-a/m-DHPBP-20 was cured at 160 °C, the Tpeak gradually shifted toward higher temperature as the curing time increased, and yet the extent of reaction reached 60% after curing at 160 °C for 7 h. Curing at 170 °C, 180 °C and 190 °C for 2 h each, caused the corresponding Tpeak to increase continuously and the extent of the reaction rose to 95% at the end of the curing process. The difference in the curing process of the two kinds of systems may be related to the curing mechanisms. For the BA-a/m-DHPBP-0 system, thermal polymerization behaviour was observed, i.e. the ring-opening reaction of BA-a slowly occurred under heat with the generation of a certain amount of phenolic hydroxyl in the early curing stage (160 °C/7 h + 170 °C/2 h), which catalyzed the ROP of the benzoxazine monomer and made the Tpeak gradually lower. Thus, when the temperature increased to 180 °C or higher, the ROP was accelerated under the effect of both heat and phenolic hydroxyl groups, and the reaction extent improved rapidly. Consequently, the number of oxazine rings decreased and the curing reaction became diffusion controlled, which made the Tpeak gradually shift to a higher temperature. For the BA-a/m-DHPBP-20 system, the phenolic hydroxyl of m-DHPBP directly catalyzed the ROP of benzoxazine, which resulted in a low Tpeak (211 °C) and a high polymerization rate at the beginning of the curing reaction. After curing at 160 °C for 7 h, a high reaction extent of 60% was produced. Similar to the BA-a/m-DHPBP-0 system, the viscosity of the BA-a/m-DHPBP-20 system increased as the reaction proceeded, and the curing reaction became diffusion controlled, resulting in the Tpeak shift to higher temperature.
Additionally, to study the difference in gel behaviours of BA-a/m-DHPBP-0 and BA-a/m-DHPBP-20, the solubility of samples at each curing stage and the molecular weights (Mn) of the corresponding soluble parts were measured; the results are shown in Fig. 5. After curing at 170 °C for 2 h, BA-a/m-DHPBP-0 was still a completely soluble oligomer and the corresponding Mn was only 2600 g mol−1 due to the low extent of the reaction. After the reaction at 180 °C for 2 h, insoluble substance appeared.
 | ||
| Fig. 5 The dissolvable mass fraction (open shapes) and the corresponding Mn (closed shapes) of BA-a/m-DHPBP-0 (squares) and BA-a/m-DHPBP-20 (circles) at each curing stage. | ||
This phenomenon suggested that BA-a/m-DHPBP-0 had turned into a sol–gel system, in which the chemical reaction was becoming diffusion controlled. These results are consistent with the increase of the Tpeak. However, for the BA-a/m-DHPBP-20 system, after it was cured at 160 °C for 5 h, a sol–gel behaviour appeared and its corresponding Mn and α were 10748 g mol−1 and 51.3%, respectively, which are obviously higher than those of the BA-a/m-DHPBP-0 system (2600 g mol−1 and 27.7%). A further study was required on the reaction mechanism and the chemical structures.
As shown as Fig. 6c, at the early reaction stages (160 °C/3 h and 160 °C/5 h), the consumption rate of the oxazine ring (namely the ROP rate, characterized by the band at 947 cm−1) in the BA-a/m-DHPBP-20 system was higher than that of the BA-a/m-DHPBP-0 system because of the catalysis by m-DHPBP, which is consistent with the change of the Tpeak observed in the DSC analysis. Actually, during the curing process of benzoxazine, the oxazine ring opens firstly and forms highly reactive intermediates, i.e. carbocations or iminium species. Then the intermediates react with aromatic rings via electrophilic substitution reactions.53 When there was no m-DHPBP in the system, the oxazine ring opened and its intermediate attacked the corresponding ortho-position of the bisphenol-A structure in BA-a. Hence, the 1,2,4-trisubstituted benzene structure of BA-a (1494 cm−1) decreased and gradually translated into the 1,2,3,5-tetrasubstituted benzene structure of PBA-a (1486 cm−1). In the whole process, the oxazine ring and 1,2,4-trisubstituted benzene structure reduced synchronously, and finally only 4.6% remained of both. However, this phenomenon was totally different for the BA-a/m-DHPBP-20 system, in which the 1,2,4-trisubstituted benzene structure (1494 cm−1) obviously increased in the early curing stage, and then decreased slowly in the mid- and late-stages. This variation could be ascribed to the reactive carbocation in the intermediates first attacking the ortho-position of terminal phenolic hydroxyls in m-DHPBP and then forming a new type of 1,2,4-trisubstituted benzene structure, which is significantly enhanced in the total contents. Furthermore, the copolymerization between BA-a and m-DHPBP resulted in the prolongation of linear chains similar to those in the “Chain-Extension Reaction” in thermosets. This result is consistent with the increase in Mn. In the mid- and late-stages of the reaction, the 1,2,4-trisubstituted benzene structure (1494 cm−1) decreased and the 1,2,3,5-tetrasubstituted benzene structure (1486 cm−1) increased gradually with the self-polymerization of residual BA-a monomer. Nevertheless, the copolymerization between BA-a and m-DHPBP led to the reduction of self-polymerization of BA-a, so the residual content of the 1,2,4-trisubstituted benzene structure was as high as 46.1%. This result is consistent with those in Fig. 2, i.e. the content of m-DHPBP influenced the contents of the 1,2,4-trisubstituted benzene structure and the 1,2,3,5-tetrasubstituted benzene structure in poly(BA-a/m-DHPBP-X). Moreover, the results further illustrated the copolymerization reaction mechanism between m-DHPBP and BA-a and the impact on polymer molecular chain structures. Therefore, we propose a curing reaction procedure of BA-a/m-DHPBP-X blends, as shown in Scheme 2.
 | ||
| Fig. 6 IR spectra of BA-a/m-DHPBP-0 (a) and BA-a/m-DHPBP-20 (b), and the variation of characteristic peaks at 1494 and 947 cm−1 for BA-a/m-DHPBP-0 and BA-a/m-DHPBP-20 (c), at each curing stage. | ||
 | ||
| Scheme 2 Polymerization reaction process of BA-a/m-DHPBP-X. | ||
Thermal properties of poly(BA-a/m-DHPBP-X)
The variation of crosslinking structures of poly(BA-a/m-DHPBP-X) will affect the thermal and mechanical properties of poly(BA-a/m-DHPBP-X). DMA curves of poly(BA-a/m-DHPBP-X) and the corresponding data are presented in Fig. 7 and Table 1. | ||
| Fig. 7 Dynamic mechanical analysis of poly(BA-a/m-DHPBP-X) (X = 0, 5, 10, 15, 20): (a) storage modulus; (b) tan delta; and (c) Tgs and crosslink densities. | ||
| System | Storage modulus (50 °C) MPa | Tg,loss modulus °C | Tg,tan delta °C | Crosslink density × 103 mol m−3 |
|---|---|---|---|---|
| Poly(BA-a/m-DHPBP-0) | 4.79 | 206.5 | 233.9 | 11.11 |
| Poly(BA-a/m-DHPBP-5) | 5.08 | 202.4 | 220.1 | 9.52 |
| Poly(BA-a/m-DHPBP-10) | 5.11 | 198.3 | 215.1 | 8.32 |
| Poly(BA-a/m-DHPBP-15) | 5.18 | 192.5 | 208.0 | 7.18 |
| Poly(BA-a/m-DHPBP-20) | 5.36 | 188.5 | 205.5 | 6.04 |
With the addition of m-DHPBP, the storage modulus (E′) at 50 °C of poly(BA-a/m-DHPBP-X) increased from 4.79 GPa to 5.36 GPa (Fig. 7a). This is because the introduction of electronegative carbonyl groups and phenolic hydroxyls can enrich the hydrogen bonded networks and reinforce the interaction between molecular chains. As the testing temperature increased, E′ of poly(BA-a/m-DHPBP-X) exhibited a sharp decrease in the range of 170–220 °C, indicating a transformation from a glassy state to a rubber state. As the content of m-DHPBP increased, the glass transition temperatures (Tgs) of poly(BA-a/m-DHPBP-X) gradually decreased as seen from Fig. 7b and c. This phenomenon may be attributed to the following two reasons: (1) m-DHPBP, as a soft segment, is inserted into the poly(BA-a/m-DHPBP-X) crosslinked network. Consequently, the three dimensional networks of poly(BA-a/m-DHPBP-X) are toughened and the corresponding Tgs diminish; (2) theoretically, the oxazine ring and the potential site of the crosslinking reaction (ortho-position of the phenolic hydroxyl) in the BA-a monomer are equimolar. After incorporating m-DHPBP, BA-a firstly reacts with the new potential site on m-DHPBP (as discussed in the section on polymerization), which results in vacancies of potential crosslinking reaction sites and the prolongation of the average molecular weights between crosslinking points (Mc) (as shown in Scheme 2). As a consequence, the crosslink density and Tgs of poly(BA-a/m-DHPBP-X) decrease. The crosslink densities of poly(BA-a/m-DHPBP-X) were estimated from the plateau of the elastic modulus in the rubbery state using eqn (2) from the statistical theory of rubber elasticity:
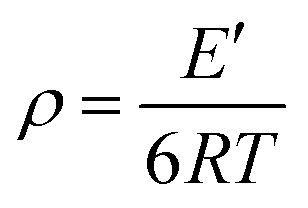 | (2) |
As shown in Fig. 7c, the crosslink densities of poly(BA-a/m-DHPBP-X) decrease linearly as m-DHPBP is increased. With the addition of 20 wt% m-DHPBP, the crosslink density decreased from 11.11 × 103 mol m−3 (poly(BA-a/m-DHPBP-0)) to 6.04 × 103 mol m−3 (poly(BA-a/m-DHPBP-20)), which confirms that the addition of m-DHPBP lowers the crosslink density of poly(BA-a), consistent with the results in the polymerization section.
Thermal stability is another important thermal property for high performance resins. The thermal stability of poly(BA-a/m-DHPBP-X) was explored by TGA, and the corresponding TGA curves are shown in Fig. 8. To some extent, the addition of m-DHPBP improved the thermal stability of poly(BA-a), which may be attributed to the high heat-resistance of the chemical structure of m-DHPBP.
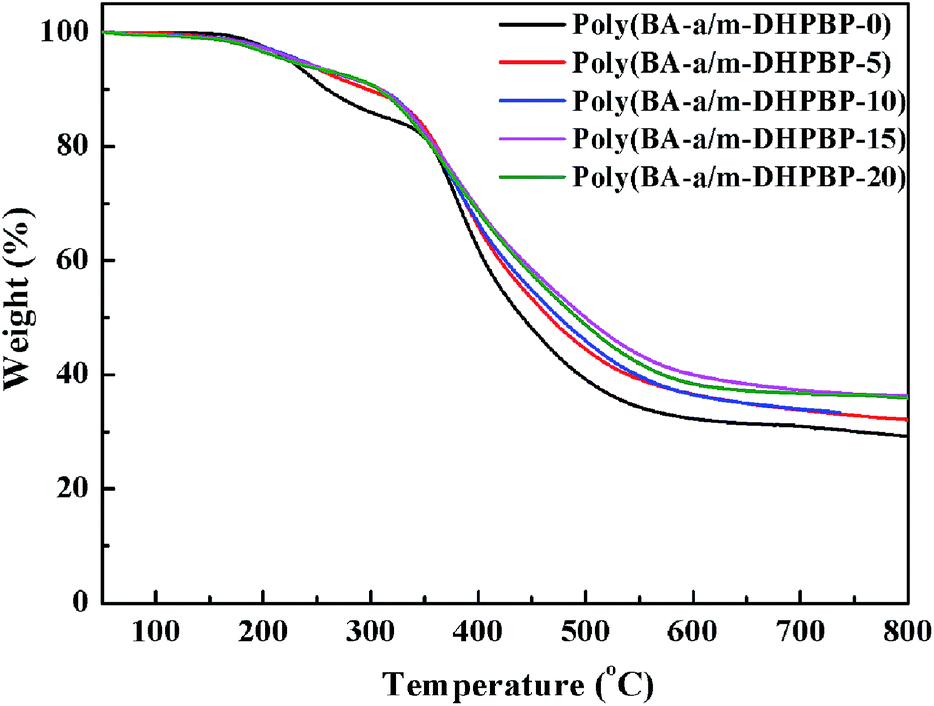 | ||
| Fig. 8 TGA curves of poly(BA-a/m-DHPBP-X). | ||
Tensile properties of poly(BA-a/m-DHPBP-X)
Another goal of this work was to improve the toughness of poly(BA-a) by introducing m-DHPBP. Therefore the tensile properties of poly(BA-a/m-DHPBP-X) films were investigated to characterize their toughness, and the results are illustrated in Fig. 9. | ||
| Fig. 9 Tensile properties of poly(BA-a/m-DHPBP-X). | ||
As shown in Fig. 9, the tensile modulus of poly(BA-a/m-DHPBP-X) gradually increased with the addition of m-DHPBP, and this is consistent with the results of the storage modulus in the DMA. It is worth noting that both the tensile strength and the elongation at break increased as the content of m-HDPBP increased. Compared with poly(BA-a/m-DHPBP-0), the tensile strength and elongation at break of poly(BA-a/m-DHPBP-20) increased by 160% and 178%, respectively. This illustrates that polybenzoxazine can be toughened effectively by adding m-DHPBP. The poly(BA-a/m-DHPBP-X) system shows excellent performance with “high modulus, high strength and high toughness”.
Conclusions
In summary, we synthesized a new modifier (m-DHPBP) with an aryl-ether-ether-ketone structure and high catalytic activity, and we prepared a series of modified benzoxazine resins with varying contents of m-DHPBP. The analysis by DSC, gel-time testing, GPC and FT-IR demonstrated that m-DHPBP not only acted as an efficient catalyst to lower the curing temperature and promote the ROP of BA-a, but was also a desired co-monomer with high reactivity, which could copolymerize with BA-a and ultimately embed itself into the three dimensional networks of poly(BA-a). With the addition of increasing m-DHPBP, the Tg and crosslink density of poly(BA-a/m-DHPBP-X) decreased. However, the tensile modulus, the tensile strength and the elongation at break increased with increasing m-DHPBP. We believe that these blends can satisfy the demands of applications relating to matrices for high-performance composites with “high modulus, high strength and high toughness”.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Project no. 51273119).References
- N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2007, 32, 1344–1391 CrossRef CAS.
- P. Yang and Y. Gu, J. Polym. Res., 2011, 18, 1725–1733 CrossRef CAS.
- Y. Zhuang and Y. Gu, J. Polym. Res., 2012, 19, 14 CrossRef.
- H. Ishida and D. J. Allen, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1019–1030 CrossRef CAS.
- H. Ishida and H. Y. Low, Macromolecules, 1997, 30, 1099–1106 CrossRef CAS.
- K. C. Chen, H. T. Li, S. C. Huang, W. B. Chen, K. W. Sun and F. C. Chang, Polym. Int., 2011, 60, 1089–1096 CrossRef CAS.
- C. F. Wang, S. F. Chiou, F. H. Ko, C. T. Chou, H. C. Lin, C. F. Huang and F. C. Chang, Macromol. Rapid Commun., 2006, 27, 333–337 CrossRef CAS.
- S. W. Kuo, Y. C. Wu, C. F. Wang and K. U. Jeong, J. Phys. Chem. C, 2009, 113, 20666–20673 CAS.
- C. S. Liao, J. S. Wu, C. F. Wang and F. C. Chang, Macromol. Rapid Commun., 2008, 29, 52–56 CrossRef CAS.
- C. H. Lin, S. L. Chang, T. Y. Shen, Y. S. Shih, H. T. Lin and C. F. Wang, Polym. Chem., 2012, 3, 935–945 RSC.
- C. F. Wang, Y. C. Su, S. W. Kuo, C. F. Huang, Y. C. Sheen and F. C. Chang, Angew. Chem., Int. Ed., 2006, 45, 2248–2251 CrossRef CAS PubMed.
- Q. C. Ran, D. X. Zhang, R. Q. Zhu and Y. Gu, Polymer, 2012, 53, 4119–4127 CrossRef CAS.
- Y. Zhu, H. Ling and Y. Gu, J. Polym. Res., 2012, 19, 9904 CrossRef.
- P. Yang and Y. Gu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1261–1271 CrossRef CAS.
- P. Yang and Y. Gu, J. Appl. Polym. Sci., 2012, 124, 2415–2422 CrossRef CAS.
- P. Yang, X. Wang, H. Fan and Y. Gu, Phys. Chem. Chem. Phys., 2013, 15, 15333–15338 RSC.
- P. Yang, X. Wang and Y. Gu, J. Polym. Res., 2012, 19, 9901 CrossRef.
- M. Nakamura and H. Ishida, Polymer, 2009, 50, 2688–2695 CrossRef CAS.
- S. Ates, C. Dizman, B. Aydogan, B. Kiskan, L. Torun and Y. Yagci, Polymer, 2011, 52, 1504–1509 CrossRef CAS.
- B. Aydogan, D. Sureka, B. Kiskan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5156–5162 CrossRef CAS.
- A. Chernykh, J. Liu and H. Ishida, Polymer, 2006, 47, 7664–7669 CrossRef CAS.
- K. D. Demir, B. Kiskan, B. Aydogan and Y. Yagci, React. Funct. Polym., 2013, 73, 346–359 CrossRef CAS.
- B. Kiskan, Y. Yagci and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 414–420 CrossRef CAS.
- J. Liu, T. Agag and H. Ishida, Polymer, 2010, 51, 5688–5694 CrossRef CAS.
- H. Ishida and Y. H. Lee, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 736–749 CrossRef CAS.
- Y. Cui, Y. Chen, X. Wang, G. Tian and X. Tang, Polym. Int., 2003, 52, 1246–1248 CrossRef CAS.
- S. Rimdusit, S. Pirstpindvong, W. Tanthapanichakoon and S. Damrongsakkul, Polym. Eng. Sci., 2005, 45, 288–296 CAS.
- H. Ishida and Y. H. Lee, J. Appl. Polym. Sci., 2002, 83, 1848–1855 CrossRef CAS.
- J. Jang and D. Seo, J. Appl. Polym. Sci., 1998, 67, 1–10 CrossRef CAS.
- Y. H. Lee, D. J. Allen and H. Ishida, J. Appl. Polym. Sci., 2006, 100, 2443–2454 CrossRef CAS.
- S. Rimdusit, C. Liengvachiranon, S. Tiptipakorn and C. Jubsilp, J. Appl. Polym. Sci., 2009, 113, 3823–3830 CrossRef CAS.
- J. Jang and D. Seo, J. Appl. Polym. Sci., 1998, 67, 1–10 CrossRef CAS.
- Y. Xia, P. Yang, Y. Miao, C. Zhang and Y. Gu, Polym. Int., 2015, 64, 118–125 CrossRef CAS.
- Y. Xia, P. Yang, R. Zhu, C. Zhang and Y. Gu, J. Polym. Res., 2014, 21, 1–8 CAS.
- P. Zhao, X. Liang, J. Chen, Q. Ran and Y. Gu, J. Appl. Polym. Sci., 2013, 128, 2865–2874 CrossRef CAS.
- Z. Wang, Q. Ran, R. Zhu and Y. Gu, RSC Adv., 2013, 3, 1350–1353 RSC.
- P. Zhao, Q. Zhou, X. Liu, R. Zhu, Q. Ran and Y. Gu, Polym. J., 2013, 45, 637–644 CrossRef CAS.
- Z. Wang, N. Cao, Y. Miao and Y. Gu, J. Appl. Polym. Sci., 2016, 133, 43259, DOI:10.1002/app.43259.
- Z. Wang, Q. Ran, R. Zhu and Y. Gu, Phys. Chem. Chem. Phys., 2014, 16, 5326–5332 RSC.
- J. Dunkers and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1913–1921 CrossRef CAS.
- H. D. Kim and H. Ishida, J. Appl. Polym. Sci., 2001, 79, 1207–1219 CrossRef CAS.
- C. Liu, D. Shen, R. M. Sebastián, J. Marquet and R. Schönfeld, Polymer, 2013, 54, 2873–2878 CrossRef CAS.
- A. Sudo, S. Hirayama and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 479–484 CrossRef CAS.
- A. W. Kawaguchi, A. Sudo and T. Endo, ACS Macro Lett., 2012, 2, 1–4 CrossRef.
- A. Musa, B. Kiskan and Y. Yagci, Polymer, 2014, 55, 5550–5556 CrossRef CAS.
- E. Semerci, B. Kiskan and Y. Yagci, Eur. Polym. J., 2015, 69, 636–641 CrossRef CAS.
- J. Sun, W. Wei, Y. Xu, J. Qu, X. Liu and T. Endo, RSC Adv., 2015, 5, 19048–19057 RSC.
- X. Liu and Y. Gu, J. Appl. Polym. Sci., 2002, 84, 1107–1113 CrossRef CAS.
- W. A. Wan Hassan, J. Liu, B. J. Howlin, H. Ishida and I. Hamerton, Polymer, 2016, 88, 52–62 CrossRef CAS.
- S. Nalakathu Kolanadiyil, M. Azechi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2811–2819 CrossRef CAS.
- A. Arnebold, O. Schorsch, J. Stelten and A. Hartwig, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1693–1699 CrossRef CAS.
- H. Oie, A. Mori, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4756–4761 CrossRef CAS.
- X. Ning and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1121–1129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27493e |
| This journal is © The Royal Society of Chemistry 2017 |