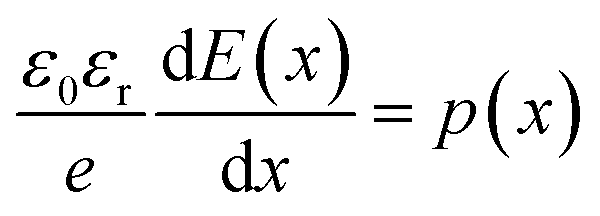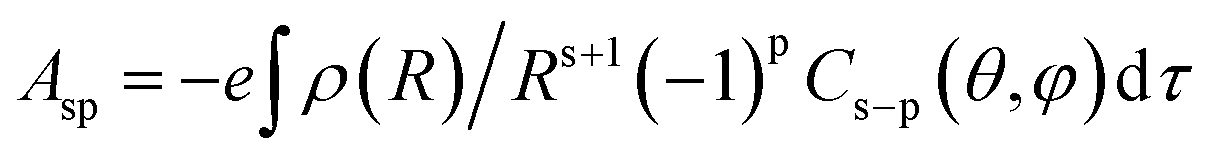DOI:
10.1039/C6RA27473K
(Paper)
RSC Adv., 2017,
7, 18239-18251
Near infrared organic light emitting devices based on a new erbium(III) β-diketonate complex: synthesis and optoelectronic investigations†
Received
28th November 2016
, Accepted 9th March 2017
First published on 27th March 2017
Abstract
An in situ reaction of two optoelectronically active organic ligands (anionic thenoyltrifluoroacetylacetone, tta−, and neutral triphenylphosphine oxide, tppo) with erbium(III) ion in the presence of a base yielded a new erbium complex, [Er(tta)3(tppo)]. The solid and solution structure of the complex was established by X-ray crystallography, NMR, ESI-MS, FTIR, TGA and Raman spectroscopy. They indicate that the Er(III) ion is coordinated to seven oxygen atoms of three tta ligands and one tppo ligand in a monocapped octahedral geometry. The Judd–Ofelt parameters of the complex were determined by a least squares fitting and dealt with its chemical structure. On UV excitation through ligand mediation, the complex shows the characteristic near-infrared emission of the corresponding Er(III) ion at 1534 nm. Furthermore, a near infra-red organic light emitting diode (NIR-OLED) was fabricated with structure: ITO/[N,N′-bis(naphthalen-2-yl)-N,N′-bis(phenyl)benzidine]/[Er(tta)3(tppo)]/[2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)]/LiF/Al. This device, with maximum applied voltage of 23 V, shows a total quenching of visible emission and electroluminescence in the C-band region (1534 nm) which is suitable for third communication window applications in fiber optics. Finally, an organic diode was fabricated to determine charge carrier mobility of the complex using a steady-state method.
Introduction
The processability and attractive luminescent properties of the lanthanide (Ln) β-diketonate complexes, like high luminescence efficiency, narrow emission bands and long luminescence lifetimes, means they find various optoelectronic applications such as organic light emitting devices (OLEDs),1 optical communication systems,2 night-vision readable displays,3 telecommunications and polymeric planar waveguides.4,1b Moreover, these complexes possess flexible coordination geometries which in turn largely affect their optoelectronic properties. Recently, chemists have used various combinations of different β-diketonates and ancillary/synergic ligands together with Ln ions to achieve coordination geometries that could lead to highly luminescent complexes of interest for visible and near infrared.5 As a result, Ln complexes with asymmetric geometries like trigonal-dodecahedron and monocapped square-antiprism were found more luminescent as compared to the symmetric ones because they facilitate the radiative transitions in the emission process.5,6 Besides them, asymmetric geometries like monocapped octahedron or monocapped trigonal prism of seven-coordinate Ln complexes are also very interesting because they are anticipated to produce highly efficient Ln complexes.5a,6f Moreover, an insight into the local structure and bonding in the vicinity of the Ln ion in such complexes can be obtained by the investigation of the Judd–Ofelt parameters (Ωt=2,4,6).7 Importantly, the Ln complexes with low symmetry structure are less reported and their development could lead to a significant contribution in the optoelectronic industry.
Among the sensitizers of Ln ions, β-diketones are the most versatile ligands which is owing to (i) their strong π–π* electronic transitions in the UV region, (ii) their strong coordination ability and (iii) the possibility of functionalization. Generally, their combination with Ln ion and O-/N-donor synergic/ancillary ligands leads to formation of fully saturated complexes leaving no space, in the vicinity of Ln ion, for high-energy O–H/C–H oscillators of solvent molecules.5,6 This, therefore, reinforces the thermal and chemical properties of their Ln complexes which are very critical for optoelectronic applications. The bidentate ancillary ligands in such complexes deliver symmetric coordination geometries (8- and 10-coordinate) which are relatively less supportive of radiative transitions in the emission process.8 It points toward the use of monodentate ligands that produce asymmetric Ln coordination geometries which are expected to give strong emission efficiency.
The O-donor triphenylphosphine oxide is a very versatile ligand because it possesses tunable excited energy states and produces strong Ln–ligand bonds. Recently, the Ln complexes of this ligand, particularly those of europium and terbium, have been exploited as regards visible light photoluminescence.5a,9 However, its Ln complexes for NIR photoluminescence (PL) and electroluminescence (EL) applications, particularly with erbium, are rarely reported. For instance, two monometallic (eight- and nine-coordinate) and one heterometallic (eight-coordinate) complexes of Er with tppo and β-diketones have been reported.10,11 The authors used perfluorinated β-diketones to avoid high energy C–H oscillators in the vicinity of the Er ion in order to get efficient emission. In contrast, we have carried out a reaction in which fluorinated β-diketone, tppo and ErCl3 in an appropriate ratio have been used to get an asymmetric seven-coordinate Er complex that is anticipated to be highly luminescent. The PL and EL of erbium are very interesting because it emits at 1.54 μm that corresponds to the amplification range of erbium-doped fiber amplifiers. The development of new erbium(III) complexes and optimization of their 1.54 μm NIR emission open up new possibilities in the fabrication of efficient photonic integrated circuits.12
In this article, a new erbium(III) complex, tris(thenoyltrifluoroacetylacetonate)mono(triphenylphosphine oxide), or [Er(tta)3(tppo)], was synthesized by an in situ method and characterized thoroughly. Single crystal X-ray diffraction and 2D-NMR spectroscopy indicate that the complex is a seven-coordinate structure arranged in a distorted monocapped octahedral geometry. It is a highly asymmetric structure that suggests efficient energy transfer from coordinated ligands to Er ion. The steady-state NIR PL spectra of the pure complex and its PMMA-doped sample were studied and are presented. The Judd–Ofelt parameters (Ωt) of the complex were calculated from spectroscopic measurements that give an insight into local structure and bonding around the Er ion. Furthermore, a NIR-OLED based on the present complex was fabricated with structure: ITO/[N,N′-bis(naphthalen-2-yl)-N,N′-bis(phenyl)benzidine] (30 nm)/[Er(tta)3(tppo)] (40 nm)/[2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)] (30 nm)/LiF (0.1 nm)/Al (100 nm). Finally, an organic diode was fabricated to determine charge carrier mobility in this novel complex by using a Mott–Gurney model.
Experimental
Materials and methods
The commercially available chemical that was used without further purification was Er2O3 (99.9%) from Aldrich. The oxide was converted to the corresponding chloride ErCl3·nH2O (n = 6–7). The tta and tppo ligands were purchased from Merck. The solvents used in this study were either AR or spectroscopic grade.
The elemental analyses were performed using Flash EA equipment (Thermo Electron, Model 1112). The infrared spectra were recorded with a PerkinElmer Spectrum 2 spectrophotometer in the range 4000–400 cm−1. The NMR spectra of the ligands and the complex in CDCl3 were recorded with a Bruker AVANCE II 400 NMR spectrometer, Department of Chemistry, PUC-Rio. Cyclic voltammetry (CV) was conducted with a COMPACTSTAT.e using the conventional three-electrode configuration consisting of a graphite working electrode, a platinum wire auxiliary electrode, and an Ag/AgCl reference electrode. The cyclic voltammograms were obtained in CH2Cl2 using tetrabutylammonium hexafluorophosphate (NBu4PF6) (0.1 M) as the supporting electrolyte at scan rate of 0.05 V s−1. Ferrocene/ferricenium (Fc/Fc+) was used as internal reference during the measurement. Raman spectroscopy was performed using a micro-Raman microscope (Horiba, model XploRA), at an excitation wavelength of 638 nm. The laser excitation beam was focused onto crystalline samples with an intensity of 25 W mm−2 by a 10× objective lens. The scan was performed using an acquisition time of 5 s with 20 accumulations with a range between 50 and 1900 cm−1 and a spectral resolution of 4 cm−1. Mass spectrometry measurement was obtained using a mass spectrometer (Thermo Scientific, model Orbitrap XL) with electrospray ionization (ESI-MS) in positive mode. The operating parameters of the ionization source were: sprayer voltage: 3 kV, capillary voltage: 42 V, capillary temperature: 275 °C, desolvation temperature: 350 °C, sheath gas flow (nitrogen): 9 (arbitrary unit). The electronic spectrum of the complex was recorded with a HP Hewlett Packard 8452 A diode array spectrophotometer, with the sample contained in a 1 cm3 stoppered quartz cell of 1 cm path length in the concentration range between 1 × 10−5 and 1 × 10−3 M. PL spectra were measured using a Model Quanta Master spectrometer with a 150 W xenon lamp as the excitation source and InGaAs photomultiplier tube as detector. The current–luminance–voltage properties were measured by using a Keithley source measurement unit (Keithley 2400) and Newport Powermeter Model 1936-C.
Single crystal X-ray diffraction
The single crystal X-ray diffraction data of the complex were collected at room temperature using a Bruker D8 Venture diffractometer equipped with Photon 100 CMOS detector and using Mo Kα radiation (0.71073 Å) from an INCOATEC micro-focus source. Final lattice parameter values and integrated intensities were obtained using SAINT software (Bruker, 2015),13 and a multi-scan absorption correction was applied with SADABS.14 The structure was solved by direct methods using Intrinsic Phasing implemented in SHELXT.15 The model was refined applying the full-matrix least-squares method using SHELXL-12 (ref. 15) with OLEX2 software.16 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed at calculated positions and refined using a riding model. Crystal data and details of measurement are listed in Tables 1 and 2.†
Table 1 Crystal data and structure refinement for [Er(tta)3(tppo)] complex
| Empirical formula |
C42H27ErF9O7PS3 |
| Formula weight |
1109.04 |
| Temperature |
293(2) K |
| Wavelength |
0.71073 Å |
| Crystal system |
Trigonal |
| Space group |
P31c |
| Unit cell dimensions |
a = 14.9084(4) Å, α = 90° |
| b = 14.9084(4) Å, β = 90° |
| c = 13.0146(3) Å, γ = 120° |
| Volume |
2505.09(15) Å3 |
| Z |
2 |
| Density (calculated) |
1.470 Mg m−3 |
| Absorption coefficient |
1.908 mm−1 |
| F(000) |
1094 |
| Crystal size |
0.37 × 0.18 × 0.13 mm3 |
| Theta range for data collection |
2.222 to 26.394° |
| Index ranges |
−18 ≤ h ≤ 18, −18 ≤ k ≤ 18, −16 ≤ l ≤ 16 |
| Reflections collected |
25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 604 604 |
| Independent reflections [R(int) = 0.0548] |
3437 [R(int) = 0.0548] |
| Completeness to theta = 25.242° |
99.9% |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3437/37/218 |
| Goodness-of-fit on F2 |
1.060 |
| Final R indices [I > 2sigma(I)] |
R1 = 0.0301, wR2 = 0.0705 |
| R Indices (all data) |
R1 = 0.0425, wR2 = 0.0752 |
| Absolute structure parameter |
−0.005(7) |
| Largest diff. peak and hole/e Å−3 |
0.617 and −0.371 |
Table 2 Selected bond lengths and bond angles of [Er(tta)3(tppo)] complexa
| Bond length/Å |
Bond angle/° |
| Symmetry transformation coordinates; (#1): −x + y − 1, −x − 1, z; (#2): − y − 1, x − y, z′′. |
| Er(1)–O1 |
2.231(7) |
O(1)–Er(1)–O(1A)#1 |
80.88(11) |
| Er(1)–O(1A)#1 |
2.284(4) |
O(1)–Er(1)–O(1A) |
80.88(11) |
| Er(1)–O(1A) |
2.284(4) |
O(1A)#1–Er(1)–O(1A) |
117.54(6) |
| Er(1)–O(1A)#2 |
2.284(4) |
O(1)–Er(1)–O(1A)#2 |
80.88(11) |
| Er(1)–O(2A)#2 |
2.284(4) |
O(1A)#1–Er(1)–O(1A)#2 |
117.54(6) |
| Er(1)–O2A |
2.284(4) |
O(1A)–Er(1)–O(1A)#2 |
117.54(6) |
| Er(1)–O(2A)#2 |
2.284(4) |
O(1)–Er(1)–O(2A)#2 |
132.74(13) |
| |
|
O(1A)#1–Er(1)–O(2A)#2 |
146.36(17) |
| |
|
O(1A)–Er(1)–O(2A)#2 |
76.14(16) |
| |
|
O(1A)#2–Er(1)–O(2A)#2 |
74.32(17) |
| |
|
O(1)–Er(1)–O(2A) |
132.74(13) |
| |
|
O(1A)#1–Er(1)–O(2A) |
76.14(16) |
| |
|
O(1A)–Er(1)–O(2A) |
74.32(17) |
| |
|
O(1A)#2–Er(1)–O(2A) |
146.36(17) |
| |
|
O(2A)#2–Er(1)–O(2A) |
79.0(2) |
| |
|
O(1)–Er(1)–O(2A)#1 |
132.74(13) |
| |
|
O(1A)#1–Er(1)–O(2A)#1 |
74.32(17) |
| |
|
O(1A)–Er(1)–O(2A)#1 |
146.36(17) |
| |
|
O(1A)#2–Er(1)–O(2A)#1 |
76.14(16) |
| |
|
O(2A)#2–Er(1)–O(2A)#1 |
79.0(2) |
| |
|
O(2A)–Er(1)–O(2A)#1 |
79.0(2) |
Device fabrication
The OLEDs were assembled using heterojunctions containing a double- and triple-layer structure using β-NPB (N,N′-bis(naphthalen-2-yl)-N,N′-bis(phenyl)benzidine) as a hole-transporting layer, the Er complex as an emitting layer, and TPBi (2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)) as a hole blocker. TcTa (tris(4-carbazoyl-9-ylphenyl)amine) and BSB (4,4′-bistriphenylsilanylbiphenyl) were used as the organic matrix to promote the energy transfer to the Er complex. Finally, a 100 nm thick aluminum film was deposited as cathode onto a 0.1 Å layer of LiF. For the multilayered device, the emitting layer was prepared by co-evaporation of TcTa (matrix) and the Er complex (dopant) from two individual thermal sources. It was followed by another co-evaporated layer of the complex doped into BSB as a second matrix. The concentration of the complex in both the matrixes is taken as the same.
All the different layers of the device were sequentially deposited in high-vacuum environment by thermal evaporation onto ITO substrates with a sheet resistance of 15 Ω square−1. The ITO substrate was initially cleaned by ultrasonification using a detergent solution followed by toluene degreasing and then cleaned again by ultrasonification with pure isopropyl alcohol. Finally, the substrate was dried using nitrogen gas. The base pressure was 6.6 × 10−5 Pa, whereas during the evaporation process the pressure was ∼10−4 Pa. The deposition rates for organic compounds were in the range of 0.3 Å s−1 to 2 Å s−1. The layer thickness was controlled in situ through a quartz crystal monitor and confirmed with a profilometer measurement. The fabricated device had an active area of around 2 mm2 and operated in forward bias voltage with ITO as the positive electrode and Al as the negative one.
Synthesis of complex
An in situ method was adopted for the synthesis of the complex, [Er(tta)3(tppo)]. An ethanol solution of Htta (1.32 g, 6 mmol) was added to 6 mL aqueous solution of 1 N NaOH (1.33 g, 6 mmol). This mixture was kept in a 50 mL beaker and stirred for 1/2 h. It gave Na(tta), sodium salt of thenoyltrifluoroacetylacetone, which was mixed with 15 mL ethanol solution each of ErCl3·6H2O (0.76 g, 2.00 mmol) and tppo (0.41 g, 2 mmol). The molar ratio between ErCl3·6H2O, Htta and tppo was taken as 1:3:1. The reaction mixture was stirred for 7 h at room temperature. During stirring a white precipitate was formed, and it was repeatedly filtered off. The filtrate was concentrated and left to slowly evaporate at room temperature. After three days, pink-coloured crystals appeared which were washed with ethanol and chloroform, and then recrystallized from an ethanol/hexane solution.
[Er(tta)3(tppo)], pink colour. Yield (82%). Elemental analysis: calcd for ErC42H27F9O7S3P1: C, 45.44; H, 2.54; N, 6.24%. Found: C, 45.82; H, 2.63; N, 6.32%. 1H NMR (400 MHz, CDCl3, 300 K; δ, ppm): 17.83 (br, 6H, tppo), 10.57 (br, 3H, tppo), 8.84 (d, 6H, tppo), 7.41 (d, 3H, tta), 6.11 (s, 3H, tta), 3.78 (br, 3H, tta) and −16.12 (s, 3H, tta, C–H). FTIR data (cm−1): 3062 (w), 1602 (vs), 1541 (s), 1509 (w), 1440 (s), 1413 (s), 1357 (s), 1310 (vs), 1249 (v), 1165 (s), 1064 (w), 1020 (w), 789 (w), 724 (s), 541 (s). Melting point 137 °C.
Results and discussion
Synthesis and characterization
The complex [Er(tta)3(tppo)] was synthesized by in situ reaction in which ErCl3·6H2O, Htta, NaOH and tppo were taken in the ratio of 1:3:3:1 (Scheme 1). The reaction gave the complex in good yield (82%). The complex is resistant to air and moisture, and highly soluble in common organic solvents.
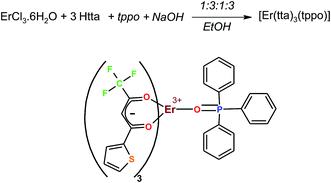 |
| | Scheme 1 Synthesis of the complex. | |
Thermogravimetric analysis
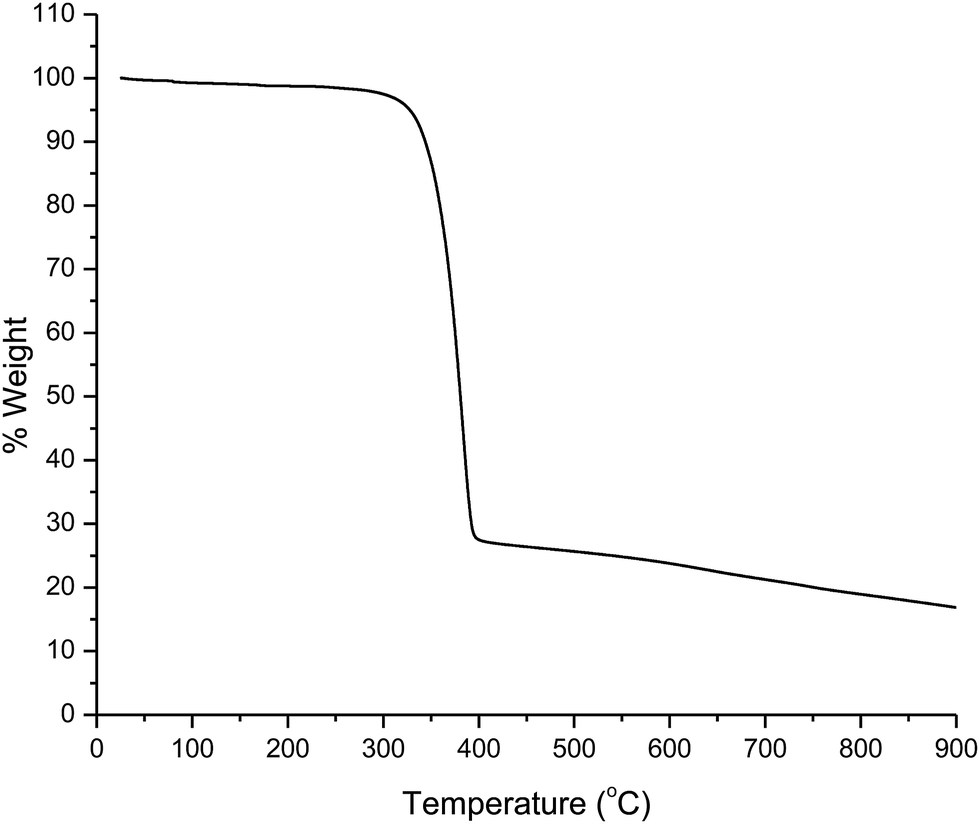
The thermogram of the complex was recorded under an N2 atmosphere. The analysis shows that the melting point of the complex is 137 °C and the decomposition temperature is 280 °C (Fig. 1). This result suggests that the complex has good thermal stability. This may be due to the presence of trifluoromethyl (–CF3) group on the tta ligand which leads to an improvement in thermal and oxidative stabilities, and volatility of the complex.17,18 These characteristics improve film-forming ability of the complex that facilitates its use for electroluminescent devices.
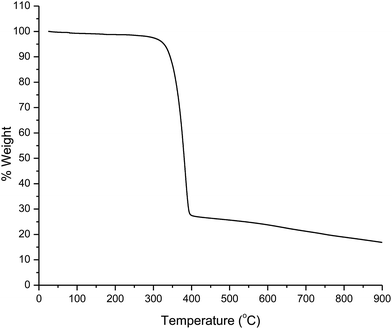 |
| | Fig. 1 TGA curve of the complex. | |
Raman spectroscopy
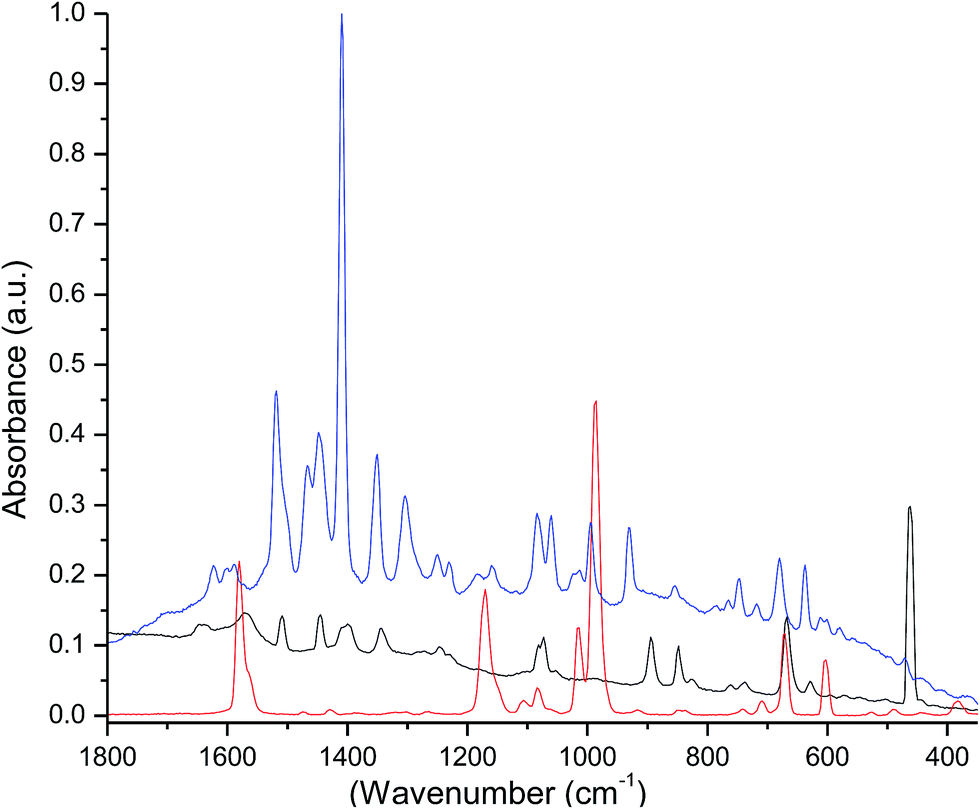
The Raman spectra of the free ligands and the complex were recorded in the solid state (Fig. 2). Their analyses were done by the direct comparison between free ligands, the complex and the respective lanthanide β-diketonate complexes.19 The spectrum of the free tppo shows bands at 1014 and 988 cm−1 that can be attributed to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations, and the bands at 600 and 671 cm−1 can be assigned to P–C6H5 vibrations.20 These bands in the spectrum of the complex have low intensity and are slightly shifted to higher frequencies relative to free tppo. This perturbation in PO bond vibrations indicates the complexation of tppo ligand to Er ion. Moreover, the multiple peaks between 1650 and 1500 cm−1 (OC–CC(str) + CF3(str)) could be attributed to the coordinated β-diketonate (tta).21
O vibrations, and the bands at 600 and 671 cm−1 can be assigned to P–C6H5 vibrations.20 These bands in the spectrum of the complex have low intensity and are slightly shifted to higher frequencies relative to free tppo. This perturbation in PO bond vibrations indicates the complexation of tppo ligand to Er ion. Moreover, the multiple peaks between 1650 and 1500 cm−1 (OC–CC(str) + CF3(str)) could be attributed to the coordinated β-diketonate (tta).21
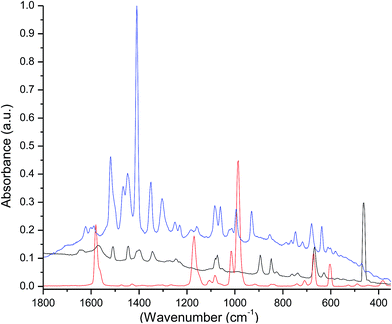 |
| | Fig. 2 Raman spectra of Htta (black), tppo (red) and the complex (blue). | |
FTIR spectroscopy
The FTIR spectrum of the complex was recorded in the solid state (Fig. S1 in ESI†) and compared with those of free ligands (tta and tppo). The spectrum of the uncoordinated tppo ligand exhibits a very strong band with a maximum at 1160 cm−1 (POstr).9 While in the spectrum of the complex, this stretching mode appears at lower frequency with very much less intensity. The shift and relatively low intensity of the (POstr) band suggest the interaction/coordination of tppo ligand to the Er ion. Moreover, the vibration bands appearing in the region of 700–400 cm−1 correspond to v(Er–Otta) and v(Er–Otppo) bands.22 This is also indicative of evidence about the existence of coordination bonds between erbium and tta, and erbium and tppo.
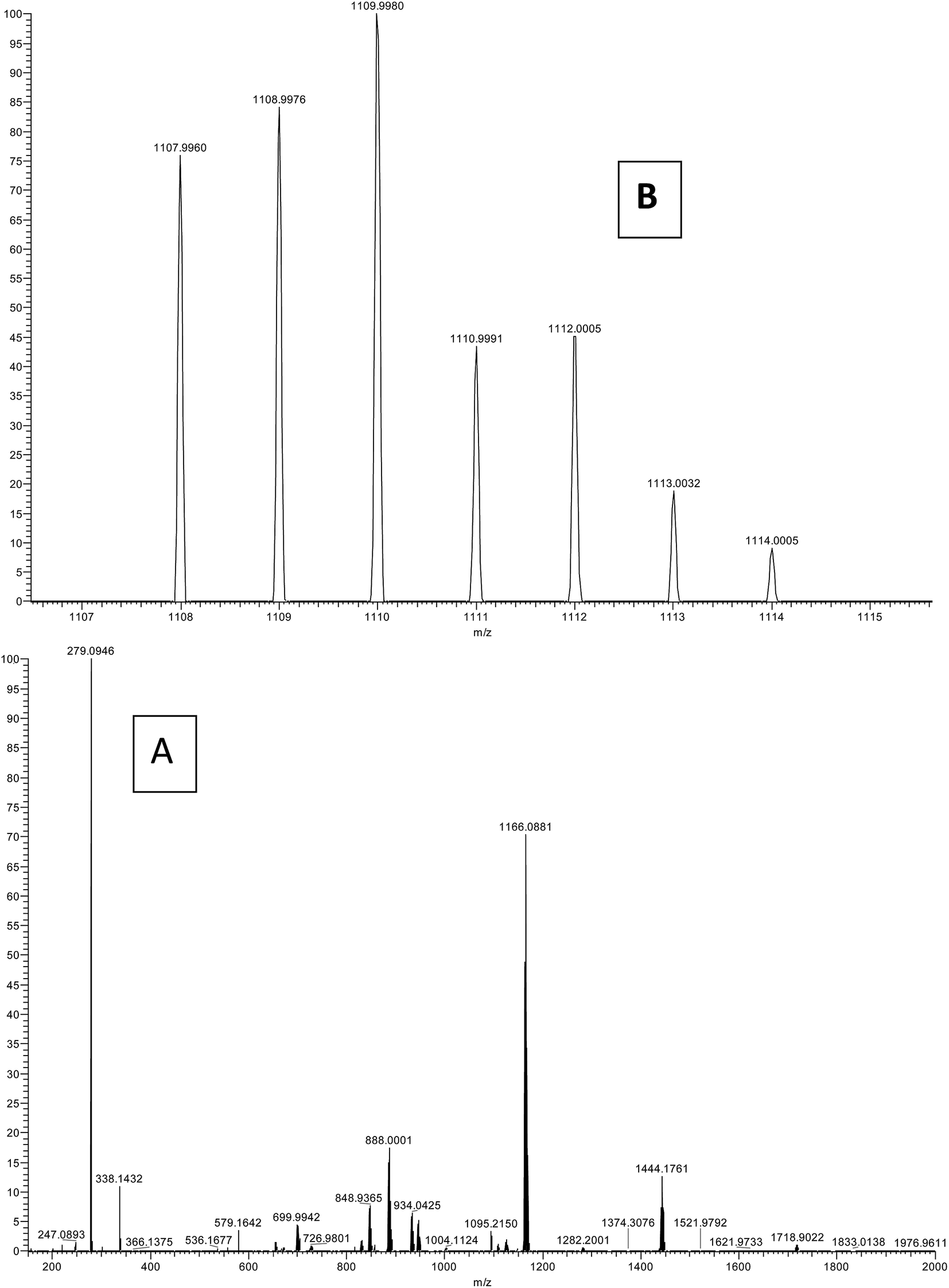
Mass spectroscopy
The ESI-MS spectrum in positive mode for the present complex was obtained in methanol solution (Fig. 3). The assignments are made on the basis of calculated m/z ratios. The spectrum of the complex displays a strong peak at m/z = 1166.088 which is assigned to [Er(tta)2(tppo)2]+. Moreover, the spectrum also shows a molecular ion peak observed at m/z = 1109.99 corresponding to [Er(tta)3(tppo) + H+]+. The other peaks observed in the spectrum may be accounted for by the diverse ionic species formed due to fragmentation, ligand exchange and redistribution.23
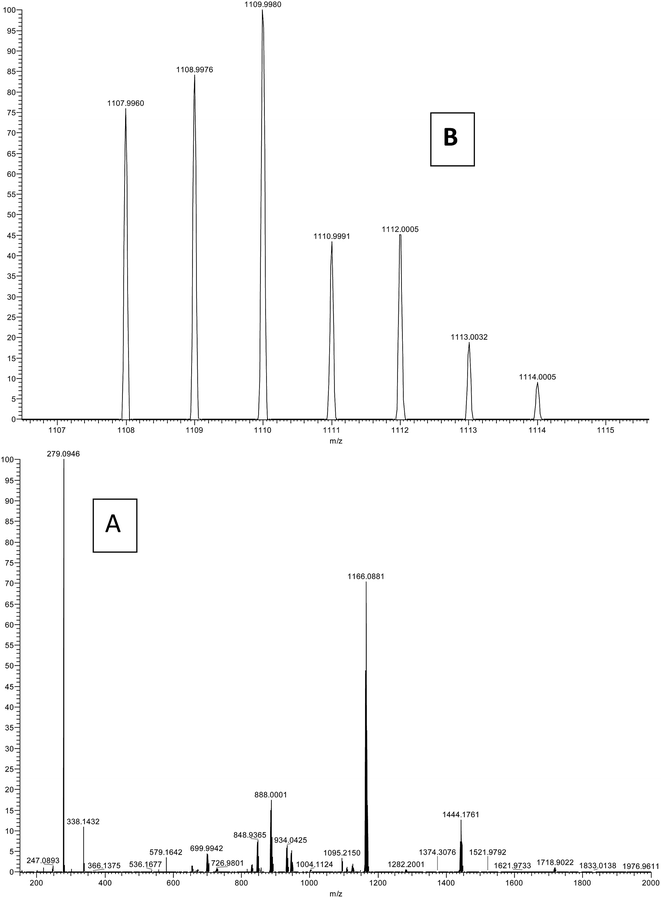 |
| | Fig. 3 ESI-MS spectrum of the complex: (A) full spectrum and (B) selected range. | |
Single crystal X-ray crystallography
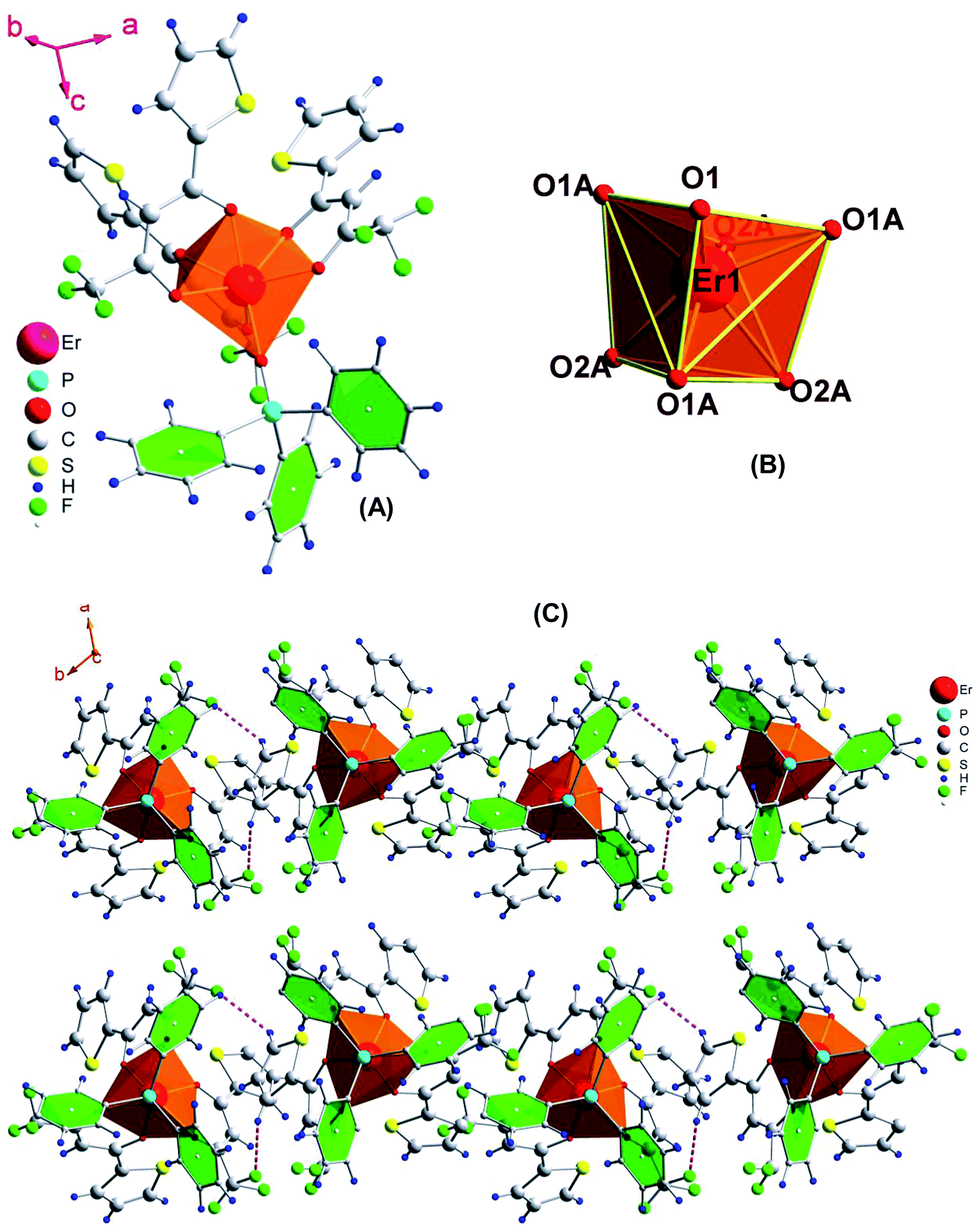
The slow evaporation of a chloroform solution of the complex gave very good crystals which were suitable for single crystal X-ray analysis (details are given in Tables 1 and 2). The unit cell consists of two units of the complex with the same formula, C42H27ErF9O7PS3, which crystallizes in the trigonal space group P31c. Furthermore, their distances (Er⋯Er) in the crystal packing are 10.79 Å. It accounts for the enhanced luminescence from Ln complexes since the energy migration between metal requires a gap less than 8.00 Å.24 The complex possesses a monocapped octahedral geometry where the Er(III) ion is coordinated to one O-atom of tppo and six O-atoms of three tta moieties, making itself a seven-coordinate structure (Fig. 4). Recently, a seven-coordinate europium β-diketonate complex with the same geometrical structure has been reported.5a It is a highly asymmetric geometry which is supportive of radiative transitions in the emission process leading to an efficient emission. The present complex shows H–F interactions occurring between the fluorine atoms of the thenoyl ring of β-diketone and the hydrogen atoms of the phenyl rings of tppo ligand. Besides this, no other interaction (π–π stacking) is found in the complex, which could account for the low melting point of the complex. Interestingly, no water molecules either coordinated or non-coordinated were found in the first coordination sphere of the complex which is a beneficial factor for achieving an enhanced emission from the complex. The average bond lengths of Er–O(tta)average and Er–O(tppo) in the present complex are 2.284(4) and 2.231(7) Å, respectively, which are in perfect agreement with those reported for [Re(CO)3Cl(μ-bppz)Er(tta)3] (Er–O(tta)average = 2.272 Å)25 and [Er(PM)3(tppo)2] (Er–O(tppo)average = 2.2920 Å)26 (bppz = 2,3-bis(2-pyridyl)pyrazine and PM = 1-phenyl-3-methyl-4-isobutyryl-5-pyrazolone).
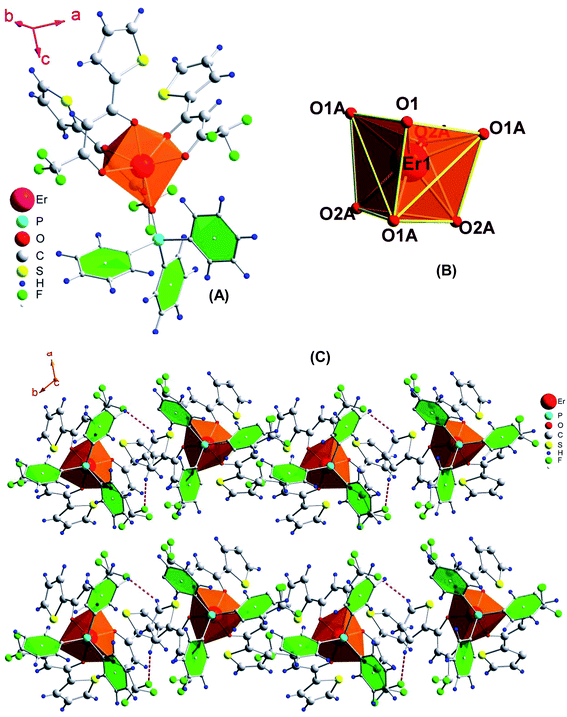 |
| | Fig. 4 (A) Molecular structure of the complex, (B) coordination geometry around Er and (C) molecular packing showing H⋯F interactions. Thermal ellipsoids are drawn at the 30% probability level. | |
1H NMR and 1H–1H COSY spectroscopy
The 1H NMR and 1H–1H COSY spectra of the complex were recorded in CDCl3 at room temperature (Fig. 5a and S2 in ESI†). The spectra of free ligands (tta and tppo) were recorded in CDCl3 and are given only for comparison (Fig. 5b and c). The assignment of the resonances of the signals is based on 2D NMR 1H–1H COSY spectroscopy.
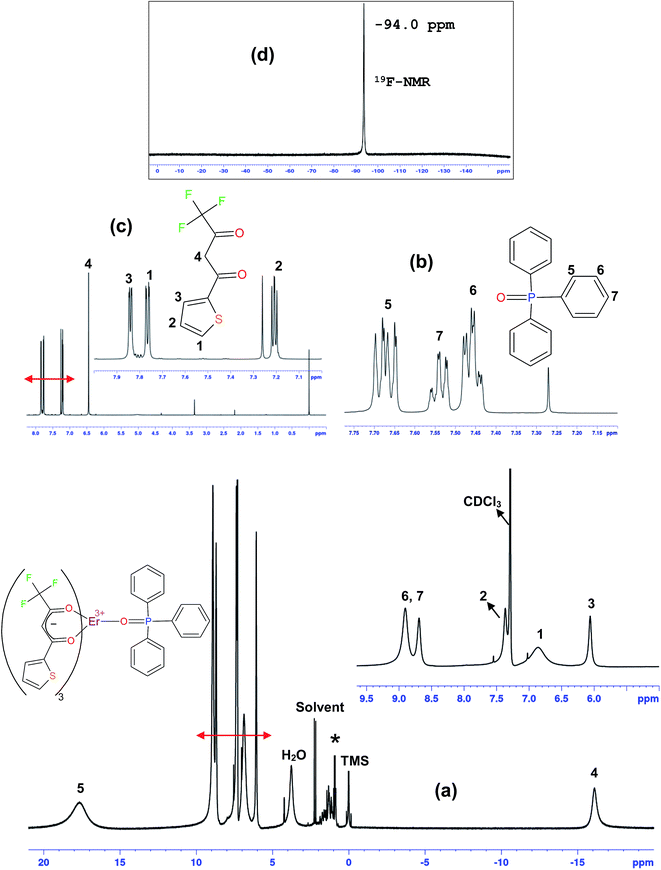 |
| | Fig. 5 1H NMR spectra of the complex (a), tppo (b), and Htta (c) and 19F NMR spectrum of the complex (d). Insets: high resolution. | |
The NMR spectrum of the Er complex is very interesting because the Er(III) ion is paramagnetic and induces sizable downfield/upfield shifts of protons of coordinated diamagnetic ligands. In the COSY spectrum, the complex shows correlations between protons 2 and 3, and the signals 6 and 7. Besides this, no correlation was found for other signals. The protons 4 of tta, being closer to metal ion, show highest upfield shift and appear at −16.01 ppm (δ). In contrast, protons 5 of tppo, being in close proximity to metal ion, show the highest downfield shift and appear at 18.02 ppm (δ). This downfield/upfield shift of protons of the coordinated ligands in opposite directions substantiates that these shifts are dipolar in nature. On the other hand, the spectrum of the complex displays 7 signals which are assigned to 27 protons; 4 signals due to tta equivalent to 12 protons and 3 signals due to tppo equivalent to 15 protons, confirming the tta/tppo ratio of 3:1. It substantiates the coordination of three tta and one tppo to Er(III) ion making the complex a seven-coordinate structure. Moreover, the spectrum covers a wide range of chemical shifts (−17 to +18 ppm (δ)) which are in good agreement with those reported for other ternary Ln(III) β-diketone complexes.5b,5c,6a
19F-NMR
The 19F-NMR spectrum of the complex was recorded in CDCl3 at room temperature (Fig. 5d). It shows only one strong signal at −94.0 ppm due to –CF3 groups of the tta ligands which is in good agreement with that reported for other Er(III) complexes.27
UV-visible and photoluminescence properties
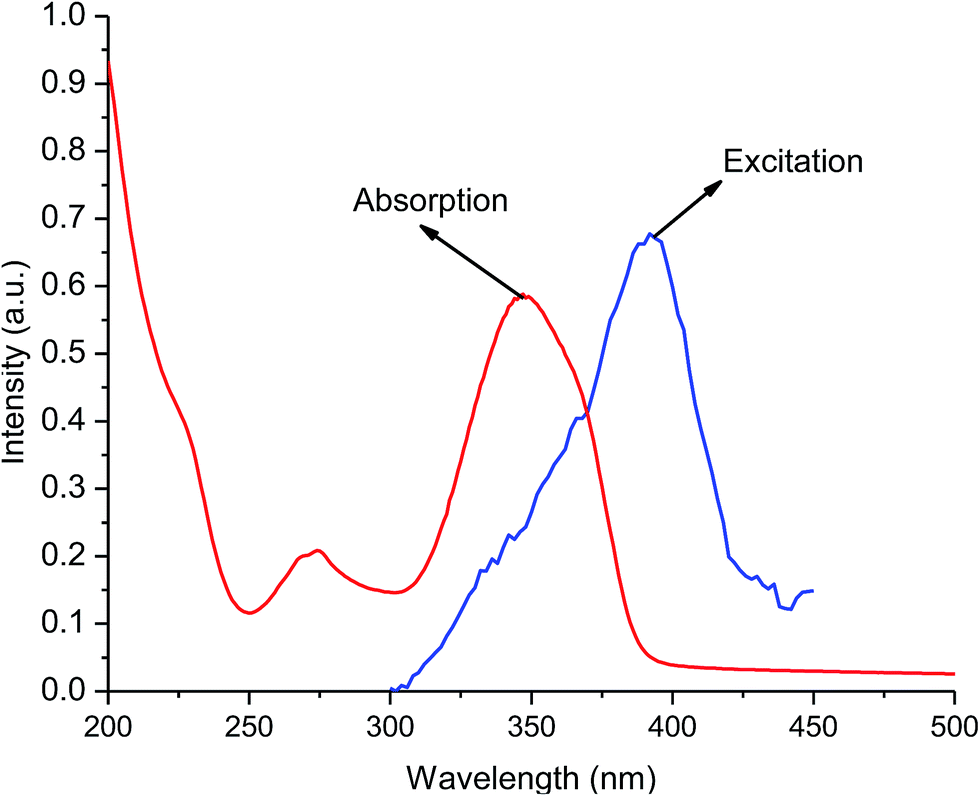
The UV-visible absorption spectra of free ligands (tta and tppo, 5 × 10−5 M) show singlet–singlet 1π–π* electronic transitions with their maxima below 280 nm. In contrast, the spectrum of the complex shows a broad absorption band at maximum wavelength, λmax = 349 nm. This shift to longer wavelength (red-shift) is suggestive of the coordination/interaction of tta and tppo ligands to the Er(III) ion. The absorption spectrum of the complex shows good overlap with of its corresponding excitation spectrum in the 200–450 nm region indicating that energy transfer occurs from the ligands to the Er ion (Fig. 6). Moreover, the excitation band of the complex and the absorption band of tta show good overlap which indicates that the tta efficiently transfers energy to Er ion.
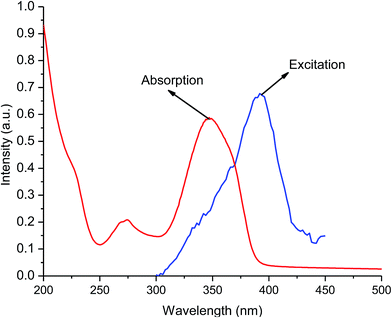 |
| | Fig. 6 Excitation and absorption spectra of the complex. | |
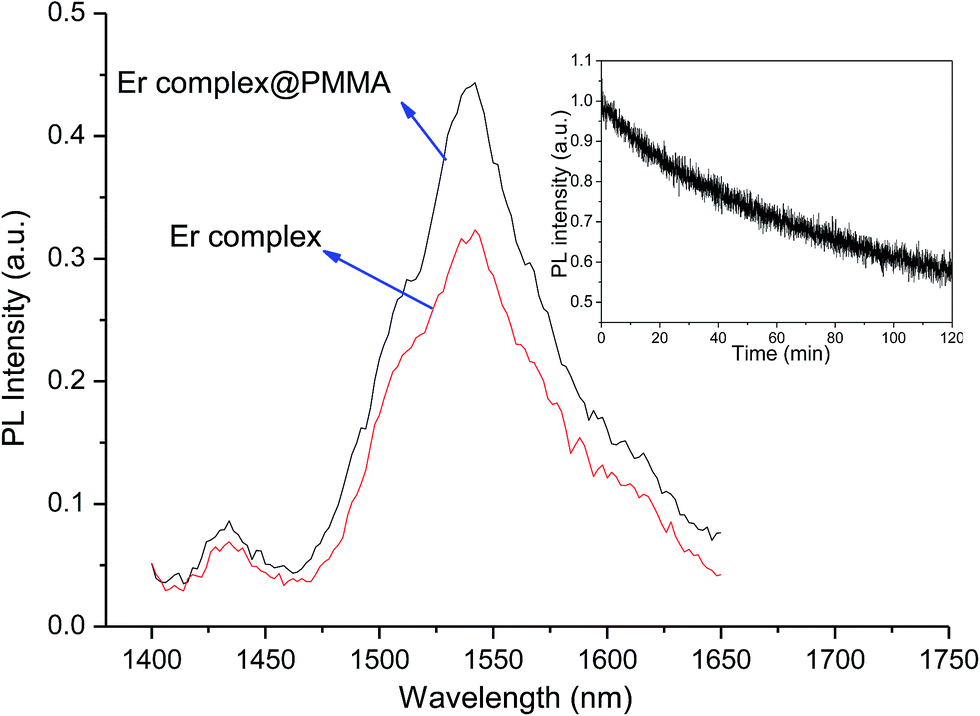
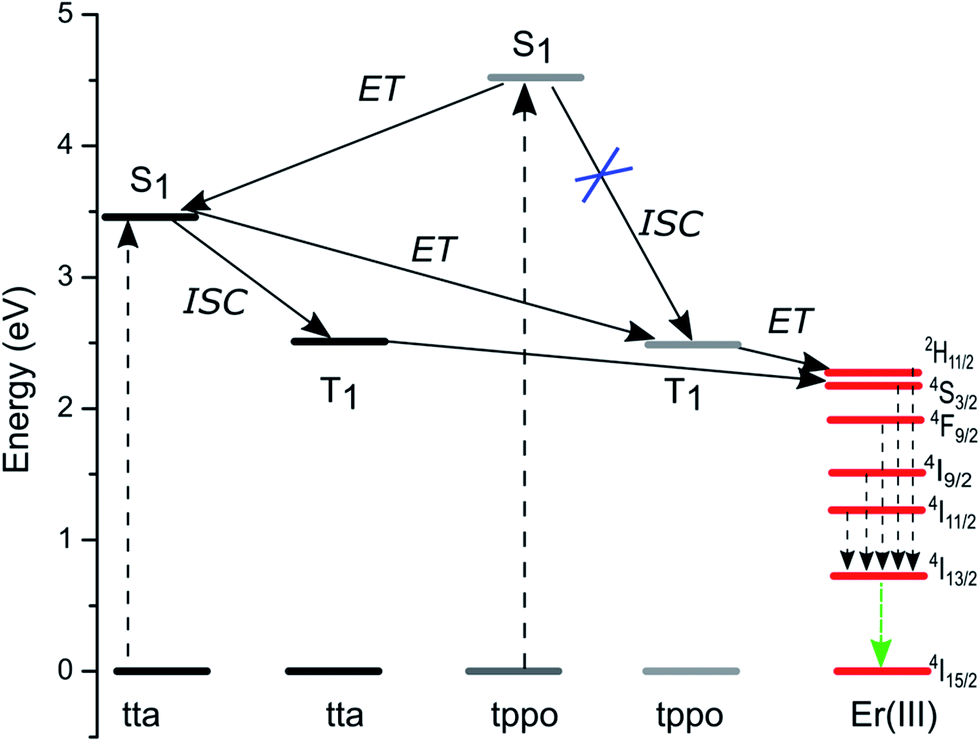
The emission spectrum of the complex was recorded in chloroform at room temperature. Upon the direct excitation of the ligands at 380 nm, the spectrum shows a peak at 1534 nm that covers a large spectral range from 1440 to 1645 nm. It is attributed to a typical 4I13/2 → 4I15/2 transition of Er(III) ion (Fig. 7). On the other hand, the singlet/triplet states of tta9,28 and tppo29 ligands are 25164/18954 and 36376/18954 cm−1, respectively, with energy differences ΔE (S1−T1) of 6982 and 17422 cm−1. The singlet/triplet states of the tta and tppo ligands were also calculated from emission spectra of the present Er complex recorded in the visible region at low and room temperatures (Fig. S3 in ESI†). The observed values (tta, 501 nm; tppo, 519 nm) are in good agreement with those reported in the literature.9,27,28 The ΔE (S1−T1) between these ligands suggests that the energy gap is more appropriate for the tta ligand since a ΔE (S1−T1) of 5000 cm−1 is generally required for efficient intersystem crossing relaxation.30 To understand the energy transfer processes in the complex, a mechanism is proposed (Fig. 8) which suggests that the tta ligand is first excited to its S1 level and then energy transfer occurs to its T1 level via intersystem crossing (ISC). At the same time, some of energy transfer takes place from S1 of tta to T1 of tppo since they lie very close to each other. Thereafter, the T1 states of tta and tppo transfer the energy to upper levels of Er(III) ion via resonant energy transfer (RET).31 The populated levels (4S3/2, 4F9/2, 4I9/2 and 4I11/2) of Er(III) ion relax the energy to lower level, 4I13/2, which upon efficient nonradiative decay gives rise to the sensitized emission at 1534 nm.
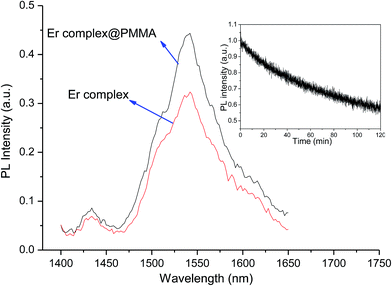 |
| | Fig. 7 NIR emission spectra of the complex in pure form and PMMA-doped sample, under excitation (λmax = 380 nm). Inset: photostability curve of the complex. | |
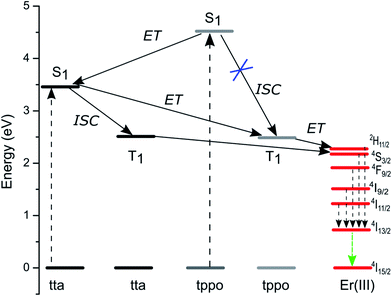 |
| | Fig. 8 Proposed energy level diagram showing Er ion excited state and the singlet/triplet states of tta and tppo sensitizers, and the energy transfer processes. | |
It is well established that polymers doped with Ln β-diketonate complexes possess enhanced electrical, optical and thermal properties since such complexes are well processable and compatible with the polymer matrixes.31 In this study, the present complex was doped into PMMA matrix and its emission spectrum was recorded and compared (Fig. 7). The spectrum of doped sample shows an enhancement in the emission intensity of 4I13/2 → 4I15/2 transition as compared to that observed for the pure complex. Furthermore, the full width at half-maximum (fwhm) of the 4I13/2 → 4I15/2 transition of doped sample is 67 nm which is higher than that observed for pure complex (52 nm). The larger fwhm for the doped sample is in agreement with other reported Er(III)-doped materials.32 It suggests that they can be used in a wide gain bandwidth for optical-amplification applications.
Photostability
To evaluate the photostability behaviour, a thin solid film of the complex was exposed to UV light for 7200 s by using a 150 W xenon lamp as excitation source under the following experimental conditions: sensitivity, 30 mV; time constant, 1 s; excitation slit, 8 nm; and emission slit, 12 nm. Upon excitation at 380 nm, the emission intensity decay curve of the complex shows photodegradation (Fig. 7, inset). Importantly, a very slow decay is observed which indicates that the complex has very good photostability. The determined decay time constant (τ) is 18 and the emission intensity of the complex remaining after 7200 s is 72%. The presence of high π-electron density of three aromatic rings in the tppo ligand accounts for the high photostability of the present complex.
Cyclic voltammetry and NIR-OLED properties
The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) levels of the complex were determined to understand its redox behaviour/carrier injection properties. The complex exhibits one irreversible oxidation peak at 1.55 V (Fig. S4 in ESI†). While it shows two irreversible reduction peaks; one at −1.50 V which is followed by a quasi-reversible redox couple with potential of −1.70 V. These peaks arise from the ligands, and no metal-centered redox processes are apparent.33 The CV of the tta ligand shows two broad irreversible peaks with potentials of −1.24 and −1.70 V.33 These peaks in the present complex are shifted to more positive potentials relative to free tta and, therefore, correspond to the reduction of tta. It is attributed to the irreversible reduction of orbitals localized on the tta ligand. In contrast, the peak at +1.55 V could be assigned to oxidation of tppo ligand. In summary, these redox processes can be attributed to the irreversible reduction of the tta ligands followed by the successive irreversible oxidation of the tppo ligand. The onset voltage was determined from the intersection point of the two tangents drawn at the rising and background currents. The HOMO and LUMO levels were estimated by the following equation:34
| EHOMO = − (1.4 ± 0.1) × (qVCV) − (4.6 ± 0.08) eV. |
The ELUMO was determined by subtracting the singlet energy gap (Eg) from the EHOMO level. The determined HOMO and LUMO levels for the complex are −2.66 and −5.92 eV.
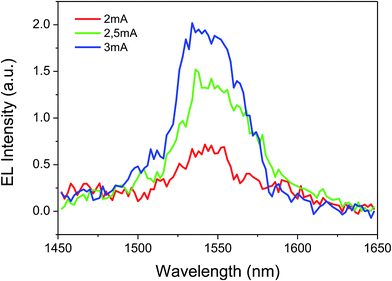
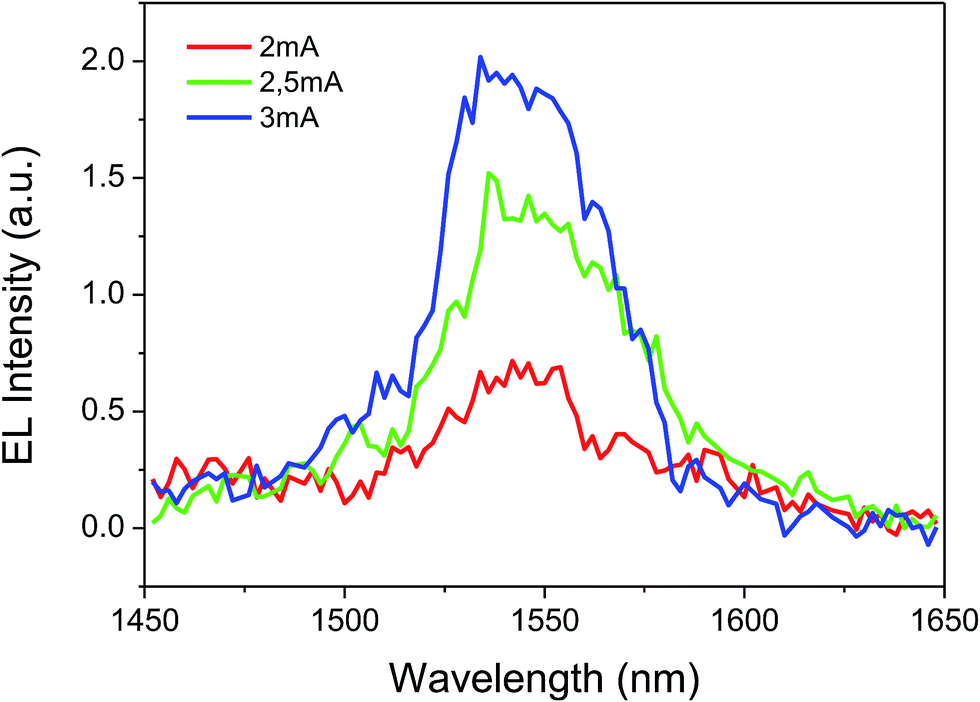
The complex shows good film-forming properties such as good volatility and transparency, light weight and easy thermal evaporation. These characteristics prompted us to use the complex in OLED fabrication. Subsequently, a triple-layered device was fabricated with the structure: ITO/β-NPB (30 nm)/[Er(tta)3(tppo)] (40 nm)/TPBi (30 nm)/LiF (0.1 nm)/Al (100 nm) (Fig. 9). The device shows EL emission at 1534 nm, measured at 2, 2.5 and 3 mA driving currents corresponding to 4I13/2 → 4I15/2 transition of the Er(III) ion (Fig. 10).
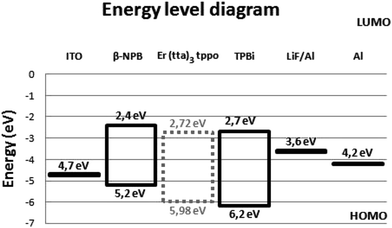 |
| | Fig. 9 Energy level diagram of device with the structure: ITO/β-NPB (30 nm)/[Er(tta)3(tppo)] (40 nm)/TPBi (30 nm)/LiF (0.1 nm)/Al (100 nm). | |
 |
| | Fig. 10 Electroluminescence spectra of the complex. | |
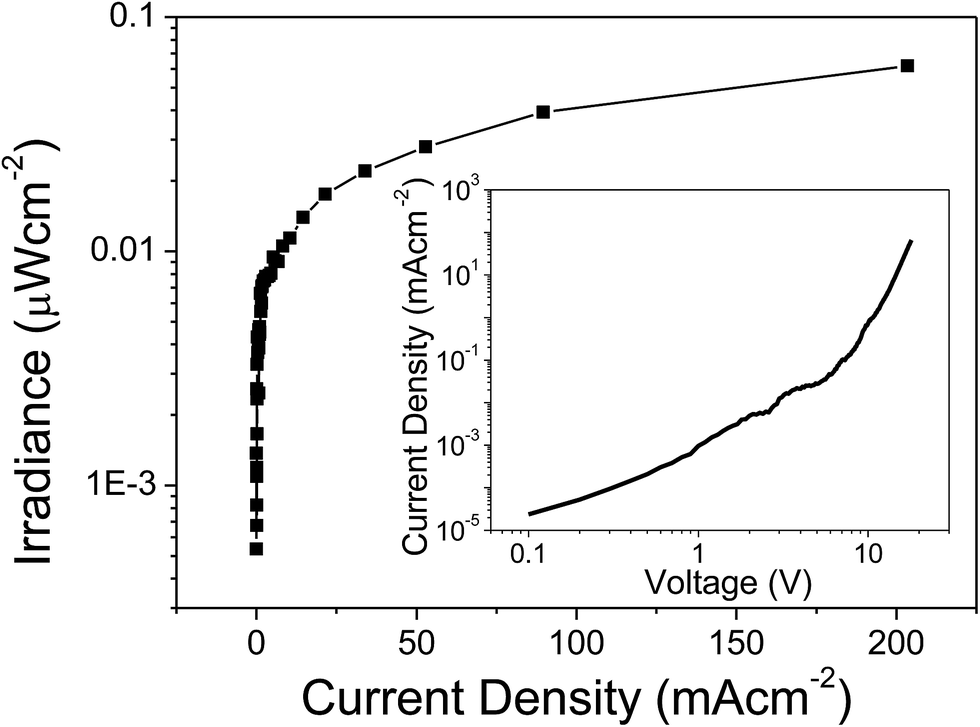
The EL emission spectrum of the device matches very well with the PL spectrum, except for a broadening at higher wavelength. This could be related to an increase in the temperature of the material, which in turn leads to the electronic population redistribution in the excited 4I13/2 multiplet. Moreover, the device did not show any ligand-associated EL emission in the visible region indicating an efficient charge transfer from the organic ligands to the Er(III) ion, i.e. exciton-harvesting processes by the ligands.27 The threshold voltage (Von) of the device was 14 V and it gave a maximum irradiance of 0.069 μW cm−2 with a current density (J) of 129 mA cm−2 at 23 V (Fig. 11). The device was resistive up to an applied voltage of 24 V. Beyond this voltage the device started to degrade. The present device shows higher EL efficiency as compared to those reported for [Er(tfac)3(bath)]-, [Er(tfac)3(5NO2phen)]- and [Er(tfac)3(bipy)]-based devices with Von values of 4, 11 and 6 V respectively.27 However, the Von value obtained for the present device is slightly higher than those for the above reported Er complex-based devices. This is owing to the use of LiF/Al electrode instead of Ca electrode. Moreover, Von of the present complex-based is lower than that reported for an ErQ-based device35 (tfac = 1,1,1-trifluoro-2,4-pentanedione, bath = bathophenanthroline, 5NO2phen = 5-nitro-1,10-phenanthroline, bipy = 2,2′-bipyridine and Q = 8-hydroxyquinoline).
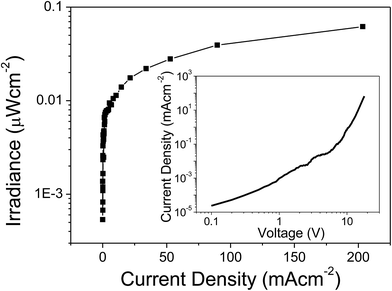 |
| | Fig. 11 Irradiance–current density curve of the complex. Inset: current density–voltage (J–V) curve. | |
Charge carrier mobility
The charge carrier mobility of an organic semiconducting material is a key parameter with which the potential application of the material can be assessed. Recently, various methods/models have been used to obtain this parameter from the current density–voltage response.36 The space charge limited current (SCLC) is one of the most convenient techniques because it reproduces the relevant conditions to OLED applications such that a 100 nm thin film of a device is sufficient to determine the mobility inside it. In this study, the Mott–Gurney eqn (1) was used to determine the carrier mobility in the present complex:37| |
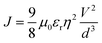 | (1) |
This equation is analytically derived from the Poisson and continuity eqn (2) and (3). This derivation assumes that the mobility μ is constant within the device and the device boundaries at x = 0 and x = L, E = 0 and 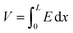 , respectively.
, respectively.
| |
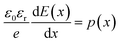 | (2) |
here
J is the steady-state current and a function of the applied voltage
V,
μ0 the steady-state charge-carrier mobility,
d the film thickness,
εr the permittivity of the material,
p(
x) the charge density,
E(
x) the electric field,
e the electronic charge and
ε0 the permittivity of free space.
The μ0 value of the complex was determined by using a single layer device which is fabricated with the following structure: ITO/[Er complex]/Al. The J–V curve of the complex gave μ0 of 6.44 × 10−9 cm2 V−1 s−1 (<5 V). It is comparable to that reported for the closely related complex [Er(tfnb)3bipy] (8.8 × 10−9 cm2 V−1 s−1) (tfnb = 4,4,4-trifluoro-1-(2-naphthyl)-1,3-butanedione; bipy = 2,2′-bipyridine).38 This is a sizable carrier mobility that could be due to the presence of electron-transporting tppo ligand. The μ0 value of the present complex represents total carrier mobility, which is due to the movement of both electrons as well as holes. The hole injection barrier between ITO (workfunction, ω = 4.7 eV) and the complex (ω = 5.98 eV) is very small, i.e. 1.28 eV. Similarly, the electron injection barrier between Al (ω = 4.2 eV) and the complex is 1.78 eV. This suggests an equal probability of hole and election injection into the device.
Oscillator strength and Judd–Ofelt parameter analysis of [Er(tta)3(tppo)]
The intensity of the absorption band can be expressed in terms of a quantity called oscillator strength, Pos. Experimentally it is related to the integrated area of the absorption band and can be expressed in terms of absorption coefficient ε(ν) and the energy of the transition “ν” (cm−1) as given in the following equation:39| |
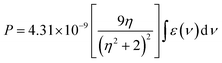 | (4) |
where η is the refractive index of the solution and ε(ν) is the molar extinction coefficient at wavelength ν. On the other hand, the Judd–Ofelt parameters, Ωt (t = 2, 4, 6), are obtained by least-squares fitting of the electric dipole contributions of Pos. In a selectively measured absorption band the magnetic dipole transitions do not contribute considerably to total Pos value.
The spectrum of [Er(tta)3(tppo)] shows eight transitions as given in Table 3. Among them, the most intense transitions are 4G11/2 (Er-VIII) and 2H11/2 (Er-IV) and are classified as hypersensitive transitions.40 Usually, the Ω2 value is closely related to the hypersensitive transitions, i.e. the larger the Pos of the hypersensitive transition is, the greater the Ω2 value. The Pos values of these hypersensitive transitions are 17- and 8-fold higher than that of Er(III) aqua-ion. Moreover, Pos of these transitions of the present complex in chloroform are higher than those reported for [Er(acac)3(H2O)] (17.61)7 and [Er(acac)3phen] (22.61)7 in methanol. The higher Pos suggests a highly asymmetric coordination environment around the Er ion in the present complex as compared to the above mentioned Er complexes.
Table 3 Oscillator strength and Judd–Ofelt parameters of [Er(tta)3(tppo)]
| S′L′J′ |
Transitions Er3+ (←4I15/2) |
Spectral ranges (cm−1) |
Er3+ aqua-ion (P × 106) |
Oscillator strength (P × 106) |
Ωt=2,4,6 (× 10−20 cm2) |
| Present complex. [Er(acac)3(H2O)]7. [Er(acac)3phen]7. |
| 4I11/2 |
Er-I |
9809–10657 |
0.19 |
0.91a, (—)b, [0.17]c |
Ω2 = 34.31a, Ω2 = 25.71b, Ω2 = 32.28c |
| 4F9/2 |
Er-II |
14657–15707 |
1.94 |
1.93a, (1.17)b, [1.07]c |
| 4S3/2 |
Er-III |
17958–18515 |
0.41 |
0.61a, (0.25)b, [0.35]c |
Ω4 = 1.45a, Ω4 = 0.95b, Ω4 = 0.91c |
| 2H11/2 |
Er-IV |
18603–19711 |
2.91 |
24.12a, (17.61)b, [22.67]c |
| 4F7/2 |
Er-V |
19923–21080 |
2.22 |
2.10a, (1.07)b, [1.54]c |
| 4F3/2, F5/2 |
Er-VI |
21765–24941 |
1.10 |
0.50a, (0.43)b, [0.36]c |
Ω6 = 1.85a, Ω6 = 0.75b, Ω6 = 0.81c |
| 2H9/2 |
Er–VII |
24100–24941 |
0.51 |
0.81a, (0.29)b, [0.45]c |
| 4G11/2 |
Er-VIII |
25610–26802 |
5.91 |
102.36a, (63.12)b, [74.98]c |
Effect of chemical environment on Ω2
The Ω2 value is very sensitive to the asymmetry of the crystal field and charge of covalence between metal ion and ligand atoms in the complex. It is largely affected by crystal field parameter Asp.41 The relation between them is given as:| | |
Ω2α(2t + 1)∑|Asp|2(2s + 1)−1
| (5) |
where Asp is the set of constants depending on the crystal field and can be expressed as
in which Cs−p is the tensor operator, ρ is the field charge density of the ligand atoms such as oxygen and nitrogen, R is the distance between the metal centre and coordinating oxygen or nitrogen atom, and θ and φ are the angular parameters of the ligands to the center ion.
The determined Ω2 value for the present complex, [Er(tta)3(tppo)], in chloroform is 34.31 which is higher than those reported for [Er(acac)3·H2O] (25.71) and [Er(acac)3phen] (32.28).7 The difference between average bond lengths of Er–O(tta) and Er–O(tppo) in the present complex is 0.053 Å, which is lower than those reported for Er–O(acac) and Er–N(phen) in [Er(acac)3phen] (0.284 Å), and Er–O(acac) and Er–O(H2O) in [Er(acac)3·H2O] (0.1 Å). This result suggests that the present complex must have high symmetry since a smaller difference in bond length leads to higher symmetry of the field charge density. However, the present complex is a seven-coordinate complex and possesses a monocapped octahedral geometry which is more asymmetric as compared to eight-coordinate [Er(acac)3phen] having symmetrical square antiprismatic geometry. On the other hand, [Er(acac)3·H2O] is also a seven-coordinate complex with a monocapped trigonal prism geometry. Nevertheless, the present complex seems more asymmetric since it possesses a tta ligand having –CF3 group (electron withdrawing) on one terminal and thiophene ring (electron donating) on another terminal, i.e. asymmetric substitution. This changes the electron density at keto–enol oxygen atoms that leads to different electron populations on the carbonyl oxygen atoms in the ligand. Moreover, the oxygen atom of tppo has different electron density from keto–enol oxygen atoms since it is attached to electronegative phosphorus atom with a PO bond. This suggests that there is a different field charge distribution around the Er(III) ion in the present complex. Consequently, it can be inferred that Asp of [Er(tta)3(tppo)] should be higher than those of [Er(acac)3·H2O] and [Er(acac)3phen], and therefore the Ω2 value of [Er(tta)3(tppo)] is larger.
Effect of chemical environment on Ω6
The Ω6 value, unlike Asp, is insensitive to the change of ligand field.7 It is largely affected by the change of radial integrals, (4f|r|nl) and (nl|rs|4f), which in turn have such a relation with electron density of 4f and 5d orbitals that their values decrease as the 5d electron density decreases. In contrast, the electron density of 5d orbitals is inversely related to 6s electron density since the 6s electrons are assumed to shield the 5d orbitals. In summary, the Ω6 value is directly related to 5d electron density or inversely to 6s electron density.42,43
The determined Ω6 value of the present complex [Er(tta)3(tppo)] (1.85) is higher than those reported for [Er(acac)3·H2O] (0.75) and [Er(acac)3phen] (0.81). In such systems, the Er(III) ion is coordinated to the ligands by σ-bonds which are formed between the filled 2p orbitals of the ligands and the empty 6s orbitals of Er(III) ion. The overlap of these orbitals leads to σ-electron donation from the ligands to Er(III) ion. It results in an increase of 6s electron density or decrease of 5d electron density and, therefore, the Ω6 value decreases. In the present complex, the Er–O(tppo) bond is less covalent as compared Er–N(phen) bonds in [Er(acac)3phen], since oxygen atom is more electronegative than nitrogen atom. While in comparison to Er–O(water) in [Er(acac)3H2O], the Er–O(tppo) bond is also assumed as less covalent because tppo has strong electron-attracting atoms/groups like phosphorus and phenyl rings on oxygen as compared to oxygen of water having only two hydrogen atoms. This result implies that σ-electron donation is less from tppo ligand to 6s orbital of Er ion in [Er(tta)3(tppo)] and, therefore, the Ω6 value is higher.
Conclusion
A new Er(III) β-diketonate complex, [Er(tta)3(tppo)] or tris(thenoyltrifluoroacetylacetonate) mono(triphenylphosphine oxide) erbium(III), was synthesized by an in situ method. Structural characterization indicates the complex is seven-coordinate where the Er(III) ion is coordinated to seven oxygen atoms of three tta ligands and one tppo ligand in a monocapped octahedral geometry. It is a highly asymmetric structure which is supportive of radiative transitions in the present complex that results in efficient 1.53 μm emission of Er. The investigation of Judd–Ofelt parameters of the complex indicates a different field charge distribution around the Er(III) ion and relatively less covalent nature of Er-ligand bonds. PMMA doped with the complex shows an enhancement in emission intensity and full width at half maximum that makes it possible for use in optical-amplification applications. Furthermore, the present complex has been successfully incorporated as emission layer into a three-layer vacuum-deposited device with structure: ITO/β-NPB/[Er(tta)3(tppo)]/TPBi/LiF/Al. This device shows an efficient electroluminescent band at 1534 nm (C-band region) with total quenching of the visible emission. Finally, an organic diode was fabricated with structure ITO/[Er(tta)3(tppo)]/Al, to determine charge carrier mobility (μ0) of the complex. The complex showed μ0 of 6.44 × 10−9 cm2 V−1 s−1 (<5 V).
Acknowledgements
The authors are grateful to the Brazilian agencies CAPES, CNPq, FAPERJ, INEO for their financial support. We also thank: the Central Analítica de Pe. Leopoldo Hainberger S. J. of PUC-Rio for Raman analysis; the Laboratório de Eletroanalítica, Espectroanalítica e Análise Elementar Aplicada (LEEA) of PUC-Rio for elemental analysis; the Laboratório de Difração de Raios X (LRDX) of Universidade Federal Fluminense for the XRD measurement; Laboratório de Tabaco e Derivados (LATAB) of Divisão of Química Analítica (DQAN) from Instituto Nacional de Tecnologia for the ESI-MS measurement. Z. Ahmed is a recipient of a CAPES/PNPD postdoctoral fellowship.
References
-
(a) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357–2368 CrossRef CAS PubMed;
(b) S. V. Eliseeva and J.-C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC;
(c) M. A. Katkova and M. N. Bochkarev, Dalton Trans., 2010, 39, 6599–6612 RSC;
(d) H. Wei, G. Yu, Z. Zhao, Z. Liu, Z. Bian and C. Huang, Dalton Trans., 2013, 42, 8951–8960 RSC.
- F. X. Zang, Z. R. Hong, L. W. Li, M. T. Li and X. Y. Sun, Appl. Phys. Lett., 2004, 84, 2679–2681 CrossRef CAS.
- K. S. Schanze, J. R. Reynolds, J. M. Boncella, B. S. Harrison, T. J. Foley and M. Bouguettaya, Synth. Met., 2003, 137, 1013–1014 CrossRef CAS.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
-
(a) K. Yanagisawa, T. Nakanishi, Y. Kitagawa, T. Seki, T. Akama, M. Kobayashi, T. Taketsugu, H. Ito, K. Fushimi and Y. Hasegawa, Eur. J. Inorg. Chem., 2015, 28, 4769–4774 CrossRef;
(b) Z. Ahmed and K. Iftikhar, Inorg. Chem., 2015, 54, 11209–11225 CrossRef CAS PubMed;
(c) Z. Ahmed and K. Iftikhar, RSC Adv., 2014, 4, 63696–63711 RSC;
(d) S. Dasari, S. Singh, S. Sivakumar and A. K. Patra, Chem.–Eur. J., 2016, 22, 17387–17396 CrossRef CAS PubMed.
-
(a) Z. Ahmed and K. Iftikhar, J. Phys. Chem. A, 2013, 117, 11183–11201 CrossRef CAS PubMed;
(b) K. Miyata, Y. Hasegawa, Y. Kuramochi, T. Nakagawa, T. Yokoo and T. Kawai, Eur. J. Inorg. Chem., 2009, 32, 4777–4785 CrossRef;
(c) K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem.–Eur. J., 2011, 17, 521–528 CrossRef CAS PubMed;
(d) M. D. Regulacio, M. H. Pablico, J. A. Vasquez, P. N. Myers, S. Gentry, M. Prushan, S. W. Tam-Chang and S. L. Stoll, Inorg. Chem., 2008, 47, 1512–1523 CrossRef CAS PubMed;
(e) A. Bellusci, G. A. Barberio, G. Crispini, M. Ghedini, M. La Deda and D. Pucci, Inorg. Chem., 2005, 44, 1818–1825 CrossRef CAS PubMed;
(f) Y. C. Miranda, L. L. A. L. Pereira, J. H. P. Barbosa, H. F. Brito, M. C. F. C. Felinto, O. L. Malta, W. M. Faustino and E. E. S. Teotonio, Eur. J. Inorg. Chem., 2015, 18, 3019–3027 CrossRef.
- H. Wang, G. Qian, Z. Wang and M. Wang, Spectrochim. Acta, Part A, 2005, 62, 146–152 CrossRef PubMed.
- Y. Hasegawa, S.-i. Tsuruoka, T. Yoshida, H. Kawai and T. Kawai, J. Phys. Chem. A, 2008, 112, 803–807 CrossRef CAS PubMed.
- H. Xu, L.-H. Wang, X.-H. Zhu, K. Yin, G.-Y. Zhong and X.-Y. Hou, J. Phys. Chem. B, 2006, 110, 3023–3029 CrossRef CAS PubMed.
-
(a) A. O. Biroli, M. Pizzotti, P. Illiano and F. Demartin, Inorg. Chim. Acta, 2011, 366, 254–261 CrossRef CAS;
(b) A. Monguzzi, R. Tubino, F. Meinardi, A. O. Biroli, M. Pizzotti, F. Demartin, F. Quochi, F. Cordella and M. A. Loi, Chem. Mater., 2009, 21, 128–135 CrossRef CAS.
- Q. Zhong, H. Wang, G. Qian, Z. Wang, J. Zhang, J. Qiu and M. Wang, Inorg. Chem., 2006, 45, 4537–4543 CrossRef CAS PubMed.
-
(a) K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 2347–2356 CrossRef CAS PubMed;
(b) L. H. Slooff, A. van Blaaderen, A. Polman, G. A. Hebbink, S. I. Klink, F. C. J. M. van Veggel, D. N. Reinhoudt and J. W. Hofstraat, J. Appl. Phys., 2002, 91, 3955–3980 CrossRef CAS;
(c) F. Artizzu, M. L. Mercuri, A. Serpe and P. Deplano, Coord. Chem. Rev., 2011, 255, 2514–2529 CrossRef CAS;
(d) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS;
(e) R. A. S. Ferreira, P. S. Andre and L. D. Carlos, Opt. Mater., 2010, 32, 1397–1409 CrossRef CAS.
- SAINT: Bruker, APEX3 and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2015 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, SADABS, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- G. M. Sheldrick, SHELXT and SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. A. Omary, M. A. Rawashdeh–Omary, H. V. K. Diyabalanage and H. V. R. Dias, Inorg. Chem., 2003, 42, 8612–8614 CrossRef CAS PubMed.
-
(a) V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 16, 1494–1495 RSC;
(b) I. R. Lasker and T. –M. Chen, Chem. Mater., 2004, 16, 111–117 CrossRef.
- P. Martín-Ramos, J. T. Coutinho, M. R. Silva, L. C. J. Pereira, F. Lahoz, P. S. P. Silva, V. Lavín and J. Martín-Gil, New J. Chem., 2015, 39, 1703–1713 RSC.
- V. V. Utochnikova, O. Pietraszkiewicz, M. Kozbial, P. Gierycz, M. Pietraszkiewicz and N. P. Kuzmina, J. Photochem. Photobiol., A, 2013, 253, 72–80 CrossRef CAS.
- A. –R. Nekoei, S. F. Tayyari, M. Vakili, S. Holakoei, A. H. Hamidian and R. E. Sammelson, J. Mol. Struct., 2009, 932, 112–122 CrossRef CAS.
- V. Tsaryuk, V. Zolin, J. Legendziewicz, R. Szostak and J. Sokolnicki, Spectrochim. Acta, Part A, 2005, 61, 185–191 CrossRef CAS PubMed.
-
(a) A. P. Hunter, A. M. J. Lees and A. W. G. Platt, Polyhedron, 2007, 26, 4865–4876 CrossRef CAS;
(b) J. Zhang, P. D. Badger, S. J. Geib and S. Petoud, Inorg. Chem., 2007, 46, 6473–6482 CrossRef CAS PubMed.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J.-C. G. Bünzli and N. P. Kuzmina, Inorg. Chem., 2011, 50, 5137–5144 CrossRef CAS PubMed.
- N. M. Shavaleev, Z. R. Bell and M. D. Ward, J. Chem. Soc., Dalton Trans., 2002, 3925–3927 RSC.
- Z. F. Li, J. B. Yu, L. Zhou, H. J. Zhang, R. P. Deng and Z. Y. Guo, Org. Electron., 2008, 9, 487–494 CrossRef CAS.
- P. Martín-Ramos, C. Coya, V. Lavín, I. R. Martín, M. R. Silva, P. S. P. Silva, M. García-Vélez, A. L. Álvarez and J. Martín-Gil, Dalton Trans., 2014, 43, 18087–18096 RSC.
- H. Xu, K. Yin and W. Huang, J. Phys. Chem. C, 2010, 114, 1674–1683 CAS.
- H. Xin, M. Shi, X. C. Gao, Y. Y. Huang, Z. L. Gong, D. B. Nie, H. Cao, Z. Q. Bian, F. Y. Li and C. H. Huang, J. Phys. Chem. B, 2004, 108, 10796–10800 CrossRef CAS.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. vandertol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS.
- P. Martín-Ramos, V. Lavín, M. R. Silva, I. R. Martín, F. Lahoz, P. Chamorro-Posada, J. A. Paixão and J. Martín-Gil, J. Mater. Chem. C, 2013, 1, 5701–5710 RSC.
- L. N. Sun, H. J. Zhang, L. S. Fu, F. Y. Liu, Q. G. Meng, C. Y. Peng and J. B. Yu, Adv. Funct. Mater., 2005, 15, 1041–1048 CrossRef CAS.
- R. Sultan, K. Gadamsetti and S. Swavey, Inorg. Chim. Acta, 2006, 359, 1233–1238 CrossRef CAS.
- B. Dandrade, S. Datta, S. Forrest, P. Djurovich, E. Polikarpov and M. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef CAS.
- R. J. Curry, W. P. Gillin, A. P. Knights and R. Gwilliam, Appl. Phys. Lett., 2000, 77, 2271–2273 CrossRef CAS.
-
(a) R. U. Khan, D. Poplavskyy, T. Kreouzis and D. D. C. Bradley, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035215–35314 CrossRef;
(b) T. Okachi, T. Nagase and T. Kobayashi, Jpn. J. Appl. Phys., 2008, 47, 8965–8972 CrossRef CAS;
(c) D. Poplavskyy and J. Nelson, J. Appl. Phys., 2003, 93, 341–346 CrossRef CAS;
(d) P. W. M. Blom, M. J. M. de Jong and M. G. van Munster, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 55, R656–R659 CrossRef.
- N. F. Mott and R. W. Gurney, Electronic processes in ionic crystals, Clarendon Press, 1948 Search PubMed.
- P. Martín-Ramos, C. Coya, A. L. Álvarez, M. R. Silva, C. Zaldo, J. A. Paixão, P. C. Posada and J. Martín-Gil, J. Phys. Chem. C, 2013, 117, 10020–10030 Search PubMed.
-
(a) W. T. Carnell, P. R. Feilds and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef;
(b) M. P. Hehlen, M. G. Brik and K. W. Krämer, J. Lumin., 2013, 136, 221–239 CrossRef CAS.
-
(a) H. A. Khan and K. Iftikhar, Polyhedron, 1994, 13, 3199–3208 CrossRef;
(b) H. A. Khan and K. Iftikhar, Polyhedron, 1997, 16, 4153–4161 CrossRef.
-
(a) M. Iwamuro, Y. Hasegawa, Y. Wada, K. Murakoshi, N. Nakashima, T. Yamanaka and S. Yanagida, J. Lumin., 1998, 79, 29–38 CrossRef CAS;
(b) R. D. Peacock, Struct. Bonding, 1975, 22, 83–122 CrossRef CAS.
- H. Ebendorff-Heidepriem, D. Ehrt, M. Bettinelli and A. Speghini, J. Non-Cryst. Solids, 1998, 240, 66–78 CrossRef CAS.
- S. Tanabe, T. Ohyagi, N. Soga and T. Hanada, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 3305–3310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1487439. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27473k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a




 , respectively.
, respectively.