
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed three-component tandem cyclization of buta-2,3-dien-1-ol, aryl iodides, and imines: an efficient protocol for the synthesis of oxazolidine derivatives†
Yunlei Hou‡
,
Mingze Qin‡,
Xiuxiu Yang,
Qi Shen,
Yanfang Zhao,
Yajing Liu* and
Ping Gong*
Key Laboratory of Structure-based Drug Design and Discovery (Shenyang Pharmaceutical University), Ministry of Education, 103 Wenhua Road, Shenhe District, Shenyang 110016, People's Republic of China. E-mail: gongpinggp@126.com; lyjpharm@126.com
First published on 23rd January 2017
Abstract
An efficient three-component tandem cyclization reaction for the synthesis of highly substituted oxazolidines was achieved through the Pd0-catalyzed cyclization of buta-2,3-dien-1-ol with aryl iodides and imines. A range of R1 and R2 functional groups is well-tolerated while affording cyclization products in moderate yields and with moderate to high diastereoselectivities.
Introduction
Multifunctional heterocyclic compounds play an essential role in organic and medicinal chemistry, as well as in drug discovery. Notably, nearly 70% of marketed drugs contain heterocycles.1 Oxazolidine rings are common pharmacophores that comprise potent antitumor tetrahydroisoquinoline-based natural products including quinocarcin (Fig. 1, I) and tetrazomine (Fig. 1, II).2 In addition, chiral oxazolidines are used as chiral auxiliaries (Fig. 1, III)3 and chiral ligands (Fig. 1, IV)4 in a variety of asymmetric transformations. Oxazolidines are also commonly utilized as prodrugs for improving the pharmacokinetic profile of certain β-amino alcohol pharmacophores.5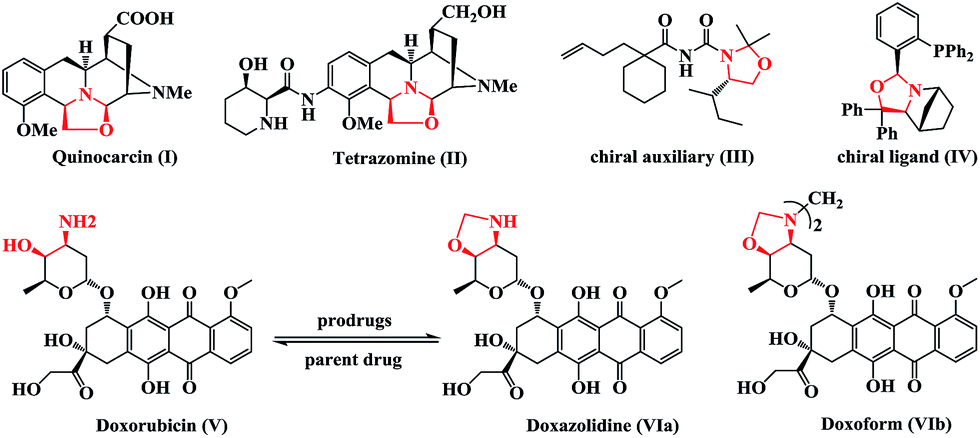 | ||
| Fig. 1 Representative medicinally relevant agents, natural products, and chiral agents containing oxazolidines. | ||
For instance, the anticancer drug doxorubicin (Fig. 1, V) could be transformed into the oxazolidine prodrug VI to increase tumor response and minimize side effects, especially the treatment-limiting cardiotoxicity associated with doxorubicin therapy.6 Accordingly, the development of new methodologies for the synthesis of oxazolidines has garnered considerable attention.
Recently, Yoon et al. developed an efficient method for synthesizing oxazolidines through the iron- or copper-catalyzed aminohydroxylation of olefins (Scheme 1, Eq (1)).7 Jarvo further demonstrated the stereoselective and stereospecific syntheses of oxazolidines using catalysts with differing rates of allylmetal isomerization (Scheme 1, Eq (2)).8 Zhang investigated the [3 + 2] cycloaddition of azomethine ylides with carbonyl compounds for the synthesis of oxazolidines (Scheme 1, Eq (3)).9 More recently, Zhang explored a new organocatalytic strategy to synthesize oxazolidines through the Pd-catalyzed asymmetric decarboxylative cycloaddition of vinylethylene carbonates (VECs) with imines in the presence of chiral ligands (Scheme 1, Eq (4)).10 Therefore, the development of operationally simple and efficient synthetic approaches towards oxazolidines using readily available and stable starting materials is desired.
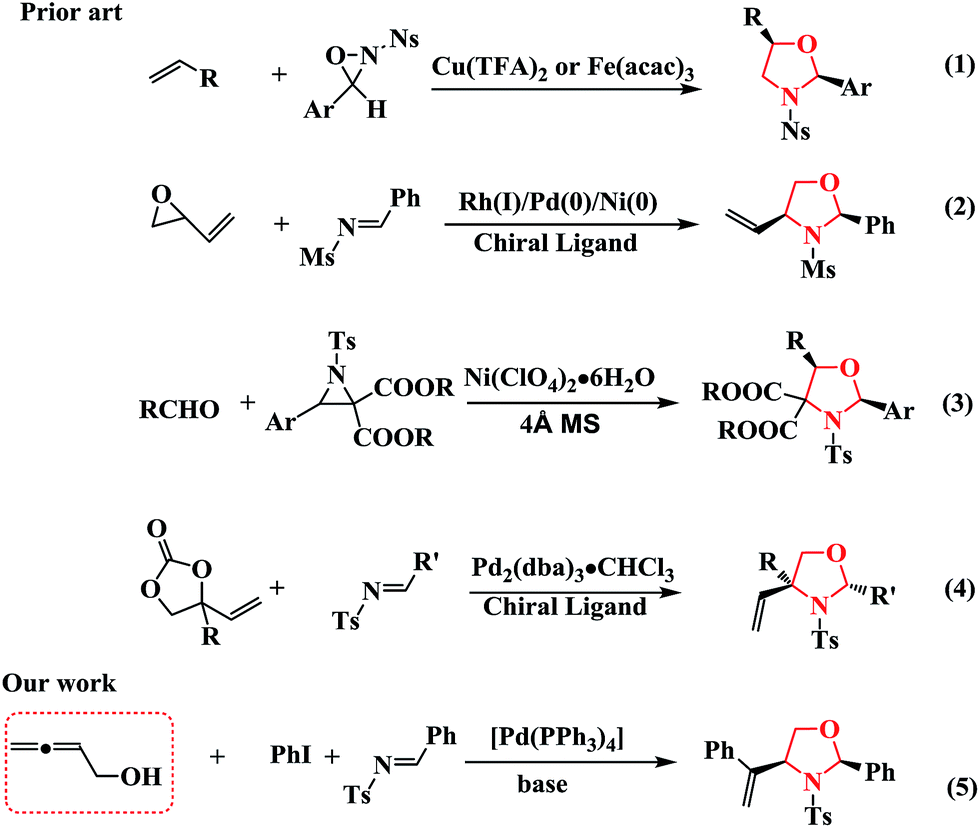 | ||
| Scheme 1 Strategies for the construction of oxazolidines. | ||
Multi-component reactions (MCRs) involving domino processes have emerged as powerful tools for the synthesis of molecules, including heterocyclic compounds, with efficiency, atom-economy, and diversity from simple substrates.11 In addition, functionalized allenes have been shown to be efficient starting materials for the synthesis of potentially useful carbo- and heterocycles.12 Inspired by the synthesis of 1,3-oxazolidines from the cycloaddition of imines and vinyl epoxides (Scheme 1, Eq (2)) or VECs (Scheme 1, Eq (4)), we hypothesized that other substrates, such as terminal β-allenols instead of vinyl epoxides or VECs, could be involved in the Pd0-catalyzed reaction of organic halides and imines without the necessity of a chiral ligand, which would allow for the formation of oxazolidine derivatives (Scheme 1, Eq (5)). Towards that end, we developed a novel and efficient route towards 1,3-oxazolidines via the cycloaddition of buta-2,3-dien-1-ol (1a) with aryl iodides and imines by varying the reaction conditions.
