
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of Mn-doped ceria oxygen-storage material on oxidation activity of diesel soot
He Huang,
Junheng Liu *,
Ping Sun,
Song Ye and
Bingxia Liu
*,
Ping Sun,
Song Ye and
Bingxia Liu
School of Automotive and Traffic Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: liujunheng365@163.com
First published on 23rd January 2017
Abstract
A Diesel Particulate Filter (DPF) is an effective device for reducing the soot emission of diesel engines. In order to realize the passive regeneration of DPF at low temperature, Ce1−xMnxO2 catalysts doped with different doses of Mn were prepared through a sol–gel method. The influence of the catalyst on soot oxidation characteristics was studied by thermogravimetric analysis (TGA). The oxygen vacancy formation energy was calculated using the first principles Perdew–Wang 1991 (PW91) method based on density functional theory (DFT) within the generalized gradient approximation (GGA). Moreover, catalytic performance was evaluated using X-ray diffraction (XRD), scanning electron microscopy (SEM), Raman spectroscopy, H2-temperature programming reduction (H2-TPR) and X-ray photoelectron spectroscopy (XPS). The results show that the ignition temperature and peak temperature of soot oxidation gradually decrease, and also the activation energy of Ce1−xMnxO2 for the catalytic soot oxidation decreases with the increase of Mn concentration. When the Mn concentration is 50%, the ignition temperature and peak temperature are decreased by 42 °C and 32 °C, respectively. The crystal structure of the prepared Ce1−xMnxO2 has a better stability, and Mn doping leads to the increase of lattice defects. Moreover, the oxygen vacancy formation energy of the Ce1−xMnxO2 catalyst decreases with increasing Mn concentrations. When Mn doping concentration is 50%, the oxygen vacancy formation energy represents a minimum value of 0.31 eV.
1. Introduction
Diesel vehicles have received lots of attention globally due to their power efficiency and fuel economy. However, the pollutants in the exhaust gas, especially the soot emission, are a threat to the environment and to human health.1–3 A Diesel Particulate Filter (DPF) is an effective means to reduce the soot emission from diesel engines. A passive regeneration technique is used to reduce the activation energy of the soot oxidation with chemocatalysts. In this way, the soot can be burnt out with energy from the diesel exhaust to realize DPF regeneration.4,5 Therefore, the selection of catalyst is the key to DPF regeneration. Rare earth based catalysts are widely used in automotive exhaust purification catalysts, ceramics, rare earth polymers, fuel cells, coatings and many other fields because of their unique chemical and physical properties, which are represented by their abundant electronic energy level structure. Cerium dioxide (CeO2), a high-performance rare earth material, is the core of the three-way catalyst used for automobile exhaust purification. CeO2 can simultaneously convert hydrocarbon (HC), carbon monoxide (CO) and nitrogen oxides (NOx) from engine exhaust gas into H2O, carbon dioxide (CO2) and nitrogen (N2). CeO2 is the core of oxygen storage/oxygen release materials, whose performance is mainly determined by the redox ability of gas molecule,6,7 oxygen storage/oxygen release performance8–11 and oxygen transfer capability.10 When the oxygen content in the exhaust gas is high, CeO2 quickly store the oxygen and also releases oxygen in a timely fashion when oxygen content is low. Therefore, it can be referred to as “oxygen buffer”. This function is essential for improving catalytic activity and prolonging the service life of the catalyst.11–17 However, pure CeO2 mainly has two defects. Firstly, there are very few oxygen defects of surface oxygen and bulk phase oxygen. Secondly, sintering occurs at high temperature, leading to the reduction in the capacity of oxygen storage and release. To increase the ability of oxygen storage/release and low temperature redox of the supported catalyst system, researchers normally doped CeO2 with a transition metal.18 Mn mainly exists in the forms of changeable valence oxides such as MnO, MnO2, Mn2O3 and Mn3O4, which is generally utilized as the electronic auxiliaries of catalysts. Ce1−xMnxO2 catalyst formed by doping Mn into CeO2 can be used to promote some reactions. Wu et al.19,20 suggested that the doping of Mn into the internal lattice of CeO2 does not only improve the migration of reactive oxygen, but also enhance the overflow ability of lattice oxygen, which could strengthen the soot catalytic oxidation. Guillén-Hurtado et al.21 studied the mechanism of soot oxidation through cerium based catalyst by isotope pulse technique, and found that gaseous phase oxygen did not directly participate in soot oxidation. The active oxygen in the catalyst was firstly released and then migrated to the soot surface for oxidization. Meantime, the oxygen vacancies generated on the catalyst surface, adsorbed the gaseous phase oxygen into vacancy to supplement the consumed active oxygen. Thus, it can be said that, the active oxygen, oxygen vacancies, oxygen migration capability and other oxygen parameters play an important role in soot catalytic oxidation. Qi et al.22 prepared MnOx–CeO2 composite oxide by doping Mn into CeO2 system, which had high NH3-SCR (selective catalytic reduction) catalytic activity at low temperature. Delimaris et al.23,24 showed that the catalytic oxidation activity of MnOx–CeO2 to chlorinated volatile organic compounds (CVOCs) and volatile organic compounds (VOCs) was better than that of pure CeO2. However, there are still some debates about the decisive factors and the reaction path of soot catalytic oxidation. Most situ detection methods are directed at reactive oxygen species, but the real-time monitoring and analysis of oxygen vacancy are scarce.Recently, new materials have been widely designed from studies at the level of atoms and molecules. This method combined with the experimental study, decreases the dependence on experimental instruments and also reduces the consumption of chemical reagents. Moreover, the catalytic reaction pathway and its mechanism can be further understood theoretically, so as to provide strong support for the designing and preparation of catalyst with good selectivity, high activity and strong anti-poisoning ability. However, there are very few reports of the influence of Ce1−xMnxO2 on diesel soot oxidation.
In the current study, firstly, Ce1−xMnxO2 catalysts doped with different Mn concentrations were prepared with sol–gel method. Secondly, X-ray diffraction (XRD), Raman spectrum, H2-temperature programming reduction (H2-TPR), X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA) were used to characterize Ce1−xMnxO2 catalyst and evaluate its oxidation activity. Finally, the first principle pseudopotential method based on the density functional theory (DFT)25–28 was adopted to further study the Ce1−xMnxO2 system doped with different Mn concentrations according to the crystal structure and oxygen vacancy. The object of this research is to systematically study the atomic structure and the oxygen vacancy formation energy of Ce1−xMnxO2 catalyst, and reveal the microscopic mechanism of the improvement of the catalytic oxidation activity with Mn-doped CeO2, which verifies experimental results.
2. Experimental
2.1. Catalyst preparation
Ce1−xMnxO2 catalyst was prepared by sol–gel method in this research. Firstly, a certain amount of Ce(NO3)3·6H2O (analytical grade, AR) and Mn(NO3)2 (AR) were obtained with an electronic balance and added to citric acid to form the mixed liquor. The mixture was subjected to a water bath at 80 °C and magnetically stirred until it forms into a gel. The gel, is then dried by air drying oven overnight, and roasted afterwards with a muffle furnace for 4 hours at 550 °C. The Mn doping concentrations in this research were 12.5%, 25% and 50%, and their corresponding chemical formulas were marked as Ce0.875Mn0.125O2, Ce0.75Mn0.25O2 and Ce0.5Mn0.5O2, respectively.2.2. Catalyst characterization
The powder X-ray diffraction (XRD) patterns were recorded by a diffractometer (D8 ADVANCE, Bruker, Germany) operated at 40 kV and 30 mA, using nickel-filtered Cu Kα radiation (λ = 0.15418 nm). The patterns were collected in a 2θ range from 20° to 80°, with a scanning step of 4° min−1.Scanning electron microscopy (SEM) image was carried out on a field emission scanning electron microscope S-4800 (Hitachi, Japan). Samples were coated with platinum to improve conductivity. The amplification range of the S-4800 was 20–8 × 105 times. The minimum resolution was 1.0 nm, and the electron acceleration voltage range was 0.5–30 kV.
The Raman spectra of the samples were obtained with a Renishaw in Viat+Reflex spectrometer at room temperature and atmospheric pressure. A wavelength of 514.5 nm was used for the exciting source from Ar+ ion laser. The power of the incident beam on the sample was 3 mW. The wave number values of the Raman spectra were accurate to 2 cm−1.
H2-Temperature programming reduction (H2-TPR) was investigated on a Micromeritics AutoChemII2920. At the beginning of the tests, the catalysts were preoxidized in 1% O2/He (50 mL min−1) at 300 °C for 10 min. Afterward they were cooled down to RT, flushed with He for 10 min, and heated from 0 °C up to 550 °C in 10% H2/Ar to finish the first TPR. The second TPR was performed subsequently, and a third TPR up to 800 °C was performed after another preoxidation process.
X-ray photoelectron spectra (XPS) were recorded using an ESCALAB 250 Xi spectrometer with Al Kα radiation (1486.6 eV). The adventitious C 1s line at 284.8 eV was used as an internal standard. The XPS core level spectra were analyzed in mixed Gaussian–Lorentzian peaks, using a Shirley background subtraction.
2.3. Catalytic testing
The evaluation indicators of the catalytic activity mainly include soot ignition temperature (Ti) and peak temperature (Tm), among which Ti is defined as the temperature during which the total mass of the sample is reduced by 5% in the reaction, and Tm is the temperature corresponding to the maximum weightlessness rate. In the catalytic activity evaluation system, lower values of Ti and Tm represent better catalytic activity.The test engine is a 4-cylinder, turbocharged, inter-cooled, electronic controlled common-rail diesel engine, and its main technical parameters are shown in Table 1. PM samples were collected under ESC 13-mode test cycle using the AVL SPC472 partial flow particle acquisition system. Thermogravimetric analysis (TGA) of particle sample was conducted on the METTLER TGA/DSC1 thermogravimetric analyzer, which has built-in high precision electronic balance and temperature sensor. In the thermogravimetric test, the sample weight was 3 mg, and the mass fraction of oxygen atmosphere was about 12%, which was close to the oxygen content in the diesel exhaust. High-purity N2 was taken as the protective gas, and its flow velocity was at a constant rate with 100 mL min−1. The programmed temperature range was 40–800 °C, and the heating rate was kept at 15 °C min−1. The collected diesel soot particles and catalyst were evenly mixed at the ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
| Sample | SOF weightlessness (°C) | Soot weightlessness (°C) | ||
|---|---|---|---|---|
| Ts | Tp | Ti | Tm | |
| CeO2 | 173 | 301 | 301 | 415 |
| Ce0.875Mn0.125O2 | 169 | 305 | 283 | 409 |
| Ce0.75Mn0.25O2 | 171 | 297 | 278 | 403 |
| Ce0.5Mn0.5O2 | 172 | 287 | 259 | 383 |
3. Results and discussion
3.1. Activity tests
Fig. 1 shows the results of the thermogravimetric (TG) experiments of diesel engine particles mixed with Ce1−xMnxO2 catalysts with four different Mn doping concentrations. Compared with the pure CeO2, the weightlessness curves of the particle shift to the low temperature region with the increase of Mn doping concentration, which indicates that the particle ignition combustion temperature decreases. There are three obvious weightlessness peaks in the particle weightlessness rate curves under Ce1−xMnxO2 catalysts, which are, in order, HC volatilization with low boiling point, HC oxidization with high boiling point and dry soot oxidative combustion. Its impact on the dry soot oxidation process is significant. With the increase of Mn in the doped proportion, the ignition temperature and peak temperature of the soot gradually decrease. | ||
| Fig. 1 TG and DTG curves of diesel particles. | ||
Table 1 lists the diesel particles weightlessness characteristic parameters under Ce1−xMnxO2 catalysis. Ts is the temperature corresponding to the peak weight loss rate of the low boiling point HC in soluble organic fraction (SOF). Tp is the temperature corresponding to the peak weight loss rate of the high boiling point HC. Ti is the ignition combustion temperature of dry soot. Tm is the peak combustion temperature of dry soot. Compared with that of pure CeO2, the soot ignition combustion temperatures are decreased by 18, 24 and 42 °C, and the peak combustion temperatures are decreased by 6, 12 and 32 °C, respectively, when the Mn concentrations are 12.5%, 25% and 50%. This indicates that the effect of CeO2 on soot catalytic oxidation is obviously improved with doping Mn, and the optimum Mn doping concentration is 50%.
The kinetic parameters such as oxidation activation energy (E) and pre-exponential factor (A) of soot particles were analyzed by Coats–Redfern integral method. In this way, the relationship between the ln[ln(1 − α)/T2] and 1/T of the diesel particles under the catalysis is obtained. The results of curve fitting are shown in Fig. 2. The linear regression coefficients of the fitted curves are all greater than 0.98, and the fitting effect is ideal.
 | ||
| Fig. 2 Fitting curves of ln[−ln(1 − α)/T2] and of 1/T particles under catalysis. | ||
The activation energy and pre-exponential factor of each fitting curve are calculated as shown in Table 2. It can be seen from Table 2 that compared with the pure CeO2, the activation energy of Ce1−xMnxO2 for the catalytic soot oxidation reaction is significantly reduced, and the pre-exponential factor is increased. This indicates that Mn doping significantly increases the number of active sites on the surface of soot and catalyst, thus enhancing the catalytic activity of the catalyst on soot oxidation reaction. With the increase of Mn concentration, the activation energy decreases gradually and reaches the minimum when the Mn concentration is 50%, which indicated that with the increase of Mn concentration, the energy required for soot oxidation reaction is gradually decreases, so that it is more easily oxidized, which is consistent with the thermogravimetric results.
| Sample | Fitting curve equation | Correlation coefficient | E/(kJ mol−1) | A/min−1 |
|---|---|---|---|---|
| CeO2 | y = −3.17x − 6.82 | 0.992 | 26.36 | 51 |
| Ce0.875Mn0.125O2 | y = −3.16x − 6.81 | 0.986 | 26.27 | 52 |
| Ce0.75Mn0.25O2 | y = −3.09x − 6.77 | 0.991 | 25.69 | 53 |
| Ce0.5Mn0.5O2 | y = −2.99x − 6.66 | 0.998 | 24.86 | 57 |
3.2. Catalyst characterization
A series of characterizations were carried out to clarify the mechanism of soot catalytic oxidation through Ce1−xMnxO2 catalyst on the microstructure. Fig. 3 gives the XRD profiles of the four Ce1−xMnxO2 samples. MnO2 will transform into Mn2O3 oxide when it is calcined at 550 °C. It can be seen that the characteristic diffraction peaks of CeO2 are detected in all samples. Compared with pure CeO2, the characteristic peaks are still the main diffraction peaks of Ce1−xMnxO2 catalyst, and the position of the peak is constant, but the intensity has been changed. It shows that the Mn doping does not change the crystal structure of cubic fluorite, and the structure is still stable. The characteristic peak of Mn2O3 also can be detected in the figure, which indicates that Mn ions have entered into CeO2 crystal lattice, forming solid solution. Due to difference in Mn electron scattering factor (fMn) and Ce electron scattering factor (fCe), the CeO2 material structure factor changes after being doped with Mn. Different Mn concentrations have different influence on the material structure factors (fhkl). The diffraction peak intensity (I) is proportional to the material structure factors squared (fhkl2), expressed as “I ∝ fhkl2”. This explains the diffraction peak intensity of the catalyst sample changes at different Mn doping concentrations. | ||
| Fig. 3 XRD profiles of Ce1−xMnxO2. | ||
The Ce1−xMnxO2 catalyst had broader diffraction peaks than the pure CeO2 catalyst. Estimation of the lattice parameter by Rietveld refinement of the main diffraction peaks of the CeO2 phase demonstrated that Ce0.875Mn0.125O2, Ce0.75Mn0.25O2 and Ce0.5Mn0.5O2 shrank slightly by 0.543, 0.534 and 0.516 nm, respectively. Slight downshift of the peaks indicated slight lattice contraction.29 The peak shift in the Ce1−xMnxO2 sample was probably due to the difference between the radii of Mn2+ (0.067 nm) and Ce4+ (0.094 m).30 Introduction of lower-valence cations generated lattice strain and facilitated defect formation within the CeO2 lattice, which eventually improved extrinsic surface defects and resulted in superior soot oxidation performance.
The particle size in the unit area was analyzed by Nano Measure software in the SEM image to calculate the average particle size. Fig. 4 shows the SEM images of Ce1−xMnxO2 samples. In Fig. 4(a), particle size range of nano-CeO2 is 30.1–55.2 nm, with an average particle size is 43 nm. In Fig. 4(b), particle size range of nano-Ce0.875Mn0.125O2 is 28.9–47.1 nm, with an average particle size is 38 nm. In Fig. 4(c), particle size range of nano-Ce0.75Mn0.25O2 is 18.5–42.3 nm, with an average particle size is 34 nm. In Fig. 4(d), particle size range of nano-Ce0.5Mn0.5O2 is 15.5–40.3 nm, with an average particle d size is 31 nm. The appearances of the three groups of particles are relatively uniform, which are mostly spherical or near spherical particles with clear boundaries. It proves that the particle aggregation is effectively controlled in the process of preparation, which makes the dispersion better.
 | ||
| Fig. 4 SEM images of Ce1−xMnxO2 sample: (a) CeO2; (b) Ce0.875Mn0.125O2; (c) Ce0.75Mn0.25O2; (d) Ce0.5Mn0.5O2. | ||
Fig. 5 shows the Raman profiles of CeO2 doped with different Mn concentrations. As shown in the figure, all the Ce1−xMnxO2 samples show a typical Raman vibration mode of CeO2. The vibration peaks of the four catalysts occur at 447 cm−1, which is the typical F2g vibration peak of CeO2 cubic fluorite structure. The offsets are very small and can be ignored. This indicates that CeO2 crystal structure does not change after being doped with Mn, which is consistent with the results of XRD. There is a weak Raman vibration peak near 641 cm−1 for the doped Mn sample, which is the characteristic Raman vibration peak of Mn species.31 It can be seen that the Raman peak intensity with different Mn doping concentrations varies. The higher Raman peak intensity indicates the better crystallinity of Ce1−xMnxO2. The peak widths at half height of the F2g Raman vibration peaks for the four catalysts are different, which corresponds to the particle size. This shows that Mn concentration affects the particle size of Ce1−xMnxO2 catalyst. In addition to the F2g mode, two weak absorption peak emerged in the spectra of the Mn-containing catalyst: one at 351 cm−1, and another between 560 cm−1 and 600 cm−1, related to oxygen vacancies.32,33 Incorporation of Mn2+ into the CeO2 lattice may have created more oxygen vacancies and enhanced the OSC, which could improve the catalytic activity of Ce1−xMnxO2 catalyst during soot oxidation.
 | ||
| Fig. 5 Raman profiles of Ce1−xMnxO2. | ||
Fig. 6 shows the H2-TPR spectrogram of Ce1−xMnxO2 catalyst. There are three obvious hydrogen consumption peaks as CeO2 catalyst is doped with Mn, marked as α, β and γ. The oxygen desorption peak area of the catalyst is greatly improved after doping Mn, changing the desorption temperature to a lower temperature. Therefore, both the oxidation reduction performance of the catalyst and the peak intensity increase with the increase of Mn content. The Ce0.5Mn0.5O2 reduction peaks at temperatures of 365 °C, 480 °C and 726 °C are the most obvious. Low temperature reduction peak (α) at 365 °C is the Ce0.5Mn0.5O2 surface adsorption oxygen reduction, the intermediate temperature reduction peak (β) at 480 °C is the Ce0.5Mn0.5O2 surface lattice oxygen reduction, and the high temperature reduction peak (γ) at 726 °C is the Ce0.5Mn0.5O2 bulk phase lattice oxygen reduction, while the pure CeO2 just has relatively weak reduction peaks of surface lattice oxygen and bulk phase lattice oxygen. The β-reduction peak areas of the three prepared Ce1−xMnxO2 catalysts are larger than their α-reduction peak areas, which indicate that they all have more surface lattice oxygen. Therefore, the Mn-doped CeO2 increases the lattice defects and enrich the oxygen species, which is favorable for adsorption, migration and transformation of oxygen. The oxygen species of Ce1−xMnxO2, especially the surface lattice oxygen, are directly related to its catalytic activity. Stronger reduction capacity of the surface oxygen species can lead to higher catalytic activity.
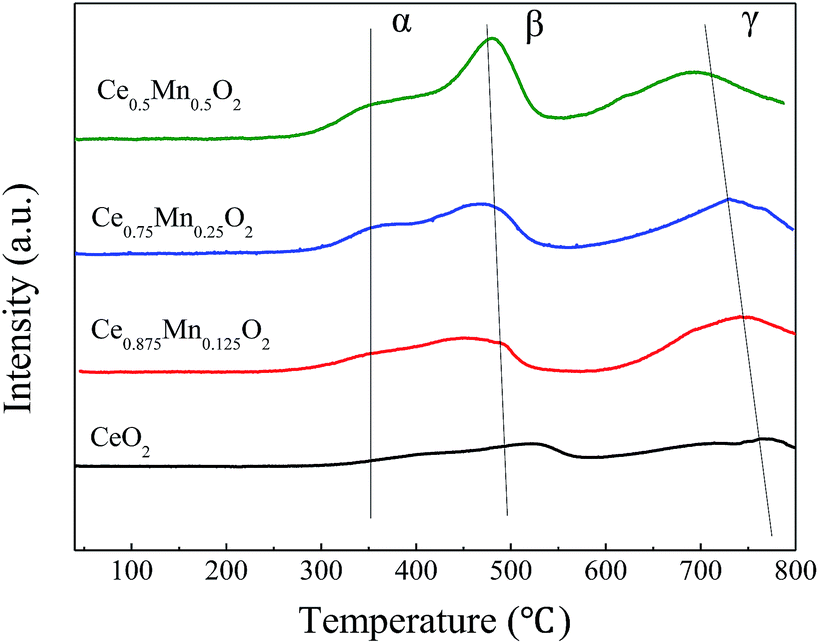 | ||
| Fig. 6 H2-TPR profiles of Ce1−xMnxO2. | ||
Fig. 7(a) shows the XPS profiles of Mn 2p orbital for Ce1−xMnxO2 catalyst. There are two peaks at 642.2 eV and 654.5 eV, which are the 2p3/2 peaks of Mn3+ and Mn4+ respectively. It indicates that Mn exists as +3 and +4 valences in the catalyst. The relative proportion of Mn4+ to Mn3+ increases with the increase of Mn content. Fig. 7(b) gives the XPS profiles of O 1s orbital for Ce1−xMnxO2 catalyst. It can be seen that each sample has a double-peak structure, which shows that the oxygen element exists on the catalytic surface in two forms. The peak with low electronic binding energy at 531.5 eV is lattice oxygen (OI), and it is related to the redox property of the metal ion. The peak with high electronic binding energy at 529.5 eV is adsorption oxygen (OII), also related to the oxygen vacancy concentration in the catalyst.
 | ||
| Fig. 7 XPS spectra of Ce1−xMnxO2, (a) Mn 2p, (b) O 1s. | ||
3.3. DFT calculations
The ideal crystal cell model of CeO2 is selected, where the Ce atom of CeO2 crystal cell is replaced by Mn atom to realize the displacement doping. For Ce1−xMnxO2 system with different doping concentrations, supercell models with different atomic numbers (6, 12, 24) are firstly established. Then, Mn atom is used to replace Ce atom in the supercell to simulate different doping concentrations. Calculations show that, the super cell atomic numbers of 6, 12 and 24 corresponds to Mn concentrations of 50%, 25% and 12.5%, respectively. Fig. 8 gives schematic cell models of CeO2 and Mn-doped with 25% concentration.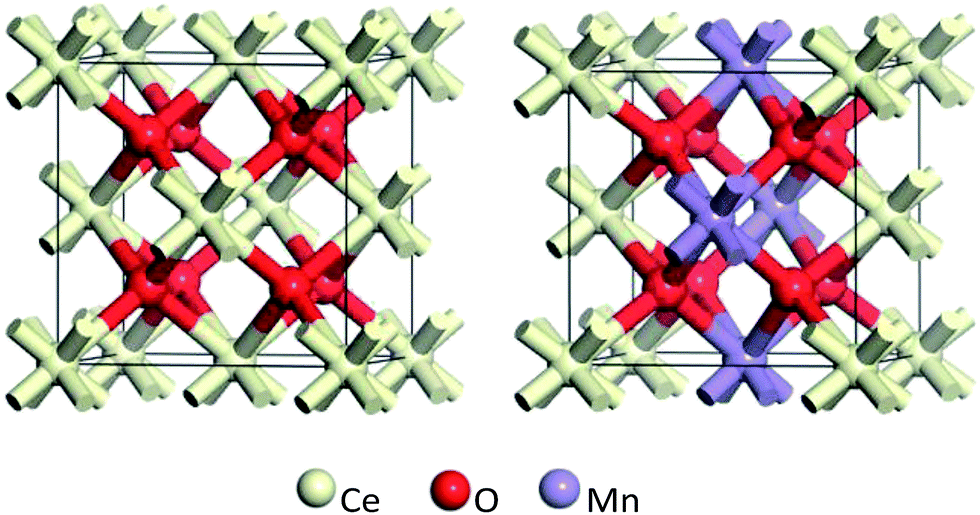 | ||
| Fig. 8 Schematic cell model of CeO2 and Mn-doped (Ce0.75Mn0.25O2). | ||
The DFT calculations corrected by on-site coulomb interaction were performed by using the Perdew–Wang 1991 (PW91) GGA approach.34 Firstly, the unit cell structure was conducted with geometry optimization. The cut-off energy was set as 360 eV, and self-consistent field energy convergence criterion was 1.0 × 10−6 eV. The optimal convergence energy was smaller than 1.0 × 10−3 eV, and the force threshold was set to 0.5 eV Å−1. The vacuum gap was set to 10 Å.
The oxygen vacancy formation energy is as an important indicator used to measure the catalytic activity of the catalyst. The decrease in oxygen vacancy formation energy shows that the oxygen release ability of the catalyst is improved. It is defined as follows:
| EV = ECeO2/Mn − (EVCeO2 + EO) | (1) |
Table 3 lists the lattice constants of the optimized pure CeO2 and the CeO2 doped with different Mn concentrations. The calculated value of 0.547 nm is in agreement with experimental value of 0.541 nm,35 which verifies the reliability of the selected model. After doping with Mn, the lattice constant of Ce1−xMnxO2 is slightly smaller than that of pure CeO2, and it monotonously decreases with the increase of Mn content. This phenomenon is due to the replacement of the ionic radius of Ce3+ (0.101 nm) or Ce4+ with the ionic radius of Mn2+, because the ionic radius of Mn2+ is smaller than that of Ce4+ and Ce3+, resulting in a smaller bond length of Mn–O than Ce–O.
| Sample | a (nm) | b (nm) | c (nm) |
|---|---|---|---|
| CeO2 | 0.547 | 0.547 | 0.547 |
| Ce0.875Mn0.125O2 | 0.542 | 0.543 | 0.544 |
| Ce0.75Mn0.25O2 | 0.534 | 0.534 | 0.532 |
| Ce0.5Mn0.5O2 | 0.516 | 0.513 | 0.513 |
Due to less Mn entering into CeO2 lattice in the case of low Mn doping concentration, the bulk oxygen concentration decrease, and the total cation concentration also decrease, which further reduce the oxygen storage capacity. It shows that there are two kinds of oxygen storage mechanisms in the Ce1−xMnxO2 system that influences each other, which are surface phase oxygen storage and bulk phase oxygen storage. Calculations indicate that Mn can better enter into CeO2 lattice when Mn concentration is 50%, forming solid solution with high structure stability, which significantly increases the oxygen storage capacity.
Fig. 9 shows the oxygen vacancy formation energy of Ce1−xMnxO2 catalyst at different Mn doping concentrations. The oxygen vacancy formation energy in pure CeO2 system is 2.99 eV, which is consistent with the research results of Yang et al.35 The oxygen vacancy formation energy in CeO2 is greatly reduced with doping Mn, showing that with Mn-doped, the oxygen release ability can be significantly improved. The oxygen vacancy formation energy of Ce1−xMnxO2 catalyst decreases with the increase of Mn doping content, which indicates that the Mn substitution eases the formation of oxygen vacancies and realizes the transformation of Ce4+ ↔ Ce3+. Fig. 9 also shows that when the Mn doping concentration is 50%, the oxygen vacancy formation energy represents a minimum value of 0.31 eV. Meantime, its oxygen storage/oxygen release capacity is the most excellent. The results are in agreement with the results of the thermogravimetric experiments shown in Fig. 1.
 | ||
| Fig. 9 Oxygen vacancy formation energy of Ce1−xMnxO2. | ||
3.4. Catalytic mechanism
The catalytic effect of Ce1−xMnxO2 catalyst is illustrated in Fig. 10, showing that the presence of cerium oxide enhances oxygen mobility in the reaction cycle. Meanwhile, small particle size also facilitates oxygen mobility from interior to surface within nano-crystallite. Interaction in Ce–Mn oxides can be regarded as an oxygen transition process from lattice oxygen (Olatt) and chemisorbed oxygen (Oads) to active O* on the active sites of catalyst surface, which is favorable for the soot oxidation. In the redox cycle, Ce4+/Ce3+ redox couple features in oxygen storage, that is, CeO2 acts as an oxygen buffer by storing/releasing oxygen species due to Ce4+/Ce3+ redox,36 which facilitates the mobility of oxygen in the redox cycle. | ||
| Fig. 10 Illustration of catalytic effect of Ce1−xMnxO2. | ||
4. Conclusions
With the increase of Mn doping concentrations, the weightlessness curves of diesel engine particles move towards low temperature regions, gradually decreasing the corresponding ignition temperature, peak temperature and the activation energy of Ce1−xMnxO2 for the catalytic soot oxidation. Compared with pure CeO2, the soot ignition combustion temperatures are decreased by 18, 24 and 42 °C, and the peak temperatures are decreased by 6, 12 and 32 °C, when the Mn concentrations are 12.5%, 25% and 50%, respectively. It shows that the oxidation activity of Ce1−xMnxO2 catalyst doped with Mn has obviously improved.Characterization results from XRD and Raman show that the three groups of the prepared Ce1−xMnxO2 catalysts still have the crystal structure of cubic fluorite and their structures are stable. Mn-doped CeO2 can improve its dispersion, increase the lattice defects and enrich the oxygen species, which is beneficial to the oxygen adsorption, oxygen migration and oxygen transformation.
The CeO2 lattice constant calculated through GGA-PW91 of DFT is close to the experimental value, which verifies the reliability of this simulation method. Calculations show that Mn can better enter into CeO2 lattice and form a solid solution with high structure stability when Mn doping concentration is 50%.
For CeO2 catalyst, Mn-doped can greatly reduce oxygen vacancy formation, improve the ability of oxygen release and enhance its redox capacity. When the Mn doping concentration is 50%, the oxygen vacancy formation energy can be minimized to 0.31 eV, and it would be very conducive to soot catalytic oxidation capacity of diesel engines.
Acknowledgements
We would like to acknowledge the financial supports from the Natural Science Major Research Project of Jiangsu province university (14KJA470001), State Key Laboratory of Engines, Tianjin University (K2016-05), Natural Science Foundation of Jiangsu Province, China (BK20160538), the Graduate Student Scientific Research Innovation Project of Jiangsu province university (KYLX_1038) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- A. Bueno-Lopez, Appl. Catal., B, 2014, 146, 1–11 CrossRef CAS.
- L. Pahalagedara, H. Sharma, C. H. Kuo, S. Dharmarathnat and A. Joshi, Energy Fuels, 2012, 26, 384–388 Search PubMed.
- Y. Sekine, H. Koyama, M. Matsukata and E. Kikuchi, Fuel, 2013, 103, 2–6 CrossRef CAS.
- S. Chatterjee, R. Conway, S. Viswanathan, M. Blomquist, B. Klueseuer and S. Anderson, NOx and PM control from heavy duty diesel engines using a combination of low pressure EGR and continuously regenerating particulate filter, SAE Paper no. 01-0048, 2003 Search PubMed.
- M. Mohr, A. M. Forss and U. Lehmann, Environ. Sci. Technol., 2006, 40, 2375–2383 CrossRef CAS PubMed.
- W. C. Zhan, X. Y. Zhang, Y. L. Guo, L. Wang and Y. Guo, J. Rare Earths, 2014, 32, 146–152 CrossRef CAS.
- T. F. Hou, Y. S. Lei, S. Y. Zhang, J. H. Zhang and W. J. Cai, J. Rare Earths, 2015, 33, 42–45 CrossRef CAS.
- Z. M. Shi, Y. Liu, W. Y. Yang, K. M. Liang, F. Pan and S. R. Gu, J. Eur. Ceram. Soc., 2002, 22, 1251–1256 CrossRef CAS.
- H. Huang, J. H. Liu, P. Sun and S. Ye, RSC Adv., 2016, 104, 28–34 Search PubMed.
- E. Aneggi, M. Boaro, C. D. Litenburg, G. Dolcetti and A. Trovarelli, J. Alloys Compd., 2006, 37, 1096–1102 CrossRef.
- H. X. Mai, L. D. Sun, Y. W. Zhang, R. Si and W. Feng, J. Phys. Chem., 2005, 109, 24380–24385 CrossRef CAS PubMed.
- F. Lin, W. Y. Huang, J. Y. Tang and P. D. Yang, Nano Res., 2011, 4, 61–71 CrossRef CAS.
- J. Kaspar, P. Fomasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef CAS.
- M. Sugiura, M. Ozawa, A. Suda, T. Suzuki and T. Kanazawa, Bull. Chem. Soc. Jpn., 2005, 78, 752–767 CrossRef CAS.
- Z. M. Shi, Y. Liu, W. Y. Yang, K. M. Liang and F. Pan, J. Eur. Ceram. Soc., 2002, 22, 1251–1256 CrossRef CAS.
- G. Dutta, T. Baidya, M. S. Hegde, K. R. Priolkar and P. R. Sarode, J. Am. Chem. Soc., 2006, 18, 3249–3256 CAS.
- E. M. And, T. Egami, R. B. And, M. Koranne and S. Tyadi, J. Phys. Chem. B, 2000, 104, 11110–11116 CrossRef.
- A. Trovarelli and P. Fornasiero, Catalysis by Ceria and Related Materials, Imperial College Press, London, 2002 Search PubMed.
- X. Wu, S. Liu, W. Duan, F. Lin and R. Ran, J. Hazard. Mater., 2011, 187, 283–290 CrossRef CAS PubMed.
- Q. Liang, X. Wu, D. Weng and H. Xu, Catal. Today, 2008, 139, 113–118 CrossRef CAS.
- N. Guillén-Hurtado, A. García-García and A. Bueno-López, J. Catal., 2013, 299, 181–187 CrossRef.
- G. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS.
- L. Shi, X. Wang, Q. Zhao, Y. Zhang and L. Zhang, Catal. Commun., 2009, 86, 166–175 Search PubMed.
- D. Delimaris and T. Ioannides, Appl. Catal., B, 2009, 89, 295–302 CrossRef CAS.
- P. Ryderg, F. S. Jorgensen and L. Olsen, Expet Opin Drug Metabol Toxicol, 2014, 10, 215–227 CrossRef PubMed.
- W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 31, 12974–12980 CrossRef.
- N. H. March, G. G. N. Angilella and R. Pucci, Int. J. Mod. Phys. B, 2013, 27, 5814–5821 CrossRef.
- P. Junell, K. Honkala, M. Hirsimaki, M. Valden and K. Laasonen, Surf. Sci., 2003, 546, 797–802 CrossRef.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. M. Reddy, RSC Adv., 2014, 4, 11322–11330 RSC.
- Y. Zuo, L. Li, X. Huang and G. Li, Catal. Sci. Technol., 2013, 4, 402–410 Search PubMed.
- Y. F. Qu, J. X. Guo, Y. H. Chu, M. C. Sun and H. Q. Yin, Appl. Surf. Sci., 2013, 282, 425–431 CrossRef CAS.
- L. F. Nascimento and O. A. Serra, Process Saf. Environ. Prot., 2016, 101, 134–143 CrossRef CAS.
- O. H. Laguna, M. A. Centeno, F. R. Sarria and J. A. Odriozola, Catal. Today, 2011, 172, 118–123 CrossRef CAS.
- M. L. Fu, J. M. Lin, W. B. Zhu, J. L. Wu and L. M. Chen, J. Rare Earths, 2014, 33, 153–158 CrossRef.
- Z. X. Yang, T. K. Woo and K. Hermansson, J. Chem. Phys., 2006, 124, 579–593 Search PubMed.
- G. Liu, R. L. Yue, Y. Jia, Y. Ni, J. Yang, H. D. Liu and Z. Wang, Particuology, 2013, 11, 454–459 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |