
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and biological evaluation of valepotriate derivatives as novel antitumor agents†
Bo Zhang‡
ab,
Ruiying Guo‡c,
Yongzhou Huc,
Xiaowu Dong c,
Nengming Linb,
Xiaoyang Daia,
Honghai Wua,
Shenglin Ma*b and
Bo Yang*a
c,
Nengming Linb,
Xiaoyang Daia,
Honghai Wua,
Shenglin Ma*b and
Bo Yang*a
aZhejiang Province Key Laboratory of Anti-Cancer Drug Research, Institute of Pharmacology and Toxicology, School of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang 310058, P. R. China. E-mail: yang924@zju.edu.cn; Tel: +86-57188208400
bTranslational Medicine Research Center, Nanjing Medical University, Affiliated Hangzhou Hospital, Hangzhou First People's Hospital, Hangzhou, Zhejiang 310006, P. R. China. E-mail: mashenglin@medmail.com.cn; Tel: +86-57156007908
cZJU-ENS Joint Laboratory of Medicinal Chemistry, Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, P. R. China
First published on 21st June 2017
Abstract
Natural products remain the largest resources of lead compounds that can be used to develop novel anticancer drug candidates. Based on deacetylisovaltratum, a natural product with promising anticancer activity, herein we designed and synthesized of a series of valepotriate derivatives with a novel skeleton from commercially available genipin. In addition, a structure–activity relationship study demonstrated the importance of an epoxy group on the C1-position and the preferable size of the sidechain ((5-methylhexanoyl)oxy) on the C-7 position of valepotriates for their cytotoxic activities. The most potent compound 1e showed moderate to good IC50 values against various cancer cells, ranging from 10.7 to 50.2 μM, which are comparable to that of deacetylisovaltratum. Additionally, we demonstrate that mitochondrion-mediated apoptosis would be its mechanism of action, thus enlightening the further development of novel valepotriate derivatives.
1 Introduction
Natural product research continues to explore a variety of lead compounds, which are potential templates for the development of new anticancer drug candidates.1–3 However, challenges remain unsolved in either the isolation or purification of bioactive natural products; thus semisynthesis becomes a preferable method to obtain bioactive compounds.4 The semisynthetic strategy is widely used when the precursor molecules are structurally complex, or difficult produce by total synthesis, such as anticancer drugs (i.e. paclitaxel, etoposide, and irinotecan), antimalarial drugs (i.e. roxithromycin) and antibiotics (i.e. cefixime) (Fig. 1).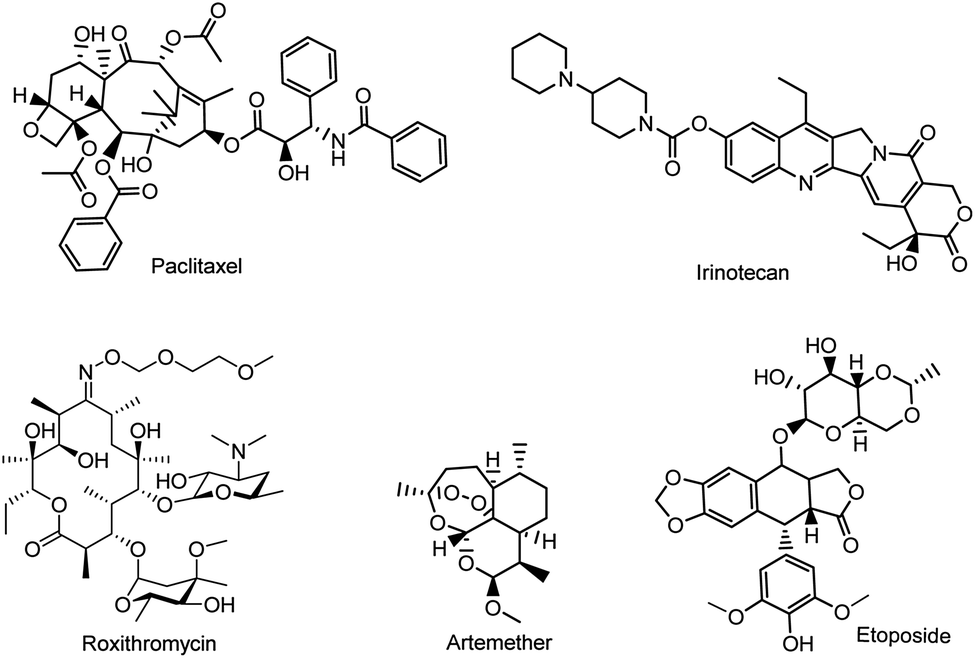 | ||
| Fig. 1 Examples of semisynthetic drugs. | ||
Valerian is a very important genus of plants used as a medicinal herb in many areas of the world, the roots and rhizomes of which have been used for the treatment of epilepsy, hysteria, nervous disorders, neurasthenia and emotional stress.5–9 In particular, Patrinia heterophylla Bunge, belonging to the genus Valerian, showed anticancer activity against metrocarcinoma and cervical cancer in ancient China, whereas the biological components responsible for its anticancer activity are not fully recognized.10,11 Indeed, a variety of pharmacologically active components (e.g. monoterpenes, valepotriates, and sesquiterpenes) have been isolated and characterized.12,13 Among them, valepotriates, belonging to the family of iridoids with a 10-carbon basic skeleton, showed their significant activity against cancer cells.1–3,14 Recently, we found that deacetylisovaltratum, a compound belonging to the class of valepotriates, could effectively cause G2/M-phase arrest in gastric cancer cells by disrupting tubulin polymerization, and inducing mitochondrion-dependent apoptosis.15,16 Considering the unique structure of epoxy group in deacetylisovaltratum, the alkylating properties could be responsible for its potent anti-cancer activity. Moreover, the structural diversity might change its physicochemical properties and also its intracellular activities. However, due to the lack of structural diversity in natural valepotriates, as well as the difficulty in synthetic procedures, the structural–activity relationship (SAR) studies of valepotriate analogues is very rare, which hampers the development of valepotriate as novel anti-cancer agents. Therefore, the exploration of the crucial structural requirement of valepotriates for anticancer activities is particularly needed. In this study, we intend to acquire series of valepotriate derivatives via semisynthesis and to study the biological activity of these compounds.
Recently, Murakami et al. reported the concise synthesis of 5,6-dihydrovaltrate from commercially available iridoid genipin, providing a novel semisynthetic route in structurally optimization of valepotriates (Fig. 2).17,18 As part of our continued interest in the area of chemical modification of natural products,19,20 herein, we designed, synthesized a series of valepotriate derivatives with novel skeleton from genipin using slightly modified Murakami's method. Noteworthy, some crucial SAR clues were found after investigating the effect of epoxy and length of aliphatic side chain on cytotoxicity against a variety of cancer cells. More significantly, some compounds showed superior cytotoxic activities than that of deacetylisovaltratum. In addition, mitochondrion-mediated apoptosis was significantly observed, which would be the exact action mechanism of its cytotoxic activities, thus enlightening the further development of novel valepotriate derivatives.
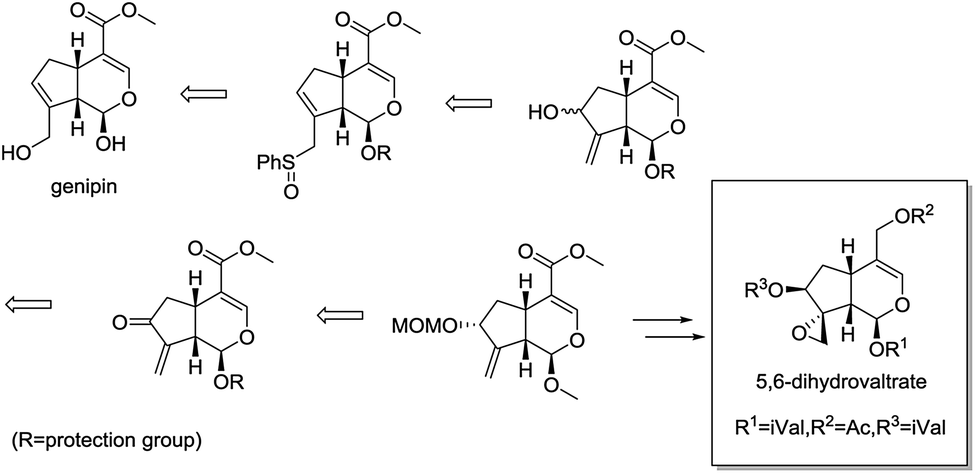 | ||
| Fig. 2 Murakami's synthetic strategy. | ||
2 Results and discussion
2.1 The synthesis of valepotriate derivatives
The synthesis of compounds 1a–h started with genipin is illustrated in Scheme 1. At first, genipin 2 was treated by Amberlyst-15 resins in methanol to afford methyl acetal 3, which was submitted to phenylsulfidation using diphenyldisulfide and tributylphosphane in toluene to give phenylsulfide 4. After acidic hydrolysis of the methyl acetal with 10% HCl–THF, the resulting hemiacetal 5 was coupled with isovaleric acid in the presence of isovaleric acid, 1,1′-carbonyldiimidazole (CDI) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to yield isovaleryl acetal 6. After oxidation of 6 with oxone in methanol, the resulting sulfoxide was subjected to Mislow–Evans rearrangement with trimethyl phosphite in MeOH to afford allyl alcohol 7. In this rearrangement procedure, the reaction time was quite crucial for the conversion, which should be carefully monitored by TLC analysis. Then, stereoselective epoxidation associated with the adjacent hydroxyl group using tert-butyl hydroperoxide (TBHP) and vanadium oxyacetylacetonate [VO(acac)2]17,18,21,22 afforded epoxyalcohols 8.18,22 Finally, desired compounds 1a–h was obtained by introduction of the appropriate aliphatic acid group to C-7 of compound 7 by Mitsunobu inversion using diethyl azodicarboxylate (DEAD) and triphenylphosphine (Fig. 3).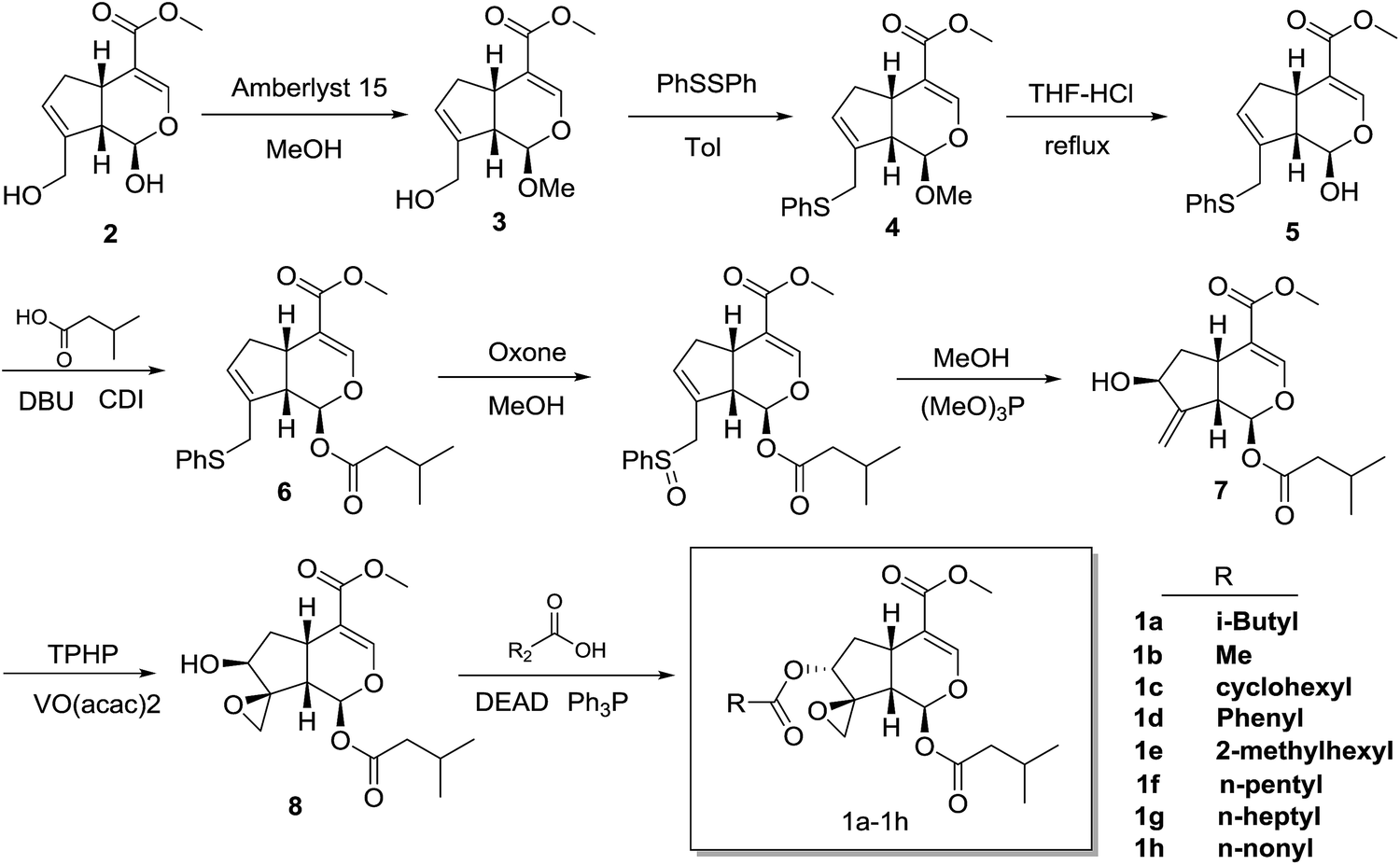 | ||
| Scheme 1 Synthesis of valepotriate derivatives. Reagents and conditions: (a) Amberlyst-15, MeOH; (b) PhSSPh, Tol; (c) 10%HCl, THF; (d) isovaleric acid, DBU, CDI; (e) oxone, MeOH; (f) (MeO)3P, MeOH; (g) TPHP, VO(acac)2, Tol; (h) appropriate acid, DEAD, Ph3P, Tol. | ||
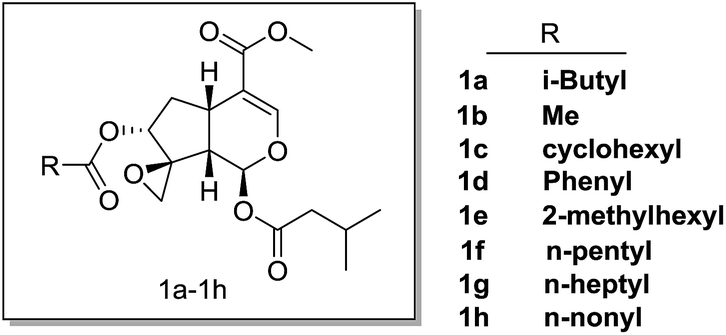 | ||
| Fig. 3 The structures of valepotriate derivatives 1a–1h in this study. | ||
2.2 Cytotoxicity of compounds 1a–h against various cancer cells
To investigate whether these compounds could suppress the growth of cancer cells, anti-proliferative effect was tested against various cancer cell lines, including pancreatic cancer cells (SW1990, BXPC3, CAPAN2, CFPAC, PANC1), breast cancer cells (BT474, MCF7, MDA-MB-231), gastric cancer cells (AGC, HGC-27, KATOIII) and lung cancer cells (H1795) (Table 1). As expected, some of these compounds (i.e. 1c, 1d, 1e) showed comparable cytotoxic activities as the parental compound deacetylisovaltratum, suggesting that the novel skeleton with methoxycarbonyl on C4-position of valepotriate remained the anticancer activities. Moreover, we found epoxy group on C1-position of valepotriate was very crucial by comparing with the anticancer activities of 7a and deacetylisovaltratum, which was in consistent with the work of Murakami et al.22 In another study, R. Bos et al. reported the cytotoxicity of valepotriates analogues against GL4 and COLO 320 cells with IC50 values ranging from 1 to 6 μM.12 Additionally, three valepotriate isomers isolated by S. Lin et al. displayed moderate cytotoxicity against various cancer cell lines with IC50 values varied from 2.8 to 8.3 μM.23 In this study, 1e showed comparable cytotoxic activity as previously reported valepotriate analogues. In further SAR analysis, either shortening or prolonging the sidechain (i.e. 1a, 1b, 1f, 1g, 1h) undermined its biological activities, revealing that (5-methylhexanoyl)oxy group on C-7 position would be preferable lipophilicity towards cancer cells. Meanwhile, the replacement of (5-methylhexanoyl)oxy group with aromatic (1d) and cyclohexyl (1c) substituents reduced its cytotoxic activity. Based on our efforts in structural optimization of valepotriate analogues, we successfully discovered a potent semisynthetic compound 1e bearing unique skeleton distinguished from deacetylisovaltratum which was previously identified as potent natural anti-cancer agent.| Cell lines | Cancer type | Cytotoxic activities, IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DI | 7a | 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | ||
| a N.T. indicates not detectable. | |||||||||||
| SW1990 | Pancreatic cancer | 34.9 | 85.2 | 59.1 | >100 | 40.2 | 54.9 | 26.1 | N.T. | N.T. | N.T. |
| BXPC3 | 14.4 | 24.6 | 48.2 | >100 | 28.1 | 37.1 | 25.1 | N.T. | N.T. | N.T. | |
| CAPAN2 | 17.5 | 63.7 | 37.4 | 53.8 | 33.9 | 68.0 | 31.6 | 40.7 | >100 | >100 | |
| CFPAC | 13.0 | 87.7 | 47.4 | 52.0 | 29.5 | 60.9 | 27.9 | 54.3 | 32.0 | 53.9 | |
| PANC1 | 14.8 | >100 | 86.9 | N.T. | 48.0 | 66.9 | 20.6 | N.T. | N.T. | N.T. | |
| BT474 | Breast cancer | 24.3 | N.T. | 97.8 | >100 | 21.4 | 54.0 | 50.2 | N.T. | N.T. | N.T. |
| MCF7 | 27.5 | 51.2 | 77.4 | >100 | 18.1 | 19.8 | 10.7 | N.T. | N.T. | N.T. | |
| MDA-MB-231 | 23.1 | 51.0 | 42.8 | >100 | 12.7 | 27.3 | 13.2 | N.T. | N.T. | N.T. | |
| AGS | Gastric cancer | 7.7 | 53.2 | 26.3 | >100 | 28.9 | 36.9 | 22.9 | 55.7 | 59.5 | >100 |
| HGC-27 | 14.8 | 25.7 | 47.5 | >100 | 20.9 | 28.2 | 21.5 | >100 | 54.0 | 26.6 | |
| KATOIII | 32.2 | N.T. | N.T. | N.T. | N.T. | N.T. | N.T. | 48.8 | 51.2 | 92.5 | |
| H1975 | Lung cancer | 14.3 | 21.0 | 25.8 | 93.0 | 28.2 | 15.9 | 11.3 | N.T. | N.T. | N.T. |
2.3 Cytotoxicity of 1e in human lung cancer H1975 cells
Human lung cancer H1975 cell harbours both L858R and T790M mutation in epidermal growth factor receptor (EGFR), thus it is resistant to first-generation EGFR tyrosine inhibitor such as gefitinib or erlotinib. As recently reported, several plants-derived compounds possessed anti-proliferation activity against H1975, and the IC50 value varied from 3 to 15 μM.24–26 In our study, the cytotoxicity of 1e was determined by CCK-8 assay in human lung cancer H1975 cells (Fig. 4). 1e exhibited both time- and concentration-dependent cytotoxicity against H1975 cells. The IC50 values of 48 and 72 h treatment with 1e were 27.4 and 13.1 μM respectively.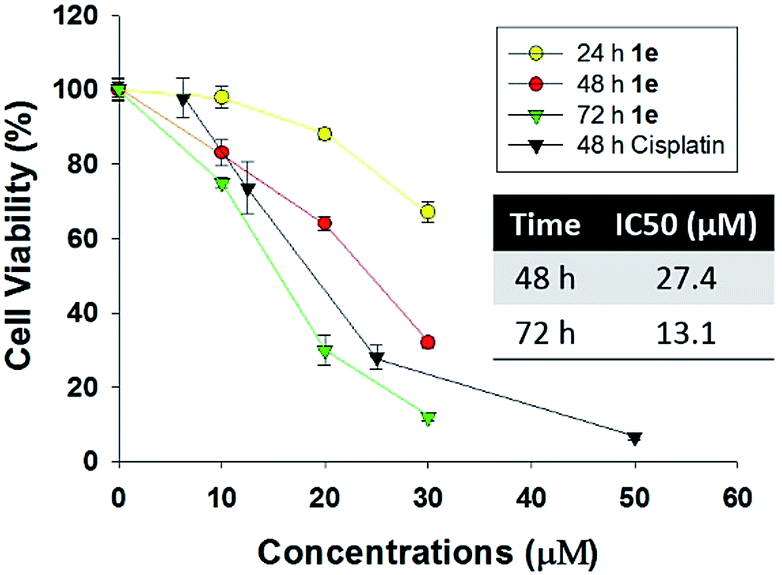 | ||
| Fig. 4 The inhibitory effect of 1e on human lung cancer cell line H1975. H1975 cells were seeded in 96-well plates and then treated with 1e as indicated. Treatment was washed off after 24, 48, 72 or 96 h and cell viability was calculated using CCK-8 assay. | ||
2.4 Compound 1e triggered apoptosis in H1975 cells
Apoptosis was usually involved in the proliferation inhibitory effect caused by natural compound.27 Annexin V/PI staining was used to characterize the early and late apoptotic cells after cells were treated with indicated concentrations of 1e for 48 h.28 In H1975 cells, treatment with 20 μM of 1e yielded a 35% apoptosis rate, which was in consistent with the IC50 value of 48 h treatment (Fig. 5A). By comparing the apoptosis induction activity of 1e with other natural derived agents in H1975 cells, 1e exhibited superior anti-cancer activity than cordycepin, but slightly less effective than shikonin.24,25 Meanwhile, depolarized mitochondria membrane potential was detected after 24 h treatment with 20 μM of 1e (Fig. 5B). In addition, DAPI stain was used to verify the occurrence of apoptosis. After 48 h treatment with 20 μM of 1e, nucleus with shrunk size and intensified fluorescence were seen under fluorescence microscopy. Moreover, apoptotic bodies could be observed at 20 μM of 1e, suggesting that H1975 cells were undergoing apoptosis (Fig. 6).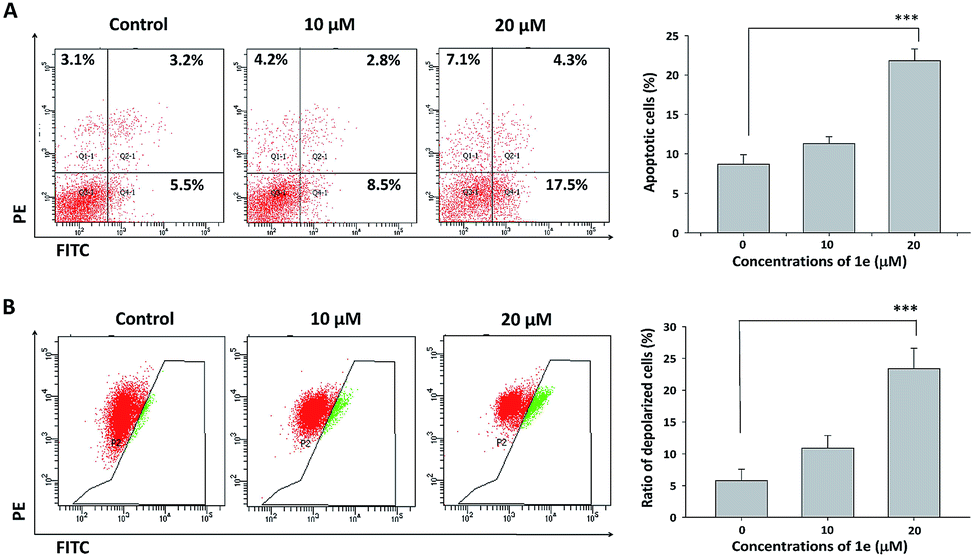 | ||
| Fig. 5 1e triggered mitochondrion-dependent apoptosis in H1975 cells. (A) H1975 cells were seeded in 6-well plate and cultured for 24 h. Cells were treated with 1e for 48 h before staining and followed by flow cytometric analysis. (B) H1975 cells were seeded in 6-well plate and cultured for 24 h. Cells were treated with 1e for 12 h before JC-1 stain and flow cytometric analysis. Three independent experiments were quantitatively analyzed. Each bar represented the mean ± SD. ***P < 0.001. | ||
 | ||
| Fig. 6 H1975 cells were seeded in 6-well plate and cultured for 24 h. Cells were treated with 1e for 48 h before DAPI stain and observed under fluorescence microscopy. Yellow arrows indicates apoptotic bodies. Scale bar = 20 μm. | ||
2.5 Compound 1e induced caspase-dependent apoptosis
Mitochondrion plays a critical role in the apoptosis caused by natural compounds. In H1975 cells, reduced expression of Bcl-2, activation of caspase-3 and cleavage of poly-(ADP-ribose)-polymerase (PARP) was closely associated with the apoptosis induced by isoliquiritigenin.29 In this study, to verify the role of mitochondrion in 1e triggered apoptosis, we performed western blots to analyze apoptosis-related proteins. As shown in Fig. 7A, 20 μM of 1e induced an elevated level of Bax, and meanwhile the expression of Bcl-2 was decreased. In addition, decreased protein level of procaspase-9 and cleaved PARP were found after treatment with 20 μM of 1e, indicating that 1e could trigger mitochondrion-dependent apoptosis. In addition, 20 μM of 1e significantly induced an increase in caspase-3 activity (Fig. 7B), which was not found in 10 μM of 1e treatment. Meanwhile, the protein expression of XIAP was found unchanged after treatment with 1e, suggesting that XIAP was not involved in 1e activated caspase-3. The anti-cancer activity of 1e was comparable with other plants derived compounds. For instance, Wang and et al. reported mitochondrion-dependent apoptosis induced by cordycepin at the concentration of 15 μM in H1975 cells.25 Lu and et al. found that treatment with 150 μg mL−1 of sulfated galactoglucan could activate caspase-3 in H1975 cells.30 These results indicated that 1e as a derivative of deacetylisovaltratum obtained from semi-synthesis, achieved superior anticancer activity than the parental compound. Moreover, the anti-proliferation ability was at a similar level of other plants derived compounds.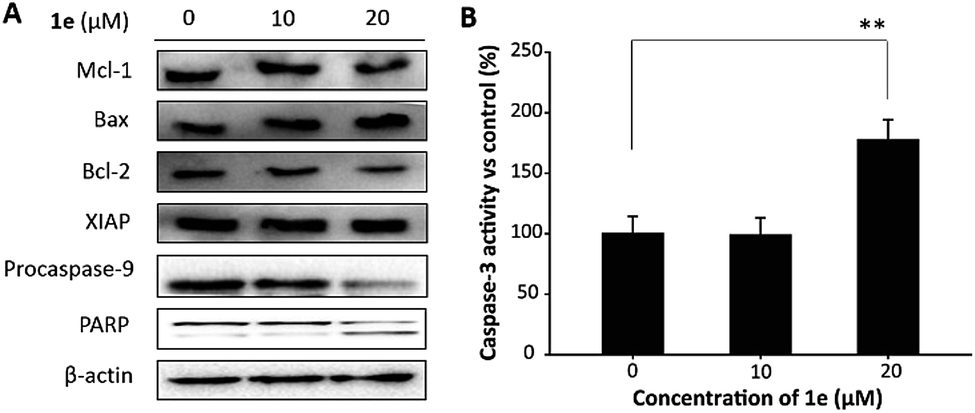 | ||
| Fig. 7 1e triggered apoptosis in H1975 cells. (A) After treated with 1e for 48 h, cells were lysed and proteins were analysed by western blot. (B) After treated with 1e for 48 h, cells were lysed and caspase-3 activity was analysed using Caspase-3 Colorimetric Assay Kit (Biovision, Milpitas, CA, US). | ||
3 Conclusion