Results and discussion
Based on literature precedent,13,14 we set out to test the model reaction of buta-2,3-dien-1-ol (1a) with iodobenzene (2a) and imine (3a) in the presence of 5 mol% of [Pd(PPh3)4] at 80 °C, as shown in Table 1. The reaction in THF with K2CO3 as the base afforded oxazolidine 4aa in 23% yield with a dr of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 1). In order to improve the yield and stereoselectivity, different bases, solvents, and additives were tested. Among the tested bases, CsF and Cs2CO3 resulted in higher efficiencies with 50% and 38% yields, respectively (Table 1, entries 4–5), while Na2CO3 only gave the product in a trace yield (Table 1, entry 3). Polar solvents such as DMSO and DMF resulted in moderate stereoselectivity with a >25:1 dr (Table 1, entries 9 and 11). Less polar solvents such as toluene decreased the diastereoselectivity to 8:1 and resulted in an 18% yield (Table 1, entry 7). When the reaction was conducted in dioxane, 4aa was obtained in 50% yield with a >30:1 dr (Table 1, entry 10). Next, the reaction temperature was examined. Upon lowering the temperature to 40 °C, the conversion dramatically dropped to only 10%, even when the reaction time was extended to 48 h. Higher temperatures (100 °C) accelerated the reaction rate without any increase in the yield (Table 1, entries 10 and 15). On the basis of previous studies, various additives were tested (Table 1, entries 12–13). Among them, the phase-transfer catalyst n-Bu4NBr had no dramatic effect on the stereoselectivity. The Lewis acid Cu(OTf)2 did not improve the yields, but the cis/trans ratio decreased to 10:1. An increase in the molar amount of imine 3a from 1.2 to 3.0 equiv. also gave a similar result (Table 1, entries 1 vs. 2, 15 vs. 16). Higher palladium concentrations (from 5 mol% to 50 mol%) was tried in order to improve the yield, but the yield did not increased (Table 1, entry 17). Overall, the best results were obtained when 3.0 equiv. of CsF was used as the base in the presence of 5 mol% [Pd(PPh3)4] in dioxane at 80 °C.
:1 (Table 1, entry 1). In order to improve the yield and stereoselectivity, different bases, solvents, and additives were tested. Among the tested bases, CsF and Cs2CO3 resulted in higher efficiencies with 50% and 38% yields, respectively (Table 1, entries 4–5), while Na2CO3 only gave the product in a trace yield (Table 1, entry 3). Polar solvents such as DMSO and DMF resulted in moderate stereoselectivity with a >25:1 dr (Table 1, entries 9 and 11). Less polar solvents such as toluene decreased the diastereoselectivity to 8:1 and resulted in an 18% yield (Table 1, entry 7). When the reaction was conducted in dioxane, 4aa was obtained in 50% yield with a >30:1 dr (Table 1, entry 10). Next, the reaction temperature was examined. Upon lowering the temperature to 40 °C, the conversion dramatically dropped to only 10%, even when the reaction time was extended to 48 h. Higher temperatures (100 °C) accelerated the reaction rate without any increase in the yield (Table 1, entries 10 and 15). On the basis of previous studies, various additives were tested (Table 1, entries 12–13). Among them, the phase-transfer catalyst n-Bu4NBr had no dramatic effect on the stereoselectivity. The Lewis acid Cu(OTf)2 did not improve the yields, but the cis/trans ratio decreased to 10:1. An increase in the molar amount of imine 3a from 1.2 to 3.0 equiv. also gave a similar result (Table 1, entries 1 vs. 2, 15 vs. 16). Higher palladium concentrations (from 5 mol% to 50 mol%) was tried in order to improve the yield, but the yield did not increased (Table 1, entry 17). Overall, the best results were obtained when 3.0 equiv. of CsF was used as the base in the presence of 5 mol% [Pd(PPh3)4] in dioxane at 80 °C.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Additive | Solvent | Temp (°C) | Time (h) | Yieldb (%) | drc (cis/trans) |
| a Reaction conditions: under a N2 atmosphere, 1a (0.35 mmol, 1 equiv.), 2a (0.42 mmol, 1.2 equiv.), 3a (0.42 mmol, 1.2 equiv.), [Pd(PPh3)4] (0.018 mmol, 5 mol%), base (1 mmol, 3 equiv.), solvent (3.0 mL).b Isolated yield.c Determined by 1H NMR.d A = n-Bu4NBr (5 mol%), B = Cu(OTf)2 (5 mol%).e Imine 3a (3.0 equiv.) was used.f [Pd(PPh3)4] (20 mol% or 50 mol%) was used. | |||||||
| 1 | K2CO3 | — | THF | Reflux | 15 | 23 | 3:1 |
| 2e | K2CO3 | — | THF | Reflux | 15 | 25 | 3:1 |
| 3 | Na2CO3 | — | THF | Reflux | 15 | Trace | — |
| 4 | Cs2CO3 | — | THF | Reflux | 12 | 38 | 10:1 |
| 5 | CsF | — | THF | Reflux | 10 | 50 | 15:1 |
| 6 | CsF | — | CH3CN | Reflux | 10 | 48 | 20:1 |
| 7 | CsF | — | Toluene | 80 | 10 | 18 | 8:1 |
| 8 | Cs2CO3 | Dioxane | 80 | 12 | 41 | >30:1 |
|
| 9 | CsF | — | DMSO | 80 | 12 | 43 | 25:1 |
| 10 | CsF | — | Dioxane | 80 | 10 | 50 | >30:1 |
| 11 | CsF | — | DMF | 80 | 10 | 45 | 25:1 |
| 12 | CsF | Ad | Dioxane | 80 | 10 | 48 | >30:1 |
| 13 | CsF | Bd | Dioxane | 80 | 10 | 50 | 10:1 |
| 14 | CsF | — | Dioxane | 40 | 48 | 10 | — |
| 15 | CsF | — | Dioxane | 100 | 8 | 45 | >30:1 |
| 16e | CsF | — | Dioxane | 80 | 10 | 49 | >30:1 |
| 17f | CsF | — | Dioxane | 80 | 10 | 49 | >30:1 |
Under the optimized reaction conditions, the scope and limitations of the transformation were evaluated for the reaction of 1a with various imines 3; the results are listed in Table 2. Tandem reactions with imines derived from arylaldehydes with different steric and electronic features proceeded smoothly to afford the corresponding oxazolidines 4 in moderate yields and diastereoselectivities (Table 2, entries 1–14). The position of the substituents on the aryl ring affected the reactivity. For example, p-fluorophenyl-substituted 3f afforded the desired cycloaddition product 4af in 48% yield with a dr of 10:1 under the optimized conditions (Table 2, entry 6). In contrast, o-fluorophenyl-substituted 3g exhibited a decreased conversion with low diastereoselectivity, indicating that o-substitution on the aryl ring is unfavorable (Table 2, entry 7). The reaction efficiency and diastereomeric ratio decreased dramatically when multiple-halogen atoms were present on the aryl ring, implying that stereospecific blockade groups have a negative effect on the cycloaddition reaction (Table 2, entries 10 and 11). Imines 3l and 3m, derived from 2-formylthiofuran and furfural, were also suitable substrates for the cycloaddition and furnished the corresponding oxazolidines in 47% and 43% yields, respectively (Table 2, entries 12–13). However, imine 3o (R1 = 2-pyrrolyl) gave the product 4ao in only a trace yield, indicating that it was significantly less reactive than 3l (R1 = 2-thienyl) and 3m (R1 = 2-furyl). Gratifyingly, (E)-N-mesyl imine 3n was a viable substrate in this transformation, affording the product in 45% isolated yield under the standard conditions. When using imines 3p with phenyl or p-methoxyl phenyl groups at nitrogen, the target product was not detected, which suggesting that imines with electron-withdrawing groups at nitrogen was necessary in this reaction. The structures and relative stereochemistries of the products were established by X-ray crystallography analysis of 4aa as the 2,4-cis-isomer (Fig. 2), and those of the other products were assigned by analogy.
|
||||
|---|---|---|---|---|
| Entry | 3 | 4 | Yieldb (%) | drc (cis/trans) |
| a Reaction conditions: under a N2 atmosphere, 1a (0.35 mmol, 1 equiv.), 2a (0.42 mmol, 1.2 equiv.), 3a (0.42 mmol, 1.2 equiv.), [Pd(PPh3)4] (0.018 mmol, 5 mol%), CsF (1 mmol, 3 equiv.), dioxane (3.0 mL).b Isolated yield.c Determined by 1H NMR. The absolute configuration of 4aa was determined by X-ray crystallography (see Fig. 2); those of the other products were assigned by analogy.d 3n = (E)-N-benzylidenemethanesulfonamide.e 3p = (E)-N,1-diphenylmethanimine or (E)-N-(4-methoxyphenyl)-1-phenylmethanimine. | ||||
| 1 | 3a (R1 = Ph) | 4aa | 50 | >30:1 |
| 2 | 3b (R1 = p-MeC6H4) | 4ab | 55 | 15:1 |
| 3 | 3c (R1 = p-MeOC6H4) | 4ac | 52 | >30:1 |
| 4 | 3d (R1 = m-MeC6H4) | 4ad | 45 | 10:1 |
| 5 | 3e (R1 = p-ClC6H4) | 4ae | 51 | 8:1 |
| 6 | 3f (R1 = p-FC6H4) | 4af | 48 | 10:1 |
| 7 | 3g (R1 = o-FC6H4) | 4ag | 30 | 3:1 |
| 8 | 3h (R1 = o-ClC6H4) | 4ah | 28 | 5:1 |
| 9 | 3i (R1 = p-BrC6H4) | 4ai | 54 | 4:1 |
| 10 | 3j (R1 = 2,4-(F)2-C6H4) | 4aj | 25 | 3:1 |
| 11 | 3k (R1 = 2-Cl,4-F-C6H4) | 4ak | 18 | 2:1 |
| 12 | 3l (R1 = 2-thienyl) | 4al | 47 | 4:1 |
| 13 | 3m (R1 = 2-furyl) | 4am | 43 | 2:1 |
| 14 | 3n (R1 = Ph)d | 4an | 45 | 4:1 |
| 15 | 3o (R1 = 2-pyrrolyl) | 4ao | Trace | — |
| 16 | 3p (R1 = Ph)e | 4ap | Trace | — |
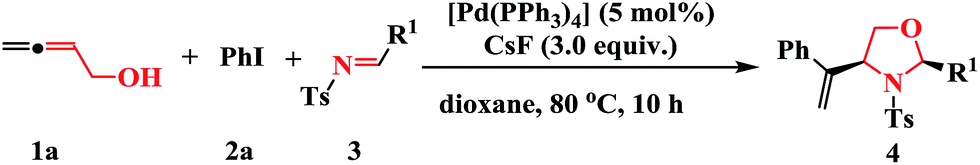
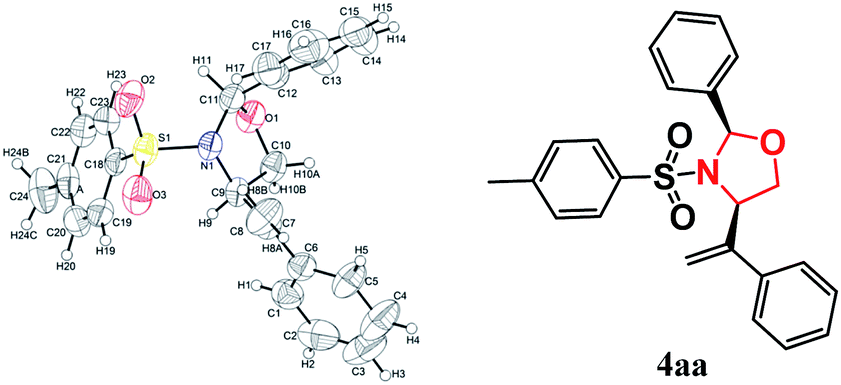 | ||
| Fig. 2 X-ray crystal structure of compound 4aa. | ||
After the successful cycloaddition of 1a and iodobenzene 2a with various imines, we subsequently turned our attention toward the cycloaddition of 1a and (E)-N-benzylidenetosylamide 3a with various aryl iodides; the results are summarized in Table 3. In general, aryl iodides bearing electron-withdrawing (Table 3, entries 4–5) or electron-donating groups (Table 3, entries 2–3) afforded the corresponding oxazolidines in moderate yields and diastereoselectivities under identical conditions. Furthermore, the incorporation of multiple-halogens or strong EWGs on the phenyl ring decreased the reaction efficiency, which suggested that the electron density of the phenyl ring played a role in the reaction efficiency (Table 3, entries 4–5, 7–8, 28–40%). Notably, when a heteroaryl iodide was employed as a substrate (Table 3, entry 9, 43%), the product yield was slightly affected, which was attributed to the electronic property of the thiophene ring. In order to investigate the limitations of aryl halides with different carbon–halogen bonds, phenyl bromide and phenyl chloride were tested. As presented in Table 3 (entries 10 vs. 1), only the carbon–iodine bond was cleaved, suggesting that aryl iodides resulted in higher efficiencies. Interestingly, compounds 4aa–4ia (dr > 10:1) in Table 3 gave a higher diastereomeric ratio than compounds 4ad–4an (dr = 2:1–10:1) in Table 2, which indicated that R2 has a more general substrate scope.
|
||||
|---|---|---|---|---|
| Entry | 2 | 4 | Yieldb (%) | drc (cis/trans) |
| a Reaction conditions: under a N2 atmosphere, 1a (0.35 mmol, 1 equiv.), 2a (0.42 mmol, 1.2 equiv.), 3a (0.42 mmol, 1.2 equiv.), [Pd(PPh3)4] (0.018 mmol, 5 mol%), CsF (1 mmol, 3 equiv.), dioxane (3.0 mL).b Isolated yield.c Determined by 1H NMR.d 2j = phenyl bromide or phenyl chloride. | ||||
| 1 | 2a (R2 = Ph) | 4aa | 50 | >30:1 |
| 2 | 2b (R2 = p-MeC6H4) | 4ba | 55 | 11:1 |
| 3 | 2c (R2 = p-MeOC6H4) | 4ca | 53 | 10:1 |
| 4 | 2d (R2 = p-MeO2CC6H4) | 4da | 40 | 10:1 |
| 5 | 2e (R2 = p-NO2C6H4) | 4ea | 38 | 12:1 |
| 6 | 2f (R2 = p-FC6H4) | 4fa | 46 | 16:1 |
| 7 | 2g (R2 = 3-Cl,4-F-C6H4) | 4ga | 33 | >30:1 |
| 8 | 2h (R2 = 3,4-(F)2-C6H4) | 4ha | 28 | >30:1 |
| 9 | 2i (R2 = 2-thienyl) | 4ia | 43 | 22:1 |
| 10 | 2jd | 4ja | Trace | — |

Based on previous studies,15 a plausible mechanism for the cross-coupling reaction is proposed in Scheme 2. The carbopalladation of PhPdI, which is formed in situ from the oxidative addition of Pd0 to iodobenzene, with buta-2,3-dien-1-ol (1a) at the center carbon atom forms the π-allylic palladium complex B. Subsequent nucleophilic attack of the oxygen nucleophile of B on the imines 3 leads to the intermediate C. Finally, the resulting intermediate C undergoes an intramolecular nucleophilic attack on the inner π-allylic carbon atom to produce the cyclized products 4, along with the regeneration of the Pd0 catalyst D.
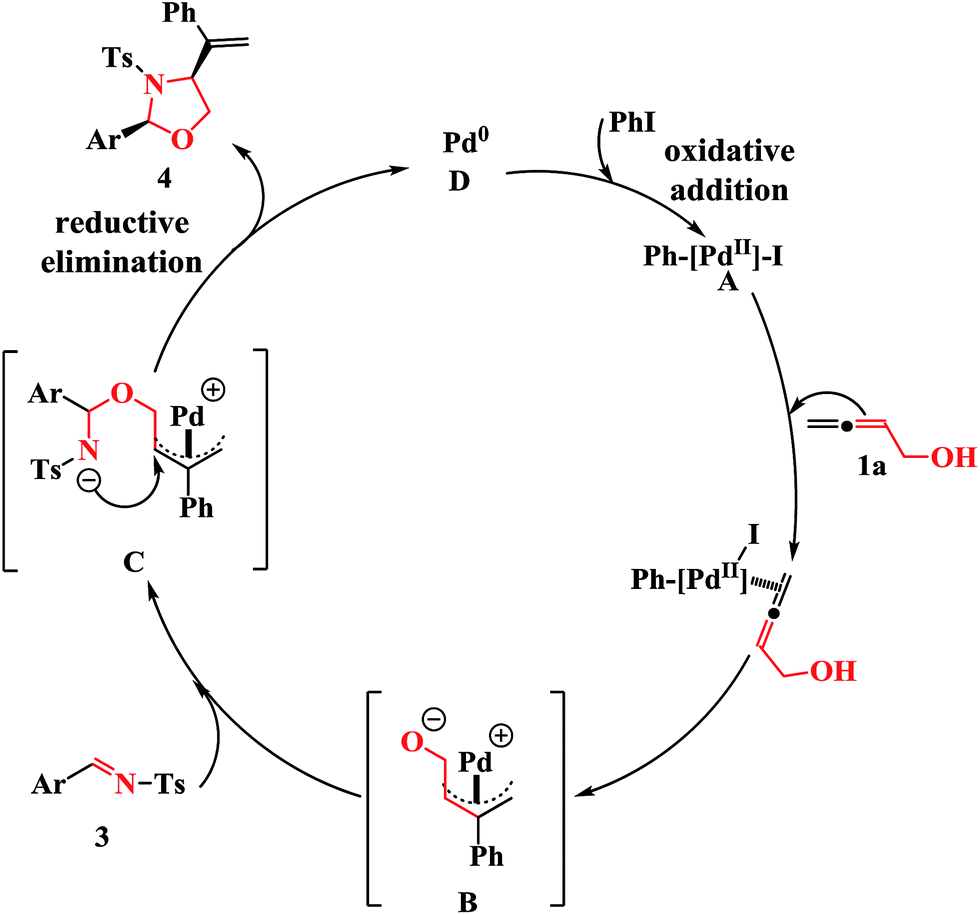 | ||
| Scheme 2 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed an efficient Pd0-catalyzed MCR for the synthesis of polysubstituted oxazolidine derivatives from buta-2,3-dien-1-ol, aryl iodides, and imines. Because of the three-component reaction concept, this cyclization could provide multiple points for diversity with excellent diastereoselectivity. As all three building blocks are readily available, this study will advance the transition-metal-catalyzed chemistry of allenes. Further studies regarding the scope and synthetic applications of this reaction are being pursued in our laboratory and will be reported in due course.Acknowledgements
We are grateful for the financial support from the Program for Innovative Research Team of the Ministry of Education and Program for Liaoning Innovative Research Team in University (IRT1073).References
- A. Barghi-Lish, S. Farzaneh and M. Mamaghani, Synth. Commun., 2016, 46, 1209 CrossRef CAS.
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (b) R. M. Williams, T. Glinka, M. E. Flanagan, R. Gallegos, H. Coffman and D. Pei, J. Am. Chem. Soc., 1992, 114, 733 CrossRef CAS; (c) F. Tomita, K. Takahashi and T. Tamaoki, Antibiotics, 1984, 37, 1268 CrossRef CAS; (d) Y.-C. Wu, M. Liron and J. Zhu, J. Am. Chem. Soc., 2008, 130, 7148 CrossRef CAS PubMed; (e) M. Yotsu-Yamashita, Y. H. Kim, S. C. Dudley Jr, G. Choudhary, A. Pfahnl, Y. Oshima and J. W. Daly, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4346 CrossRef CAS PubMed.
- (a) A. Pastor, W. Adam, T. Wirth and G. Tóth, Eur. J. Org. Chem., 2005, 14, 3075 CrossRef; (b) I. Hoppe, H. Hoffmann, I. Grtner, T. Krettek and D. Hoppe, Synthesis, 1991, 12, 1157 CrossRef; (c) C. Scolastico, Pure Appl. Chem., 1988, 60, 1689 CrossRef CAS; (d) D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS; (e) S. Kanemasa and K. Onimura, Tetrahedron, 1992, 48, 8631 CrossRef CAS; (f) N. Hiroto, O. Yuko and K. Eunsang, Heterocycles, 2014, 89, 1 CrossRef.
- (a) C. Wolf and H. Xu, Chem. Commun., 2011, 47, 3339 RSC; (b) C. Agami and F. Couty, Eur. J. Org. Chem., 2004, 4, 677 CrossRef; (c) A. Tessier, N. Lahmar, J. Pytkowicz and T. Brigaud, J. Org. Chem., 2008, 73, 3970 CrossRef CAS PubMed; (d) C. A. Caputoand and N. D. Jones, Dalton Trans., 2007, 41, 4627–4640 RSC.
- (a) G. P. Moloney, D. J. Craik, M. N. Iskander and T. L. Nero, J. Chem. Soc., Perkin Trans. 2, 1998, 2, 199 RSC; (b) J. Danielsson, L. Toomand and P. Somfai, Eur. J. Org. Chem., 2011, 3, 607 CrossRef; (c) B. Seashore-Ludlowand and P. Somfai, Eur. J. Org. Chem., 2010, 20, 3927 CrossRef; (d) T. H. Fife and L. Hagopian, J. Am. Chem. Soc., 1968, 90, 10074 Search PubMed; (e) F. Gosselin, A. Roy, P. D. O'Shea, C. Chen and R. P. Volante, Org. Lett., 2004, 6, 641 CrossRef CAS PubMed.
- (a) G. C. Post, B. L. Barthel, D. J. Burkhart, J. R. Hagadorn and T. H. Koch, J. Med. Chem., 2005, 48, 7648 CrossRef CAS PubMed; (b) D. J. Burkhart, B. L. Barthel, G. C. Post, B. T. Kalet, J. W. Nafie, R. K. Shoemaker and T. H. Koch, J. Med. Chem., 2006, 49, 7002 CrossRef CAS PubMed; (c) G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185 CrossRef CAS PubMed; (d) D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727 CrossRef CAS PubMed.
- (a) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129, 1866–1867 CrossRef CAS PubMed; (b) D. J. Michaelis, M. A. Ischay and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 6610–6615 CrossRef CAS PubMed; (c) K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 4570–4571 CrossRef CAS PubMed.
- M. B. Shaghafi, R. E. Grote and E. R. Jarvo, Org. Lett., 2011, 13, 5188 CrossRef CAS PubMed.
- X. Wu, L. Li and J. Zhang, Chem. Commun., 2011, 47, 7824 RSC.
- L. Yang, A. Khan, R. Zheng, L. Y. Jin and Y. J. Zhang, Org. Lett., 2015, 17, 6230 CrossRef CAS PubMed.
- (a) X. Chu, X. Xu and S. Ji, Chem.–Eur. J., 2016, 22, 14181 CrossRef CAS PubMed; (b) M. Ghorbani, B. Mohammadi, M. Saraii, B. Masoumi, M. Abbasian, A. Ramazani, K. Slepokura and T. Lis, Org. Lett., 2016, 18, 4759 CrossRef CAS PubMed; (c) L. G. Voskressensky, T. N. Borisova, M. D. Matveeva, V. N. Khrustalev, A. V. Aksenov, A. A. Titov, A. E. Vartanova and A. V. Varlamova, RSC Adv., 2016, 6, 74068 RSC; (d) H. Dong, L. Xu, S. Li, L. Wang, C. Shao and J. Xiao, ACS Comb. Sci., 2016, 18, 604 CrossRef CAS PubMed; (e) X. Lian, J. Meng and Z. Han, Org. Lett., 2016, 18, 4270 CrossRef CAS PubMed; (f) J. Chu, B. Hu, Z. Liao and X. Zhang, J. Org. Chem., 2016, 81, 8647 CrossRef CAS PubMed.
- (a) B. Miao, S. Li, G. Li and S. Ma, Org. Lett., 2016, 18, 2556 CrossRef CAS PubMed; (b) T. Cao and S. Ma, Org. Lett., 2016, 18, 1510 CrossRef CAS PubMed; (c) W. Yuan and S. Ma, Org. Lett., 2014, 16, 193 CrossRef CAS PubMed; (d) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS PubMed; (e) X. Huang, W. Wu, S. Song, C. Fu and S. Ma, Adv. Synth. Catal., 2016, 358, 2791 CrossRef CAS; (f) Y. Shigeo, K. Yasuaki, O. Yuta and M. Chisato, Chem.–Eur. J., 2016, 22, 12181 CrossRef PubMed; (g) T. Aurelien, B. Aurelie, N. W. Vijay and L. Benjamin, Angew. Chem., Int. Ed., 2016, 55, 8962 CrossRef PubMed; (h) G. Charlotte, M. Veronique and T. Y. Patrick, Org. Lett., 2016, 18, 676 CrossRef PubMed.
- S. Ma and N. Jiao, Angew. Chem., Int. Ed., 2002, 41, 4737 CrossRef CAS PubMed.
- X. Cheng and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 4581 CrossRef CAS PubMed.
- (a) A. Khan, J. Xing, J. Zhao, Y. Kan, W. Zhang and Y. Zhang, Chem.–Eur. J., 2015, 21, 120 CrossRef CAS PubMed; (b) J. Cheng, X. Jiang, C. Zhu and S. Ma, Org. Lett., 2011, 353, 1676 CAS; (c) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef CAS PubMed; (d) Q. Xiao, B. Wang, L. Tian, Y. Yang, J. Ma, Y. Zhang, S. Chen and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 9305 CAS; (e) P. Miao, H. Wang, L. Liu, W. Chang and J. Li, Asian J. Org. Chem., 2015, 4, 1050 CrossRef CAS; (f) J. Le Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 3003 RSC; (g) R. W. Bates and V. atcharoen, Chem. Soc. Rev., 2002, 31, 12 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1516742. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27993g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |