
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cytotoxicity study of polyethylene glycol derivatives
Guoqiang Liua,
Yongsan Liab,
Lei Yangd,
Yen Weia,
Xing Wangb,
Zhiming Wangc and
Lei Tao *a
*a
aKey Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: leitao@mail.tsinghua.edu.cn
bThe State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
cCollege of Pharmaceutical Science, Zhejiang Chinese Medical University, Hangzhou, Zhejiang 311400, P. R. China
dCancer Institute & Hospital, Peking Union Medical College & Chinese Academy of Medical Science, Beijing 100021, P. R. China
First published on 27th March 2017
Abstract
Polyethylene glycol (PEG) derivatives have been widely used in bio-related research. However, PEG oligomers (with different molecular weights) or PEG based monomers (with different chain end groups) actually have different chemical and physical properties, which might lead to potential toxicity. In this work, the cytotoxicity of a series of PEG derivatives (oligomers and monomers) has been measured using human cervical cancer cells (HeLa) and a cell line of fibroblasts derived from mice (L929) as model cells. Most of the PEG oligomers are safe to both types of cells except triethylene glycol (TEG), which is toxic at high concentrations to L929 cells. On the other hand, PEG-based monomers including poly(ethylene glycol) methyl ether acrylate (mPEGA) and poly(ethylene glycol) methyl ether methacrylate (mPEGMA) showed obvious cytotoxicity. Subsequently, those toxic PEG derivatives have been studied to reveal the different mechanisms of their toxicity. This current research evaluated the cytotoxicity of PEG derivatives and pointed out the potential hazard of ‘safe’ biomaterials, which might offer a useful reference for people to use the PEG derivatives in future biomedical research.
1. Introduction
Polyethylene glycol (PEG) has been widely applied in various medical fields due to its outstanding properties such as satisfactory safety, biocompatibility, hydrophilicity etc. For example, PEG has been used in some Food and Drug Administration approved laxatives because of its excellent moisture retention and lubrication ability.1–3 In another example, modifying drugs/proteins with PEG (PEGylation) can effectively improve their water-solubility and circulation half-life of medicines,4,5 resulting in enhanced safety and therapy effects. Actually, PEGylation is becoming one of the most attractive biotechnologies and achieving huge success in fundamental research and in the market.6–11 By now, many PEG derivatives have been used as excellent starting materials to synthesize a number of new polymeric materials for bioapplications.12 For example, a linear PEG has been modified as a difunctionalized PEG (DF-PEG) which could be used to quickly generate an interesting self-healing hydrogel.13,14 Some PEG oligomers have been used to modify the inorganic nano-particle surface to improve the water-solubility and reduce the toxicity of those inorganic materials.15–17 Some commercial PEG-based monomers such as poly(ethylene glycol) methyl ether acrylate (mPEGA) and poly(ethylene glycol) methyl ether methacrylate (mPEGMA) have been used to prepare branched polymers for subsequent protein conjugation, self-assembly, drug delivery etc.18–27Although PEG is normally considered as almost non-toxic, some unsafe problems of PEG with low molecular weights have been noticed by researchers. Smyth et al. reported the chronic oral toxicity of PEG oligomer (Mn ∼ 200) in rats,28,29 the adverse results have also be observed in monkeys,30 suggesting potential safety problems of these ‘safe’ materials. Therefore, considered the broad utilization of PEG derivatives in research areas, the systematic cytotoxicity study of those PEG oligomers and PEG-based monomers is necessary and important for their further application in bio-related fields (Scheme 1).
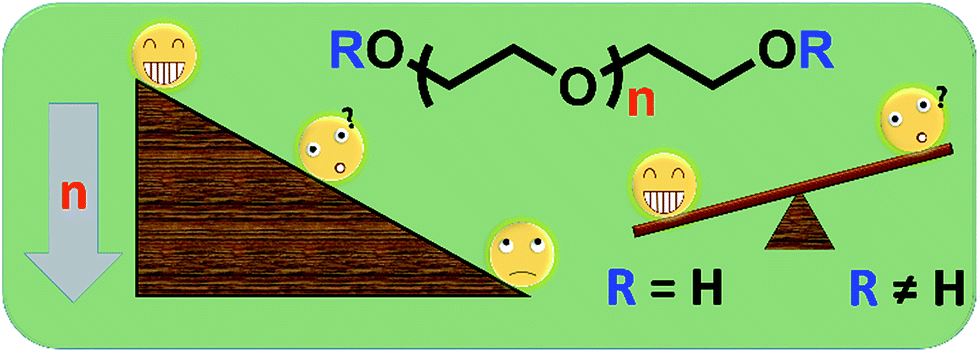 | ||
| Scheme 1 The possible cytotoxicity of PEG derivatives with different molecular weights and end groups. | ||
In the current work, cytotoxicity of some PEG oligomers (with different molecular weights) and commercial PEG-based monomers (with different chain end groups) to human cervical cancer cells (HeLa) and a cell line of fibroblast derived from mice (L929) was evaluated. The molecular weight-dependent cytotoxicity of PEG oligomers has been observed, and the different cytotoxicity mechanisms of PEG oligomers and PEG-based monomers were revealed for the first time. Those cytotoxicity information of PEG derivatives might be valuable references for people to choose PEG as starting materials and design purification routes to prepare biocompatible PEG-based materials for further biomedical applications.
2. Materials and methods
2.1 Chemicals
Triethylene glycol (TEG, 97%, HEOWNS), PEG-400 (Mn ∼ 400, TCI), PEG-1000 (Mn ∼ 1000, Jiangsu Haitian Petrochemical Works), PEG-2000 (Mn ∼ 2000, SCRC), PEG-4000 (Mn ∼ 4000, SCRC), poly(ethylene glycol) methyl ether acrylate 480 (mPEGA-480, Mn ∼ 480, Sigma-Aldrich), poly(ethylene glycol) methyl ether methacrylate 500 (mPEGMA-500, Mn ∼ 500, Sigma-Aldrich) and poly(ethylene glycol) methyl ether methacrylate 950 (mPEGMA-950, Mn ∼ 950, Sigma-Aldrich) were used directly without further purification.Human cervical cancer cells (HeLa) and a cell line of fibroblast derived from mice (L929) were acquired from American type culture collection (ATCC). Dulbecco's modified eagle medium (DMEM, Corning-Cellgro), Phosphate Buffered Saline (PBS, pH ∼ 7.2–7.4, 0.01 M, Solarbio), fetal bovine serum (FBS, Gibco), Roswell Park Memorial Institute 1640 culture medium (RPMI 1640 culture medium, Gibco), penicillin–streptomycin solution (Gibco), trypsin–EDTA (Gibco, 0.25%), 4-(3-(2-methoxy-4-nitrophenyl)-2-(4-nitrophenyl)-2H-tetrazol-3-ium-5-yl)benzene-1,3-disulfonate (CCK-8, Beyotime), 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, Beyotime), oxidized glutathione (GSSG, Beyotime), nicotinamide adenine dinucleotide phosphate (NADPH, Beyotime), 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB, Beyotime), fluorescein diacetate (FDA, Sigma) and propidium iodide (PI, 94%, Sigma) were used as purchased.
2.2 Cell culture
Cell culture was maintained in a 37 °C incubator with 5% CO2, culture medium was changed every one or two days for maintaining the exponential growth of the cells. HeLa cells were derived from cervical cancer cells and cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. L929 cells were derived from mice and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin.2.3 Cytotoxicity analysis
The results were presented as mean ± standard deviation (SD), and the half maximal inhibitory concentration 50% (IC50) values of PEG derivatives were calculated by SPSS 15.0.
According to our previous work,32,33 fluorescein diacetate (FDA) and propidium iodide (PI) could be used as a rapid and convenient double-staining method to observe the cell viability. L929 cells were seeded in a 24-well plate at a density of ∼5 × 104 cells per mL. After attachment, cells were washed with PBS and cultured with different concentration of PEG derivatives in culture medium for 24 h. Then, the medium was removed, the cells were washed with PBS. A PBS–FDA–PI mixed solution (FDA: 3 μg mL−1; PI: 3 μg mL−1) was subsequently added and the 24-well plate was incubated at 37 °C for 15 min. A fluorescence microscope (Leica Germany) was used to observe the live and dead cells under 450–490 nm and 515–560 nm band-pass excitation filters (I3 and N2.1), respectively (100 W mercury lamp).
Briefly, L929 cells were seeded in a 24-well plate and incubated in 0.5 mL of culture medium with different concentration of TEG (A, A′) for 1 h (Fig. 1), A1–A6 wells were used to measure the total amount of glutathione (GSH + GSSG) while A′1–A′6 wells were used to measure the amount of oxidized glutathione (GSSG) in the presence of TEG. After 1 h, cells were collected by trypsin digestion and centrifugation (1000 rpm, 5 min). Then, treble volume protein removal solution (50 mg mL−1) was added to remove the noisy proteins. After thrice quick freezing–thawing cycles in liquid nitrogen and a 37 °C water-bath, the suspension were placed in a refrigerator (4 °C) for 5 min, and the samples of cell supernatant were obtained after centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min). Samples from A1–A6 wells were used to measure the total glutathione (GSH + GSSG) level. NADPH solution (0.5 mg mL−1) was added to reduce all of glutathione to GSH, then, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) was used to oxidize the GSH, the changed absorbance at 405 nm was recorded by a microplate reader (VICTOR™ X3 PerkinElmer 2030 Multilabel Plate Reader).35,36 Samples from A′1–A′6 wells were used to measure the oxidized glutathione (GSSG) level. By using the GSH remover solution, only GSSG was left in the sample solution. The samples were analysed through same method as above to measure the GSSG's level. The total glutathione amount and the oxidized glutathione amount were collected to calculate the level of reduced glutathione while the GSH level of cells in pure culture medium was used as the control. The results were presented as mean ± SD.
000 rpm, 10 min). Samples from A1–A6 wells were used to measure the total glutathione (GSH + GSSG) level. NADPH solution (0.5 mg mL−1) was added to reduce all of glutathione to GSH, then, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) was used to oxidize the GSH, the changed absorbance at 405 nm was recorded by a microplate reader (VICTOR™ X3 PerkinElmer 2030 Multilabel Plate Reader).35,36 Samples from A′1–A′6 wells were used to measure the oxidized glutathione (GSSG) level. By using the GSH remover solution, only GSSG was left in the sample solution. The samples were analysed through same method as above to measure the GSSG's level. The total glutathione amount and the oxidized glutathione amount were collected to calculate the level of reduced glutathione while the GSH level of cells in pure culture medium was used as the control. The results were presented as mean ± SD.
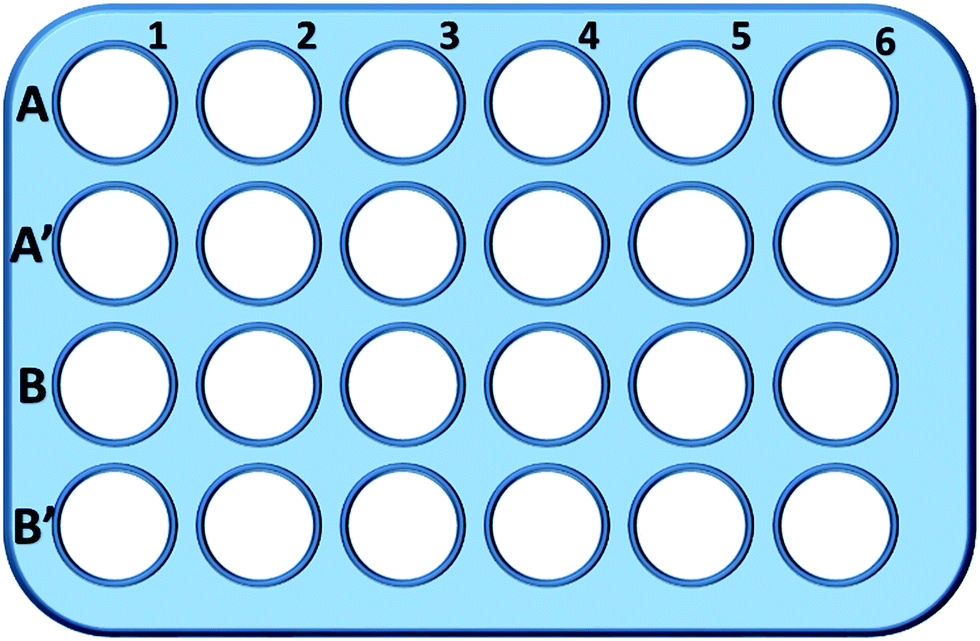 | ||
| Fig. 1 The configuration of the 24-well plate for GSH analysis. Cells in line 1 were cultured in pure culture medium and used as the control; cells in A2–A6 and A′2–A′6 wells were cultured with 1, 2, 5, 10 and 20 mg mL−1 of TEG, respectively; cells in B2–B6 and B′2–B′6 wells were cultured with 1, 2, 5, 10 and 20 mg mL−1 of mPEGA-480, respectively. | ||
Wells in line B and B′ were used to test the total amount of GSH and GSSG in the presence of mPEGA-480, respectively, through the same procedure.
3. Results and discussion
3.1 Cell viability study
When HeLa cell (a model of cancer cells) was used (Fig. 2A), the cytotoxicity of all PEG oligomers at low concentrations (≤5 mg mL−1) could be neglected. With increased concentration (≥10 mg mL−1), the molecular weight-dependent cytotoxicity of PEG samples was observed. The triethylene glycol (TEG) showed obvious cytotoxicity over 10 mg mL−1 (p < 0.05; contrast with any other oligomer when concentration is over 5 mg mL−1) and the IC50 value of the TEG to HeLa cells was calculated as 19.8 mg mL−1. Meanwhile, other PEG oligomers (Mn ∼ 400–4000) also showed tiny cytotoxicity.
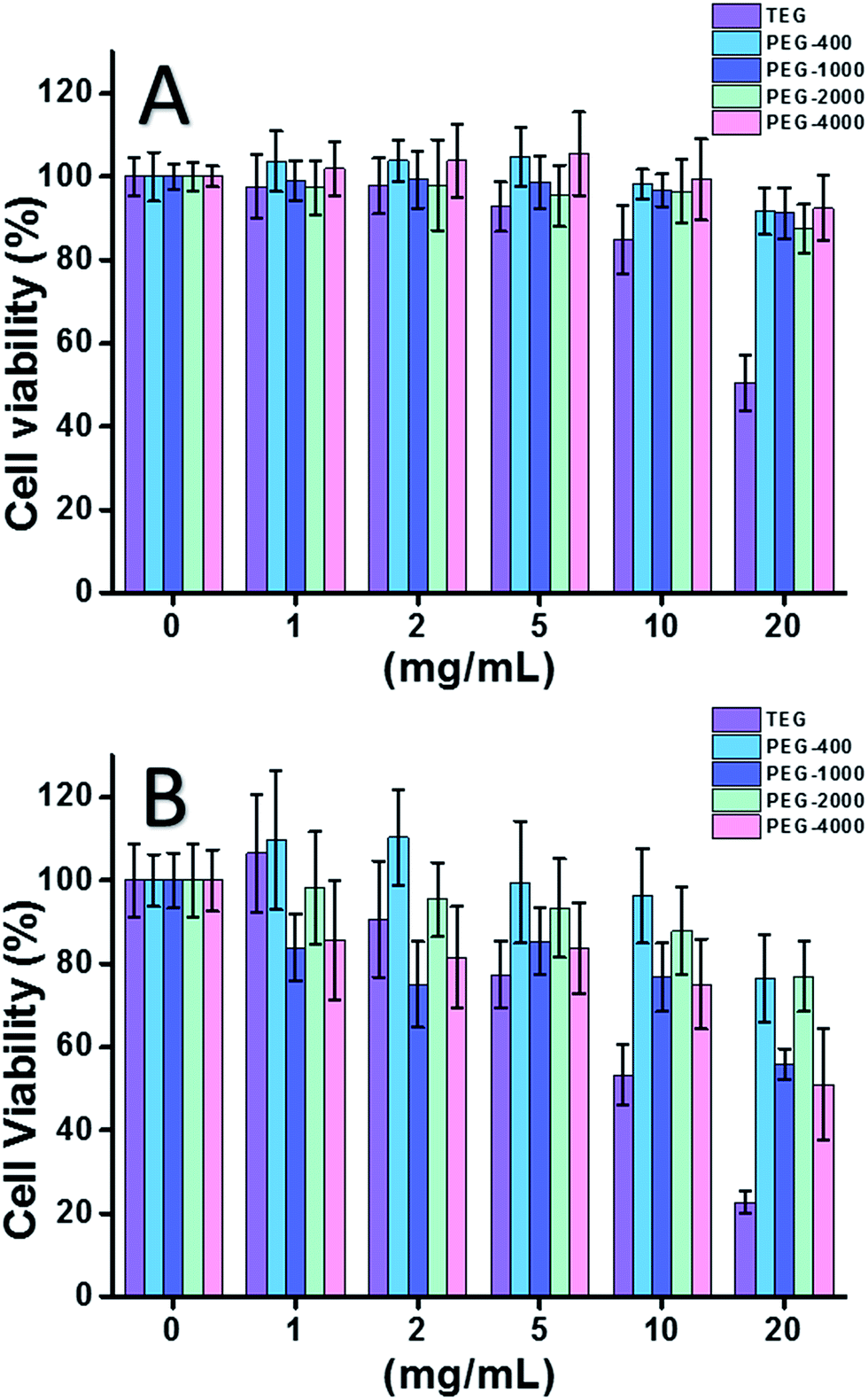 | ||
| Fig. 2 Concentration-dependent cytotoxicity of PEG oligomers to HeLa cells (A) and L929 cells (B), the concentrations of PEG range from 1 to 20 mg mL−1. Cells in pure culture medium served as the positive control and no cells as negative control. | ||
The cytotoxicity of PEG oligomers to L929 cells (a model of normal cells) was also evaluated (Fig. 2B). The cell viability decreased with increased PEG concentration as expected. Besides TEG, some PEG oligomers (PEG-1000, PEG-4000) which are safe to HeLa cells also showed detectable cytotoxicity, the IC50 values of TEG, PEG-1000 and PEG-4000 were calculated as 12.4, 22.5, and 20.0 mg mL−1 to L929 cells, respectively, also indicating the molecular weight-dependent cytotoxicity of PEG oligomers.
Above results suggested that HeLa cells have better tolerance to PEG oligomers than L929 cells due to the tenacious vitality of cancer cells, and the TEG has much higher cytotoxicity than other PEG oligomers with higher molecular weights (p < 0.05; contrast with any other oligomer when concentration is over 5 mg mL−1). It is noticed the current research demonstrated the moderate cytotoxicity of PEG-1000 and PEG-4000 to L929 cells, and the nearly non-cytotoxicity of PEG-400 and PEG-2000, which is different with the common impression that high molecular weight PEGs should be more biocompatible than their low molecular weight counterparts. Although the reason of PEG's cytotoxicity is still not clear, PEG-400 and PEG-2000 seem to be better choices for people to synthesize biocompatible materials.
When mPEGMA-500 was tested, the cell viability of both HeLa cells and L929 cells decreased with increased concentration of mPEGMA-500 (Fig. 3A), and no apparent difference between two types of cells was observed. The IC50 values of mPEGMA-500 are 4.7 mg mL−1 to HeLa cells, and 5.3 mg mL−1 to L929 cells after 24 h culture, indicating more significant cytotoxicity of mPEGMA-500 than abovementioned PEG oligomers, and implying the chain end groups of PEG derivatives are crucial to their toxicity. When mPEGMA-950, another PEG-based methacrylate with same chain end group but higher molecular weight was tested through same procedure (Fig. 3B), much lower cytotoxicity of mPEGMA-950 to HeLa cells or L929 cells was observed (p < 0.05; when concentration is over 2 mg mL−1), the IC50 values of mPEGMA-950 are 20.8 mg mL−1 to HeLa cells, and 21.7 mg mL−1 to L929 cells after 24 h culture, also demonstrating the molecular weight-dependent cytotoxicity of PEG-based methacrylate.
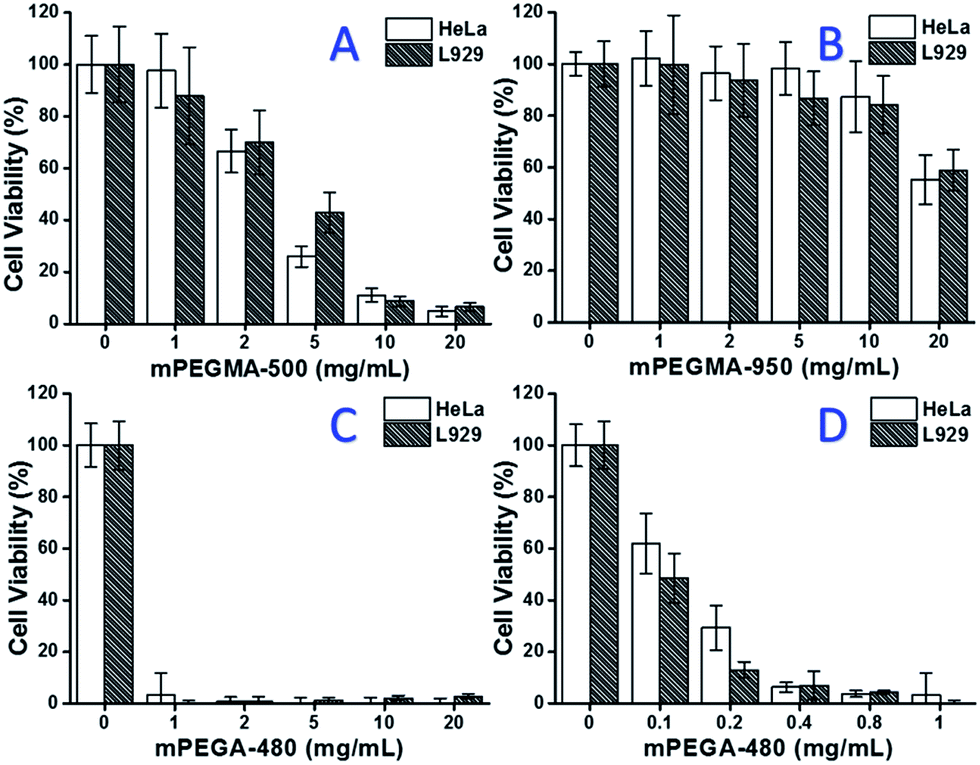 | ||
| Fig. 3 Concentration-dependent cytotoxicity of mPEGMA-500 (A), mPEGMA-950 (B) and mPEGA-480 (C and D) to HeLa cells and L929 cells, the concentrations of mPEGMA range from 1 to 20 mg mL−1, the concentrations of mPEGA range from 0.1 to 20 mg mL−1. Cells without PEG-based monomers served as the positive control and no cells as negative control. | ||
Furthermore, PEG-based acrylate mPEGA-480 was also tested. Although the difference between mPEGA-480 and mPEGMA-500 is only a methyl group at chain end, mPEGA-480 showed acute cytotoxicity (Fig. 3C). More than 95% cells lost viability even with 1 mg mL−1 mPEGA-480 in the culture medium. Thus, additional experiments have to be carried out with lower concentrations to determine its cytotoxicity (Fig. 3D), and the IC50 values of mPEGA-480 were calculated as 0.2 mg mL−1 to HeLa cells and 0.1 mg mL−1 to L929 cells after 24 h culture.
The cell viability experiments confirmed that both chain end groups and molecular weight are important influence elements to the cytotoxicity of PEG derivatives, and the PEG-based monomers showed much more noticeable cytotoxicity compared with PEG oligomers.
Meanwhile, the cytotoxicity of inhibitors in PEG-based monomers, hydroquinone methyl ether (MEHQ, 100 ppm in mPEGA) and butylated hydroxytoluene (BHT, 300 ppm in mPEGMA), were also evaluated (Fig. 4), and the L929 cells remained high viability (≥90%) at low concentrated inhibitors (≤1 μg mL−1). There are 0.1 μg mL−1 of MEHQ in 1 mg mL−1 of mPEGA-480, and 3 μg mL−1 of BHT in 10 mg mL−1 of mPEGA-500, while mPEG-480 and mPEGMA-500 demonstrated obvious cytotoxicity at those concentrations. Thus, the trace amount inhibitors in PEG-based monomers seemed have negligible influence on the cytotoxicity of monomers.
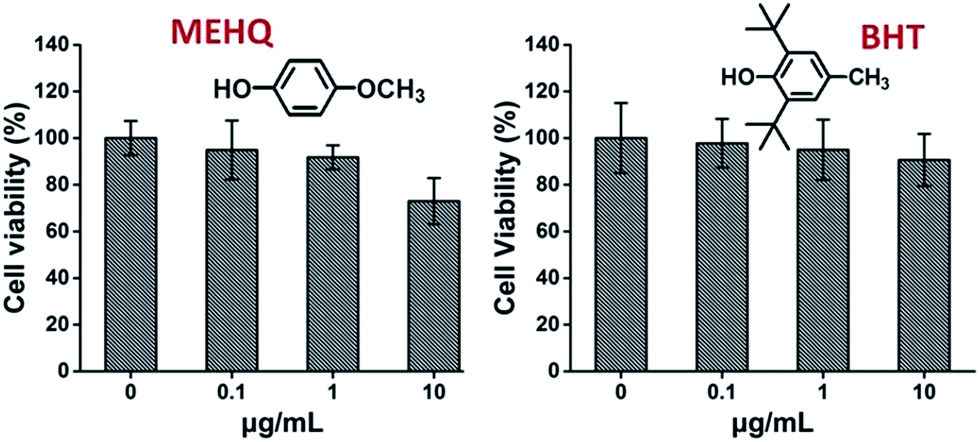 | ||
| Fig. 4 Concentration-dependent cytotoxicity of two inhibitors include MEHQ and BHT to L929 cells, the concentrations of inhibitors range from 0.1 to 10 μg mL−1. Cells without inhibitor served as the positive control and no cells as negative control. | ||
3.2 Cell morphology observation
Optical microscope observation was used to directly evaluate the cytotoxicity of PEG derivatives to L929 cells. The optical images of L929 cells exposed to 20 mg mL−1 TEG and 0.1 mg mL−1 mPEGA for 24 h were shown in Fig. 5A and B, respectively.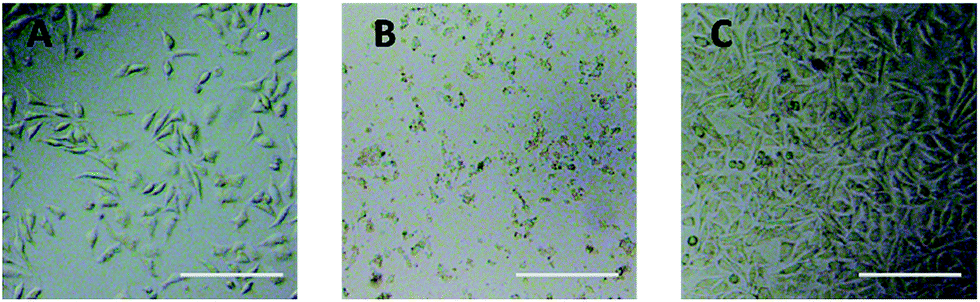 | ||
| Fig. 5 Optical microscope images of L929 cells incubated with 20 mg mL−1 TEG (A) and 0.1 mg mL−1 mPEGA-480 (B) for 24 h, and the cells in pure culture medium (C, control group). Scale bar = 200 μm. | ||
After incubation with 20 mg mL−1 TEG for 24 h, the cells kept the similar morphology (Fig. 5A) with the control group (Fig. 5C, cells in pure culture medium), but the cell number dramatically decreased, indicating the cytotoxicity of TEG at high concentration, which led to the death and followed detachment of cells from the microplate bottom. Fig. 5B showed the morphology of L929 cells after incubation with 0.1 mg mL−1 mPEGA-480. Almost all cells lost normal morphology, suggesting the acute cytotoxicity of mPEGA-480 even at such a low concentration.
Additionally, the cells were also stained using fluorescein. FDA/PI double-staining is a rapid, simple and simultaneous procedure to observe the living and dead cells.43 FDA can enter the complete cells and accumulate in cells, therefore, the complete cell membrane is necessary to prevent the leaking of fluorescein from the cell. In contrast, PI can't pass through the intact cell membrane, but can stain the nucleus of dead cells by passing through the damaged cell membrane. As the Fig. 6 showed, L929 cells incubated with 20 mg mL−1 TEG have less viable cells after the FDA staining (Fig. 6b) compared with the control (Fig. 6a), and more dead cells can be observed after the PI staining (Fig. 6b′), identical with the result by observation through an optical microscopy. mPEGA-480's acute cytotoxicity was also be observed after the FDA/PI double-staining procedure, no green signal could be identified after incubating cells with 0.1 mg mL−1 mPEG-480 for 24 h (Fig. 6c and c′′).
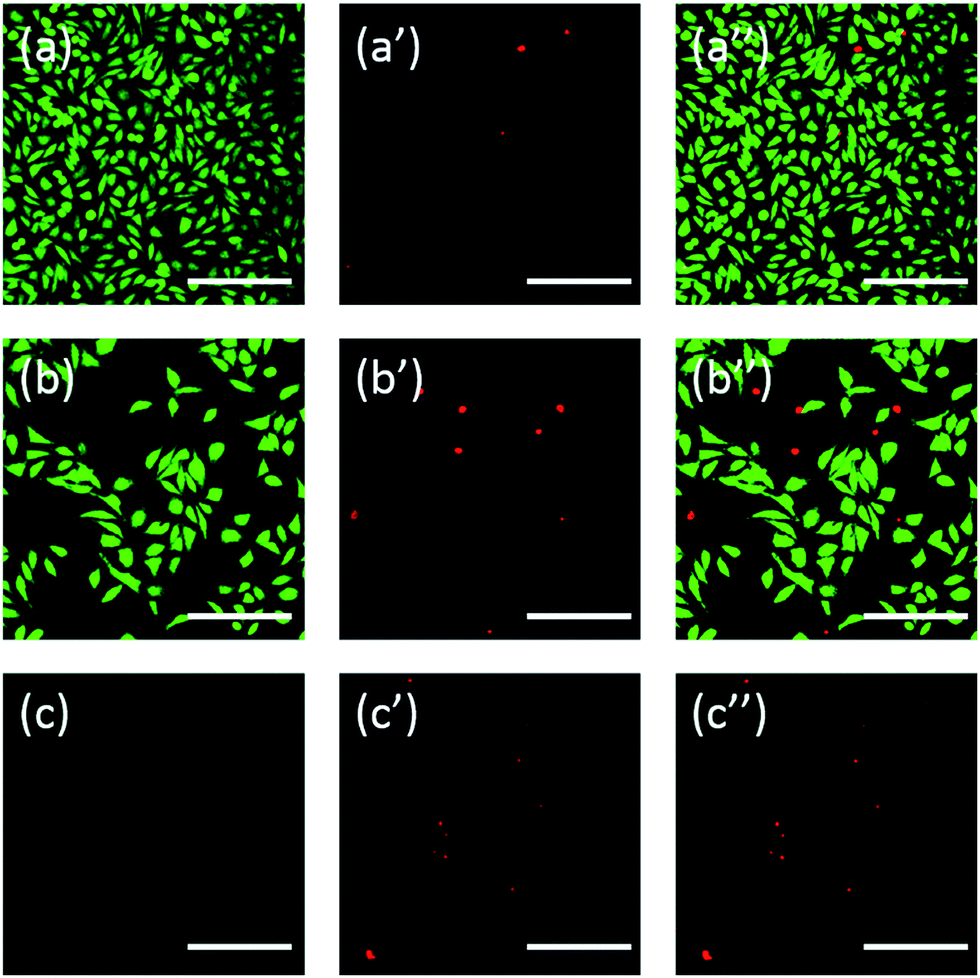 | ||
| Fig. 6 Fluorescent images of L929 cells incubated with complete culture medium as control group (a, a′ and a′′), 20 mg mL−1 TEG (b, b′ and b′′) and 0.1 mg mL−1 mPEGA-480 (c, c′ and c′′) for 24 h. Live cells showed green by FDA (excited by blue light (a, b and c)), dead cells showed red by PI (excited by green light (a′, b′ and c′)), mixed images were excited by blue light and green light (a′′, b′′ and c′′). Scale bar = 200 μm. | ||
Both optical and fluorescent images intuitively demonstrated the cytotoxicity of those PEG derivatives, further supporting the conclusion obtained through CCK-8 assays. It is noticed the mPEGA-480 seemed more toxic by the FDA/PI double staining assay, which is attributed to the FDA/PI double staining distinguishes the completeness of live/dead cell membrane while the CCK-8 assay detects the activity of the dehydrogenase in mitochondria, suggesting FDA/PI staining an excellent supplement of CCK-8 assay to offer more details of the cell status.
3.3 Mechanism study
According to the cell viability experiments, the IC50 values of the PEG oligomers and PEG-based monomers to HeLa and L929 cells after 24 h culture were calculated and summarized (Table 1), suggesting that the PEG-based monomers are much more toxic to cells than the PEG oligomers, and the mPEGA has the highest cytotoxicity. The generation of ROS and decline of GSH in cells, two main mechanisms of cytotoxicity were therefore analysed to probe the possible reason of cytotoxicity.| TEG | PEG-400 | PEG-1000 | PEG-2000 | PEG-4000 | mPEGMA-500 | mPEGMA-950 | mPEGA-480 | |
|---|---|---|---|---|---|---|---|---|
| HeLa cells | 19.8 | 32.5 | 36.2 | 38.2 | 29.6 | 4.7 | 20.8 | 0.2 |
| L929 cells | 12.4 | 24.7 | 22.5 | 28.7 | 20.0 | 5.3 | 21.7 | 0.1 |
L929 cells were incubated with TEG, and the ROS levels at different time points (0, 0.5, 1.0, 1.5, 2 h) were detected according to our previous reports and shown in the Fig. 7. As an example, the ROS levels after incubation with different concentration TEG for 2 h were shown in Fig. 7A. It is clearly to see the ROS levels kept stable with low TEG concentration (≤2 mg mL−1), but significantly increased when TEG concentration is higher than 2 mg mL−1, consistent with the result of cell viability experiment in Fig. 2B. Similarly, the figures of ROS level vs. TEG concentration at different time points were drawn and merged to get a 3D map (Fig. 7B). The maximum ROS levels were found at 2 h with 20 mg mL−1 TEG, an approximately 400% increased ROS level over control (ROS level = 100) was detected, resulting in the brightest fluorescence intensity. Those results suggest the generation of ROS is the possible mechanism of the cytotoxicity of TEG, and the high level of ROS induced by TEG might be the main reason for cell apoptosis.
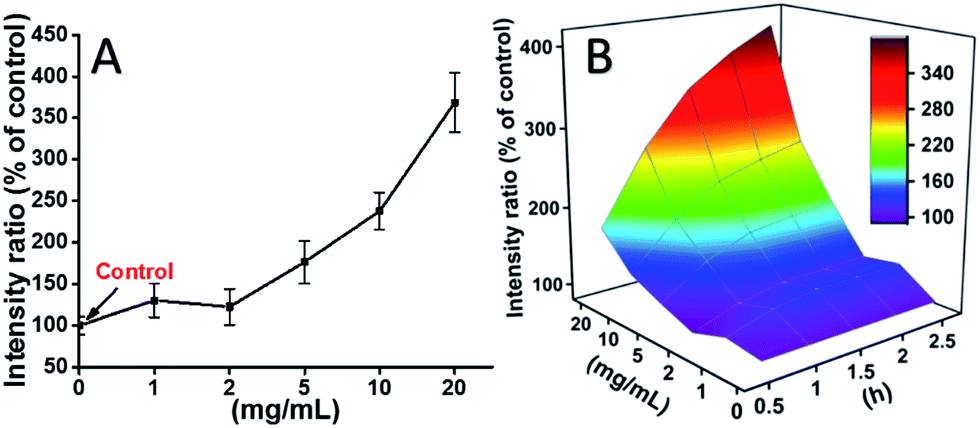 | ||
| Fig. 7 Generation of ROS from L929 cells by the hydrolysis of DCFH-DA after incubation with the TEG for 2 h (A) and for multiple time (B). | ||
The ROS levels induced by mPEGA-480 after 2 h were also tested (Fig. 8A). Different from TEG, mPEGA did not cause the quick increase of ROS in cells, only ∼170% ROS increase compared with the control was observed as the maximum. The 3D ROS level vs. mPEGA & time (Fig. 8B) showed the ROS level induced by mPEGA-480 as a wave and the crest appeared at moderate concentration of mPEGA (5 mg mL−1), no significant generation of ROS can be observed, suggesting that the generation of ROS might only partly contribute to the cytotoxicity of mPEGA-480.
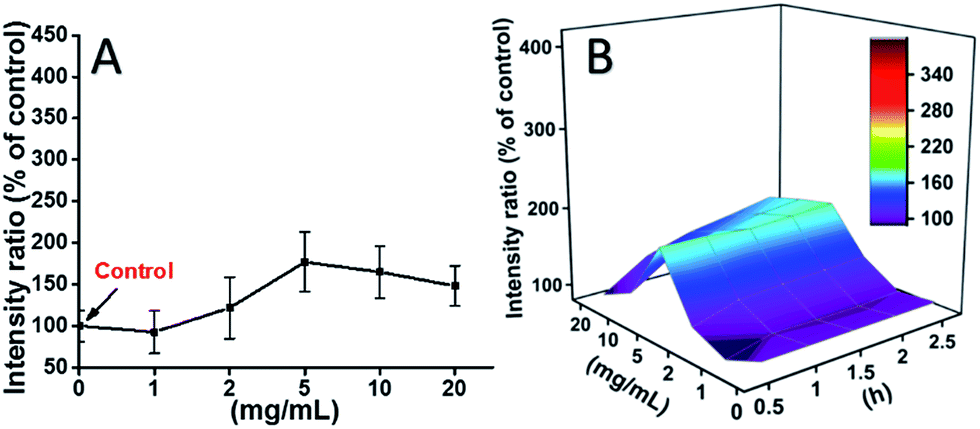 | ||
| Fig. 8 Generation of ROS from L929 cells by the hydrolysis of DCFH-DA after incubation with mPEGA-480 for 2 h (A) and for multiple time (B). | ||
 | ||
| Fig. 9 Decline of GSH in L929 cells by the DNTB assay after incubating cells with the TEG and mPEGA-480. | ||
The reducing thiol group in GSH is an important anti-oxidation source in organisms. A possible mechanism for the cytotoxicity of mPEGA-480 is thereof attributed to the Michael addition reaction between thiol group and vinyl bond, leading to the broken intracellular redox equilibrium and cell apoptosis. Previous research suggested that the Michael addition between thiol and acrylate is much faster than that between thiol and methacrylate,52 which might be a reasonable cause for the different acute cytotoxicity between mPEGA-480 and mPEGMA-500.
4. Conclusions
The cytotoxicity of PEG oligomers and PEG-based monomers to HeLa cells and L929 cells were evaluated for the first time. Concentration-dependent cytotoxicity of PEG oligomers and PEG-based monomers has been confirmed, and presented as IC50 values to HeLa cells and L929 cells. PEG-400, PEG-2000 seem almost non-cytotoxic in the current research. PEG-1000, PEG-4000 and mPEGMA-950 showed moderate cytotoxicity especially at high concentration. TEG and mPEGMA-500 showed significant cytotoxicity, and mPEGA-480 showed acute cytotoxicity. Moreover, cell types also influence the results of the cytotoxicity study, HeLa cells are more robust than L929 cells to tolerate PEG derivatives. These results about PEG derivatives cytotoxicity are preliminary but fundamental for their future biomedical application, more information about the cell response and behaviour to other PEG derivatives will be further investigated.Acknowledgements
This research was supported by the National Science Foundation of China (21574073, 21534006).References
- M. V. Cleveland, D. P. Flavin, R. A. Ruben, R. M. Epstein and G. E. Clark, South. Med. J., 2001, 94, 478–481 CrossRef CAS PubMed.
- J. A. DiPalma, M. V. B. Cleveland, J. McGowan and J. L. Herrera, Am. J. Gastroenterol., 2007, 102, 1436–1441 CrossRef CAS PubMed.
- B. A. Erickson, J. C. Austin, C. S. Cooper and M. A. Boyt, J. Urol., 2003, 170, 1518–1520 CrossRef CAS PubMed.
- J. L. Cleland, S. E. Builder, J. R. Swartz, M. Winkler, J. Y. Chang and D. I. C. Wang, Bio/Technology, 1992, 10, 1013–1019 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS PubMed.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS PubMed.
- H. Zhang, T. Zhao, P. Duffy, Y. Dong, A. Ni Annaidh, E. O'Cearbhaill and W. Wang, Adv. Healthcare Mater., 2015, 4, 2260–2268 CrossRef CAS PubMed.
- Y. Dong, Y. Qin, M. Dubaa, J. Killion, Y. Gao, T. Zhao, D. Zhou, D. Duscher, L. Geever, G. C. Gurtner and W. Wang, Polym. Chem., 2015, 6, 6182–6192 RSC.
- T. M. Allen, C. Hansen, F. Martin, C. Redemann and A. Yauyoung, Biochim. Biophys. Acta, Biomembr., 1991, 1066, 29–36 CrossRef CAS.
- Y. L. Zhang, L. Tao, S. X. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- Y. L. Zhang, B. Yang, L. X. Xu, X. Y. Zhang, L. Tao and Y. Wei, Acta Chim. Sin., 2013, 71, 485–492 CrossRef CAS.
- C. C. Luo, Y. H. Zhang, X. W. Zeng, Y. W. Zeng and Y. G. Wang, J. Colloid Interface Sci., 2005, 288, 444–448 CrossRef CAS PubMed.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS PubMed.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS.
- X. Y. Zhang, C. K. Fu, L. Feng, Y. Ji, L. Tao, Q. Huang, S. X. Li and Y. Wei, Polymer, 2012, 53, 3178–3184 CrossRef CAS.
- X. Y. Zhang, J. F. Hui, B. Yang, Y. Yang, D. D. Fan, M. Y. Liu, L. Tao and Y. Wei, Polym. Chem., 2013, 4, 4120–4125 RSC.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS PubMed.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772–4779 CrossRef CAS.
- H. Hussain, K. Y. Mya and C. He, Langmuir, 2008, 24, 13279–13286 CrossRef CAS PubMed.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS PubMed.
- F. J. Xu, Y. L. Li, E. T. Kang and K. G. Neoh, Biomacromolecules, 2005, 6, 1759–1768 CrossRef CAS PubMed.
- L. L. Yu, L. Yao and K. Yang, J. Porous Mater., 2016, 23, 1581–1589 CrossRef CAS.
- S. Yuan, D. Wan, B. Liang, S. O. Pehkonen, Y. P. Ting, K. G. Neoh and E. T. Kang, Langmuir, 2011, 27, 2761–2774 CrossRef CAS PubMed.
- S. A, Q. Xu, D. Zhou, Y. Gao, J. M. Vasquez, U. Greiser, W. Wang, W. Liu and W. Wang, Polym. Chem., 2017, 8, 1283–1287 RSC.
- H. F. Smyth, C. P. Carpenter and C. S. Weil, J. Am. Pharm. Assoc., 1950, 39, 349–354 CrossRef CAS.
- H. F. Smyth, C. P. Carpenter and C. S. Weil, J. Am. Pharm. Assoc., 1955, 44, 27–30 CrossRef CAS.
- D. E. Prentice and S. K. Majeed, Toxicol. Lett., 1978, 2, 119–122 CrossRef CAS.
- H. Tominaga, M. Ishiyama, F. Ohseto, K. Sasamoto, T. Hamamoto, K. Suzuki and M. Watanabe, Anal. Commun., 1999, 36, 47–50 RSC.
- Y. Li, Y. Zhang, F. Shi, L. Tao, Y. Wei and X. Wang, Colloids Surf., B, 2016, 149, 168–173 CrossRef PubMed.
- B. Yang, Y. Zhang, X. Zhang, L. Tao, S. Li and Y. Wei, Polym. Chem., 2012, 3, 3235–3238 RSC.
- H. Wang and J. A. Joseph, Free Radical Biol. Med., 1999, 27, 612–616 CrossRef CAS PubMed.
- I. K. Smith, T. L. Vieweller and C. A. Thorne, Anal. Biochem., 1988, 175, 408–413 CrossRef CAS PubMed.
- I. Rahman, A. Kode and S. K. Biswas, Nat. Protoc., 2006, 1, 3159–3165 CrossRef CAS PubMed.
- R. Jevprasesphant, J. Penny, R. Jalal, D. Attwood, N. B. McKeown and A. D'Emanuele, Int. J. Pharm., 2003, 252, 263–266 CrossRef CAS PubMed.
- A. K. Gupta and S. Wells, IEEE Trans. Nanobioscience, 2004, 3, 66–73 CrossRef PubMed.
- D. Luo, K. Haverstick, N. Belcheva, E. Han and W. M. Saltzman, Macromolecules, 2002, 35, 3456–3462 CrossRef CAS.
- C. W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef CAS PubMed.
- T. Zhou, Y. Zhu, X. Li, X. Liu, K. W. K. Yeung, S. Wu, X. Wang, Z. Cui, X. Yang and P. K. Chu, Prog. Mater. Sci., 2016, 83, 191–235 CrossRef CAS.
- K. H. Jones and J. A. Senft, J. Histochem. Cytochem., 1985, 33, 77–79 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- A. Lankoff, W. J. Sandberg, A. Wegierek-Ciuk, H. Lisowska, M. Refsnes, B. Sartowska, P. E. Schwarze, S. Meczynska-Wielgosz, M. Wojewodzka and M. Kruszewski, Toxicol. Lett., 2012, 208, 197–213 CrossRef CAS PubMed.
- Y. Chang, S.-T. Yang, J.-H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- X. Cai, J. Hao, X. Zhang, B. Yu, J. Ren, C. Luo, Q. Li, Q. Huang, X. Shi, W. Li and J. Liu, Toxicol. Appl. Pharmacol., 2010, 243, 27–34 CrossRef CAS PubMed.
- S. J. Park, Y. C. Park, S. W. Lee, M. S. Jeong, K.-N. Yu, H. Jung, J.-K. Lee, J. S. Kim and M.-H. Cho, Toxicol. Lett., 2011, 207, 197–203 CrossRef CAS PubMed.
- K. Pulskamp, S. Diabate and H. F. Krug, Toxicol. Lett., 2007, 168, 58–74 CrossRef CAS PubMed.
- A. M. Studer, L. K. Limbach, L. Van Duc, F. Krumeich, E. K. Athanassiou, L. C. Gerber, H. Moch and W. J. Stark, Toxicol. Lett., 2010, 197, 169–174 CrossRef CAS PubMed.
- T. Thurnherr, C. Brandenberger, K. Fischer, L. Diener, P. Manser, X. Maeder-Althaus, J.-P. Kaiser, H. F. Krug, B. Rothen-Rutishauser and P. Wick, Toxicol. Lett., 2011, 200, 176–186 CrossRef CAS PubMed.
- G. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
| This journal is © The Royal Society of Chemistry 2017 |