In this study, we designed and synthesized of a series of valepotriate derivatives from genipin using slightly modified Murakami's method. Among these derivatives, 1e which contained a novel skeleton with methoxycarbonyl on C4-position and (5-methylhexanoyl)oxy group on C-7 position possessed stronger anticancer activity than its parental compound deacetylisovaltratum. Noteworthy, specific SAR has been found: (1) natural derived group at 1st position is better than others; (2) different substituents at the 7th position can be critical for its bioactivity; (3) the length of aliphatic side chain is crucial to its anticancer activity. Finally, 1e has been found with superior potency than lead compound deacetylisovaltratum. In addition, 1e induced mitochondrion-mediated apoptosis was significantly observed, which might be the action mechanism of its cytotoxic activities, thus enlightening the further development of novel valepotriate derivatives.4 Experimental section
4.1 Chemistry
Proton NMR spectra were obtained on a Bruker AVII 500 or 400 NMR spectrometer (500 or 400 MHz) by use of CDCl3, CD3OD, or DMSO-d6 as solvent. Carbon-13 NMR spectra were obtained a Bruker spectrometer (125 MHz) by use of CDCl3 CD3OD or DMSO-d6 as solvent. Carbon-13 chemical shifts are referenced to the central peak of CDCl3 (d 77.0 ppm) or DMSO-d6 (d 39.5 ppm). Multiplicities are recorded by the following abbreviations: s, singlet; d, double; t, triplet; q, quartet; m, multiplet; J, coupling constant (Hz). ESI-MS spectra were obtained from Shimadzu LCMS-2020 mass spectrometer. Unless otherwise noted, reagents and solvent were obtained from commercial suppliers and without further purification.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford 3 (3.2 g, 84%).1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 5.81 (s, 1H), 4.46 (d, J = 8.1 Hz, 1H), 4.22 (s, 2H), 3.70 (s, 3H), 3.56 (s, 3H), 3.17 (q, J = 8.4 Hz, 1H), 2.85 (ddd, J = 16.5, 8.5, 1.3 Hz, 1H), 2.58 (t, J = 8.0 Hz, 1H), 2.09–2.02 (m, 1H). LC-MS: 241 (M + H).:1) to afford 4 (13.88 g, 94%). 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 0.9 Hz, 1H), 7.33–7.30 (m, 2H), 7.29–7.26 (m, 2H), 7.19 (d, J = 7.3 Hz, 1H), 5.68 (s, 1H), 4.51 (d, J = 7.7 Hz, 1H), 3.71 (s, 3H), 3.62–3.58 (m, 1H), 3.56 (s, 3H), 3.12 (td, J = 8.2, 0.9 Hz, 1H), 2.77 (dd, J = 16.4, 8.5 Hz, 2H), 2.05 (dd, J = 8.2, 1.4 Hz, 1H). LC-MS: 333 (M + H).:1 to PE/EtOAc = 2:1) to afford 5 (1.98 g, 41.3%, 40% recovery of 4). 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.32 (d, J = 7.5 Hz, 2H), 7.29–7.24 (m, 2H), 7.18 (t, J = 7.3 Hz, 1H), 5.71 (s, 1H), 4.85 (d, J = 8.3 Hz, 1H), 3.87 (d, J = 14.4 Hz, 1H), 3.71 (s, 3H), 3.58 (t, J = 6.6 Hz, 1H), 3.12 (q, J = 8.2 Hz, 1H), 2.79 (dd, J = 16.5, 8.6 Hz, 1H), 2.69 (t, J = 7.5 Hz, 1H), 2.03–1.97 (m, 1H), 1.87 (dd, J = 14.8, 6.8 Hz, 1H), 1.76–1.68 (m, 1H). LC-MS: 319 (M + H).:1) to afford 6a (2.8 g, 80%). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 1.1 Hz, 1H), 7.33–7.26 (m, 4H), 7.20 (ddd, J = 7.2, 3.9, 1.3 Hz, 1H), 5.95 (d, J = 6.9 Hz, 1H), 5.67 (s, 1H), 3.76 (d, J = 14.2 Hz, 1H), 3.71 (s, 3H), 3.49–3.44 (m, 1H), 3.19 (dd, J = 11.4, 4.5 Hz, 1H), 2.97 (t, J = 7.3 Hz, 1H), 2.76 (dd, J = 16.7, 8.2 Hz, 1H), 2.31–2.07 (m, 4H), 0.96 (dd, J = 6.6, 4.2 Hz, 6H). LC-MS: 425 (M + Na+).:1) to afford 7 (430 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 7.56–7.53 (m, 1H), 7.42 (d, J = 1.2 Hz, 1H), 7.35 (dd, J = 4.3, 3.1 Hz, 1H), 5.89 (d, J = 6.7 Hz, 1H), 5.43–5.40 (m, 1H), 5.31 (t, J = 1.8 Hz, 1H), 4.48 (t, J = 4.2 Hz, 1H), 4.10 (q, J = 7.1 Hz, 1H), 3.81 (d, J = 12.7 Hz, 3H), 3.72 (s, 3H), 3.25 (d, J = 7.4 Hz, 1H), 2.95–2.89 (m, 1H), 2.31–2.21 (m, 2H), 2.18 (ddd, J = 13.5, 6.7, 4.4 Hz, 1H), 2.12 (dt, J = 13.7, 6.8 Hz, 1H), 2.03 (s, 2H), 1.89 (ddd, J = 13.7, 8.0, 6.0 Hz, 1H), 1.24 (t, J = 7.1 Hz, 2H), 0.96 (dd, J = 6.6, 2.6 Hz, 7H), 0.90–0.78 (m, 1H). LC-MS: 311 (M + H).:1) to afford the target compounds.
:1) to afford 3 (3.2 g, 84%).1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 5.81 (s, 1H), 4.46 (d, J = 8.1 Hz, 1H), 4.22 (s, 2H), 3.70 (s, 3H), 3.56 (s, 3H), 3.17 (q, J = 8.4 Hz, 1H), 2.85 (ddd, J = 16.5, 8.5, 1.3 Hz, 1H), 2.58 (t, J = 8.0 Hz, 1H), 2.09–2.02 (m, 1H). LC-MS: 241 (M + H).:1) to afford 4 (13.88 g, 94%). 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 0.9 Hz, 1H), 7.33–7.30 (m, 2H), 7.29–7.26 (m, 2H), 7.19 (d, J = 7.3 Hz, 1H), 5.68 (s, 1H), 4.51 (d, J = 7.7 Hz, 1H), 3.71 (s, 3H), 3.62–3.58 (m, 1H), 3.56 (s, 3H), 3.12 (td, J = 8.2, 0.9 Hz, 1H), 2.77 (dd, J = 16.4, 8.5 Hz, 2H), 2.05 (dd, J = 8.2, 1.4 Hz, 1H). LC-MS: 333 (M + H).:1 to PE/EtOAc = 2:1) to afford 5 (1.98 g, 41.3%, 40% recovery of 4). 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.32 (d, J = 7.5 Hz, 2H), 7.29–7.24 (m, 2H), 7.18 (t, J = 7.3 Hz, 1H), 5.71 (s, 1H), 4.85 (d, J = 8.3 Hz, 1H), 3.87 (d, J = 14.4 Hz, 1H), 3.71 (s, 3H), 3.58 (t, J = 6.6 Hz, 1H), 3.12 (q, J = 8.2 Hz, 1H), 2.79 (dd, J = 16.5, 8.6 Hz, 1H), 2.69 (t, J = 7.5 Hz, 1H), 2.03–1.97 (m, 1H), 1.87 (dd, J = 14.8, 6.8 Hz, 1H), 1.76–1.68 (m, 1H). LC-MS: 319 (M + H).:1) to afford 6a (2.8 g, 80%). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 1.1 Hz, 1H), 7.33–7.26 (m, 4H), 7.20 (ddd, J = 7.2, 3.9, 1.3 Hz, 1H), 5.95 (d, J = 6.9 Hz, 1H), 5.67 (s, 1H), 3.76 (d, J = 14.2 Hz, 1H), 3.71 (s, 3H), 3.49–3.44 (m, 1H), 3.19 (dd, J = 11.4, 4.5 Hz, 1H), 2.97 (t, J = 7.3 Hz, 1H), 2.76 (dd, J = 16.7, 8.2 Hz, 1H), 2.31–2.07 (m, 4H), 0.96 (dd, J = 6.6, 4.2 Hz, 6H). LC-MS: 425 (M + Na+).:1) to afford 7 (430 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 7.56–7.53 (m, 1H), 7.42 (d, J = 1.2 Hz, 1H), 7.35 (dd, J = 4.3, 3.1 Hz, 1H), 5.89 (d, J = 6.7 Hz, 1H), 5.43–5.40 (m, 1H), 5.31 (t, J = 1.8 Hz, 1H), 4.48 (t, J = 4.2 Hz, 1H), 4.10 (q, J = 7.1 Hz, 1H), 3.81 (d, J = 12.7 Hz, 3H), 3.72 (s, 3H), 3.25 (d, J = 7.4 Hz, 1H), 2.95–2.89 (m, 1H), 2.31–2.21 (m, 2H), 2.18 (ddd, J = 13.5, 6.7, 4.4 Hz, 1H), 2.12 (dt, J = 13.7, 6.8 Hz, 1H), 2.03 (s, 2H), 1.89 (ddd, J = 13.7, 8.0, 6.0 Hz, 1H), 1.24 (t, J = 7.1 Hz, 2H), 0.96 (dd, J = 6.6, 2.6 Hz, 7H), 0.90–0.78 (m, 1H). LC-MS: 311 (M + H).:1) to afford the target compounds.Compound 1a (26.6 mg, 43.2%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 1.0 Hz, 1H), 6.11 (d, J = 5.6 Hz, 1H), 5.03 (dd, J = 7.7, 4.1 Hz, 1H), 3.73 (d, J = 3.1 Hz, 3H), 3.25 (d, J = 7.4 Hz, 1H), 3.06 (d, J = 4.6 Hz, 1H), 2.95 (d, J = 4.6 Hz, 1H), 2.86–2.75 (m, 1H), 2.37 (dd, J = 7.7, 5.6 Hz, 1H), 2.24 (t, J = 6.8 Hz, 2H), 2.10 (t, J = 6.1 Hz, 3H), 2.04–1.98 (m, 1H), 1.89 (dd, J = 14.8, 1.4 Hz, 1H), 0.96 (dd, J = 6.6, 1.8 Hz, 6H), 0.91 (d, J = 6.6 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 170.92, 170.04, 165.64, 150.33, 109.57, 87.30, 76.21, 75.26, 64.40, 50.41, 47.01, 42.25, 42.04, 41.74, 36.07, 29.85, 24.61, 24.59, 21.30, 21.24. LC-MS: 411 (M + H).
Compound 1b (28.5 mg, 51.6%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 1.3 Hz, 1H), 6.12 (d, J = 5.8 Hz, 1H), 5.00 (dd, J = 7.7, 4.4 Hz, 1H), 3.74 (s, 3H), 3.25 (d, J = 7.7 Hz, 1H), 3.05 (d, J = 4.6 Hz, 1H), 2.97 (d, J = 4.6 Hz, 1H), 2.82 (dd, J = 7.8, 7.0 Hz, 1H), 2.59 (s, 3H), 2.34 (dd, J = 7.6, 5.9 Hz, 1H), 2.24 (dd, J = 7.1, 5.9 Hz, 2H), 2.15–2.06 (m, 1H), 1.90–1.83 (m, 1H), 0.96 (dd, J = 6.6, 1.9 Hz, 6H). LC-MS: 369 (M + H).
Compound 1c (27 mg, 41.2%): 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 1.2 Hz, 1H), 6.10 (d, J = 5.3 Hz, 1H), 5.01 (dd, J = 7.6, 3.8 Hz, 1H), 3.73 (s, 3H), 3.25 (dd, J = 13.5, 6.6 Hz, 1H), 3.06 (d, J = 4.6 Hz, 1H), 2.93 (d, J = 4.6 Hz, 1H), 2.80–2.73 (m, 1H), 2.39 (dd, J = 7.6, 5.3 Hz, 1H), 2.23 (dd, J = 15.4, 8.6 Hz, 3H), 2.10 (m, 1H), 1.91 (ddd, J = 14.9, 5.3, 3.9 Hz, 1H), 1.79 (s, 2H), 1.70 (s, 2H), 1.62 (d, J = 9.5 Hz, 1H), 1.57 (d, J = 4.1 Hz, 3H), 1.40–1.31 (m, 2H), 0.96 (dd, J = 6.6, 1.9 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 174.83, 171.08, 166.65, 151.35, 110.59, 88.27, 77.23, 76.24, 65.55, 51.41, 48.01, 43.06, 42.85, 37.02, 30.93, 28.79, 28.75, 25.66, 25.62, 25.35, 25.31, 22.26. LC-MS: 437 (M + H).
Compound 1d (24 mg, 37.2%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.94 (dd, J = 8.3, 1.2 Hz, 2H), 7.56 (s, 1H), 7.49 (d, J = 1.3 Hz, 1H), 7.43 (t, J = 7.8 Hz, 2H), 6.20 (d, J = 5.1 Hz, 1H), 5.30 (s, 1H), 3.67 (s, 3H), 3.33 (dt, J = 7.7, 6.4 Hz, 1H), 3.12 (d, J = 4.5 Hz, 1H), 3.04 (d, J = 4.5 Hz, 1H), 2.90–2.81 (m, 1H), 2.48 (dd, J = 7.6, 5.2 Hz, 1H), 2.25 (t, J = 6.9 Hz, 2H), 2.18–2.07 (m, 2H), 0.96 (dd, J = 6.6, 3.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 171.14, 166.60, 165.41, 151.32, 133.31, 129.67, 128.43, 110.70, 88.25, 77.43, 77.23, 65.72, 51.40, 48.20, 43.07, 37.02, 31.17, 25.65, 22.26. LC-MS: 431 (M + H).
Compound 1e (30 mg, 44.2%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.42 (s, 1H), 6.10 (d, J = 5.3 Hz, 1H), 5.05 (dt, J = 7.5, 3.8 Hz, 1H), 3.73 (d, J = 2.4 Hz, 3H), 3.26 (dd, J = 13.7, 7.3 Hz, 1H), 3.06 (dd, J = 4.5, 2.2 Hz, 1H), 2.91 (d, J = 4.6 Hz, 1H), 2.78 (dtd, J = 10.6, 7.8, 2.9 Hz, 1H), 2.41–2.36 (m, 1H), 2.28–2.20 (m, 2H), 2.20–2.14 (m, 1H), 2.10 (dt, J = 13.6, 6.8 Hz, 1H), 1.93–1.86 (m, 1H), 1.56–1.36 (m, 4H), 1.30–1.25 (m, 2H), 1.21–1.12 (m, 2H), 0.99–0.94 (m, 6H), 0.89–0.78 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 175.15, 175.07, 171.07, 166.64, 151.38, 110.60, 88.29, 88.28, 77.25, 76.28, 76.22, 65.62, 51.42, 48.08, 48.05, 47.35, 47.19, 43.06, 42.88, 37.17, 31.58, 31.51, 31.05, 29.53, 29.52, 25.61, 25.32, 25.27, 22.57, 22.51, 22.27. LC-MS: 453 (M + H).
Compound 1f (30 mg, 47.2%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 1.3 Hz, 1H), 6.11 (d, J = 5.6 Hz, 1H), 5.02 (dd, J = 7.7, 4.2 Hz, 1H), 3.74 (s, 3H), 3.25 (d, J = 7.6 Hz, 1H), 3.05 (d, J = 4.6 Hz, 1H), 2.95 (dd, J = 9.8, 5.2 Hz, 2H), 2.85–2.75 (m, 1H), 2.36 (dd, J = 7.7, 5.7 Hz, 1H), 2.26–2.19 (m, 4H), 2.14–2.06 (m, 1H), 1.87 (ddd, J = 14.8, 5.9, 4.3 Hz, 1H), 1.68 (s, 1H), 1.57–1.52 (m, 2H), 1.34 (dd, J = 7.4, 3.6 Hz, 2H), 0.96 (dd, J = 6.6, 2.0 Hz, 6H), 0.89–0.86 (m, 3H). LC-MS: 425 (M + H).
Compound 1g (15 mg, 22.1%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 1.1 Hz, 1H), 6.11 (d, J = 5.6 Hz, 1H), 5.01 (dd, J = 7.7, 4.2 Hz, 1H), 3.74 (s, 3H), 3.25 (d, J = 7.5 Hz, 1H), 3.05 (d, J = 4.6 Hz, 1H), 2.95 (d, J = 4.7 Hz, 2H), 2.85–2.76 (m, 1H), 2.36 (dd, J = 7.6, 5.8 Hz, 1H), 2.28–2.18 (m, 4H), 2.14–2.07 (m, 1H), 1.87 (ddd, J = 14.8, 5.8, 4.4 Hz, 1H), 1.67 (td, J = 14.5, 7.0 Hz, 1H), 1.58–1.50 (m, 2H), 1.25 (d, J = 4.0 Hz, 7H), 0.96 (dd, J = 6.6, 2.0 Hz, 6H), 0.87 (t, J = 6.9 Hz, 3H). LC-MS: 453 (M + H).
Compound 1h (18 mg, 25.0%): colorless oil 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 1.1 Hz, 1H), 6.11 (d, J = 5.7 Hz, 1H), 5.01 (dd, J = 7.7, 4.2 Hz, 1H), 3.73 (s, 3H), 3.29–3.19 (m, 1H), 3.05 (d, J = 4.6 Hz, 1H), 2.95 (d, J = 4.6 Hz, 1H), 2.81 (dt, J = 15.4, 7.9 Hz, 1H), 2.36 (dd, J = 7.6, 5.8 Hz, 1H), 2.26–2.19 (m, 4H), 2.10 (dt, J = 13.6, 6.8 Hz, 1H), 1.87 (ddd, J = 14.8, 5.8, 4.4 Hz, 1H), 1.57–1.50 (m, 2H), 1.25 (s, 12H), 0.96 (dd, J = 6.6, 2.0 Hz, 6H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 172.67, 171.06, 166.68, 151.33, 110.64, 88.36, 77.23, 76.28, 65.38, 51.45, 47.97, 43.08, 42.74, 37.10, 34.25, 31.85, 30.82, 29.40, 29.25, 29.22, 29.09, 25.62, 24.79, 22.66, 22.27, 22.25, 14.10. LC-MS: 481 (M + H).
4.2 Pharmacological assay
RPMI-1640 medium and fetal bovine serum (FBS) were purchased from Gibco, BRL (Grand Island, NY, USA). Human pancreatic cancer cell lines (SW1990, BXPC3, CAPAN2, CFPAC, PANC1), human breast cancer cell lines (BT474, MCF7, MDA-MB-231), human lung cancer cell line (H1975) and human gastric cancer cell lines (AGC, HGC-27, KATOIII) were purchased from Shanghai Institutes for Biological Sciences, CAS (Shanghai, China). DAPI was purchased from Sigma-Aldrich (St Louis, MO, US). The Annexin V-FITC Apoptosis Kit was purchased from BD (Franklin Lakes, NJ, US). The primary antibodies against Bax, Bcl-2, Mcl-1, procaspase-9, poly-ADP-ribose polymerase (PARP), XIAP and β-actin were purchased from Abcam Inc. (Cambridge, MA, US). Caspase-3 Colorimetric Assay Kit was purchased from Biovision (Milpitas, CA, US). Human lung cancer cell line H1975 was purchased from Shanghai Institutes for Biological Sciences, CAS (Shanghai, China). Cells were cultured with RPMI-1640 medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C, 5% CO2 humidified atmosphere. 1e was dissolved in DMSO at the concentration of 100 mM.Acknowledgements
This work was supported by Hangzhou Major Science and Technology Project (20112312A01, Shenglin Ma; 20172016A01, Nengming Lin), National Natural Science Foundation of China (81603144, Bo Zhang), Natural Science Foundation of Zhejiang Province (LQ15H310001, Bo Zhang), Scientific Research Foundation of Zhejiang Health and Family Planning Commission (2015KYA177, Bo Zhang), Fundamental Research Funds for the Central Universities (2016QNA7030, Xiaoyang Dai), Science and Technology Project of Zhejiang Province (2016C33067, Xiaowu Dong).References
- Y. Fukuyama, Y. Minoshima, Y. Ishimoto, I. S. Chen, H. Takahashi and T. Esumi, J. Nat. Prod., 2004, 67, 1833–1838 CrossRef CAS PubMed
.
- W. Hui-lian, Z. Dong-fang, L. Zhao-feng, L. Yang, L. i. Qian-rong and W. Yu-zhen, Toxicol. Appl. Pharmacol., 2003, 188, 36–41 CrossRef PubMed
- C. Bounthanh, C. Bergmann, J. P. Beck, M. Haagberrurier and R. Anton, Planta Med., 1981, 41, 21–28 CrossRef CAS PubMed
- N. T. Doan, F. Crestey, C. E. Olsen and S. B. Christensen, J. Nat. Prod., 2015, 78, 1406–1414 CrossRef CAS PubMed
- L. D. Kapoor, Handbook of Ayurvedic medicinal plants, CRC Press, Boca Raton, 2001 Search PubMed
- B. P. Mikhova, N. V. Handjieva, S. S. Popov and S. L. Spassov, J. Nat. Prod., 1987, 50, 1141–1145 CrossRef CAS
- H. Becker, S. Chavadej, P. W. Thies and E. Finner, Planta Med., 1984, 50, 245–248 CrossRef CAS PubMed
- W. Kucaba, P. W. Thies and E. Finner, Phytochemistry, 1980, 19, 575–577 CrossRef CAS
- N. Handjieva and V. G. Zaikin, Planta Med., 1978, 34, 203–206 CrossRef
- B. M. Dietz, A. Hajirahimkhan, T. L. Dunlap and J. L. Bolton, Pharmacol. Rev., 2016, 68, 1026–1073 CrossRef PubMed
- X. Li, T. Chen, S. Lin, J. Zhao, P. Chen, Q. Ba, H. Guo, Y. Liu, J. Li, R. Chu, L. Shan, W. Zhang and H. Wang, Curr. Cancer Drug Targets, 2013, 13, 472–483 CrossRef CAS PubMed
- R. Bos, H. Hendriks, J. J. C. Scheffer and H. J. Woerdenbag, Phytomedicine, 1998, 5, 219–225 CrossRef CAS PubMed
- S. Lin, T. Chen, P. Fu, J. Ye, X.-W. Yang, L. Shan, H. L. Li, R. H. Liu, Y. H. Shen, X. K. Xu and W. D. Zhang, J. Asian Nat. Prod. Res., 2015, 17, 455–461 CrossRef CAS PubMed
- O. Kelber, T. Wegener, B. Steinhoff, C. Staiger, J. Wiesner, W. Knoess and K. Kraft, Phytomedicine, 2014, 21, 1124–1129 CrossRef PubMed
- D. Zhang, B. Zhang, L. X. Zhou, J. Zhao, Y. Y. Yan, Y. L. Li, J. M. Zeng, L. L. Wang, B. Yang and N. M. Lin, Acta Pharmacol. Sin., 2016, 37, 1597–1605 CrossRef CAS PubMed
- B. Yang, N. Li, Y. Q. Wang, J. Chen and R. S. Zhang, Rev. Bras. Farmacogn., 2011, 21, 471–476 CrossRef CAS
- S. Tamura, K. Fujiwara, N. Shimizu, S. Todo and N. Murakami, Bioorg. Med. Chem., 2010, 18, 5975–5980 CrossRef CAS PubMed
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed
- X. W. Dong, J. Chen, C. Y. Jiang, T. Liu and Y. Z. Hu, Arch. Pharm., 2009, 342, 428–432 CrossRef CAS PubMed
- H. Jing, X. L. Zhou, X. W. Dong, J. Cao, H. Zhu, J. S. Lou, Y. Z. Hu, Q. J. He and B. Yang, Cancer Lett., 2010, 294, 167–177 CrossRef CAS PubMed
- K. B. Sharpless and R. F. Lauer, J. Am. Chem. Soc., 1973, 95, 2697–2699 CrossRef CAS
- S. Tamura, K. Fujiwara, N. Shimizu, S. Todo and N. Murakami, Bioorg. Med. Chem., 2010, 18, 5975–5980 CrossRef CAS PubMed
- S. Lin, P. Fu, T. Chen, J. Ye, X. W. Yang and W. D. Zhang, J. Asian Nat. Prod. Res., 2017, 19, 15–21 CrossRef CAS PubMed
- X. Li, X. X. Fan, Z. B. Jiang, W. T. Loo, X. J. Yao, E. L. Leung, L. W. Chow and L. Liu, Pharmacol. Res., 2017, 115, 45–55 CrossRef CAS PubMed
- Z. Wang, X. Wu, Y. N. Liang, L. Wang, Z. X. Song, J. L. Liu and Z. S. Tang, Molecules, 2016, 21(10), 1267 CrossRef PubMed
- L. H. Li, P. Wu, J. Y. Lee, P. R. Li, W. Y. Hsieh, C. C. Ho, C. L. Ho, W. J. Chen, C. C. Wang, M. Y. Yen, S. M. Yang and H. W. Chen, PLoS One, 2014, 9, e104203 Search PubMed
- N. Khan, F. Jajeh, M. I. Khan, E. Mukhtar, S. M. Shabana and H. Mukhtar, Carcinogenesis, 2017, 38, 184–195 Search PubMed
- Y. Du, W. Chen, X. Fu, H. Deng and J. Deng, RSC Adv., 2016, 6, 109718–109725 RSC
- S. K. Jung, M. H. Lee, D. Y. Lim, J. E. Kim, P. Singh, S. Y. Lee, C. H. Jeong, T. G. Lim, H. Chen, Y. I. Chi, J. K. Kundu, N. H. Lee, C. C. Lee, Y. Y. Cho, A. M. Bode, K. W. Lee and Z. Dong, J. Biol. Chem., 2014, 289, 35839–35848 CrossRef CAS PubMed
- M. K. Lu, T. Y. Lin, C. H. Hu, C. H. Chao, C. C. Chang and H. Y. Hsu, Carbohydr. Polym., 2017, 167, 229–239 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of the synthesized compounds. See DOI: 10.1039/c6ra27478a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |