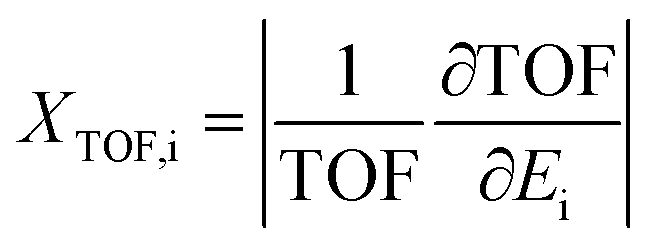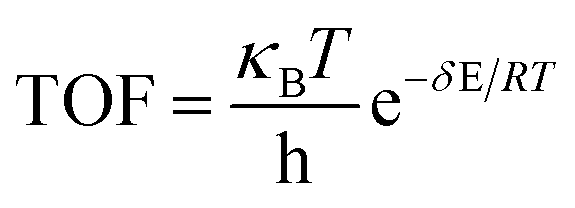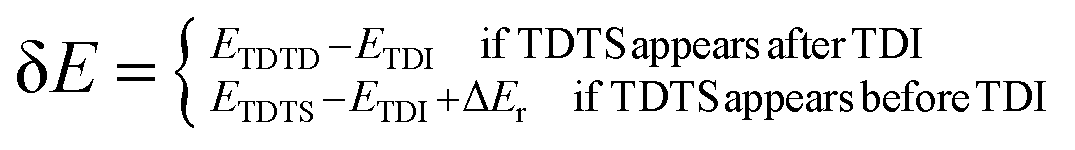DOI:
10.1039/C6RA27207J
(Paper)
RSC Adv., 2017,
7, 13473-13486
Theoretical insights into ω-alkynylfuran cycloisomerisation catalyzed by Au/CeO2(111): the role of the CeO2(111) support†
Received
23rd November 2016
, Accepted 20th February 2017
First published on 1st March 2017
Abstract
Understanding the role of CeO2(111) supports is meaningful for designing high-performance metal oxide-supported gold nanoparticle catalysts. Here, density functional theory calculations were employed to study ω-alkynylfuran cycloisomerisation on CeO2(111)-supported Au clusters. The various reactive sites, including the atop, interface and edge sites, and the role of oxygen vacancies on the defective CeO2(111) surface were taken into account. On the basis of the computed results and energetic analysis, it was found that the atop and interface sites show high catalytic activity toward ω-alkynylfuran cycloisomerisation, and the edge site of Au10/CeO2(111)-s (stoichiometric) exhibits similar catalytic activity. Meanwhile, the surface oxygen vacancies of CeO2(111) can cause some negative and positive effects on the adsorption energy and the catalytic activity. Furthermore, to shed light on the role of the CeO2(111) support, an analysis of ω-alkynylfuran cycloisomerisation catalyzed by Au3–4/CeO2(111) and free Au3–4 clusters was performed as well. Our results indicate that the presence of the CeO2(111) support can not only change the orbital levels of the gold clusters, but also cause charge recombination and decrease the positive charge on the Au (top) atom. These virtues can effectively lead to a decrease in the adsorption energy, facilitating electrophilic attack of the C atom on the furan ring.
Introduction
Metal-oxide-supported gold nanoparticle (NP) catalysts have attracted extensive attention in both theory and experiments owing to their extraordinary catalytic activities for numerous reactions, including the water–gas shift reaction,1,2 CO oxidation,3,4 and so on. Among the various metal oxides, due to its high oxygen storage and release capacity, and the fact that it contains highly localized 4f orbitals,5–7 cerium oxide/ceria (CeO2) has been recognized as an excellent supporting material. In addition to the above-mentioned properties, the formation of oxygen vacancies on the surface of CeO2 is easy, which has been demonstrated via experimental studies.8,9 The presence of these oxygen vacancies can lead to strong binding between Au nanoparticles and the CeO2 surface.10 In addition, oxygen vacancy formation on the CeO2 surface can result in the reduction of adjacent Ce4+ ions to Ce3+ ions, so the concentration of Ce3+ ions is closely related with the number of oxygen vacancies, exhibiting a proportional relationship.9 Meanwhile, according to previous reports,11 oxygen vacancies on the CeO2 surface can play a curial role in the catalytic process. In a nutshell, CeO2-based catalysts have attracted wide attention due to the unique characteristics of the CeO2 surface.
CeO2-supported Au catalysts have been regarded as promising catalytic systems because they include two highly active components, namely, the CeO2 surface and Au nanoparticles.12,13 Ciftci et al. prepared a set of catalysts with Au and Pt supported on CeO2 nanorods and nanocubes, which were applied as catalysts in the decomposition of ethanol and formic acid.14 They found that Au catalysts are more active and selective toward H2 than Pt catalysts in formic acid decomposition. Fu and coworkers reported that nanostructured gold–cerium oxide catalysts are active for the water–gas shift reaction.15 Based on the studied results, they found that the metal nanoparticles do not take part in the reaction, and that the reaction is actually catalyzed by non-metallic gold species strongly associated with surface cerium oxygen groups. Han and colleagues prepared low Au-loaded CeO2 nanoparticle catalysts with high activity via several solution chemistries, and used them as catalysts for low-temperature CO oxidation.16 They discovered that the Au-loaded CeO2 nanoparticle catalysts exhibit high catalytic activity because of their large surface area as well as the coexistence of Auδ+ (Au+ and Au3+) and Au0 species, in which Au+ plays a predominant role. Therefore, in order to recognize and design a highly active ceria-based catalyst, it is extremely important to illustrate the nature of the active sites and the catalytic activity of CeO2-supported Au nanoparticles in detail.
Lattice oxygen atoms17 and under-coordinated Au atoms in Au nanoparticles18 have both been reported as reactive species for CO oxidation catalyzed by CeO2-supported Au nanoparticles. CO oxidation, as a prototypical reaction in heterogeneous catalysis,19 has attracted enormous attention and has been extensively studied. Kim et al. compared CO oxidation by Au13 nanoparticles supported on stoichiometric and partially reduced CeO2 with the help of DFT+U calculations, and confirmed the critical role of oxygen vacancies in the CeO2 surface. They also suggested that lowering the vacancy formation energy of the CeO2 surface is a promising method for enhancing the CO catalytic activity.11 Additionally, analysis shows that CO oxidation by the Mars–van Krevelen mechanism at the interface between doped CeO2 and supported Au nanoparticles is more effective than that at undoped CeO2 surfaces.20 In general, for nanocatalysts, it is meaningful to understand the interface effects of metal/support catalysts. The interface between the metal nanoparticles and the oxide supporting materials can cause a pronounced effect on the catalytic activity.21–23 Recently, on the basis of a theoretical study, Bing Liu et al. explored the three reaction mechanisms for CO oxidation on CeO2-supported Pd nanorods, that is, the Pd-Ce3+ dual site mechanism, M–vK mechanism and Pd-only mechanism.24 By analyzing the calculated results, they found that interface effects play a crucial role and can strongly affect the catalytic activity, which can be understood by considering the structural and electronic properties of the materials. In subsequent work, CO oxidation on CeO2-supported Pd bimetallic nanorods was investigated by means of a theoretical method and it was found that CO and O2 can bind at different sites on Pd-Ag/CeO2 and Pd-Cu/CeO2.25 The results suggested that both the Pd-Ag and Pd-CeO2 interfaces in Pd-Ag/CeO2 can play a vital effect on CO oxidation. However, according to some previous reports,4,26 the catalytic activity of oxide-supported Au nanoparticles originates predominantly from the Au nanoparticles themselves, and the oxide surface is regarded as a stabilizer for the Au nanoparticles. Therefore, without doubt, knowledge of the reactive species, including the oxygen vacancies and under-coordinated Au atoms, and the interface between the Au nanoparticles and the supporting material, is meaningful for designing high-performance catalysts and understanding the origin and nature of the catalytic activity.27–30
In order to achieve this goal, the mechanism of ω-alkynylfuran cycloisomerisation on CeO2(111)-supported subnanometer gold clusters is investigated via a theoretical study in this article. Cycloisomerisation is an effective strategy used to obtain phenols and their derivatives, which is extensively used to synthesize natural products, polymers and pharmaceuticals.31–36 According to a previous study, Au/CeO2 can efficiently catalyze cycloisomerisation, leading to a high conversion ratio.35 However, to date, the mechanism of ω-alkynylfuran cycloisomerisation catalyzed by CeO2-supported gold nanoparticles has not been elaborated or unveiled in detail. Herein, ω-alkynylfuran cycloisomerisation catalyzed by CeO2-supported gold nanoparticles (Au3, Au4, Au10, Au11) is explored with the help of density functional theory (DFT), as an expansion of our prior research.37–39 Gold nanoparticles (Au3, Au4, Au10, Au11) were employed for consistence with experimental results reported by Oliver-Meseguer et al. and others.40–42 In these studies, it was found that small gold clusters (only 3 to 10 atoms in size) can be formed during the catalytic processes, which can catalyze the ω-alkynylfuran cycloisomerisation reaction. In this paper, only the 5-exo Friedel–Crafts-type reaction pathway was investigated, and other possible reaction pathways were not considered. The 5-exo Friedel–Crafts-type mechanism (FCT) is the most feasible pathway of ω-alkynylfuran cycloisomerisation to produce the targeted phenol derivative product, catalyzed by subnanometer gold clusters and planar gold clusters, based on our previous reports.37–39 In order to illustrate the catalytic role of the supporting material, stoichiometric CeO2 and partially reduced CeO2 with one vacancy are also considered here. In addition, the possible catalytic sites of Aun/CeO2(111), including the atop, edge and interface sites, are investigated, aiming to illustrate the catalytic activity of the catalytic sites and interfacial effects. Other clusters, such as Pd, Pt or Ag clusters, supported on the surface of CeO2 will be employed in future work to explore the effect of different clusters on the catalytic properties for ω-alkynylfuran cycloisomerisation.
Computational models and methods
In this work, all of the results were obtained with the help of spin-unrestricted density functional theory (DFT) in a generalized gradient approximation of the PBE functional.43 For geometric optimization, all-electron scalar relativity was chosen, along with the double numerical plus p-polarization (DNP) basis set.44 Dmol3 software was employed, which uses a local pseudopotential acting on all electrons, including those in the core, to get scalar relativistic corrections for the relevant valence orbitals. In order to accelerate convergence, a thermal smearing of 0.005 Hartree was applied to the orbital occupation, but all energies were extrapolated to 0 K. Additionally, the transition states were calculated using the linear and quadratic synchronous transit (LST/QST) method, and confirmed by nudged elastic band calculations and relaxed potential energy surface scans.45,46 The solvent effect of acetonitrile (dielectric constant: 37.50) was evaluated via the conductor-like screening model (COSMO) solvation procedure.47–49 All the calculations mentioned above were realized using the DMol3 software package.50,51
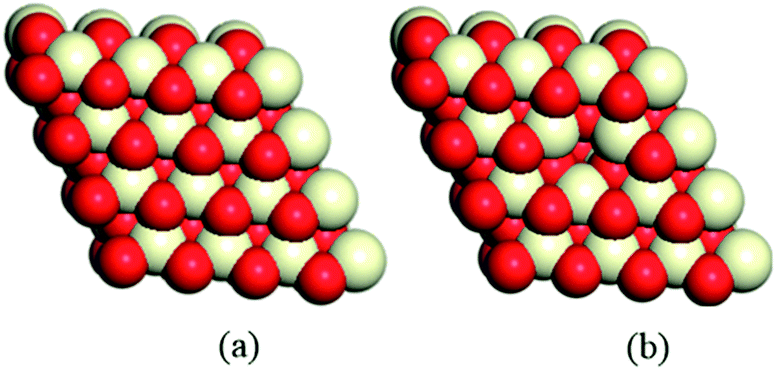
It is well-known that the CeO2(111) surface is the most thermodynamically stable among the three low-index surfaces of ceria, that is, CeO2(111), CeO2(110) and CeO2(100), implying that the CeO2(111) surface is the most commonly exposed surface of ceria.8,52 Therefore, a 4 × 4 CeO2(111) slab with 6 atomic layers53,54 was employed to describe the stoichiometric CeO2 surface (CeO2-s) and the reduced surface (with gold clusters on an oxygen vacancy, CeO2-v), and 30 Å of vacuum (Fig. 1) was used in order to minimize the interaction between cells. During the geometry optimization, the three upper atomic layers of the CeO2 slab were relaxed, while the other remaining atomic layers were fixed. Consideration of the enough large surface cells for all models, only a gamma-point mesh was chosen for Brillouin zone integration. In addition, the bulk equilibrium lattice constant (5.44 Å) was calculated by means of the PBE/DNP method with 8 × 8 × 8 k-points, which is consistent with the experimental values55 and previous theoretical studies.56–58
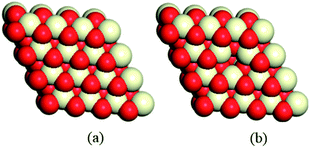 |
| | Fig. 1 A 4 × 4 CeO2(111) slab is used to model (a) stoichiometric CeO2 and (b) partially reduced CeO2 with one vacancy. | |
Usually, to correct the resulting electronic structure of reduced ceria, the Hubbard-U term in the DFT+U approach is employed. However, according to a paper reported by Da Silva et al., a reasonable description of the structural parameters, relative energies of different oxides and spectroscopic properties cannot be given at the same time using a unique value of U; therefore, it is not clear whether the DFT+U method provides a systematic improvement of the energetics.59 In addition, in Bernard Delley and coworkers' reports, the stability and morphology of cerium oxide surfaces in an oxidizing environment, the adsorption and thermodynamic stability of CHx (x = 1–4) on a CeO2(111) surface, and water adsorption on stoichiometric and reduced CeO2(111) surfaces were explored by employing the DFT-GGA approach using Dmol3 software and the results were reliable and comparable.60–62 The same choice was made to study CO adsorption on CeO2 and investigate the interaction of water with stoichiometric and reduced CeO2 surfaces by Yang et al. and Kumar et al., respectively.63,64 Hence, in view of these considerations, the DFT-GGA approach is also adopted in this article.
Additionally, herein, the turnover frequency (TOF) was employed to evaluate the efficiency of a catalytic cycle. The energetic span model can be used to predict the TOF from theoretically obtained free energy profiles, which was proposed by Shaik and co-workers.65–68 The Gibbs free energy is related to the corrected enthalpy (ΔHcorrect) and entropy (ΔS). These two parameters were not considered because numerical frequency computations are difficult for large system in Dmol3. On the basis of this model, the TOF-determined transition state (TDTS) and intermediate (TDI) can be determined by evaluating the degree of TOF control (XTOF,i), as shown in eqn (1):
| |
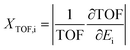 | (1) |
Ei denotes the energy of the transition state or intermediate. The larger the
XTOF value, the higher the influence of the corresponding state (transition state or intermediate) on the TOF. The TOF can be calculated using
eqn (2):
| |
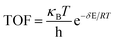 | (2) |
where δ
E is the energetic span, which can be defined as shown in
eqn (3):
| |
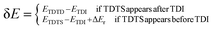 | (3) |
All of the kinetic information was obtained by applying the energetic span model using the AUTOF program.67
Results and discussion
Morphology of gold clusters on CeO2(111)
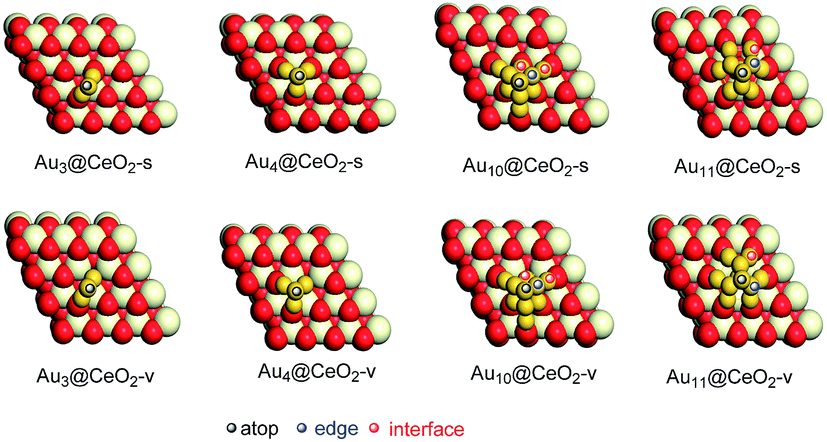
Before the reaction mechanism of ω-alkynylfuran cycloisomerisation catalyzed by Aun/CeO2 was unveiled, the atomic structures of the gold clusters on the CeO2(111) surface were investigated. For Au3 and Au4 clusters supported on CeO2(111) surfaces, all possible configurations are taken into account. However, for Au10 and Au11 clusters supported on CeO2(111) surfaces, only the fcc-like and hcp-like clusters are considered, as these have been proven to be the stable structures of gold clusters on CeO2(111) surfaces.69 Three structures for Au3 clusters on stoichiometric CeO2(111) surfaces, including triangle perpendicular to the surface, triangle parallel to the surface and linear parallel to the surface, are considered. According to the calculated results, it is demonstrated that the triangle perpendicular to the CeO2(111) surface structure is the most stable among the three configurations. Nevertheless, only two configurations, namely, triangle perpendicular to and linear parallel to the surface Au3 clusters on reduced CeO2(111) surfaces, were found, because during optimization the triangle parallel to the surface configuration converged to the triangle perpendicular to the surface configuration. Similarly, the triangle structure is also more stable than the linear structure. For Au4 clusters on both stoichiometric and reduced CeO2(111) surfaces, three different structures were revealed based on the theoretical calculations. The computed results show that the tetrahedral Au4 cluster is the most stable structure on both stoichiometric and reduced CeO2(111) surfaces. The stable structures of Au3 and Au4 clusters on CeO2(111) surfaces are in line with previous theoretical studies using PBE+U (U = 5.0 eV).69 In addition, the tetrahedral Au10 cluster is more stable than the hemispherical structures, which is also in agreement with the PBE+U (U = 5.0 eV) study.69 It is well known that, for Au4–Au10 clusters, the most stable isomers are planar structures in the gas phase, indicating that the CeO2 support results in structural changes from planar to three-dimensional. The corresponding optimized structures are shown in Fig. S1† and the most stable structures are also exhibited in Fig. 2, together with the possible catalytic sites.
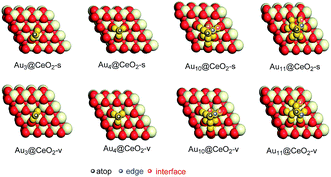 |
| | Fig. 2 The most stable structures of gold clusters on CeO2(111) and possible catalytic sites of Aun/CeO2(111). | |
Electronic properties of Aun/CeO2(111)
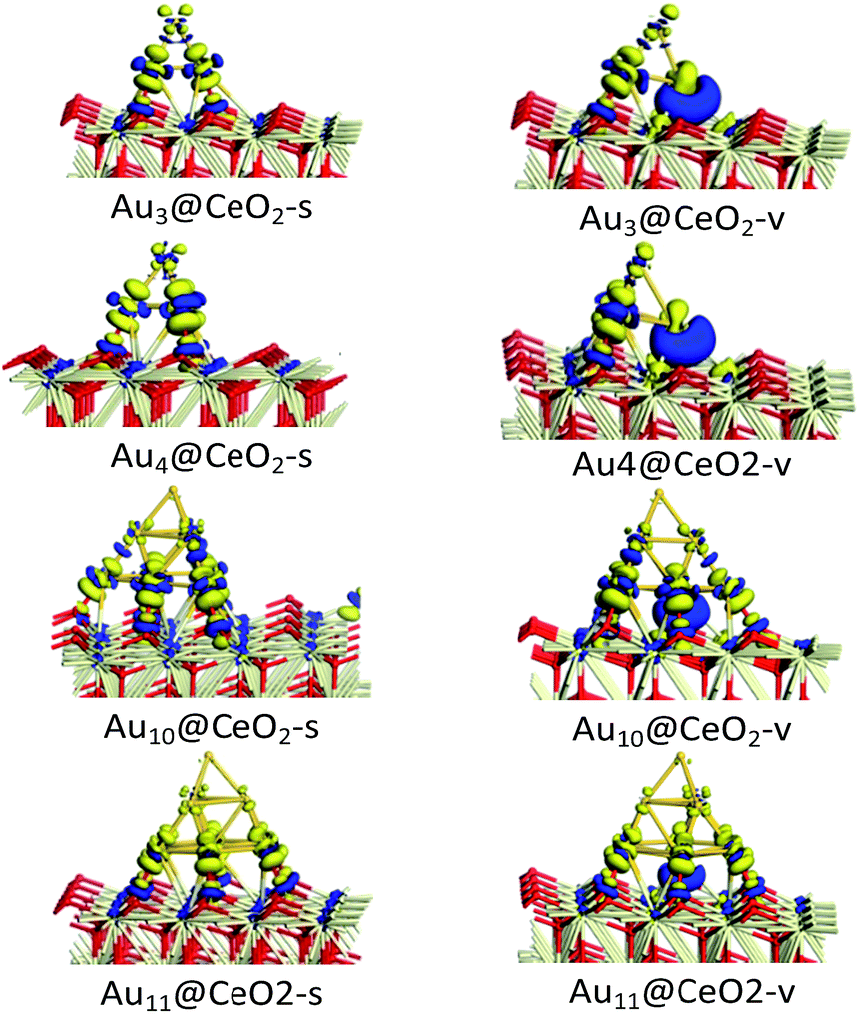
To illustrate the electronic properties of the interactions between Au clusters and CeO2 surfaces, the charge density differences of Aun/CeO2(111) were calculated, as shown in Fig. 3. According to Fig. 3, it can be concluded that significant charge transfer occurs between the Au cluster and the CeO2 support. The electronic interaction is largely restricted to the bottom Au atoms of the cluster, which are in direct contact with the CeO2 support, indicating that these atoms have been oxidized. In contrast, little decrease in the electron density is observed for the top Au layer, illustrating that this layer is much less affected electronically by the interaction with the CeO2 surface. In addition, analysis of charge transfer between the Au cluster and the CeO2 support indicates that the increase in electron density for Aun@CeO2-v mainly occurs near the oxygen vacancy, and is obviously different to that seen for Aun@CeO2-s. These results indicate that charge transfer happens at the interface between the Au cluster and the CeO2 support, and the presence of an oxygen vacancy can have a large effect on charge transfer.
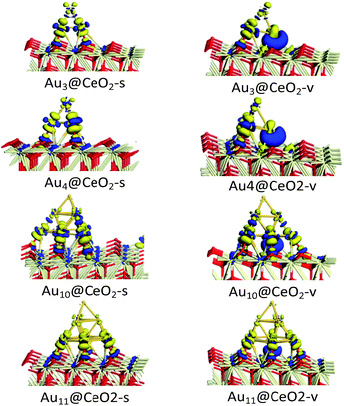 |
| | Fig. 3 Charge density difference of Aun/CeO2(111) (blue and yellow isosurfaces represent increasing and decreasing electron densities, respectively; isovalue = 0.03). | |
The possible mechanisms for ω-alkynylfuran cycloisomerisation catalyzed by Au3/CeO2
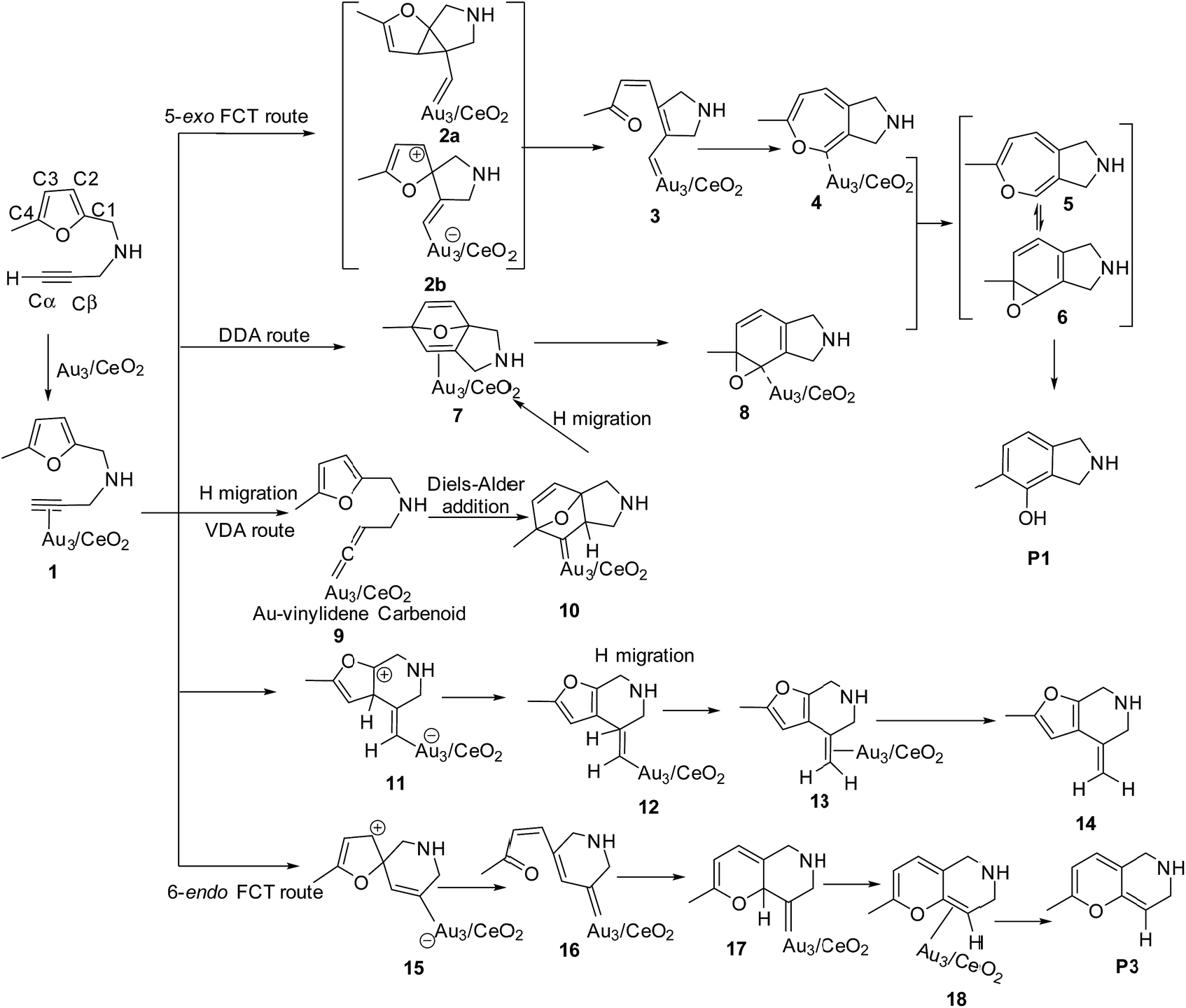
To illustrate the most feasible pathway for ω-alkynylfuran cycloisomerisation, Au3/CeO2 was selected as a model catalyst to simulate the detailed mechanism of ω-alkynylfuran cycloisomerisation. The possible pathways of ω-alkynylfuran cycloisomerisation catalyzed by Au3/CeO2 were designed and are shown in Scheme 1. As shown in Scheme 1, after the adsorption of the substrate on Au3/CeO2, there are three possible pathways to produce phenol derivatives. The first one is the 5-exo Friedel–Crafts-type reaction route starting from the nucleophilic addition of a Cβ atom to the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 bond or C1 site of the furan ring (abbreviated as 5-exo FCT). The second pathway is [4 + 2] cycloaddition between the C
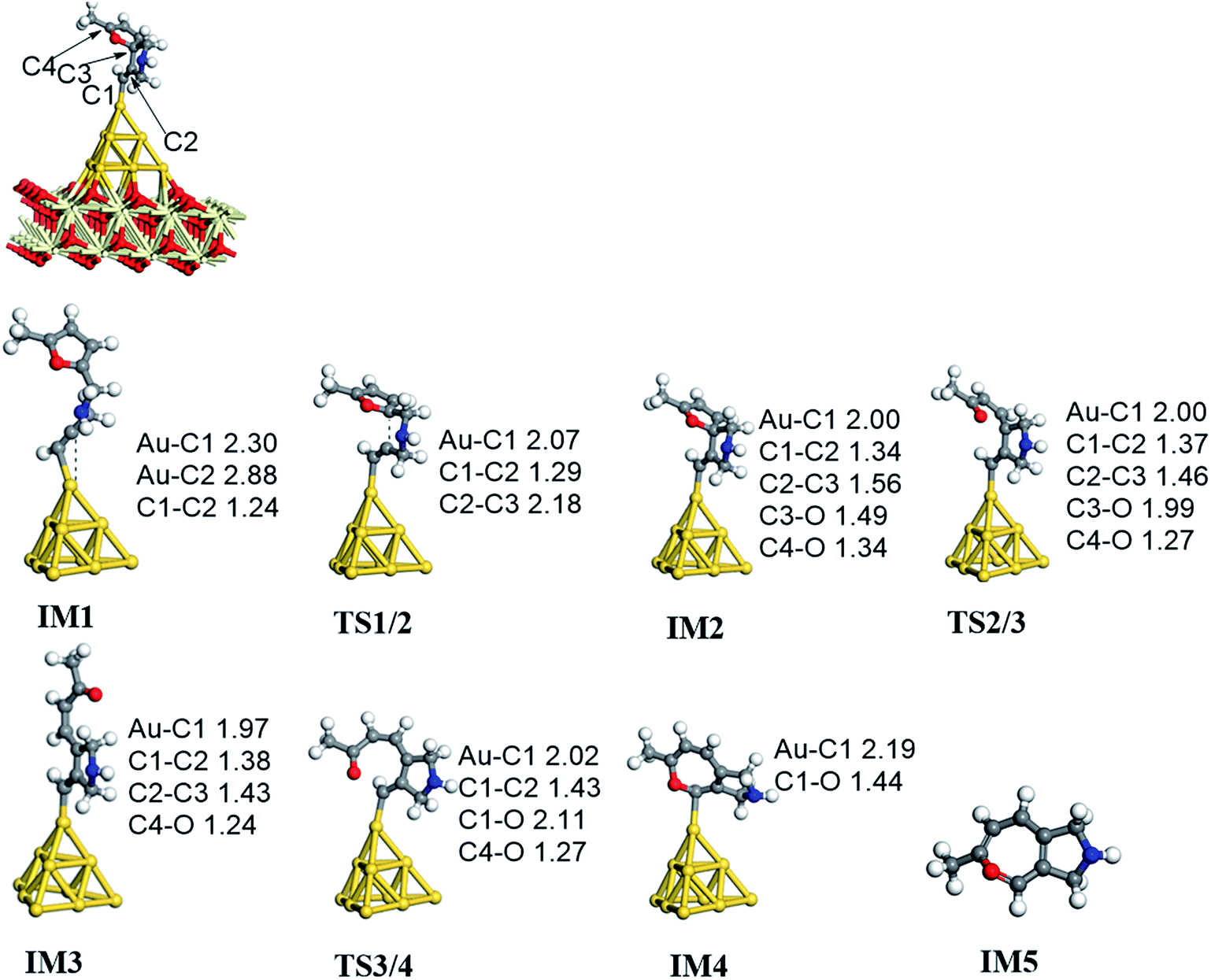
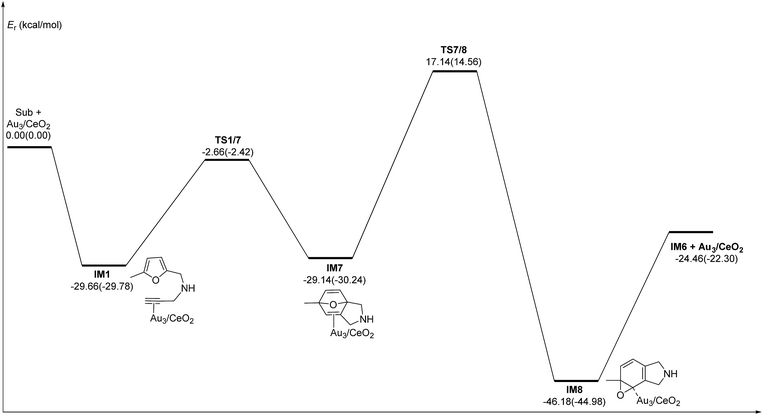
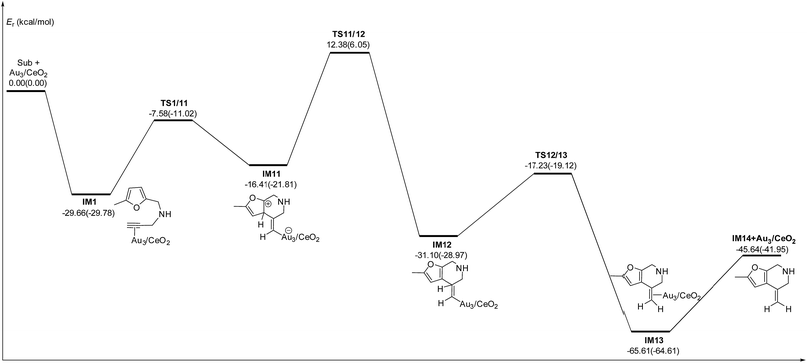
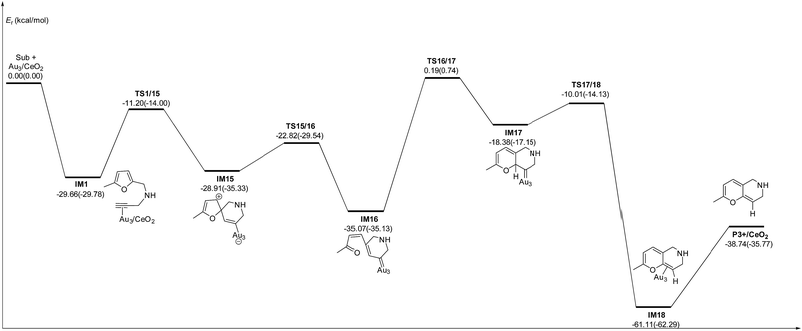
C2 bond or C1 site of the furan ring (abbreviated as 5-exo FCT). The second pathway is [4 + 2] cycloaddition between the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond and the C1C2–C3C4 of the furan ring to produce the final product (abbreviated as DDA). The last pathway is direct hydrogen transfer from the Cα atom to the Cβ atom to form an Au-vinylidene carbenoid intermediate, and subsequent Diels–Alder addition of the Cα–Cβ double bond to the furan ring (abbreviated as VDA). Additionally, there is a 6-endo FCT pathway, which involves the nucleophilic addition of a Cα atom to the furan ring in IM1 in competition with the 5-exo FCT pathway, finally leading to a pyran derivative. Moreover, the Cβ atom can attack the C2 atom of the furan ring, finally obtaining a β-alkenylated furan via H migration. The energy profiles for all of the possible mechanisms are depicted in Fig. 4–7. For the VDA route, H migration requires a large energy barrier to be overcome, and therefore it is not taken into account. According to Fig. 4–7, the rate-limiting step of the 5-exo FCT reaction route in the gas phase is the nucleophilic addition of the Cβ atom to the C1 site of the furan ring to form the spiro-intermediate IM2, which has an energy barrier of 13.66 kcal mol−1, while the formation of IM4 is the rate-determining step in acetonitrile. However, for the DDA pathway, the formation of the β-alkenylated furan and the 6-endo FCT pathway, the energy barriers of the rate-limiting steps are 46.28 (44.80), 28.79 (27.86) and 35.26 (35.87), respectively. Obviously, the 5-exo FCT reaction route is the most feasible pathway for ω-alkynylfuran cycloisomerisation to produce the targeted phenol derivative product.
C bond and the C1C2–C3C4 of the furan ring to produce the final product (abbreviated as DDA). The last pathway is direct hydrogen transfer from the Cα atom to the Cβ atom to form an Au-vinylidene carbenoid intermediate, and subsequent Diels–Alder addition of the Cα–Cβ double bond to the furan ring (abbreviated as VDA). Additionally, there is a 6-endo FCT pathway, which involves the nucleophilic addition of a Cα atom to the furan ring in IM1 in competition with the 5-exo FCT pathway, finally leading to a pyran derivative. Moreover, the Cβ atom can attack the C2 atom of the furan ring, finally obtaining a β-alkenylated furan via H migration. The energy profiles for all of the possible mechanisms are depicted in Fig. 4–7. For the VDA route, H migration requires a large energy barrier to be overcome, and therefore it is not taken into account. According to Fig. 4–7, the rate-limiting step of the 5-exo FCT reaction route in the gas phase is the nucleophilic addition of the Cβ atom to the C1 site of the furan ring to form the spiro-intermediate IM2, which has an energy barrier of 13.66 kcal mol−1, while the formation of IM4 is the rate-determining step in acetonitrile. However, for the DDA pathway, the formation of the β-alkenylated furan and the 6-endo FCT pathway, the energy barriers of the rate-limiting steps are 46.28 (44.80), 28.79 (27.86) and 35.26 (35.87), respectively. Obviously, the 5-exo FCT reaction route is the most feasible pathway for ω-alkynylfuran cycloisomerisation to produce the targeted phenol derivative product.
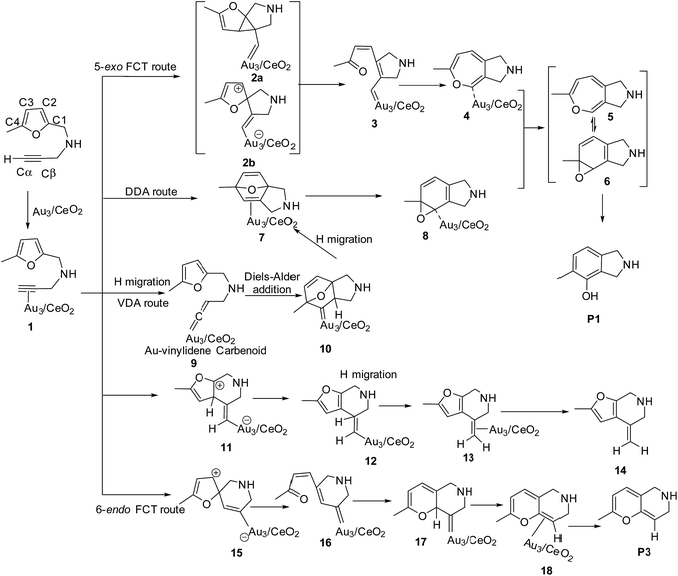 |
| | Scheme 1 The 5-exo Friedel–Crafts-type (FCT) route, direct Diels–Alder-type (DDA) route and Au-vinylidene carbenoid (VDA) route to form the phenol derivative P1, the formation of β-alkenylated furan P2, and the 6-endo Friedel–Crafts-type (FCT) route toward the pyran derivative P3. | |
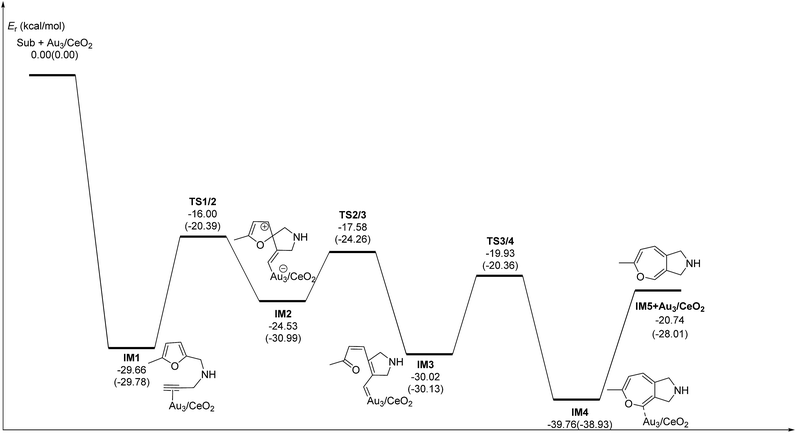 |
| | Fig. 4 Energy profile of the 5-exo FCT pathway that was used to form the phenol derivative P1 in the gas phase and acetonitrile solvent (parentheses). | |
 |
| | Fig. 5 Energy profile of the DDA pathway that was used to form the phenol derivative P1 in the gas phase and acetonitrile solvent (parentheses). | |
 |
| | Fig. 6 Energy profile for the formation of the β-alkenylated furan P2 in the gas phase and acetonitrile solvent (parentheses). | |
 |
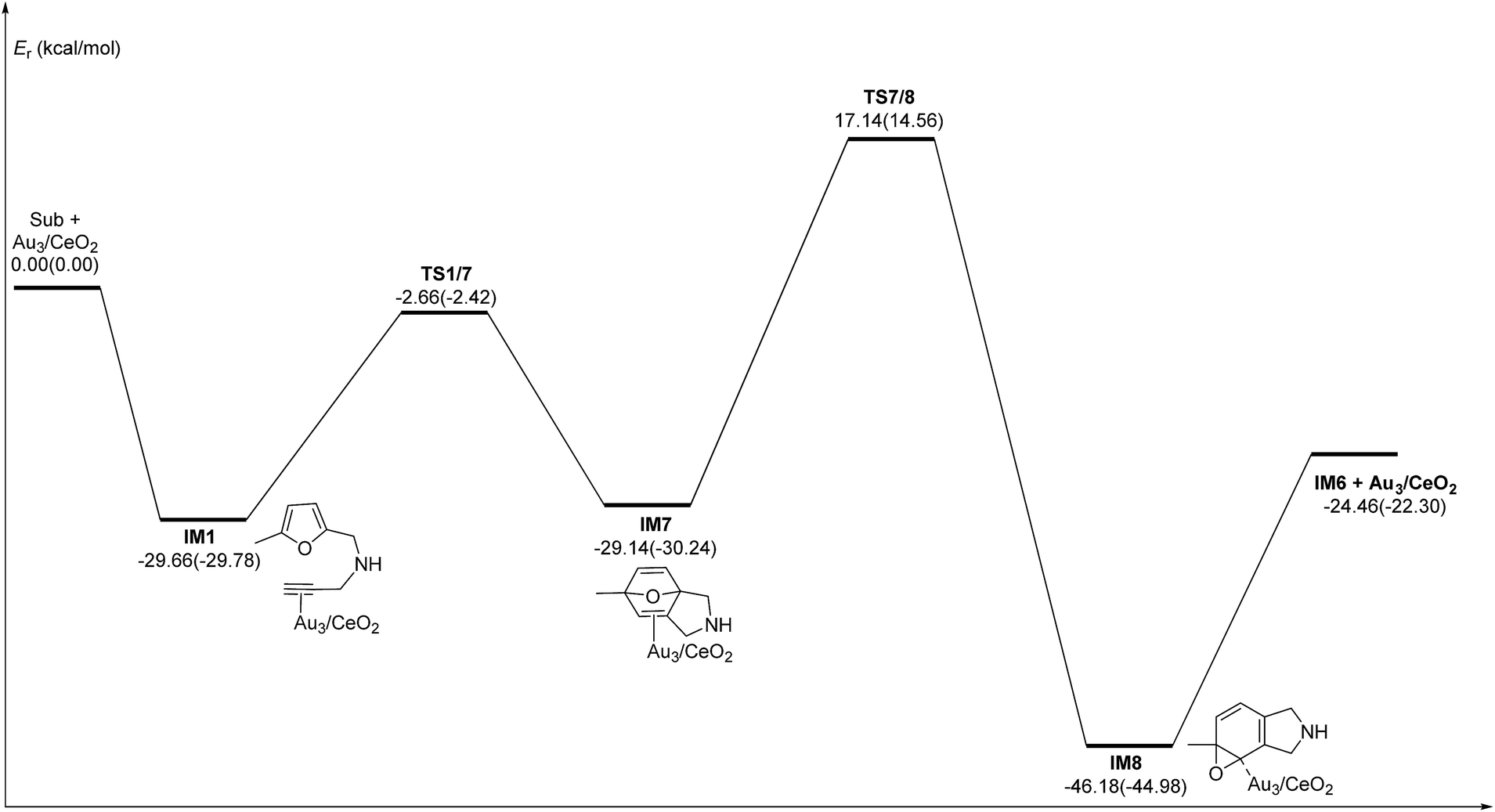
| | Fig. 7 Energy profile of the 6-endo Friedel–Crafts-type (FCT) pathway that was used to form the pyran derivative P3 in the gas phase and acetonitrile solvent (parentheses). | |
Mechanism of ω-alkynylfuran cycloisomerisation catalyzed by Au10/CeO2(111) and Au11/CeO2(111)
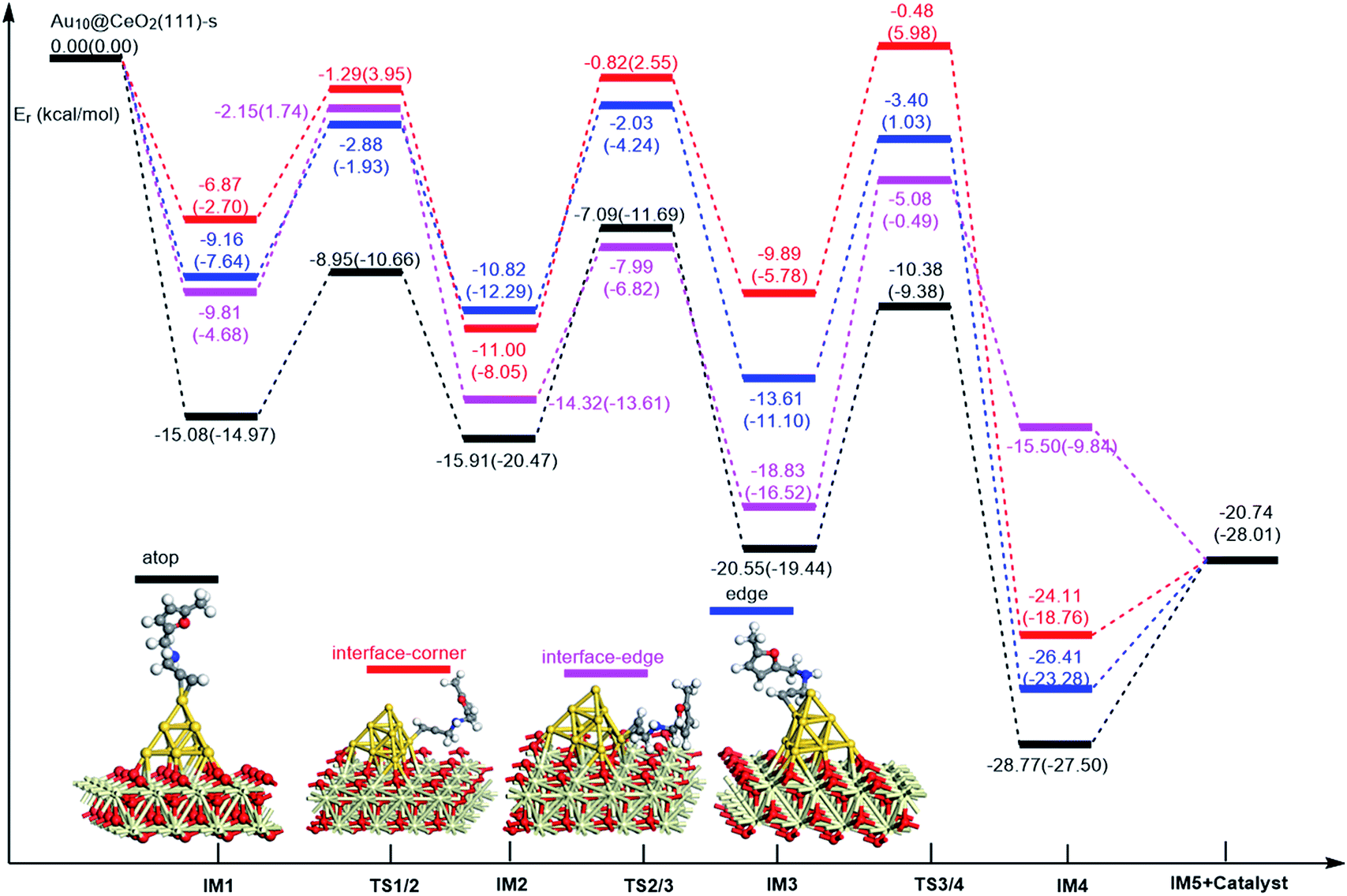
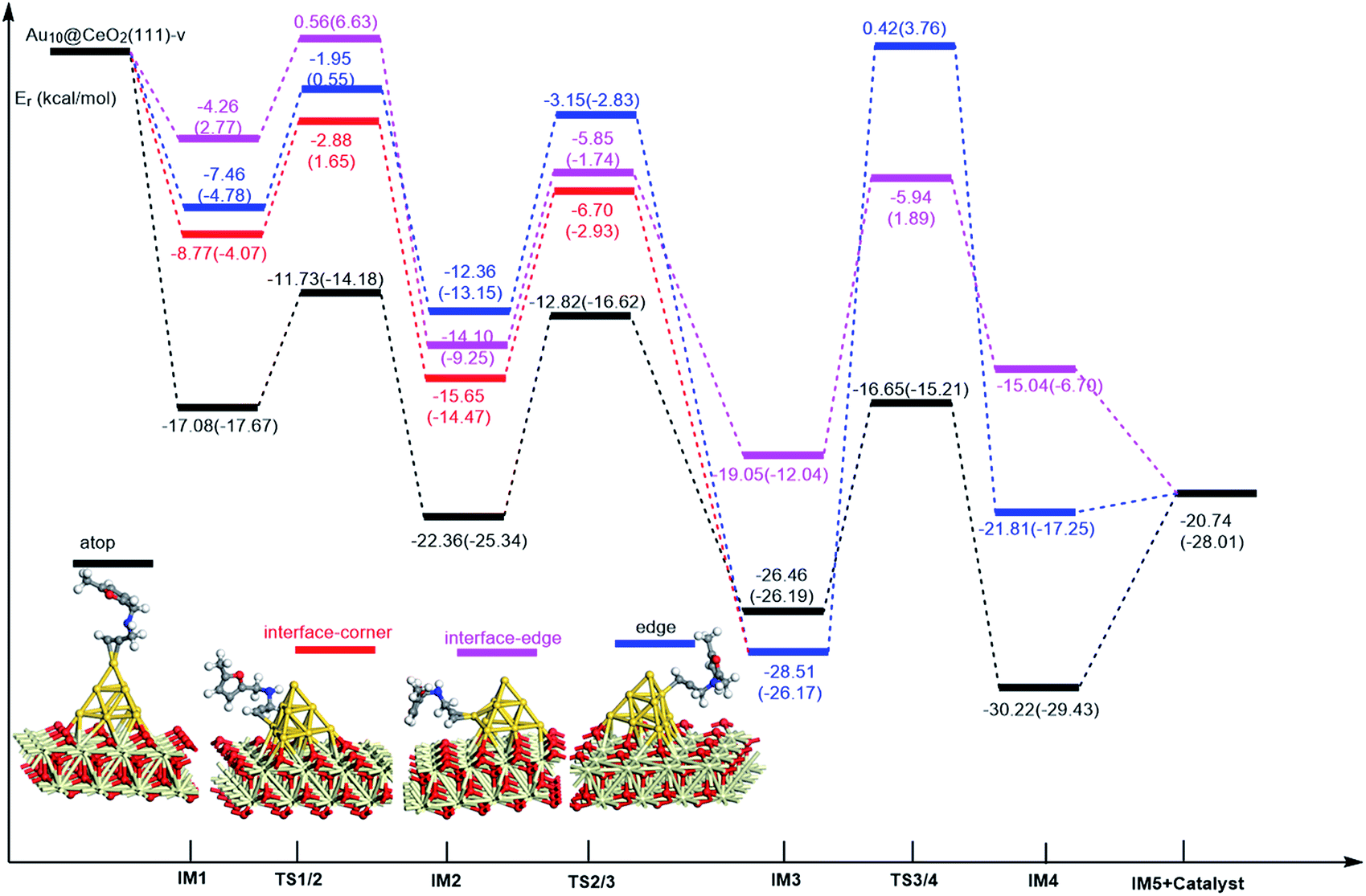
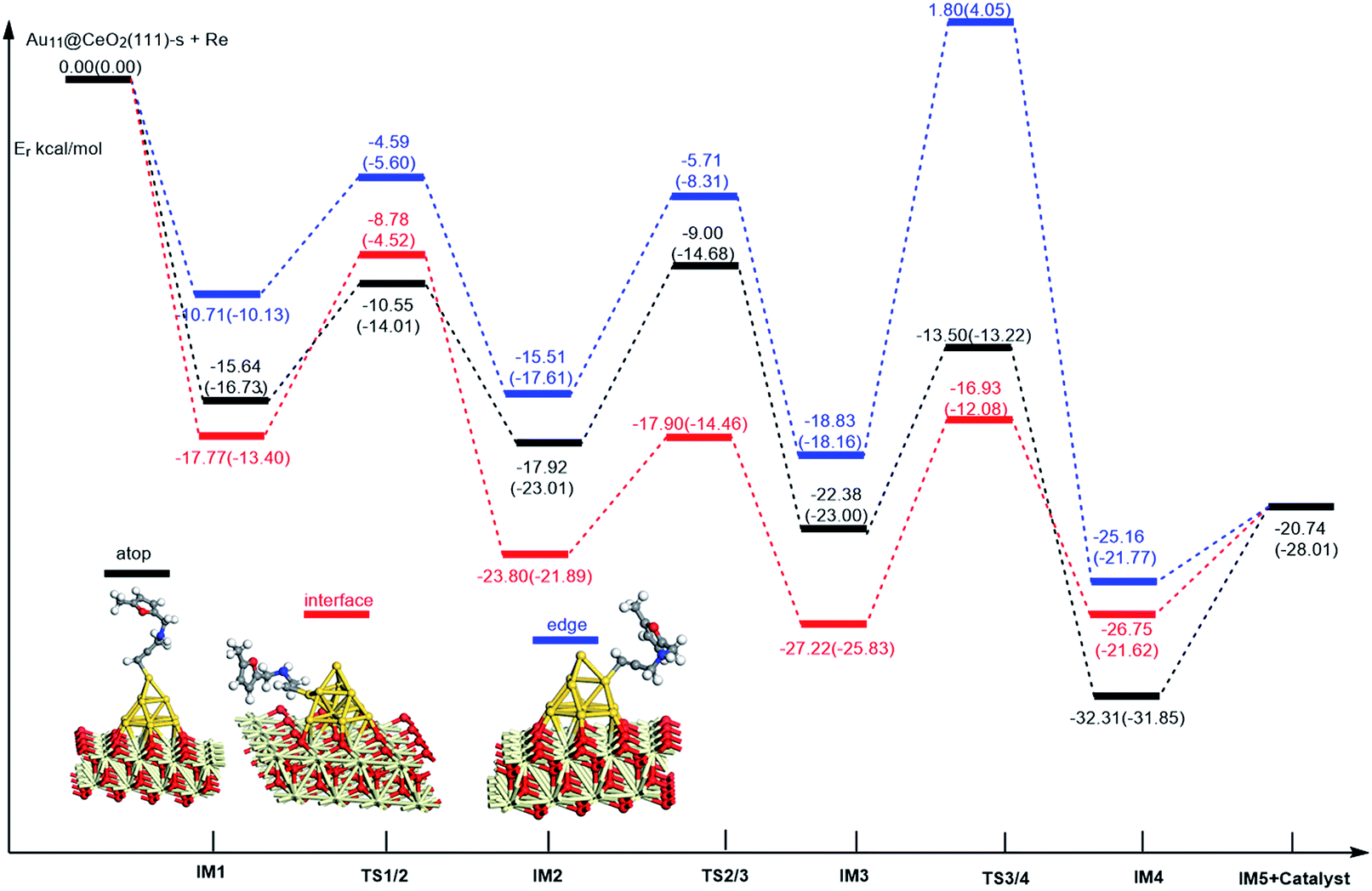
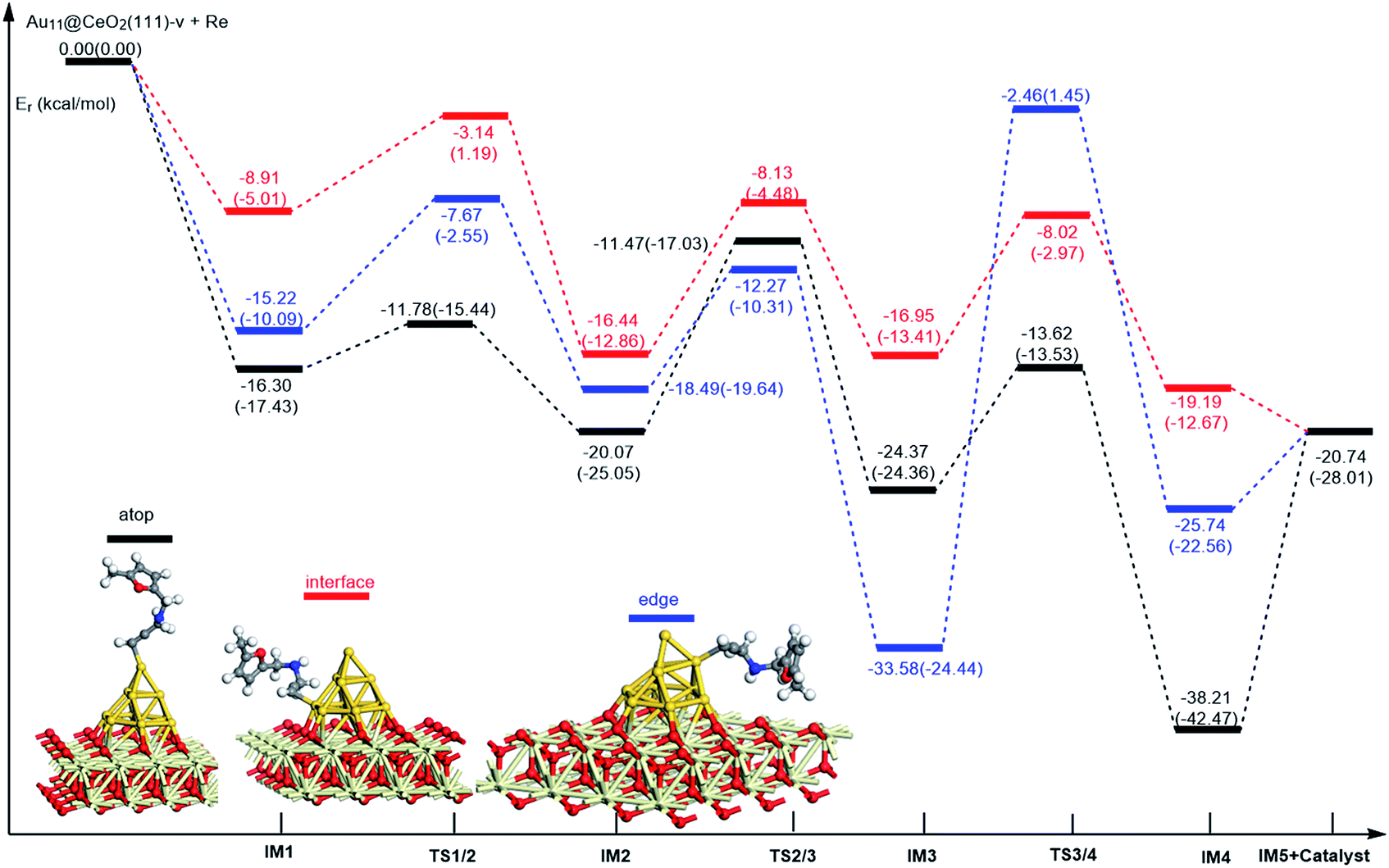
On the basis of our previous reports37–39 and the above discussions, the 5-exo Friedel–Crafts-type mechanism (FCT) is the most feasible pathway for ω-alkynylfuran cycloisomerisation. Accordingly, the 5-exo FCT mechanism of ω-alkynylfuran cycloisomerisation on the various reaction sites of Au10/CeO2(111) and Au11/CeO2(111) were investigated. The corresponding reaction sites are exhibited in Fig. 2. The optimized structures and related parameters of the reaction catalyzed by Au10/CeO2(111)-s are shown in Fig. 8, and the corresponding structures and parameters for the other Aun/CeO2 materials are plotted in Fig. S2–S14 (ESI†). Moreover, the potential energy profiles are presented in Fig. 9–12.
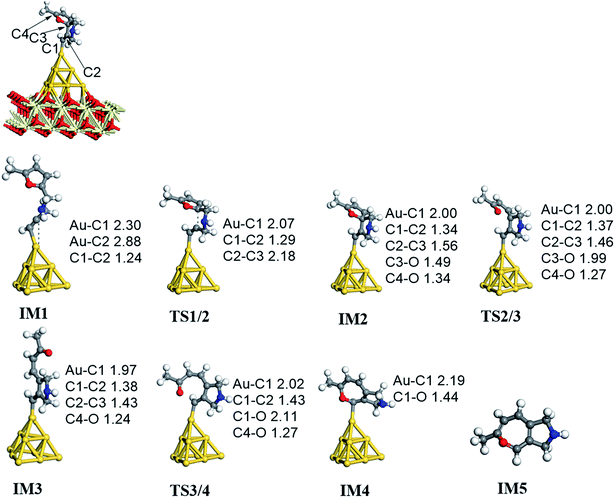 |
| | Fig. 8 The optimized structures of ω-alkynylfuran cycloisomerisation catalyzed by the atop site of Au10/CeO2(111)-s. | |
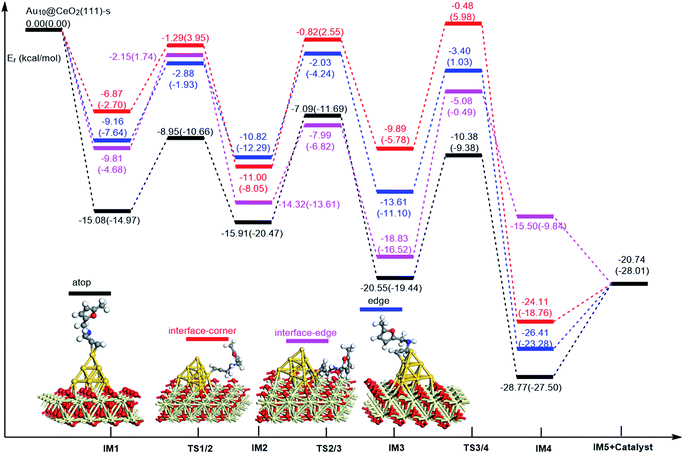 |
| | Fig. 9 Energy profile for ω-alkynylfuran cycloisomerisation on stoichiometric CeO2-supported Au10. | |
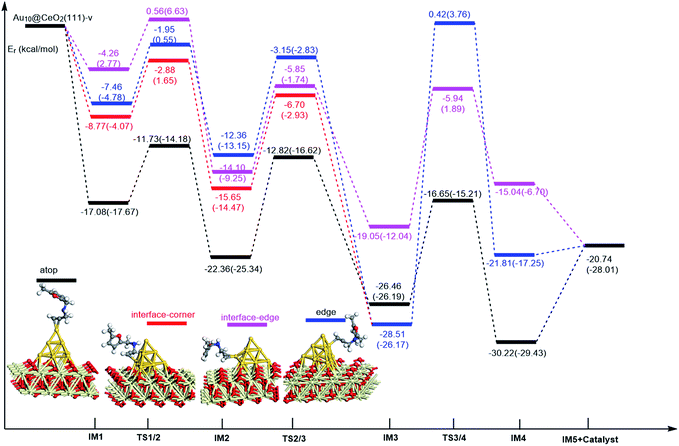 |
| | Fig. 10 Energy profile for ω-alkynylfuran cycloisomerisation on reduced CeO2-supported Au10. | |
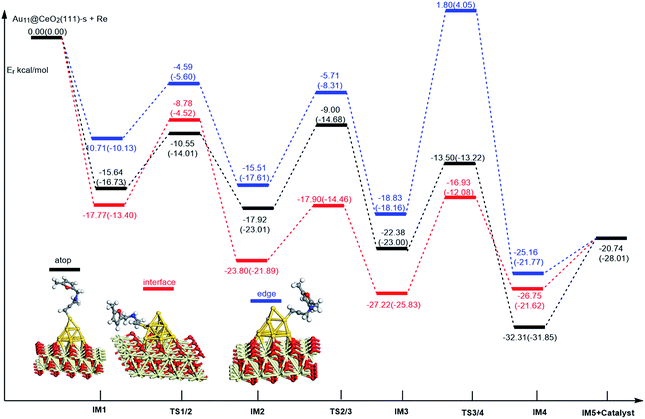 |
| | Fig. 11 Energy profile for ω-alkynylfuran cycloisomerisation on stoichiometric CeO2-supported Au11. | |
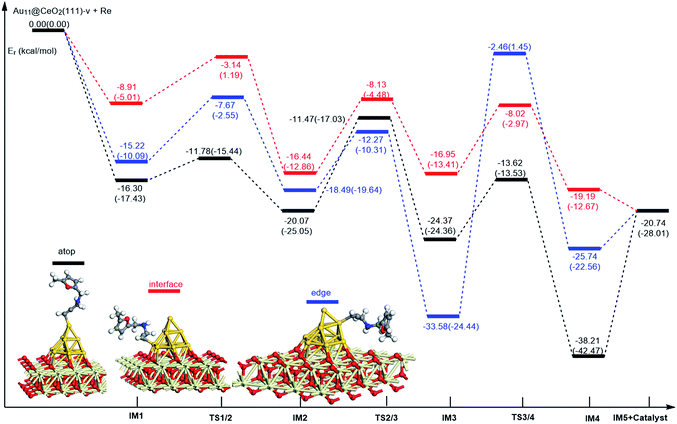 |
| | Fig. 12 Energy profile for ω-alkynylfuran cycloisomerisation on reduced CeO2-supported Au11. | |
Firstly, ω-alkynylfuran cycloisomerisation on the atop sites of Au10/CeO2(111) and Au11/CeO2(111) is discussed. As shown in Fig. 5–8, the adsorption of ω-alkynylfuran on the atop sites is exothermic by about 15–18 kcal mol−1 in the gas phase and acetonitrile solvent (Ca (catalyst) + Sub (substrate) → IM1 (intermediate)). The adsorption energies of the substrates on the atop sites of the gold clusters supported on the CeO2(111) surfaces are comparable with those on subnanometer gold clusters (Au38, Au27, Au28, Au30, Au35 and Au55).38 In addition, analysis of the computed adsorption energies exhibited in Fig. 5–8 suggests that the surface O vacancy of CeO2(111) only has a slight influence on the adsorption of the substrate on the atop sites. Once the ω-alkynylfuran is adsorbed on the atop sites of the gold clusters supported on the CeO2(111) surfaces, the Cβ atom electrophilically attacks the furan ring to give rise to a spiro-intermediate, IM2 (IM1 → IM2). This step is kinetically feasible, with an energy barrier of about 5 kcal mol−1. In addition, the IM1 → IM2 conversion step is exothermic by about 5–8 kcal mol−1 in acetonitrile solvent. Subsequently, the cleavage of the C–O bond of the furan ring in IM2 produces a linear conjugate triene ketone intermediate (IM3) through transition state TS2/3 with an energy barrier of 8–9 kcal mol−1. This step is nearly thermoneutral in acetonitrile solvent. In the next step, an oxygen atom from a carbonyl group approaches the Au–C bond yielding an oxepine intermediate (IM4), which is exothermic in both the gas phase and acetonitrile solvent with an energy barrier of about 8–11 kcal mol−1. Finally, oxepine is desorbed from the oxepine intermediate to recover the catalyst. This step is endothermic by about 8–18 kcal mol−1. The oxepine then quickly converts into the phenol product with an exothermicity of about 40 kcal mol−1 upon heating or exposing to acid.33,34,70 Additionally, on the basis of the energetic span model by Shaik and coworkers, the TOF values of ω-alkynylfuran cycloisomerisation on the atop, interface-corner, interface-edge and edge sites are calculated and shown in the Table 1. Compared with ω-alkynylfuran cycloisomerisation on the interface-corner, interface-edge and edge sites, the atop site has larger TOF values of about 105 to 106 s−1, indicating that the atop site possesses high catalytic activity. Clearly, the catalytic activity of the atop sites is similar to that of subnanometer gold clusters, due to the similar binding characters of the gold atoms. Acetonitrile solvent can have a slight influence on the energy barriers and the reaction rates.
Table 1 The energetic span (δE) and TOF values for ω-alkynylfuran cycloisomerisation catalyzed by Au10/CeO2(111) and Au11/CeO2(111)
| |
TDI/TDTS |
δE (kcal mol−1) |
TOF (s−1) |
| Au10/CeO2(111)-s |
| Atop |
IM2/TS3/4 |
11.09 |
2.9 × 105 |
| Interface-corner |
IM2/TS3/4 |
14.03 |
4.1 × 103 |
| Interface-edge |
IM3/TS3/4 |
16.03 |
4.3 × 102 |
| Edge |
IM2/TS3/4 |
13.32 |
2.0 × 104 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Au10/CeO2(111)-v |
| Atop |
IM3/TS3/4 |
10.98 |
3.3 × 105 |
| Interface-corner |
IM3/TS3/4 |
29.93 |
1.6 × 10−7 |
| Interface-edge |
IM3/TS3/4 |
13.93 |
4.9 × 103 |
| Edge |
IM3/TS3/4 |
29.93 |
1.6 × 10−7 |
|
| Au11/CeO2(111)-s |
| Atop |
IM2/TS3/4 |
9.79 |
1.2 × 106 |
| Interface |
IM3/TS3/4 |
13.75 |
6.5 × 103 |
| Edge |
IM3/TS3/4 |
22.21 |
1.3 × 10−2 |
|
| Au11/CeO2(111)-v |
| Atop |
IM4/IM5 |
14.46 |
1.1 × 103 |
| Interface |
IM3/TS3/4 |
10.44 |
6.6 × 105 |
| Edge |
IM3/TS3/4 |
25.89 |
7.1 × 10−5 |
Subsequently, ω-alkynylfuran cycloisomerisation on the edge sites of Au10/CeO2(111) and Au11/CeO2(111) were investigated and discussed. The adsorption energies are smaller as compared to that of the atop sites because of the higher coordination numbers of the edge sites. Interestingly, a distinctive substrate adsorption behavior is observed for the edge site of Au11/CeO2(111)-v. As shown in Fig. S14,† the adsorption of the substrate on the edge site of Au11/CeO2(111)-v induces strong structural deformation of the Au11 fragment due to the strong interaction between the adsorbed alkyne group and the Au atoms. The corresponding structural deformation raises the energy barrier for the electrophilic attack of the C atom on the furan ring. The electrophilic attack of the C atom on the furan ring (IM1 → IM2) and the cleavage of C–O in the furan ring (IM2 → IM3) steps on the edge sites have similar energy barriers. The closure of the ring to form the oxepine intermediate on the edge site of Au10/CeO2(111)-s needs to overcome an energy barrier of 10.21 (12.12) kcal mol−1 in the gas phase (acetonitrile solvent), which is comparable to the situation on the atop site. However, the closure of the ring to form the oxepine intermediate on the edge site of Au10/CeO2(111)-v and Au11/CeO2(111)-v entails a large energy barrier of about 29 kcal mol−1 due to the μ2-coordination in IM3. Simultaneously, the closure of the ring to form the oxepine intermediate on the edge site of Au11/CeO2(111)-s must overcome an energy barrier of about 20.00 kcal mol−1 due to the large deformation of the substrate fragment. Hence, only the edge sites of Au10/CeO2(111)-s show similar catalytic activity toward ω-alkynylfuran cycloisomerisation to the atop sites.
Finally, the 5-exo FCT mechanism can be extended to the interface sites of Au10/CeO2(111) and Au11/CeO2(111), so ω-alkynylfuran cycloisomerisation on the interface sites of Au10/CeO2(111) and Au11/CeO2(111) is also explored. As seen from Fig. 9–12, there are two types of interface sites for Au10/CeO2(111)-s and Au10/CeO2(111)-v. In contrast, the interface sites of Au11/CeO2(111)-s and Au11/CeO2(111)-v have similar bonding character, and hence there is only one type of interface site. The adsorption of the substrate on the interface sites causes a large structural deformation of the supported gold clusters due to the strong interaction between the alkyne group and gold atoms. The large structural deformation of the supported gold clusters counteracts the partial stabilizing interaction between the alkyne group and the gold atoms, which results in lower adsorption energies of the substrate on the interface sites. For the interface sites, the closure of the ring to form the oxepine intermediate is the rate-limiting step in both the gas phase and acetonitrile solvent. The interface-corner sites of Au10/CeO2(111)-v may be viewed as inactive toward ω-alkynylfuran cycloisomerisation compared to the other interface sites, due to the formation of μ2-coordination in IM3. The catalytic activity of the interface sites of Au10/CeO2(111) is lower than that of the atop sites. Acetonitrile solvent enhances the energy barrier of the closure of the ring to form the oxepine intermediate and reduces the reaction rate.
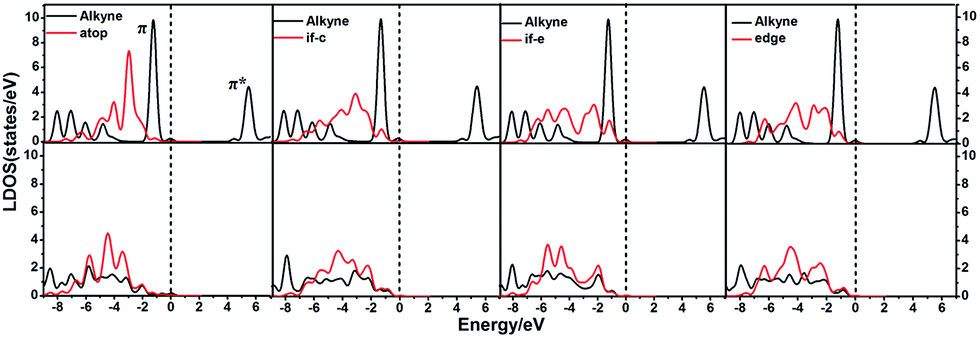
In order to illustrate the adsorption properties of ω-alkynylfuran on the different sites of Au10/CeO2(111), the corresponding local density of states (LDOS) are calculated and shown in Fig. 13 and S15.† In this article, the LDOS discussion only focuses on the d-band information because the valence orbitals are the s and d orbitals of gold. Moreover, the interaction between the clusters and the ω-alkynylfuran depends on π-back-donation and σ-coordinate bonding, that is, charge transfer between the d and π* orbitals or π and d* orbitals. Hence, the d-band information is mainly investigated in this article.71 According to Fig. 13 and S15,† the bands of the adsorbed alkyne and Au10/CeO2(111) moved to the left toward the lower energy region, compared with the free substrate and Au10/CeO2(111). The main peak of the alkyne overlapped with those of the different sites of Au10/CeO2(111), indicating chemical bond formation between the substrate and Au10/CeO2(111). Compared with the interface-corner, interface-edge and edge sites, the band of the adsorbed alkyne is more down-shifted for the atop site and the main peaks of the alkyne are more effectively overlapped with the atop site, leading to a larger adsorption energy. In addition, the adsorption energies of ω-alkynylfuran on the interface-edge sites of Au10/CeO2(111) (stoichiometric and reduced) exhibit obvious differences, which can be attributed to the intensity and overlap degree of the bands.
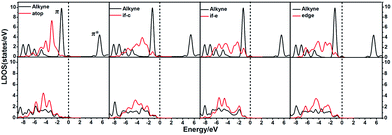 |
| | Fig. 13 The local density of states (LDOS) projected on triplet bonds on Au10/CeO2(111)-s for the adsorption of the substrate at different sites, together with the d-projected LDOS of Au10/CeO2(111)-s of the Au atom at different sites. Free C–C triplet bond and CeO2(111) (top), and adsorption (bottom). The Fermi level is set to zero (if-c = interface-corner, if-e = interface-edge). | |
Similarly, the local density of states (LDOS) are calculated again to elaborate the adsorption of ω-alkynylfuran on Au11/CeO2(111) (stoichiometric and reduced). Based on the computed results, the adsorption energy of ω-alkynylfuran on the interface sites is larger than that on the atop and edge site owing to the better overlap of the peaks, which can be seen from Fig. S16.† Compared with the edge sites, the band of the absorbed alkyne is more down-shifted and there is better overlap of the peaks for the alkyne and the atop Au atoms, leading to larger adsorption energies. However, for the adsorption of ω-alkynylfuran on Au11/CeO2(111) (vacancy, Fig. S17†), the adsorption energy is the smallest at the interface sites due to the weak d peak at around 8 eV and the relatively weak overlap of the bands.
In summary, according to the above-mentioned analyses, the atop and interface sites exhibit high catalytic activity toward ω-alkynylfuran cycloisomerisation via the FCT mechanism. The edge sites of Au10/CeO2(111)-s show similar catalytic activity toward ω-alkynylfuran cycloisomerisation to the atop sites. The adsorption energies of the substrate on the atop sites of the gold clusters supported by the CeO2(111) surfaces are comparable with those on subnanometer gold clusters. Due to the higher coordination number of the edge and interface sites, lower adsorption energies of the substrate on the edge and interface sites were identified. In addition, based on the calculated results, it is found that the surface O vacancy of CeO2(111) has some negative and positive influences on the adsorption energies of the substrate and the catalytic activity.
Comparison of the catalytic activity of free gold clusters and supported gold clusters
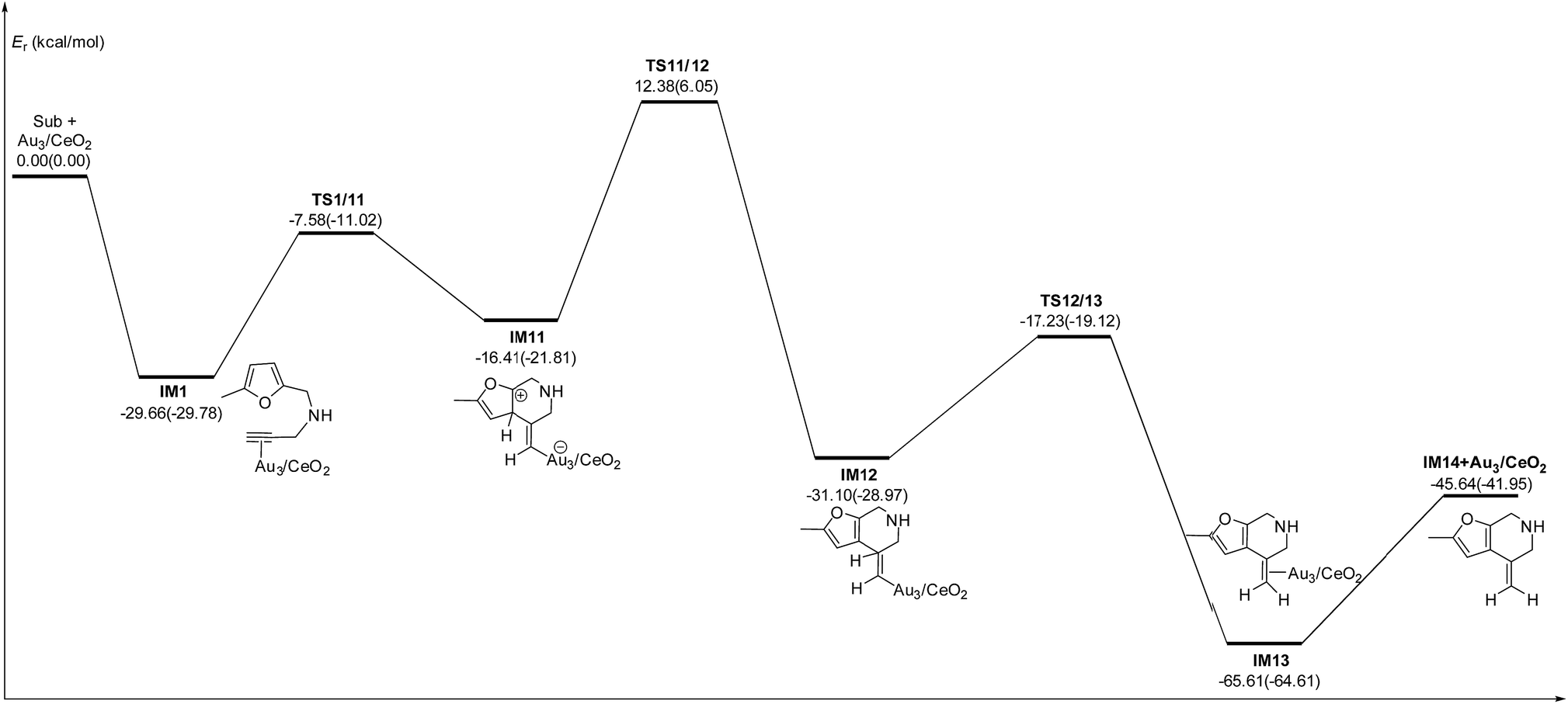
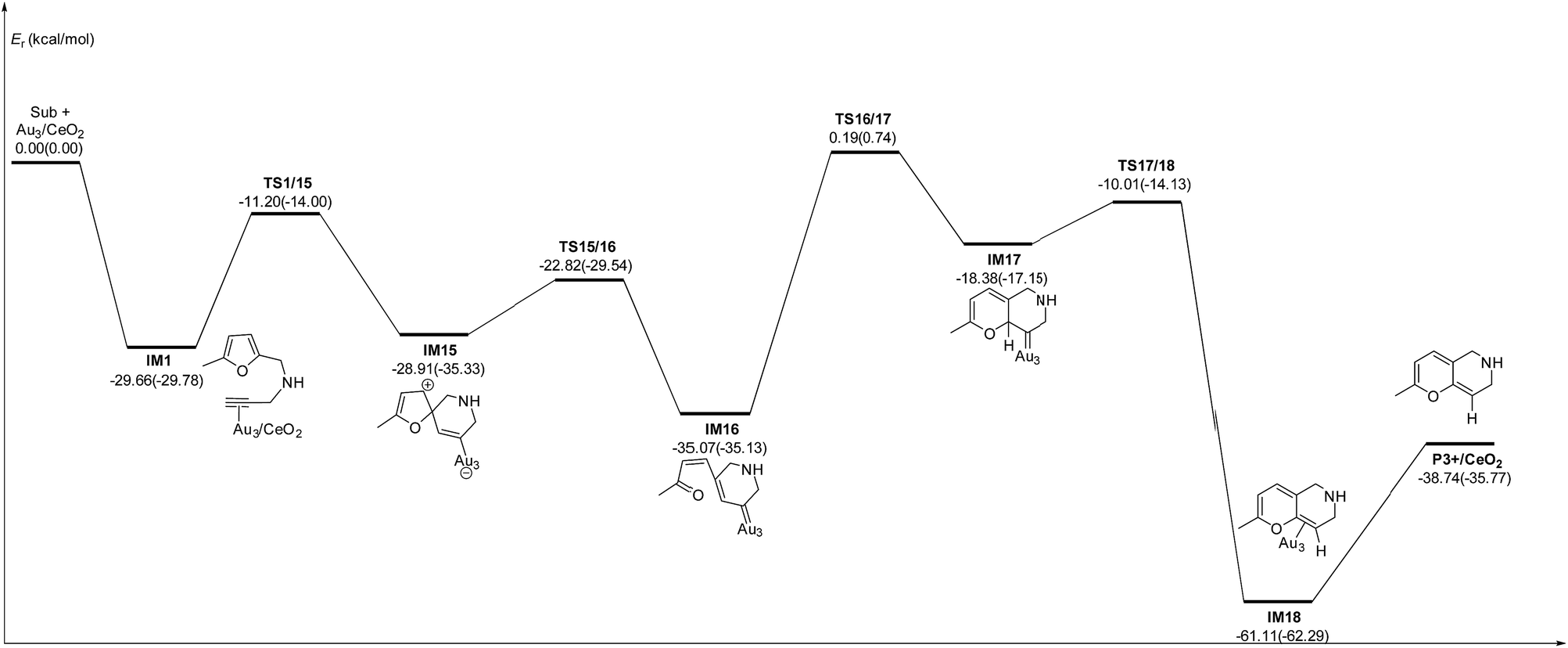
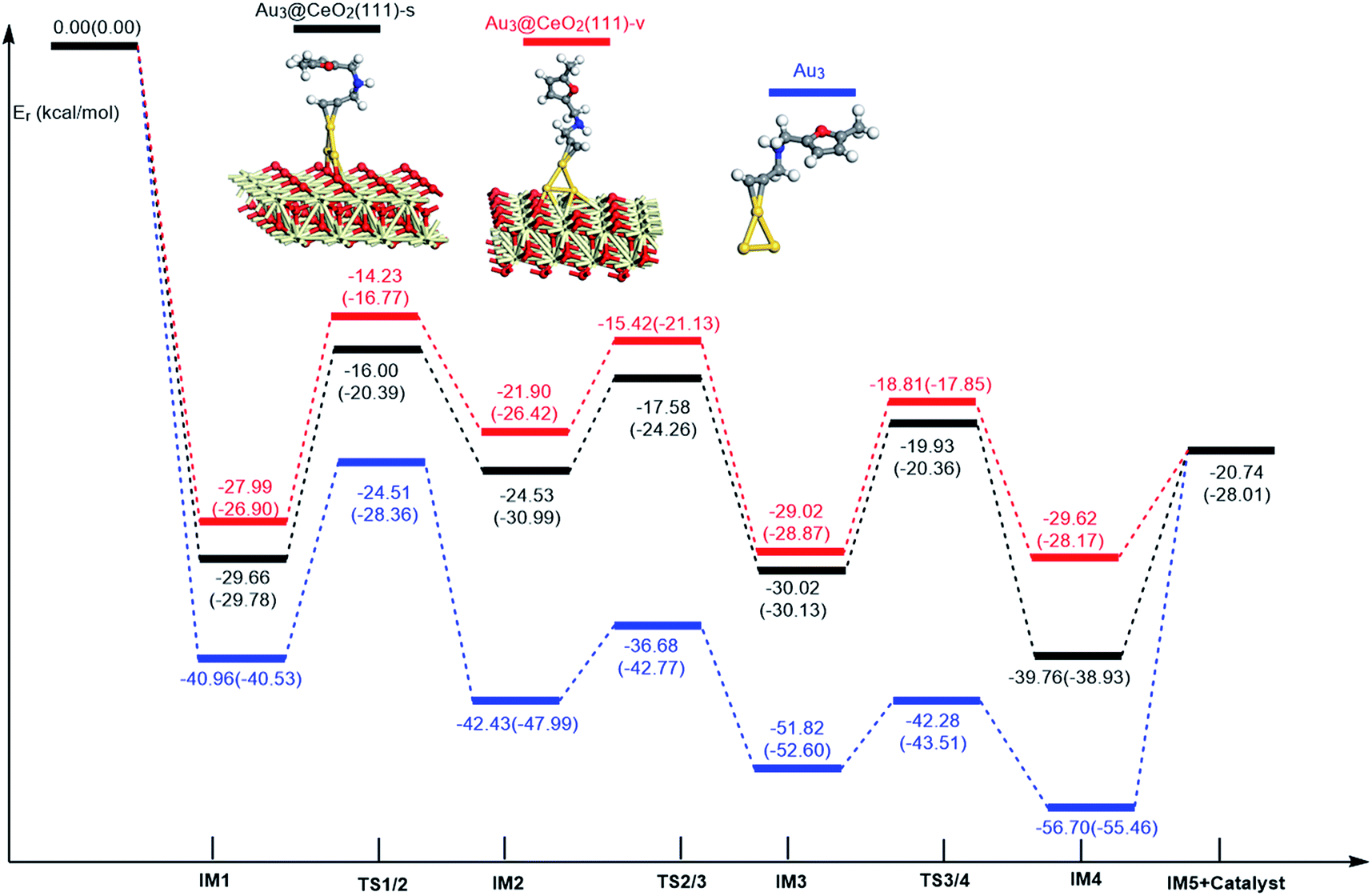
In order to clarify the role of the CeO2 support, the 5-exo FCT pathway was studied on free and supported Au3–4 clusters. As plotted in Fig. 14 and 15, the reaction schemes of ω-alkynylfuran cycloisomerisation on CeO2-supported Au3–4 and free Au3–4 are similar to those on Au10/CeO2(111) and Au11/CeO2(111). The adsorption of ω-alkynylfuran on the CeO2-supported Au3–4 and free Au3–4 clusters generates intermediate IM1. An analysis of the adsorption of ω-alkynylfuran on the CeO2-supported Au3–4 and free Au3–4 clusters suggests that the adsorption energies are much larger as compared to those of ω-alkynylfuran adsorption on Au10/CeO2(111) and Au11/CeO2(111), due to the lower coordination numbers of the Au3–4 clusters. The large adsorption energies have a pronounced effect on the Cβ atom of the alkynyl group, which moves close to the C1 atom of the furan ring. For ω-alkynylfuran cycloisomerisation on Au3/CeO2(111)-s, Au3/CeO2(111)-v and free Au3 clusters, overcoming the energy barriers of 13.66 (9.39), 13.76 (10.13) and 16.45 (12.17) kcal mol−1 in the gas phase (acetonitrile solvent), the Cβ–C1 bond is formed in the spiro-intermediate IM2. These corresponding energy barriers are larger than those on Au10/CeO2(111) and Au11/CeO2(111). Furthermore, this elementary reaction on free Au3 needs to overcome the highest energy barrier. Next, the C1–O bond of the furan ring is broken to form the Au-vinylidene intermediate IM3. The corresponding energy barriers are 6.95 (6.73), 6.48 (5.29) and 5.75 (5.22) kcal mol−1 in the gas phase (acetonitrile solvent) on Au3/CeO2(111)-s, Au3/CeO2(111)-v and free Au3 clusters, respectively. The reaction that breaks the furan ring is exothermic by about 5.49 (−0.86), 7.12 (2.45) and 9.39 (4.61) kcal mol−1 in the gas phase (acetonitrile solvent) on Au3/CeO2(111)-s, Au3/CeO2(111)-v and free Au3. Then, the O atom of the carbonyl group attacks the Cα atom of the Au-vinylidene with energy barriers of 10.09 (9.77), 10.21 (11.02) and 9.54 (9.09) kcal mol−1 in the gas phase (acetonitrile solvent) on Au3/CeO2(111)-s, Au3/CeO2(111)-v and free Au3 clusters, leading to the formation of a seven-membered ring intermediate (IM4).
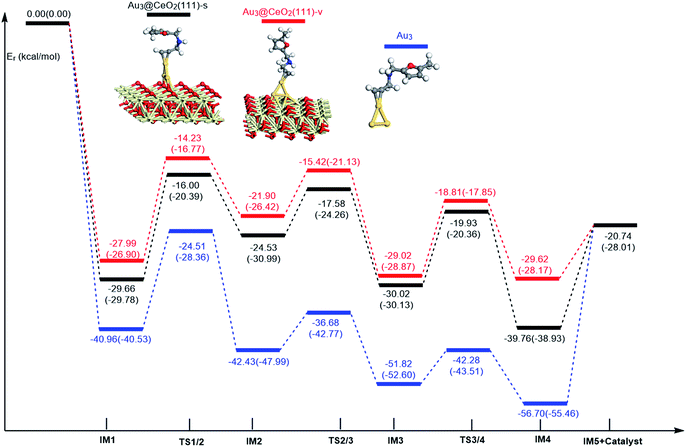 |
| | Fig. 14 Energy profile for ω-alkynylfuran cycloisomerisation on CeO2-supported Au3 and free Au3. | |
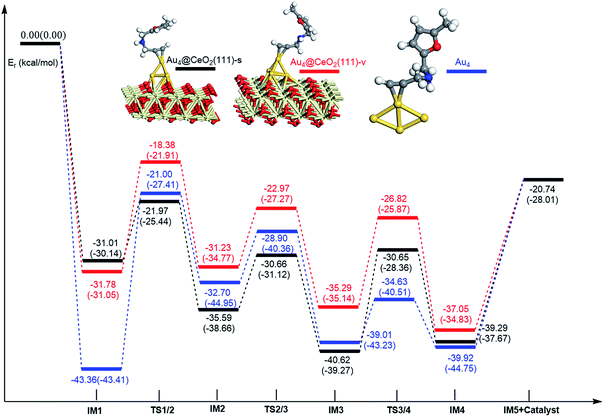 |
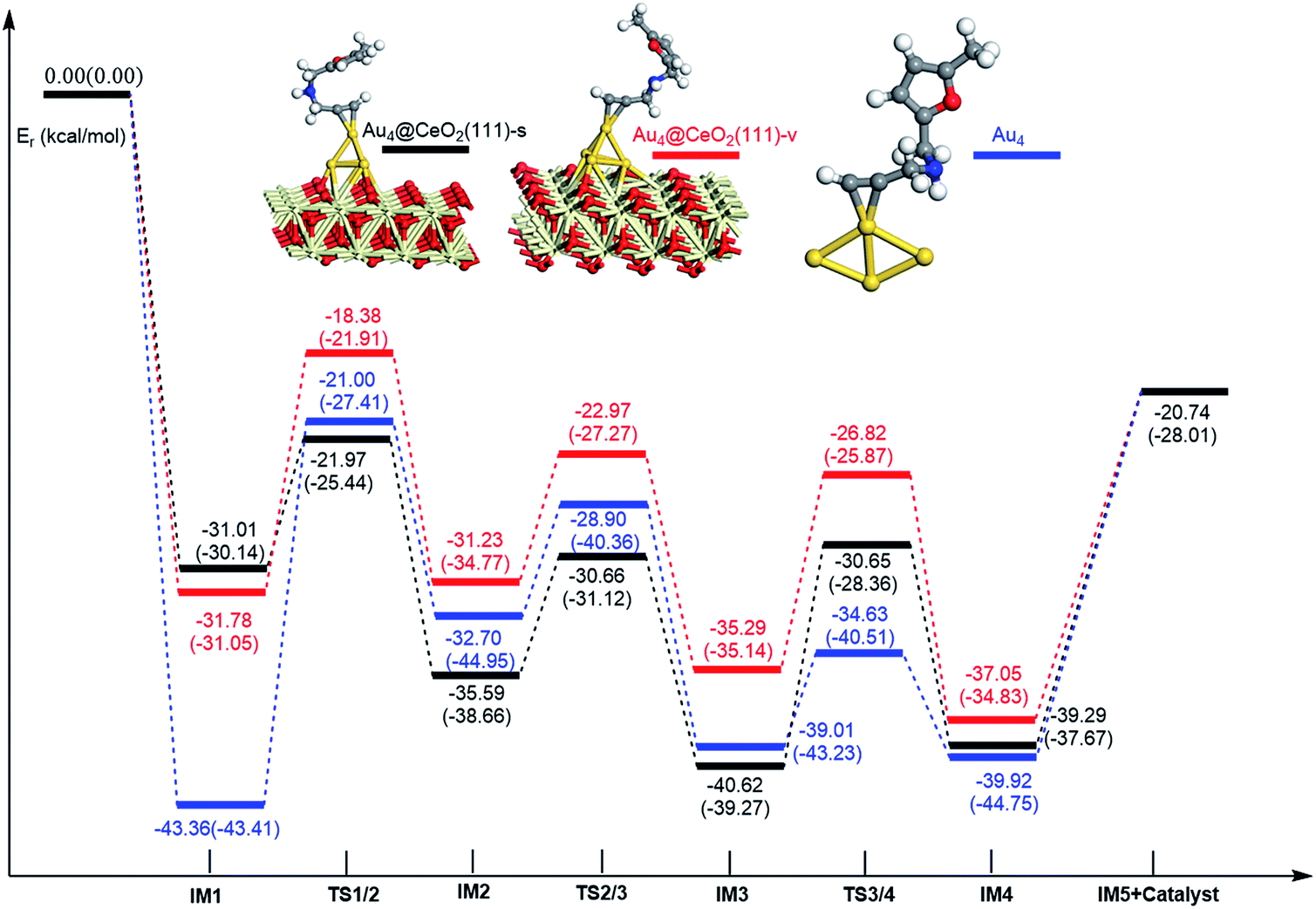
| | Fig. 15 Energy profile for ω-alkynylfuran cycloisomerisation on CeO2-supported Au4 and free Au4. | |
Additionally, the mechanisms of ω-alkynylfuran cycloisomerisation catalyzed by Au4/CeO2(111)-s, Au4/CeO2(111)-v and free Au4 clusters are also unveiled. As shown in Fig. 15, the catalytic cycles of ω-alkynylfuran cycloisomerisation on Au4/CeO2(111)-s, Au4/CeO2(111)-v and free Au4 clusters are similar to those on Au10/CeO2(111), Au11/CeO2(111), Au3/CeO2(111) and free Au3, including the adsorption of the ω-alkynylfuran, the formation of the Cβ–C1 bond, the breakage of the C1–O bond in the furan ring, and the formation of the seven-membered ring. On the basis of the energy profiles, the supported Au4 clusters are clearly more active than the free Au4 clusters toward ω-alkynylfuran cycloisomerisation.
According to the calculated results, the rate-determining steps of ω-alkynylfuran cycloisomerisation on CeO2-supported Au3/Au4 and free Au3/Au4 are the formation of the Cβ–C1 bond, that is, the IM1 → IM2 conversion. Compared with free Au3, the CeO2 supporting material can efficiently facilitate ω-alkynylfuran cycloisomerisation, decreasing the energy barriers for IM1 → IM2 conversion to 2.69–2.79 (2.04–2.78) kcal mol−1 in the gas phase (acetonitrile solvent). In addition, the oxygen vacancy can have a minor effect on the energy barriers for ω-alkynylfuran cycloisomerisation on Au3/CeO2 in the gas phase (acetonitrile solvent). Similarly, compared with free Au4, CeO2-supported Au4 can decrease the energy barriers of the rate-determining step to 8.96–13.28 (6.86–11.30) kcal mol−1 in the gas phase (acetonitrile solvent), indicating the active role of CeO2 in ω-alkynylfuran cycloisomerisation.
In order to better illustrate the catalytic reactivity of the free gold clusters and supported gold clusters, the corresponding energetic span (δE) and TOF values were calculated and the values are given in Table 2. As shown in Table 2, the TOF values for ω-alkynylfuran cycloisomerisation catalyzed by free Au3–4 are smaller than those for Au3–4/CeO2(111). Therefore, the presence of CeO2(111) can effectively facilitate ω-alkynylfuran cycloisomerisation.
Table 2 The energetic span (δE) and TOF values for ω-alkynylfuran cycloisomerisation catalyzed by free gold clusters and supported gold clusters
| |
TDI/TDTS |
δE (kcal mol−1) |
TOF (s−1) |
| Au3 |
IM4/IM5 |
27.45 |
3.3 × 10−6 |
| Au3/CeO2(111)-s |
IM4/TS3/4 |
10.92 |
2.3 × 105 |
| Au3/CeO2(111)-v |
IM3/TS3/4 |
11.02 |
3.0 × 105 |
| Au4 |
IM2/IM5 |
16.94 |
13 × 101 |
| Au4/CeO2(111)-s |
IM3/IM5 |
11.26 |
7.4 × 104 |
| Au4/CeO2(111)-v |
IM3/TS3/4 |
9.27 |
2.2 × 106 |
The role of CeO2 surface
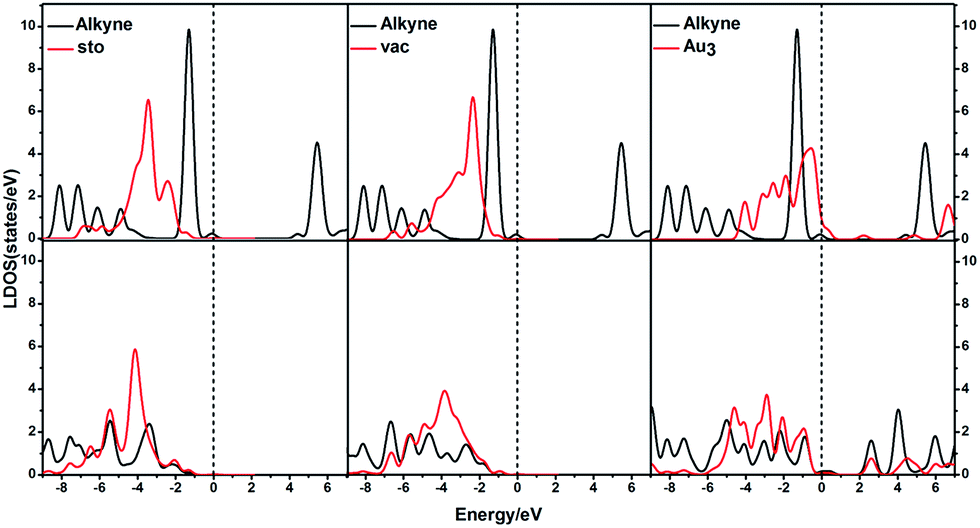
Based on the calculated results, the presence of the CeO2(111) surface can largely decrease the adsorption energies. In order to illustrate this interesting phenomenon, the corresponding local density of states (LDOS) were computed and plotted in Fig. 16. As shown in Fig. 16, for the adsorption of ω-alkynylfuran on Au3 clusters, there is better peak overlap between the Au3 cluster and the alkyne as compared to that for Au3/CeO2(111) (stoichiometric and reduced) and Au3 clusters, leading to a larger adsorption energy. The different orbital interactions can be attributed to the charge transfer between the CeO2(111) surface and the Au3 cluster. Similarly, for Au4/CeO2(111) (stoichiometric and reduced) and Au4, the local density of states (LDOS) shown in Fig. S24† clearly show that the peak overlap plays a crucial role in determining the adsorption energies. For free Au4 clusters, the main peak of the alkyne can effectively overlap with the d-band of the Au4 cluster, facilitating the interaction between the alkyne and the Au4 cluster. Further, the interactions between the Au clusters and CeO2 surface are taken into account via calculating the adsorption energies without the CeO2 surface, that is, by removing the CeO2 surface from Aun/CeO2 (the resulting free Au3 and Au4 are represented as Au3-re and Au4-re). The computed results are collected in Table S1† along with the initial values. Compared with the initial adsorption energies, the new ones possess larger values, implying that the strong interaction between the CeO2(111) surface and the substrate decreases the adsorption energies. To interpret the inherent reason for this, the d-projected LDOS were plotted in Fig. S25.† Based on Fig. S25,† near the Fermi level, the intensities of the d-bands of the Au3-re and Au4-re clusters are larger than those of Au3/CeO2(111) and Au4/CeO2(111), indicating that Au3-re and Au4-re clusters allow more opportunity for π-back-donation and σ-coordinate bonding with the substrate. In addition, compared with the Au3-re and Au4-re clusters, the d-bonds of Au3/CeO2(111) and Au4/CeO2(111) are downshifted, which further illustrates that there is less opportunity to form σ-coordinate bonds with the substrate. Additionally, the Mulliken charges of the Au atoms for the free Au3/Au4 clusters, Au3/Au4/CeO2 and Au3/Au4/CeO2 were also calculated and the corresponding values are plotted in Fig. S26.† As shown in Fig. S26,† the Mulliken charges of Au (top) are 0.04, −0.02, −0.09, 0.10, 0.03 and −0.01, respectively, and the values of total charge transfer between the Au clusters and the support are 0.10, 0.43, 0.27, and 0.28. More positive charge indicates more unoccupied or partial occupied orbitals for the corresponding Au atom, which is beneficial for forming σ-coordinate bonds. Based on the computed results, the CeO2 support can not only change the orbital levels of the gold clusters, but also cause charge recombination and decrease the positive charge on the Au (top) atom. These significant roles lead to the elevation of the adsorption energy, facilitating ω-alkynylfuran cycloisomerisation via the 5-exo FCT mechanism.
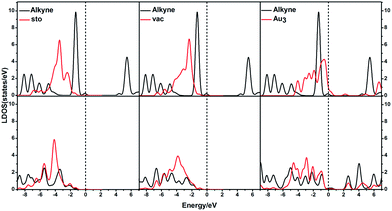 |
| | Fig. 16 The local density of states (LDOS) projected on triplet bonds on Au3/CeO2(111) for the adsorption of the substrate at different sites, together with the d-projected LDOS of Au3/CeO2(111) (stoichiometric and reduced) of the Au atom at different sites. Free C–C triplet bond and CeO2(111) (top), and adsorption (bottom). The Fermi level is set to zero. | |
Conclusions
In summary, the catalytic properties of Au10–11/CeO2, Au3–4/CeO2, and free Au3 and Au4 clusters toward ω-alkynylfuran cycloisomerisation via the 5-exo FCT mechanism were systematically investigated based on density functional theory. The calculated results show that the atop and interface sites of Au10–11/CeO2 can effectively facilitate ω-alkynylfuran cycloisomerisation via the FCT mechanism, indicating that the atop and interface sites possess excellent catalytic activity. Meanwhile, a surface oxygen vacancy on Au10–11/CeO2 can have a minor effect on the adsorption energies. In addition, for the sake of illustrating the role of the CeO2 support, free Au3 and Au4 clusters were employed. By comparing the present results, supporting the Au3 and Au4 clusters on CeO2 can decrease the energy barriers for the rate-determining step. For ω-alkynylfuran cycloisomerisation catalyzed via Au3–4/CeO2, the oxygen vacancy plays an active role. Overall, this study shows the catalytic activities of different reaction species on Aun/CeO2 and the role of the CeO2 support, which is beneficial for providing some valuable information for the design of highly active metal/oxide catalysts for ω-alkynylfuran cycloisomerisation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21573030), the Chongqing Science & Technology Commission, China (Grant No. CSTC2013JCYJA50028) and the Scientific Research Foundation of Chongqing University of Arts and Sciences (R2013CJ03). The calculations were performed at the National Supercomputing Centre in Shenzhen (Shenzhen Cloud Computing Centre).
Notes and references
- F. Boccuzzi, A. Chiorino, M. Manzoli, D. Andreeva and T. Tabakova, J. Catal., 1999, 188, 176–185 CrossRef CAS.
- W. D. Williams, M. Shekhar, W.-S. Lee, V. Kispersky, W. N. Delgass, F. H. Ribeiro, S. M. Kim, E. A. Stach, J. T. Miller and L. F. Allard, J. Am. Chem. Soc., 2010, 132, 14018–14020 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 2, 405–408 CrossRef.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- H. Y. Kim and G. Henkelman, J. Phys. Chem. Lett., 2012, 4, 216–221 CrossRef PubMed.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- C. Zhang, A. Michaelides and S. J. Jenkins, Phys. Chem. Chem. Phys., 2011, 13, 22–33 RSC.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- N. J. Lawrence, J. R. Brewer, L. Wang, T.-S. Wu, J. Wells-Kingsbury, M. M. Ihrig, G. Wang, Y.-L. Soo, W.-N. Mei and C. L. Cheung, Nano Lett., 2011, 11, 2666–2671 CrossRef CAS PubMed.
- M. Baron, O. Bondarchuk, D. Stacchiola, S. Shaikhutdinov and H.-J. Freund, J. Phys. Chem. C, 2009, 113, 6042–6049 CAS.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed.
- W. Song and E. J. M. Hensen, Catal. Sci. Technol., 2013, 3, 8 Search PubMed.
- L. C. Wang, D. Widmann and R. J. Behm, Catal. Sci. Technol., 2014, 5, 925–941 Search PubMed.
- A. Ciftci, D. M. Ligthart, P. Pastorino and E. J. Hensen, Appl. Catal., B, 2013, 130, 325–335 CrossRef.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- M. Han, X. Wang, Y. Shen, C. Tang, G. Li and R. L. Smith Jr, J. Phys. Chem. C, 2009, 114, 793–798 Search PubMed.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed.
- H. Falsig, B. Hvolbæk, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Nørskov, Angew. Chem., 2008, 120, 4913–4917 CrossRef.
- W. Song and E. J. Hensen, Catal. Sci. Technol., 2013, 3, 3020–3029 CAS.
- H. Y. Kim and G. Henkelman, J. Phys. Chem. Lett., 2012, 3, 2194–2199 CrossRef CAS PubMed.
- M. Cargnello, V. V. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- Z. Zhou, S. Kooi, M. Flytzani-Stephanopoulos and H. Saltsburg, Adv. Funct. Mater., 2008, 18, 2801–2807 CrossRef CAS.
- B. Liu, J. Liu, T. Li, Z. Zhao, X.-Q. Gong, Y. Chen, A. Duan, G. Jiang and Y. Wei, J. Phys. Chem. C, 2015, 119, 12923–12934 CAS.
- B. Liu, Z. Zhao, G. Henkelman and W. Song, J. Phys. Chem. C, 2016, 120, 5557–5564 CAS.
- N. Lopez, T. Janssens, B. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- K. Zhao, J. Qi, H. Yin, Z. Wang, S. Zhao, X. Ma, J. Wan, L. Chang, Y. Gao and R. Yu, J. Mater. Chem. A, 2015, 3, 20465–20470 CAS.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 CAS.
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC.
- H. Yin, H. Tang, D. Wang, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. C. Blanco, E. Kurpejović, W. Frey and J. W. Bats, Adv. Synth. Catal., 2006, 348, 709–713 CrossRef CAS.
- J. H. Tyman, Synthetic and natural phenols, Elsevier, 1996 Search PubMed.
- A. S. K. Hashmi, T. M. Frost and J. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, Org. Lett., 2001, 3, 3769–3771 CrossRef CAS PubMed.
- S. Carrettin, M. C. Blanco, A. Corma and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283–1288 CrossRef CAS.
- A. S. K. Hashmi, J. Hofmann, S. Shi, A. Schütz, M. Rudolph, C. Lothschütz, M. Wieteck, M. Bührle, M. Wölfle and F. Rominger, Chem.–Eur. J., 2013, 19, 382–389 CrossRef CAS PubMed.
- M. Yang, Z. Chen, Y. Luo, J. Zhang, D. Tang, R. He, W. Shen and M. Li, RSC Adv., 2016, 6, 22709–22721 RSC.
- M. Yang, Z. Chen, Y. Luo, J. Zhang, R. He, W. Shen, D. Tang and M. Li, ChemCatChem, 2016, 14, 2367–2375 CrossRef.
- D. Tang, M. Yang, J. Zhang, R. He, W. Shen and M. Li, ChemCatChem, 2016, 8, 461–470 CrossRef CAS.
- J. Oliver-Meseguer, A. Leyva-Pérez and A. Corma, ChemCatChem, 2013, 5, 3509–3515 CrossRef CAS.
- J. Olivermeseguer, J. R. Cabreroantonino, I. Domínguez, A. Leyvapérez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- J. Oliver-Meseguer, A. Leyva-Pérez, S. I. Al-Resayes and A. Corma, Chem. Commun., 2013, 49, 7782–7784 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Delley, Int. J. Quantum Chem., 1998, 69, 423–433 CrossRef CAS.
- S. Bell and J. S. Crighton, J. Chem. Phys., 1984, 80, 2464–2475 CrossRef CAS.
- S. Fischer and M. Karplus, Chem. Phys. Lett., 1992, 194, 252–261 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- B. Delley, Mol. Simul., 2006, 32, 117–123 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- A. Trovarelli, Catalysis by ceria and related materials, World Scientific, 2002, pp. 1–501 Search PubMed.
- H. Y. Kim and G. Henkelman, J. Phys. Chem. Lett., 2012, 3, 2194–2199 CrossRef CAS PubMed.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed.
- G. Vilé, P. Dähler, J. Vecchietti, M. Baltanás, S. Collins, M. Calatayud, A. Bonivardi and J. Pérez-Ramírez, J. Catal., 2015, 324, 69–78 CrossRef.
- A. D. Mayernick and M. J. Janik, J. Phys. Chem. C, 2008, 112, 14955–14964 CAS.
- L. C. Hsu, M. K. Tsai, Y. H. Lu and H. T. Chen, J. Phys. Chem. C, 2013, 117, 367–371 Search PubMed.
- Y. Zhao, C. Cui, J. Han, H. Wang, X. Zhu and Q. Ge, J. Am. Chem. Soc., 2016, 138, 10191–10198 CrossRef CAS PubMed.
- J. L. F. D. Silva, V. M. O. N. Ganduglia-Pirovano, J. Sauer, V. Bayer and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 33–39 Search PubMed.
- M. Fronzi, S. Piccinin, B. Delley, E. Traversa and C. Stampfl, Phys. Chem. Chem. Phys., 2009, 11, 9188–9199 RSC.
- M. Fronzi, S. Piccinin, B. Delley, E. Traversa and C. Stampfl, RSC Adv., 2013, 4, 12245–12251 RSC.
- M. Fronzi, A. Soon, B. Delley, E. Traversa and C. Stampfl, J. Chem. Phys., 2009, 131, 104701–104716 CrossRef.
- S. Kumar and P. K. Schelling, J. Chem. Phys., 2006, 125, 261–288 Search PubMed.
- Z. Yang, T. K. Woo, M. Baudin and K. Hermansson, J. Chem. Phys., 2004, 120, 7741–7749 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2010, 44, 101–110 CrossRef PubMed.
- S. Kozuch and J. M. Martin, ACS Catal., 2011, 1, 246–253 CrossRef CAS.
- A. Uhe, S. Kozuch and S. Shaik, J. Comput. Chem., 2011, 32, 978–985 CrossRef CAS PubMed.
- S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 795–815 CrossRef CAS.
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, J. Am. Chem. Soc., 2010, 132, 2175–2182 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Rudolph, J. P. Weyrauch, M. Wölfle, W. Frey and J. W. Bats, Angew. Chem., Int. Ed., 2005, 44, 2798–2801 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27207j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab