
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient Pd@MIL-101(Cr) hetero-catalysts for 2-butyne-1,4-diol hydrogenation exhibiting high selectivity†
Dongdong Yina,
Chuang Lia,
Hangxing Rena,
Osama Shekhahb,
Jinxuan Liu *c and
Changhai Liang*a
*c and
Changhai Liang*a
aLaboratory of Advanced Materials and Catalytic Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: changhai@dlut.edu.cn
bAdvanced Membranes and Porous Materials Center, 4700 King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia
cInstitute of Artificial Photosynthesis, State Key Laboratory of Fine Chemicals, Dalian University of Technology, 116024 Dalian, China. E-mail: jinxuan.liu@dlut.edu.cn
First published on 5th January 2017
Abstract
Pd@MIL-101(Cr) hetero-catalysts have been successfully prepared using the metal–organic chemical vapour deposition (MOCVD) approach, by choosing [Pd(η3-C3H5)(η5-C5H5)] as a volatile precursor, and the hydrothermally stable metal–organic framework, MIL-101(Cr) as a support. The prepared Pd@MIL-101(Cr) hetero-catalysts characterized with various analytical techniques, exhibited highly monodispersed immobilized Pd nanoparticles in the MIL-101(Cr) cavities, while retaining the pristine crystallinity and porosity. The intact hybrid Pd@MIL-101(Cr) has been demonstrated to be an efficient catalyst for 2-butyne-1,4-diol hydrogenation with excellent activity, stability and selectivity (2-butene-1,4-diol (>94%)).
1. Introduction
Metal–organic frameworks (MOFs), a new class of porous crystalline materials, also called porous coordination polymers (PCPs), are constructed by the coordination of metal ions/clusters and organic bridging ligands.1,2 The versatility of a vast number of organic and inorganic building struts and the possibility of modifying and functionalizing the frameworks by conventional chemistry strategies imbue the MOFs with diversity and enhanced capabilities for a plethora of applications.3–6 A key structural feature of MOFs is the ultrahigh porosity (up to 90% free volume) and high internal surface areas, with values up to 7000 m2 g−1 for NU-109 and NU-110.7 As a result of these outstanding structural features, i.e. high porosity and tunability, MOFs have emerged as a class of promising materials with regards to applications in gas storage/separation,8 luminescence,9 sensing,10 drug delivery,11 catalysis,12,13 and photoelectric conversion.14,15The encapsulation of metal nanoparticles (MNPs) into MOFs has been demonstrated to be an efficient method to impede MNP aggregation and benefit from their activity for heterogeneous catalysis.16,17 Among the well-established methods for fabrication of MNPs@MOFs, such as incipient wetness infiltration,18 and double solvent method,19 direct diffusion/adsorption of the volatile metal precursors into MOFs cavities is considered as one of the most appropriate strategies for fabrication of MNPs@MOF composites. Through which, the problem of solvent-induced competitive adsorption with metal precursors and structure destruction can be substantially suppressed. Pre-capped MNPs with surfactants have been demonstrated to be loaded into MOFs with a controlled dispersion such as Ru@MOF-5 and Au@ZIF-8.20,21 However, the catalytic performances are severely affected by the capped surfactants.22
Solvent-free gas-phase infiltration, also known as metal–organic chemical vapour deposition (MOCVD), has been shown to be an effective approach to incorporate Pd NPs into MOF-5 by Fischer and co-workers.23,24 Compared to solution-based impregnation of MOFs with MNPs, MOCVD exhibit advantages in avoiding MOF destruction, competitive adsorption between solvents and metal precursors.25
The production of 2-butene-1,4-diol (BED) and 1,4-butanediol (BDO) with hydrogenation of 2-butyne-1,4-diol (BYD) has an important industrial use.26 As an essential starting material with a worldwide production ≥5000 tons per year, BED is used for the synthesis of vitamins and insecticides.27 BDO has also numerous applications in manufacturing polymer and chemicals such as tetrahydrofuran and γ-butyrolactone. The hydrogenation of BYD has been widely studied using Pd-/Ni-based catalysts supported on CaCO3,28,29 carbon30 and alumina.31 However, further hydrogenation of BED is ineluctably accompanied with side reactions including double bond migration, cyclization, hydrogenolysis and isomerization forming various by-products as shown in Fig. 1, which leads to an extremely low selectivity.32 While the use of Pd composites with Zn, Cu, Pb33 and the introduction of basic reaction modifiers such as pyridine, quinoline or isoquinoline34 has been carried out to improve the selectivity of BED, the conversion efficiency of the hydrogenation of BYD was significantly decreased.
 | ||
| Fig. 1 Reaction network for 2-butyne-1,4-diol hydrogenation. | ||
It has been demonstrated previously that MOFs can modulate the catalytic properties of the encapsulated NPs.35,36 Very recently, Tang and co-workers have demonstrated that MOFs can be used as selectivity regulators for hydrogenation reactions.37 The porous structure of MOFs allow volatile metalorganic precursors to diffuse into the pores, adsorb on the interior of MOFs, facilitate reactants to reach the metal sites in the pores and products to release from the active sites. Among the huge numbers of reported MOFs, MIL-101(Cr) exhibit, in addition to the high surface area (up to 4000 m2 g−1), excellent thermal and chemical stability to solvents than other MOFs.38 The pore size of MIL-101(Cr) amounts to be 2.9–3.4 nm with large window sizes of 1.2–1.4 nm. These features make MIL-101(Cr) an excellent support for fabrication of metal NPs@MOF hetero-catalysts using volatile metalorganic as precursors.23,24
Herein, we successfully demonstrate the immobilization of uniform and highly dispersed palladium nanoparticles into the MIL-101(Cr) using the MOCVD approach at low temperature using volatile Pd(η3-C3H5)(η5-C5H5) precursor. The catalytic performance of Pd@MIL-101(Cr) towards the selective hydrogenation of BYD to BED were examined. To best of our knowledge, this is the first demonstration of Pd@MIL-101(Cr) for 2-butyne-1,4-diol hydrogenation with excellent activity and selectivity of 2-butene-1,4-diol (>94%).
2. Experimental
2.1 Preparation of Pd@MIL-101(Cr)
The metal–organic precursor [Pd(η3-C3H5)(η5-C5H5)] has been synthesized according to previously reported literature procedures.39,40 MIL-101(Cr) was prepared using a hydrothermal method with some minor modifications.38 In brief, terephthalic acid (0.82 g, 5 mmol), Cr(NO3)3·9H2O (2.0 g, 5 mmol), HF (0.2 g) and H2O (24 ml, 1333 mmol) were mixed and stirred for 15 minutes. After that, the mixture was introduced into a Teflon-lined steel autoclave and heated at 220 °C for 8 h. After cooling to room temperature, the obtained green mixture was filtrated using a large pore fritted glass filter (G2) to separate the free recrystallized terephthalic acid. The green products were collected by centrifuging and washed with H2O, DMF and ethanol for three times, respectively. Finally, the recovered sample was dried under vacuum at 150 °C for 12 h to remove the adsorbed water molecules.The gas-phase loading was performed in a specially designed CVD apparatus, where Pd(η3-C3H5)(η5-C5H5) and MIL-101(Cr) were placed separately on the glass sieve plate, allowing the gas flow to pass through them sequentially. The temperature was controlled by a CVD oven, coupled with two mass flow controllers to monitor the flow of Ar and H2. The Pd(η3-C3H5)(η5-C5H5) vapour was carried with Ar and spread, followed by adsorption on the pre-activated MIL-101(Cr) at 30 °C for 12 hours. Subsequently, the reduction of Pd ions to Pd was performed by introduction of H2 at 30 °C for 2 hours. Finally, the products were further treated with Ar for 12 hours to get rid of the hydrocarbon residues.
2.2 Characterization of Pd@MIL-101(Cr)
Powder X-ray diffraction (PXRD) patterns were recorded at ambient temperature with D/MAX-2400 diffractometer at 40 kV, 100 mA for Cu Kα (λ = 1.5418 Å). The Brunauer–Emmett–Teller (BET) surface area, pore volume and pore size distribution of the samples were measured with N2 adsorption–desorption isotherms at 77 K using a Quantachrome Autosorb-IQ apparatus. Thermogravimetric (TG) experiments were carried out on a Mettler Toledo TGA/SDTA851° thermogravimetric analyzer. As-prepared MIL-101(Cr) was heated from 25 °C to 600 °C in air at a constant rate of 5 °C min−1. The Pd contents in the Pd@MIL-101(Cr) catalysts were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (Perkin-Elmer Optima 2000DV). Fourier transform infrared (FT-IR) spectra were collected at room temperature using a Thermo fisher Nicolet 6700 spectrometer with a resolution of 0.09 cm−1. Transmission electron microscopy (TEM) measurement was done using a JEOL model JEM-2000EX at 120 kV to examine the Pd NPs encapsulated into MIL-101(Cr).2.3 Characterization of catalytic performance
The catalytic performance of Pd@MIL-101(Cr) catalysts for selective hydrogenation of 2-butyne-1,4-diol was examined in a 50 ml batch reactor using H2O as solvent. Typically, the catalysts were reduced at 200 °C for 2 h under Ar and H2 (Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 2:1), followed by passivation under Ar overnight. The reaction mixture which contained 1,2-propanediol (1 g) and 2-butyne-1,4-diol (1 g) dissolving in H2O (18 g) and freshly passivated catalyst (0.03 g) was added into the batch reactor. After purging with H2 for 5 times, the reactor was heated to the desired temperature and then filled with a particular H2 pressure under stirring (600 rpm). After the reaction was completed, the reactor was cooled to room temperature, and the products were separated by centrifuging and subsequently analysed by gas chromatograph (GC-7890F, FID, FFAP column 30 m × 0.32 mm × 0.5 μm). In order to test the recycling sustainability, the Pd@MIL-101(Cr) after hydrogenation reaction was separated by centrifuging followed by washing with water, and reused for a new batch of hydrogenation reactions. The Pd@MIL-101(Cr) catalysts were reused for four times.
:H2 = 2:1), followed by passivation under Ar overnight. The reaction mixture which contained 1,2-propanediol (1 g) and 2-butyne-1,4-diol (1 g) dissolving in H2O (18 g) and freshly passivated catalyst (0.03 g) was added into the batch reactor. After purging with H2 for 5 times, the reactor was heated to the desired temperature and then filled with a particular H2 pressure under stirring (600 rpm). After the reaction was completed, the reactor was cooled to room temperature, and the products were separated by centrifuging and subsequently analysed by gas chromatograph (GC-7890F, FID, FFAP column 30 m × 0.32 mm × 0.5 μm). In order to test the recycling sustainability, the Pd@MIL-101(Cr) after hydrogenation reaction was separated by centrifuging followed by washing with water, and reused for a new batch of hydrogenation reactions. The Pd@MIL-101(Cr) catalysts were reused for four times.
3. Results and discussion
3.1 Characterization of MIL-101(Cr) and Pd@MIL-101(Cr)
In order to characterize the crystalline structure of as-synthesized MIL-101(Cr) and Pd@MIL-101(Cr), X-ray diffraction (XRD) experiments have been carried out. The XRD patterns of the as-prepared MIL-101(Cr) and Pd@MIL-101(Cr) with various amounts of Pd loadings are depicted in Fig. 2. The XRD pattern of as-synthesized MIL-101(Cr) is in excellent agreement with the simulated XRD pattern, presenting the characteristic diffraction peaks at 2θ = 2.81°, 3.29°, 3.98°, 5.19°, 5.92°, 9.62°, which confirms the successful construction of MOF supports for subsequent Pd NPs loading. A close look at the peaks at 2.81° and 3.29°, the relative peak intensity changes for MIL-101(Cr) and Pd@MIL-101(Cr) compared with simulated one. This phenomenon results from the small amounts of residual terephthalic acid and the introduction of Pd NPs into the MIL-101(Cr) pores.41–43 The MIL-101(Cr) didn't show apparent loss of crystallinity after deposition of Pd precursor and reduction treatment, which confirms the high stability of this MOF. It should be noted that the diffraction peaks from Pd NPs were not observed in the wide-angle XRD patterns (Fig. S1†), which is attributed to the small Pd loading amounts. The existence of Pd species is revealed by inductively coupled plasma (ICP) for three different batches of Pd@MIL-101(Cr). The analysis of ICP results, show that Pd loadings of 0.38 wt%, 0.62 wt% and 1.82 wt% were obtained via changing the mass ratio of Pd(η3-C3H5)(η5-C5H5) in MIL-101(Cr). | ||
| Fig. 2 Powder X-ray diffraction patterns of: (a) simulated MIL-101(Cr), (b) MIL-101(Cr), (c) 0.38 wt% Pd@MIL-101(Cr), (d) 0.62 wt% Pd@MIL-101(Cr), (e) 1.82 wt% Pd@MIL-101(Cr), (f) recovered 0.62 wt% Pd@MIL-101(Cr) after reaction. | ||
N2 physisorption experiments were performed to study the porosity of the parent MIL-101(Cr) and Pd@MIL-101(Cr) with different Pd loadings. All the samples exhibit the typical type I isotherms as shown in Fig. 3a. The pore size distribution curves are displayed in Fig. 3b, which shows the presence of two different pore sizes with values of 1.4 nm and 1.7 nm, confirming the formation of micropores of MIL-101(Cr). The Brunauer–Emmett–Teller (BET) surface area, micropore area and volume are listed in Table 1, which are well agreement with the reported values.44,45 In comparison with pristine MIL-101(Cr), the Pd@MIL-101(Cr) exhibits a slight decrease of SBET and total volume for the low Pd content of 0.38 wt% and 0.62 wt%. However with increasing the Pd content up to 1.82 wt%, the SBET and total volume dramatically decrease from 3099 m2 g−1 and 1.59 cm3 g−1 to 1833 m2 g−1 and 1.16 cm3 g−1, respectively, as a result of the occupation of cavities or block of outer surfaces of MIL-101(Cr) with Pd NPs.
 | ||
| Fig. 3 N2 adsorption–desorption isotherms at 77 K (a) and pore size distribution curves (b) for MIL-101(Cr), Pd@MIL-101(Cr) (Pd 0.38 wt%, 0.62 wt% and 1.82 wt%) and recovered Pd@MIL-101(Cr) after reaction. | ||
| Sample | SBET (m2 g−1) | MAa (m2 g−1) | EAb (m2 g−1) | MVc (cm3 g−1) | TVd (cm3 g−1) |
|---|---|---|---|---|---|
| a Micropore area.b External surface area.c Micropore volume.d Total volume. | |||||
| MIL-101 | 3099 | 2878 | 221 | 1.32 | 1.59 |
| 0.38 wt% Pd@MIL-101 | 2711 | 2406 | 305 | 1.13 | 1.56 |
| 0.62 wt% Pd@MIL-101 | 2805 | 2565 | 240 | 1.20 | 1.56 |
| 1.82 wt% Pd@MIL-101 | 1833 | 1521 | 312 | 0.72 | 1.16 |
| 0.62 wt% Pd@MIL-101 after reaction | 1963 | 1659 | 304 | 0.74 | 1.22 |
The stability of MIL-101(Cr) was investigated using a thermal gravimetric analysis (TGA) in the temperature range from 25 °C to 600 °C under atmosphere conditions. Two distinct weight-loss steps are clearly seen from TGA curves as displayed in Fig. S2.† The first weight-loss of 46% occurs in the range 25–250 °C, which is corresponds to the release of guest water molecules from framework. The second weight-loss of 32% starts from 300 °C with obvious drop, until a constant weight is reached after the temperature is up to 380 °C, which is mainly attributed to the departure of OH/F groups and the decomposition of the framework. These results indicate that the as-synthesized MIL-101(Cr) is stable below 300 °C for the catalytic reactions.38
The IR spectra of MIL-101(Cr), Pd(η3-C3H5)(η5-C5H5)@MIL-101(Cr) and Pd@MIL-101(Cr) are presented in Fig. 4. The characteristic bands at 1401 cm−1 and 1617 cm−1 are observed for all the samples, which are assigned to the symmetric and asymmetric vibrational stretching of COO− groups,46 respectively. The observed weak and narrow bands at 749 cm−1, 1017 cm−1 and 1157 cm−1 can be assigned to δ(C–H) and γ(C–H) vibrations of the benzene rings.47 The presences of the above vibrational bands strongly confirm the successful formation of MIL-101(Cr) framework. Weak bands at 582 cm−1 are likely due to the in-plane and out-of-plane bending modes of the COO– groups. The weak band appeared at 2929 cm−1 for Pd(η3-C3H5)(η5-C5H5)@MIL-101(Cr) is ascribed to vibrations of allyl, identifying that the successful deposition of Pd(η3-C3H5)(η5-C5H5) in MIL-101(Cr). The other features of Pd(η3-C3H5)(η5-C5H5) are not seen due to the low loading content or the overlap with the bands from host MIL-101(Cr) material. The intense and broad bands around 3426 cm−1 can be attributed to the OH vibrations, which suggest the existence of a large amount of water molecules within these samples. As expected, the band at 2929 cm−1 for Pd@MIL-101(Cr) disappeared after hydrogen reduction indicating the successful occurrence of reduction of the Pd(η3-C3H5)(η5-C5H5) precursor to the desired Pd NPs.
 | ||
| Fig. 4 FT-IR spectra of (a) MIL-101(Cr), (b) Pd(η3-C3H5)(η5-C5H5)@MIL-101(Cr), and (c) Pd@MIL-101(Cr). | ||
The X-ray photoelectron spectroscopy (XPS) was used to study the chemical state of Pd during preparation of Pd@MIL-101(Cr) catalyst. The peaks appeared at 337.4 eV and 342.7 eV in Fig. S3a† for Pd(η3-C3H5)(η5-C5H5)@MIL-101(Cr) correspond to Pd 3d5/2 and Pd 3d3/2 of Pd(II) existed in the Pd(η3-C3H5)(η5-C5H5) respectively, which confirms the successful deposition of Pd precursor on MIL-101(Cr). After treated with H2/Ar, the sample color changes from reddish brown to black, the 3d5/2 and 3d3/2 peaks of Pd(0) appeared at 335.6 eV and 340.9 eV (Fig. S3b†) indicates Pd(II) has been reduced to Pd(0).48 While the two bigger peaks at 337.3 eV and 342.6 eV observed for Pd@MIL-101(Cr) corresponds to the Pd(II) of PdO due to the oxidation of metallic Pd in Pd@MIL-101(Cr) in air during the transport for XPS measurement.41
The TEM images of the as-prepared Pd@MIL-101(Cr) with different Pd loadings are shown in Fig. 5. The typical cubic structure for pristine MIL-101(Cr) is clearly seen,49 which indicates that the intact structure is preserved after Pd incorporation. In addition to that, the Pd nanoparticles are obviously seen and are uniformly dispersed within MIL-101(Cr) with an average nanoparticle sizes of 6.49 nm and 6.53 nm for Pd content of 0.38 wt% and 0.62 wt%, respectively, which is substantially larger than the pore size of MIL-101(Cr) with a value of 1.2 nm. Therefore, the Pd nanoparticles occupied several adjacent pores instead of single pore of MIL-101(Cr).17 Upon increasing the Pd loading content up to 1.82 wt% the average nanoparticle size of Pd has increased to 9.21 nm, which is due to the aggregation of Pd nanoparticles at high loading amounts. The formation of these agglomerations leads to the occupation of several adjacent pores beyond MOF cavity by these larger Pd nanoparticles. The obtained TEM images strongly support the formation of highly dispersed Pd nanoparticles within the MIL-101(Cr) abundant cavities.17 The catalytic activity of as-prepared Pd@MIL-101(Cr) samples were further investigated in the selective hydrogenation of 2-butyne-1,4-diol.
 | ||
| Fig. 5 TEM images of (a) 0.38 wt% Pd@MIL-101(Cr), (b) 0.62 wt% Pd@MIL-101(Cr), (c) 1.82 wt% Pd@MIL-101(Cr). | ||
3.2 Catalytic performances
Palladium based catalysts have been proven to be one of the most effective catalysts for achieving the transformation of alkynes, in particular, for the catalytic partial hydrogenation (semi-hydrogenation) of alkynes to alkenes.50,51 As shown in Fig. 1, in first step, the 2-butene-1,4-diol (cis-BED and/or trans-BED) were generated through the hydrogenation of BYD, the produced BED adsorbed on the Pd sites to be further hydrogenated and yield γ-hydroxybutyraldehyde, n-butyraldehyde, n-butanol, crotyl alcohol and acetal due to the double bond migration and hydrogenolysis of BED, which are treated as side products in this work. In this study the selective hydrogenation of 2-butyne-1,4-diol (BYD) was chosen as a model system for the evaluation of the catalytic performance of the prepared Pd@MIL-101(Cr) samples.The catalytic performance of the as-prepared Pd@MIL-101(Cr) samples, such as catalytic activity, selectivity and reaction pathways with respect to hydrogenation of BYD has been evaluated under mild conditions at 50 °C and 0.5 MPa using water as solvent. Fig. 6a displays the changes of reactant and products concentration as a function of time and Fig. 6b shows the BED selectivity versus BYD conversion.26,52 It can be seen that the conversion of BYD increased with prolonging the reaction time, and reached 93.3% after 5 h, suggesting the high activity of small amount of Pd@MIL-101(Cr) under these mild conditions. This extremely high selectivity of about 99.0% remained constant until the BYD conversion increased to 72.5%, and the formation of BED was mainly from BYD hydrogenation without being further catalysed to yield BDO and other side products. This is correlated to that the majority of the unconverted BYD are absorbed on Pd NPs, due to the stronger adsorption affinity on the metal surface compared with BED, which therefore hindered further hydrogenation of BED and the formation of side products. When the conversion of BYD was above 72.5%, the selectivity of BED slightly decreased, as a result of the further reaction of BED, which leads to the formation of BDO and other side products. This leads to the increase in the reaction selectivity toward BDO and the other side products. It should be noted that the content of BDO was higher than that of side products during the first 4 h of the reaction, which suggests that hydrogenation was easier than the double bond migration and hydrogenolysis for BED.
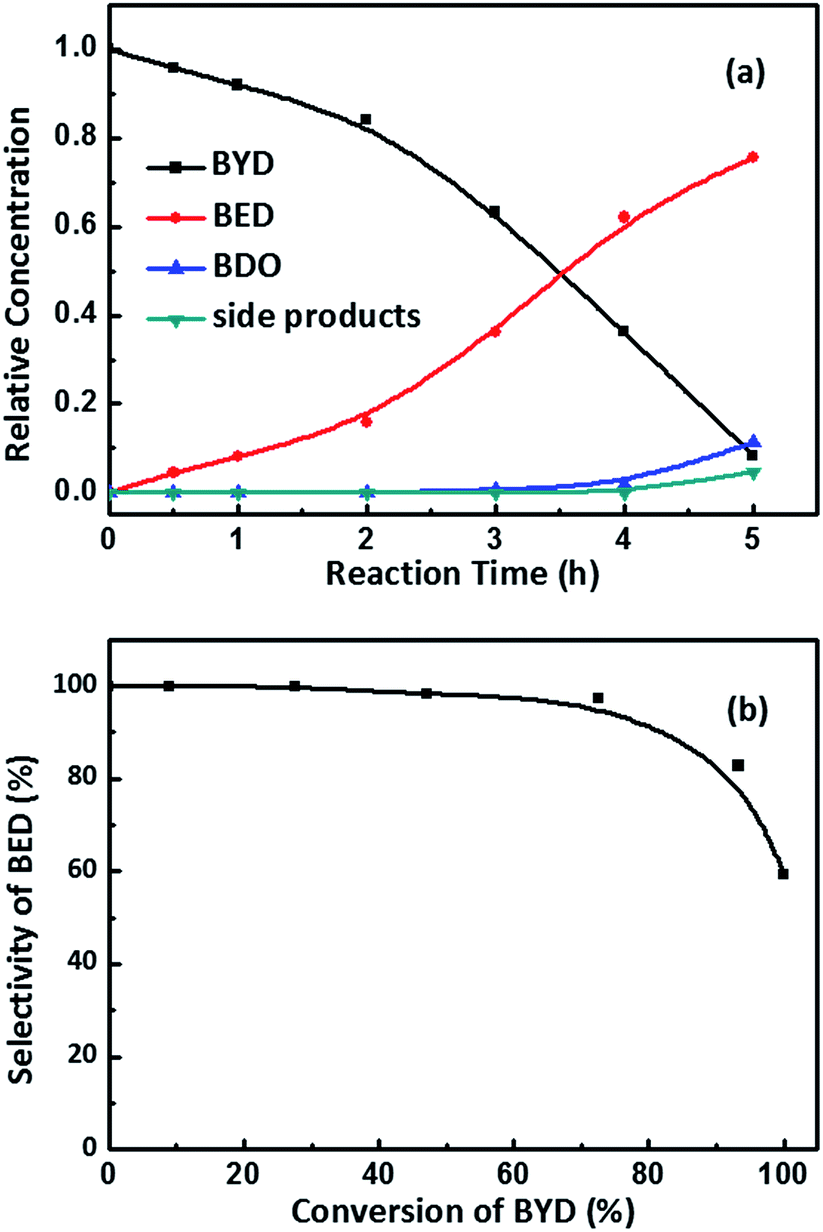 | ||
| Fig. 6 Changes of reactant and products concentration as a function of time (a) and BED selectivity versus BYD conversion (b). Reaction conditions: Pd@MIL-101(Cr) (0.03 g, Pd 0.62 wt%), temperature (50 °C), H2 (0.5 MPa). | ||
The influence of temperature on the conversion of BYD and selectivity of the different products was also investigated, by variation of reaction temperature from 40 °C to 80 °C at the same pressure of 0.5 MPa. In Fig. 7a, the conversion of BYD increased from 42.0% to 100% with increasing temperature from 40 °C to 70 °C, which can be well explained by significantly impact of temperature on the reaction kinetics.53 In contrast, the slightly decreased selectivity of BED and increased selectivity of BDO and side products, which are mainly caused by the consumption of BYD, and the BED being activated on Pd NPs leading to the formation of other side products. It can be seen that the selectivity of BED still remain at 93.2% when BYD was completely converted at 70 °C, this value is higher than 82.8% when 93.3% of BYD were converted at 50 °C. This result clearly indicates that higher temperature is in favour of obtaining high selectivity of BED until the BYD were completely consumed due to the more obvious accelerative effect in the hydrogenation of BYD.
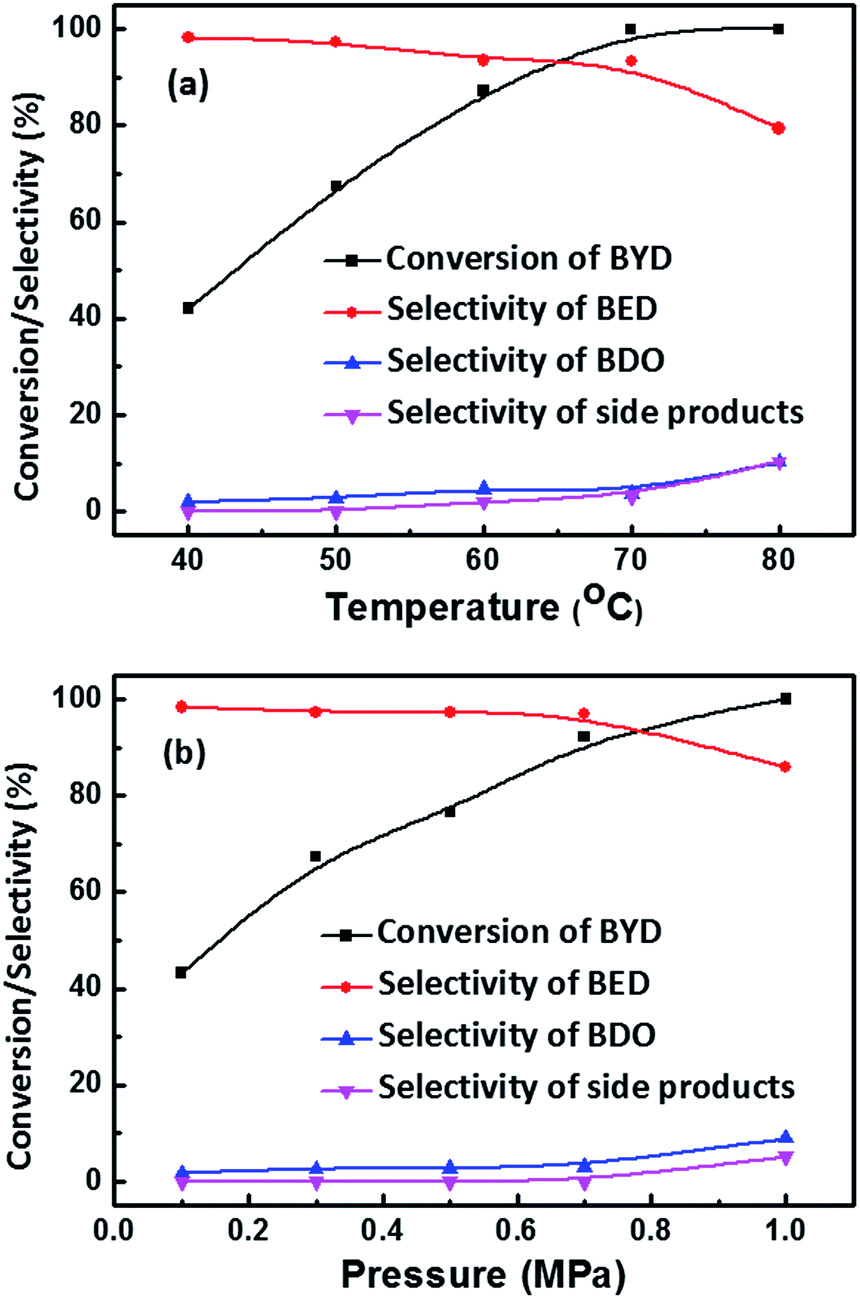 | ||
| Fig. 7 Effect of temperature (a) and H2 pressure (b) on the conversion of BYD and selectivity of different products. Reaction conditions: Pd@MIL-101(Cr) (0.03 g, Pd 0.62 wt%), temperature [50 °C for (b)], H2 [0.5 MPa for (a)]. | ||
Fig. 7b depicts the conversion of BYD and the selectivity of BED and other products versus hydrogen pressure from 0.1 MPa to 1.0 MPa. With the increase of hydrogen pressure the conversion of BYD increases almost linearly, which is due to the improved gas–liquid and liquid–solid mass transfer coefficient and acceleration of conversion rate.54 The selectivity of BED show a slight decrease from 98.3% at 0.1 MPa to 96.9% at 0.7 MPa, and further decreased to 85.6% when the pressure reached 1 MPa. As discussed above, the majority of BYD was consumed under high pressure within 4 h, which explains well the variation in selectivity towards BED and the formation of undesired side products. In addition, the influence of different Pd loadings (0.38 wt%, 0.62 wt%, and 1.82 wt%) of Pd@MIL-101(Cr) on the activity and selectivity for BYD hydrogenation was examined. The results reveal that with increasing Pd loading content, the conversion of BYD increases (Fig. S4†) due to the existence of more active sites in Pd@MIL-101(Cr) catalysts with higher Pd content.
The different catalytic tests have shown that the selectivity of BED can reach 94% with complete consumption of BYD by using the as-prepared Pd@MIL-101(Cr) as catalysts for hydrogenation reaction under appropriate conditions. The Pd@MIL-101(Cr) catalysts exhibit excellent performance for BYD hydrogenation compared to the commercial Pd/C catalysts (5 wt% Pd/C (Johnson Matthey)) with 72.5% of selectivity towards BED when 19.1% of BYD were consumed,55 and comparable performance of Nanoselect or Lindlar catalysts with the selectivity of 97%.56,57 In addition to that the selectivity of BED is lower than 10% until the complete consumption of BYD for the 0.5 wt% Pd/C with the commercial activated carbon (Chemviron SCXII) used as a support,58 and the Pd@MIL-101(Cr) shows higher selectivity for BED than Bio-Pd59 and Bio-Pt26 with the same conversion of BYD. The comparison of the Pd@MIL-101(Cr) with other reported catalysts for 2-butyne-1,4-diol hydrogenation can be found in Table S1.† It is well known that metal nanoparticles terraces are favourable for alkyne hydrogenation, while the vertex and edge sites are favourable for alkene hydrogenation or isomerization.60 The excellent catalytic performance of high selectivity of BED can be well explained as follows: (i) the Pd nanoparticles with irregular shape were encapsulated in the cavities of MIL-101(Cr) resulting in the block of the parts of the vertex and edge sites by the framework, therefore, further hydrogenation and isomerization of BED is substantially suppressed; (ii) alkene hydrogenation may take place on terraces with the absence of starting alkyne; (iii) the first stage of BYD hydrogenation proceeds much faster than the second stage of BED hydrogenation due to stronger adsorption of BED on the metal–organic coordination polymers than that on activated carbon.26
To evaluate the stability of Pd@MIL-101(Cr), which is an essential requirement for industrial catalysts, XRD and N2 adsorption experiments were conducted to examine the structural stability before and after catalytic reactions. As shown in Fig. 2f, the main characteristic diffraction peaks of the Pd@MIL-101(Cr) have virtually no change after reaction, strongly confirms the high stability of the used Pd@MIL-101(Cr) catalysts. The dramatic decrease of the surface area (SBET) for 0.62 wt% Pd@MIL-101(Cr) after reaction (Fig. 3a and Table 1) and the slight shift in the pore size distribution (Fig. 3b) are caused by partial-collapsed frameworks during the reaction. By comparison of the TEM image of Pd@MIL-101(Cr) obtained after reaction with the fresh catalysts (Fig. S5†), the octahedral structure of MIL-101(Cr) remains unchanged and no occurrence of Pd nanoparticles aggregation.
Recyclability, the other essential requirement for industrial catalysts in industrial applications, was evaluated for Pd@MIL-101(Cr) with BYD hydrogenation. The recyclability of 1.82 wt% Pd@MIL-101(Cr) for BYD hydrogenation is shown in Fig. 8, which shows that the Pd@MIL-101(Cr) exhibits excellent recyclability up to 5 successive cycles. The conversion of BYD shows almost no decrease after the different cycles, with nearly 100% conversion during hydrogenation reaction. The selectivity of BED was above 86% in first run, and remained above 90% in the later four runs. The slightly improved selectivity of BED can be due to the partial collapse of frameworks during reaction, which could lead to the blocking of more vertex and edge sites, which will favour the hydrogenation or isomerization of BED.
 | ||
| Fig. 8 Catalyst recycle studies for 1.82 wt% Pd@MIL-101(Cr). Reaction conditions: Pd@MIL-101(Cr) (0.03 g, Pd 1.82 wt%), temperature (50 °C), H2 (0.5 MPa). | ||
4. Conclusions
In summary, we have successfully demonstrated the preparation of Pd@MIL-101(Cr) hetero-catalyst using the MOCVD approach, by employing Pd(η3-C3H5)(η5-C5H5) as volatile precursor and MIL-101(Cr) as support. TEM confirms the presence of the Pd NPs and their high dispersion within the cages of MIL-101(Cr) without affecting the MOF structure, crystallinity and porosity compared to pristine MIL-101(Cr). The high catalytic activity and excellent selectivity of BED was achieved for the prepared Pd@MIL-101(Cr) in the partial hydrogenation of BYD, due to the high dispersion and well confined Pd NPs within MIL-101(Cr), which suppresses further hydrogenation and isomerization of BED. The recyclability of the Pd@MIL-101(Cr) hetero-catalyst were also evaluated, and has shown that they possess excellent stability. Our results have revealed that the hybrid Pd@MIL-101(Cr) is a promising hetero-catalyst for BYD hydrogenation towards industrial applications. Further improvements of the selectivity of BYD hydrogenation by optimizing the Pd@MIL-101(Cr) catalyst and developments of other metal NPs@MOFs hetero-catalysts are undergoing in our lab.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (21673032, 21573031 and 21428301) and the Fundamental Research Funds for the Central Universities (DUT15ZD106 and DUT15RC(4)09).References
- M. Eddaoudi, D. F. Sava, J. F. Eubank, K. Adil and V. Guillerm, Chem. Soc. Rev., 2015, 44, 228–249 RSC.
- Z. Zhang, L. Zhang, L. Wojtas, P. Nugent, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 924–927 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444–1230455 CrossRef PubMed.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. Allan, G. Maurin and P. Couvreur, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- L. Ye, J. Liu, Y. Gao, C. Gong, M. Addicoat, T. Heine, C. Wöll and L. Sun, J. Mater. Chem. A, 2016, 4, 15320–15326 CAS.
- J. Liu, W. Zhou, J. Liu, Y. Fujimori, T. Higashino, H. Imahori, X. Jiang, J. Zhao, T. Sakurai, Y. Hattori, W. Matsuda, S. Seki, S. K. Garlapati, S. Dasgupta, E. Redel, L. Sun and C. Wöll, J. Mater. Chem. A, 2016, 4, 12739–12747 CAS.
- J. Liu, W. Zhou, J. Liu, I. Howard, G. Kilibarda, S. Schlabach, D. Coupry, M. Addicoat, S. Yoneda, Y. Tsutsui, T. Sakurai, S. Seki, Z. Wang, P. Lindemann, E. Redel, T. Heine and C. Wöll, Angew. Chem., Int. Ed., 2015, 54, 7441–7445 CrossRef CAS PubMed.
- J. Li, Q. L. Zhu and Q. Xu, Chem. Commun., 2014, 50, 5899–5901 RSC.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 2010, 3701–3714 CrossRef.
- M. Sabo, A. Henschel, H. Fröde, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827 RSC.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Ronnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- F. Schröder, D. Esken, M. Cokoja, M. W. E. v. d. Berg, O. I. Lebedev, G. V. Tendeloo, B. Walaszek, G. Buntkowsky, H.-H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119–6130 CrossRef PubMed.
- D. Esken, S. Turner, O. I. Lebedev, G. V. Tendeloo and R. A. Fischer, Chem. Mater., 2010, 22, 6393–6401 CrossRef CAS.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- S. Hermes, M. K. Schroter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed.
- M. Zhang, J. Guan, B. Zhang, D. Su, C. T. Williams and C. Liang, Catal. Lett., 2012, 142, 313–318 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- V. I. Isaeva, O. P. Tkachenko, E. V. Afonina, L. M. Kozlova, G. I. Kapustin, W. Grünert, S. E. Solov’eva, I. S. Antipin and L. M. Kustov, Microporous Mesoporous Mater., 2013, 166, 167–175 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Ind. Org. Chem., John Wiley & Sons, 2008 Search PubMed.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS.
- M. Telkar, C. Rode, V. Rane and R. Chaudhari, Catal. Commun., 2005, 6, 725–730 CrossRef CAS.
- I. T. Duncanson, I. W. Sutherland, B. Cullen, S. D. Jackson and D. Lennon, Catal. Lett., 2005, 103, 195–199 CrossRef CAS.
- H. Li, Y. Zhao, C. Gao, Y. Wang, Z. Sun and X. Liang, Chem. Eng. J., 2012, 181–182, 501–507 CrossRef CAS.
- C. Berguerand, I. Yuranov, F. Cárdenas-Lizana, T. Yuranova and L. Kiwi-Minsker, J. Phys. Chem. C, 2014, 118, 12250–12259 CAS.
- H.-U. Blaser, A. Baiker and R. Prins, Heterogeneous catalysis and fine chemicals IV, Elsevier, 1997 Search PubMed.
- M. Telkar, C. Rode, V. Rane, R. Jaganathan and R. Chaudhari, Appl. Catal., A, 2001, 216, 13–22 CrossRef CAS.
- M. Zhang, Y. Yang, C. Li, Q. Liu, C. T. Williams and C. Liang, Catal. Sci. Technol., 2014, 4, 329–332 CAS.
- I. Luz, C. Rösler, K. Epp, F. X. Llabrés i Xamena and R. A. Fischer, Eur. J. Inorg. Chem., 2015, 2015, 3904–3912 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- B. Shaw, Proc. Chem. Soc., 1960, 7, 247 Search PubMed.
- W. T. Dent, R. Long and A. J. Wilkinson, J. Chem. Soc., 1964, 8, 1585 RSC.
- H. Li, Z. Zhu, F. Zhang, S. Xie, H. Li, P. Li and X. Zhou, ACS Catal., 2011, 1, 1604–1612 CrossRef CAS.
- A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192–4194 RSC.
- Y. Pan, B. Yuan, Y. Li and D. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- G. Chen, S. Wu, H. Liu, H. Jiang and Y. Li, Green Chem., 2013, 15, 230–235 RSC.
- W. Qin, W. Cao, H. Liu, Z. Li and Y. Li, RSC Adv., 2014, 4, 2414–2420 RSC.
- N. Maksimchuk, M. Timofeeva, M. Melgunov, A. Shmakov, Y. A. Chesalov, D. Dybtsev, V. Fedin and O. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS.
- K. Yang, Q. Sun, F. Xue and D. Lin, J. Hazard. Mater., 2011, 195, 124–131 CrossRef CAS PubMed.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef CAS.
- P. A. Szilagyi, E. Callini, A. Anastasopol, C. Kwakernaak, S. Sachdeva, R. van de Krol, H. Geerlings, A. Borgschulte, A. Zuttel and B. Dam, Phys. Chem. Chem. Phys., 2014, 16, 5803–5809 RSC.
- M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2013, 114, 1783–1826 CrossRef PubMed.
- N. Semagina, E. Joannet, S. Parra, E. Sulman, A. Renken and L. Kiwi-Minsker, Appl. Catal., A, 2005, 280, 141–147 CrossRef CAS.
- X. Chen, M. Zhang, K. Yang, C. T. Williams and C. Liang, Catal. Lett., 2014, 144, 1118–1126 CrossRef CAS.
- C. Rode, P. Tayade, J. Nadgeri, R. Jaganathan and R. Chaudhari, Org. Process Res. Dev., 2006, 10, 278–284 CrossRef CAS.
- C. W. A. Chan, Y. Xie, N. Cailuo, K. M. K. Yu, J. Cookson, P. Bishop and S. C. Tsang, Chem. Commun., 2011, 47, 7971–7973 RSC.
- G. Vile, N. Almora-Barrios, S. Mitchell, N. Lopez and J. Perez-Ramirez, Chemistry, 2014, 20, 5926–5937 CrossRef CAS PubMed.
- D. Albani, G. Vilé, S. Mitchell, P. T. Witte, N. Almora-Barrios, R. Verel, N. López and J. Pérez-Ramírez, Catal. Sci. Technol., 2016, 6, 1621–1631 CAS.
- M. Musolino, C. Cutrupi, A. Donato, D. Pietropaolo and R. Pietropaolo, J. Mol. Catal. A: Chem., 2003, 195, 147–157 CrossRef CAS.
- J. Wood, L. Bodenes, J. Bennett, K. Deplanche and L. E. Macaskie, Ind. Eng. Chem. Res., 2010, 49, 980–988 CrossRef CAS.
- J. A. Bennett, G. A. Attard, K. Deplanche, M. Casadesus, S. E. Huxter, L. E. Macaskie and J. Wood, ACS Catal., 2012, 2, 504–511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25722d |
| This journal is © The Royal Society of Chemistry 2017 |