DOI:
10.1039/C6RA27003D
(Paper)
RSC Adv., 2017,
7, 4320-4328
Synthesis, mesomorphic and photophysical properties of novel triads and pentads of perylene liquid crystals with cholesterol units at the bay-position†
Received
18th November 2016
, Accepted 29th December 2016
First published on 16th January 2017
Abstract
Series of triads and pentads of perylene derivatives with two cholesterol units at the bay-position or four cholesterol units on both bay-position and imide position were designed and synthesized in yields of 55–65%. The influence of the different number and position of cholesterol substituents on mesomorphic properties and photophysical properties was studied. The results suggested that, in comparison with similar perylene derivatives with alkyl units at the imide position, the perylene liquid crystals with alkyl units at the bay-positions not only maintained excellent mesomorphic properties but also enhanced the fluorescence greatly. More cholesterol units resulted in a lower mesophase temperature, wider phase transfer temperature range, and higher fluorescence. A long spacer between perylene and cholesterol units led to more excellent mesomorphic properties and higher fluorescence.
Introduction
During past decades, perylene bisimide (PBI) derivatives have been not only considered as important semiconductor materials with high electron affinity and strong fluorescence,1–6 but also recognized as excellent building platforms to construct well-defined supramolecular structures based on their unique π–π stacking.7–9 Among all kinds of PBI aggregates, PBI liquid crystals (LC) have attracted much attention due to their excellent π–π stacking properties for supramolecular self-assembling processes.10,11 By modifying the imide groups with siloxane,12,13 polyoxyethylene chain,14 fluoro-pentenyl groups,15 alkyl esters,16 organosilica,17 alkoxyl/alkyl groups,18,19 dendritic peptides,20 phenyl substituents,21,22 3,4,5-trialkoxyphenyl,23,24 or triphenylene groups,25–27 different series of perylene columnar liquid crystals have been synthesized and their mesomorphic properties investigated. A survey of relevant literature implied that all perylene liquid crystals were prepared by modifying the two imide positions with two alkyl groups. Recently, we studied systematically perylene liquid crystals with two different bay-substituents and found that suitable substituents at the bay-positions were favorable for the hexagonal columnar liquid crystalline phase and led to wider phase transfer temperature ranges.28 Inspired by this research, we are further interested in whether it is possible to construct perylene liquid crystals by introducing alkyl groups onto the bay-position or onto both the imide position and bay-position. Moreover, what is the influence of different numbers and substitution positions of the alkyl groups on the mesomorphic and photophysical properties? However, no paper has studied this so far.
Based on the above considerations, in the present research, series triads and pentads of perylene derivatives with two cholesterol units at the bay-position or four cholesterol units on both bay-position and imides position were purposely designed and synthesized. The detailed structures and synthetic routes of these compounds were exhibited in Scheme 1. Also, the influence of the different number and diverse position of alkyl substituents on mesomorphic properties and photophysical properties were firstly investigated. The results suggested that the perylene liquid crystals with cholesterol units at the bay-position not only maintained excellent mesomorphic properties but also enhanced greatly the fluorescence in solution. The more cholesterol units leaded to lower mesophase temperature, wider scopes of phase transfer temperatures, and higher fluorescence.
 |
| | Scheme 1 The synthetic routes for perylene liquid crystals 9a, 9b, 10a and 10b. | |
Experimental
Instrumentation and methods
The organic solvents were purified according to standard anhydrous methods before use. All other chemical reagents were used directly without further purification. TLC analysis was carried out by pre-coated glass plates. Column chromatography was performed by using silica gel (200–300 mesh) as absorbing column. All NMR spectra were done on a Bruker-ARX 400 instrument at 25 °C, using tetramethylsilane (TMS) as internal standard. MS spectra were obtained on Bruker MALDI-TOF mass spectrometer. UV-vis and fluorescence spectra were performed on Varian spectrometer. Fluorescence spectra were measured on a Hitachi F-4500 spectrometer equipped with excitation and emission slits 10 nm wide. The fluorescence absolute ΦF values were obtained on an Edinburgh Instruments FLS920 Fluorescence Spectrometer with a 6-inch integrating sphere. POM (Leica DMRX) was used with the help of a hot stage (Linkam THMSE 600) to examine phase transitions. Thermal analysis was performed by using DSC (Thermal Analysis Q100), with the scanning rate of 10 °C min−1 under N2 atmosphere. XRD experiments were done on SEIFERT-FPM (XRD7), using Cu Kα 1.5406 Å as the radiation source with 40 kV, 30 mA power. Compounds 2 and 5 were synthesized by the literature methods,29,30 respectively.
Synthesis of compounds 3a and 3b
Under N2 atmosphere, the mixture of compound 2a or 2b (1 mmol), hydroquinone (0.55 g, 5 mmol) and K2CO3 (0.69 g, 5 mmol) was stirred in DMF (50 mL) at 110 °C for 20 h. The reaction was monitored by TLC. After cooling, 40 mL of HCl solution (1 M) was added slowly in the reaction system, and the white precipitates appeared. Then the precipitates were filtered and purified by recrystallization with (CH2Cl2/CH3OH (1/1, v/v)) for three times. The compound 3a and 3b were obtained as white solids. The yields were 63% and 67%, respectively. Compound 3a: 1H NMR (400 MHz, CDCl3) δppm: 0.68–2.35 (m, 43H of cholesterol), 4.55 (s, 2H, OCH2), 4.76 (bs, 1H, OCH on cholesterol), 5.40 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C on cholesterol), 6.77 (d, J = 8.0 Hz, 2H, ArH), 6.82 (d, J = 8.0 Hz, 2H, ArH), 6.86 (s, 1H, OH). Compound 3b: 1H NMR (400 MHz, CDCl3) δppm: 0.68–2.35 (m, 45H of cholesterol), 2.50 (t, 2H, J = 7.6 Hz, CH2CO), 3.51 (t, 2H, J = 7.6 Hz, OCH2), 3.96 (bs, 1H, OCH on cholesterol), 4.60 (bs, 1H, OH), 5.38 (s, 1H, CHC on cholesterol), 6.76 (d, J = 8.0 Hz, 2H, ArH), 6.78 (d, J = 8.0 Hz, 2H, ArH).
C on cholesterol), 6.77 (d, J = 8.0 Hz, 2H, ArH), 6.82 (d, J = 8.0 Hz, 2H, ArH), 6.86 (s, 1H, OH). Compound 3b: 1H NMR (400 MHz, CDCl3) δppm: 0.68–2.35 (m, 45H of cholesterol), 2.50 (t, 2H, J = 7.6 Hz, CH2CO), 3.51 (t, 2H, J = 7.6 Hz, OCH2), 3.96 (bs, 1H, OCH on cholesterol), 4.60 (bs, 1H, OH), 5.38 (s, 1H, CHC on cholesterol), 6.76 (d, J = 8.0 Hz, 2H, ArH), 6.78 (d, J = 8.0 Hz, 2H, ArH).
Synthesis of compound 6
Under N2 atmosphere, the mixture of 1,7-dibromoperylene tetracarboxylic anhydride 5 (0.55 g, 1 mmol) and octylamine (0.27 g, 2.1 mmol) was stirred in the solution of NMP (50 mL) at 80 °C for 10 h. Then 50 mL of distilled water was poured in the reaction mixture. The obtained red precipitates were filtered and washed by 20 mL of methanol. The crude product was purified by column chromatography (CH2Cl2/ethyl acetate (5/1, v/v)). The compound 6 were collected as red solid in yield of 86%. Compound 6: 1H NMR (400 MHz, CDCl3) δppm: 9.34 (d, J = 8.0 Hz, 2H, ArH), 8.78 (s, 2H, ArH), 8.57 (d, J = 8.0 Hz, 2H, ArH), 4.16 (t, J = 8.0 Hz, 4H, NCH2), 1.73 (bs, 4H, CH2), 1.22–1.50 (m, 20H, CH2), 0.88 (t, J = 8.0 Hz, 6H, CH3); MALDI-TOF-MS (C40H40Br2N2O4) [M]+: calcd: 772.1. Found: 772.8.
Synthesis of compound 7
Under N2 atmosphere, 1,7-dibromoperylene tetracarboxylic anhydride (0.5 g, 0.9 mmol) was reacted with 6-amino-1-hexanol (0.27 g, 2.3 mmol) in NMP (15 mL) at 95 °C for 6 h. After reaction, 50 mL of distilled water was added in the solution, and the red precipitates were obtained. Then the red precipitates were separated out. The crude product was washed by 30 mL of methanol. The yield was 85%. Because of the crude product had poor solubility in all kinds of solvents, the NMR spectrum was not done for the crude product, which was used directly for the next step without further purification. Compound 7: FT-IR(KBr), ν/cm−1: 3392, 2937, 1698, 1658, 1591, 1435, 1337, 1241, 1052, 808, 746. MALDI-TOF-MS: calculated for C36H32Br2N2O6 771.0503[M]+; found: 771.0525.
Synthesis of compound 8
Under N2 atmosphere, the mixture of compound 7 (0.38 g, 0.05 mmol) and chloracetyl chloride (0.5 mL) were stirred in CH2Cl2 (10 mL) at 35 °C for 6 h. TLC detection implied the disappearance of reactant. Then, 10 mL of distilled water was added and the organic layer was separated. The organic layer was washed by brine solution and dried over anhydrous MgSO4. The solution was concentrated and the residue was treated with 10 mL of MeOH. The obtained red precipitate was collected and further purified by column chromatography (CH2Cl2/ethyl acetate (10/1, v/v)). The compound 8 were obtained as red solid in the yield of 75%. Compound 8: 1H NMR (400 MHz, CDCl3) δppm: 9.44 (d, J = 8.0 Hz, 2H, ArH), 8.87 (s, 2H, ArH), 8.66 (d, J = 8.0 Hz, 2H, ArH), 4.16–4.22 (m, 8H, OCH2 and NCH2), 4.06 (s, 4H, ClCH2CO), 1.40–1.80 (m, 16H, CH2); MALDI-TOF-MS (C40H34Br2Cl2N2O8) [M]+: calcd: 900.0. Found: 899.5.
Synthesis of compounds 9a and 9b
Under N2 atmosphere, the mixture of compound 6 (0.39 g, 0.5 mmol), compound 3a or 3b (1.3 mmol), anhydrous K2CO3 (0.18 g, 1.3 mmol) were stirred in 30 mL of DMF at 95 °C for 10 h. The reaction process was detected by TLC till the disappearance of compound 6. Then the solution was cooled to room temperature and 50 mL of HCl solution (1 M) was added in it. The solution was extracted with 40 mL of CHCl3. The organic layer was partitioned, washed with brine solution and dried over anhydrous MgSO4. After filtration, the solution was concentrated and further purified by column chromatography (CH2Cl2/ethyl acetate (6/1, v/v)). The compounds 9a and 9b were collected as red solids in the yields of 60% and 65%, respectively. Compound 9a: 1H NMR (400 MHz, CDCl3) δppm: 9.60 (d, J = 8.0 Hz, 2H, ArH), 8.61 (d, J = 8.0 Hz, 2H, ArH), 8.29 (s, 2H, ArH), 7.12 (d, J = 8.0 Hz, 4H, ArH), 7.01 (d, J = 8.0 Hz, 4H, ArH), 5.40 (bs, 2H, CCH), 4.77 (bs, 2H, OCH), 4.63 (s, 4H, OCH2CO), 4.13 (t, 4H, J = 8.0 Hz, NCH2), 0.64–2.41 (m, 116H, CH, CH2 and CH3); 13C NMR (100 MHz, CDCl3) δppm: 168.14, 163.28, 162.86, 155.79, 155.38, 149.10, 139.25, 124.82, 123.70, 123.59, 123.33, 123.12, 123.04, 122.14, 121.04, 119.68, 116.73, 115.85, 75.41, 66.21, 56.68, 56.14, 50.01, 42.32, 39.72, 39.52, 36.59, 36.19, 35.80, 31.83, 29.23, 23.84, 22.57, 19.33, 18.72, 14.11, 11.87; MALDI-TOF-MS calcd for m/z = 1684.1, found: m/z = 1683.1 (M−). HR-MS (ESI) (C110H142N2O12) [M]+: calcd: 1684.0591. Found: 1684.0636. Compound 9b: 1H NMR (400 MHz, CDCl3) δppm: 9.63 (d, J = 8.0 Hz, 2H, ArH), 8.61 (d, J = 8.0 Hz, 2H, ArH), 8.29 (s, 2H, ArH), 7.11 (d, J = 8.0 Hz, 4H, ArH), 6.98 (d, J = 8.0 Hz, 4H, ArH), 5.38 (bs, 2H, CCH), 4.65 (bs, 2H, OCH), 4.14 (t, 4H, J = 7.2 Hz, NCH2), 4.05 (t, 4H, J = 7.2 Hz, OCH2), 0.64–2.57 (m, 124H, CH, CH2 and CH3); 13C NMR (100 MHz, CDCl3) δppm: 172.57, 163.37, 162.99, 157.20, 156.42, 148.24, 139.72, 133.51, 130.03, 1224.74, 124.51, 123.61, 123.21, 122.98, 122.71, 121.26, 121.15, 116.31, 116.26, 74.23, 67.39, 56.59, 56.08, 50.03, 42.32, 40.63, 39.73, 39.42, 38.17, 36.92, 36.68, 36.19, 35.80, 32.63, 31.82, 31.04, 29.71, 28.23, 28.02, 27.11, 24.72, 24.29, 23.83, 22.57, 20.98, 19.27, 18.65, 14.10, 11.79; MALDI-TOF-MS calcd for m/z = 1740.1, found: m/z = 1740.8 (M+). HR-MS (ESI) (C114H150N2O12) [M]+: calcd: 1740.1217. Found: 1740.1227.
Synthesis of compounds 10a and 10b
Under N2 atmosphere, the mixture of compound 8 (0.27 g, 0.3 mmol), compound 3a or 3b (1 mmol), anhydrous K2CO3 (0.21 g, 1.5 mmol) was stirred in DMF (30 mL) at 105 °C for 20 h. Then the solution was treated by the similar synthetic procedure of compound 9, and the products were purified by column chromatography (CH2Cl2/ethyl acetate (8/1, v/v)). The compounds 10a and 10b were collected as red solids in the yields of 55% and 58%, respectively. Compound 10a: 1H NMR (400 MHz, CDCl3) δppm: 9.55 (d, J = 8.0 Hz, 2H, ArH), 8.55 (d, J = 8.0 Hz, 2H, ArH), 8.23 (s, 2H, ArH), 7.09 (d, J = 8.0 Hz, 4H, ArH), 6.98 (d, J = 8.0 Hz, 4H, ArH), 6.80 (s, 8H, ArH), 5.39 (bs, 2H, CCH), 5.34 (bs, 2H, CCH), 4.83 (bs, 4H, OCH), 4.63 (s, 4H, OCH2CO), 4.55 (s, 4H, OCH2CO), 4.50 (s, 4H, OCH2CO), 4.06–4.19 (m, 8H, OCH2 and NCH2), 0.64–2.47 (m, 188H, CH, CH2 and CH3); 13C NMR (100 MHz, CDCl3) δppm: 169.19, 168.45, 168.13, 162.94, 162.61, 159.08, 155.74, 154.52, 152.61, 139.24, 133.50, 133.32, 131.98, 130.47, 130.15, 128.87, 124.86, 124.76, 123.13, 123.00, 122.15, 121.74, 121.05, 116.73, 116.07, 115.87, 115.78, 75.40, 75.03, 71.88, 66.28, 66.19, 65.27, 56.54, 56.05, 49.86, 42.43, 39.66, 39.43, 37.95, 36.88, 36.54, 36.27, 35.86, 31.85, 29.71, 28.24, 27.93, 27.73, 26.56, 25.52, 24.17, 23.85, 22.77, 22.50, 20.90, 19.41, 18.72, 11.97; MALDI-TOF-MS calcd for m/z = 2812.7, found: m/z = 2812.3 (M+). HR-MS (ESI) (C180H238N2O24) [M]+: calcd: 2813.7526. Found: 2813.7513. Compound 10b: 1H NMR (400 MHz, CDCl3) δppm: 9.64 (d, J = 8.0 Hz, 2H, ArH), 8.61 (d, J = 8.0 Hz, 2H, ArH), 8.29 (s, 2H, ArH), 7.11 (d, J = 8.4 Hz, 4H, ArH), 6.98 (d, J = 8.4 Hz, 4H, ArH), 6.80 (s, 8H, ArH), 5.37 (bs, 4H, CCH), 4.64 (bs, 4H, OCH), 4.12–4.19 (m, 8H, OCH2CO and ArOCH2), 4.02–4.08 (m, 8H, OCH2), 3.94 (t, J = 7.2 Hz, 4H, NCH2), 0.62–2.59 (m, 204H, CH, CH2 and CH3); 13C NMR (100 MHz, CDCl3) δppm: 169.19, 168.53, 168.23, 163.26, 162.85, 159.03, 155.93, 154.58, 152.68, 139.28, 133.60, 132.02, 130.50, 128.92, 124.86, 123.71, 123.13, 123.00, 122.15, 121.74, 121.05, 116.73, 116.10, 115.87, 115.78, 75.53, 75.03, 71.76, 66.33, 66.03, 65.20, 56.54, 56.02, 49.99, 42.43, 40.40, 39.71, 39.58, 38.00, 36.91, 35.80, 31.91, 31.85, 29.71, 28.38, 28.24, 28.03, 27.73, 27.66, 26.69, 25.57, 24.29, 23.85, 22.84, 22.50, 21.05, 19.33, 18.72, 11.87; MALDI-TOF-MS calcd for m/z = 2926.0, found: m/z = 2925.0 (M+). HR-MS (ESI) (C188H254N2O24) [M]+: calcd: 2925.8778. Found: 2925.8809.
Results and discussion
Synthesis and characterization
The previous reports had shown that the four bay-substituents on perylene derivatives might destroy the π–π stacking of perylene core and resulted in no liquid crystalline behaviors due to the strong steric hindrance.31–33 However, two bay-substituents might be favorable for the wide scopes of phase transfer temperatures.28,31–33 Thus, the 1,7-bay-substituted perylene derivative 5 was chosen as building platform to construct the triads and pentads of perylene with multi-cholesterol units. The synthetic routes were illustrated in Scheme 1. Firstly, by esterifying reaction with chlorinated carboxylic acid and subsequently etherifying reaction with excess p-benzenediol, cholesterol was conveniently transferred to cholesterol derivatives 3a and 3b with terminal phenolic hydroxyl group, which easily react with halogen group on perylene. In this reaction, two and four carbon atom chains for chlorinated carboxylic acid were used to investigate the influence of chain length on mesomorphic and photophysical property. On the other hand, the 1,7-dibrominated perylene bisanhydrides 5 was prepared by the previous literature.30 By ammonolysis procedure of compound 5 with n-octylamine or 6-amino-1-hexanol, the corresponding perylene bisimides 6 and 7 with two halogen groups were obtained in yields of 85% and 86%, respectively. Furthermore, compound 7 was reacted with chloroacetyl chloride to afford perylene imides 8 containing four halogen groups in yield of 75%. Due to the poor dissolubility, compounds 5 and 7 was not purified and used directly in next step. Compounds 6 and 8 were purified by column chromatography and some by-products in the previous steps, such as the complicated isomers in bromination reaction, were separated out. The pure compounds 6 and 8 were obtained in ideal yields of 75% and 85%, respectively. Finally, by treating compound 3a(b) with perylene imides 6 or 8 in K2CO3/DMF system, the triads 9a(b) with two cholesterol units at the bay-position, and the pentads 10a(b) with four cholesterol units on both bay-position and imides position were prepared in yields of 55–65% after column chromatography, respectively.
All new compounds were characterized by 1H NMR, 13C NMR, MALDI-TOF-MS and HR-MS spectral analysis. Their mass spectrometry spectra showed corresponding molecular ion peaks for M+ or MNa+. The deviations of HR-MS were less than 5 ppm. In the 1H NMR spectra, all protonic signals were well assigned for the corresponding structures (see ESI†). Especially, the protons of perylene core of compounds 9 and 10 showed two symmetrical doublets and one singlet, suggesting the 1,7-bay-positions of perylene skeleton. The signals of 13C NMR also supported the 1,7-bay-substituents structures with two singlets for CO of imides. These data implied that the small quantities of isomers or by-products (such as 1,6-dibrominated isomer in preparing for compound 5) were removed completely by column chromatography. All these characteristic data were in accordance with the proposed structures of target compounds 9a, 9b, 10a and 10b. Although compounds 5 and 7 showed the poor dissolubility, the target perylene derivatives exhibited excellent dissolubility in common organic solvents such as CH2Cl2, CHCl3, toluene, THF and DMF.
Mesomorphic properties
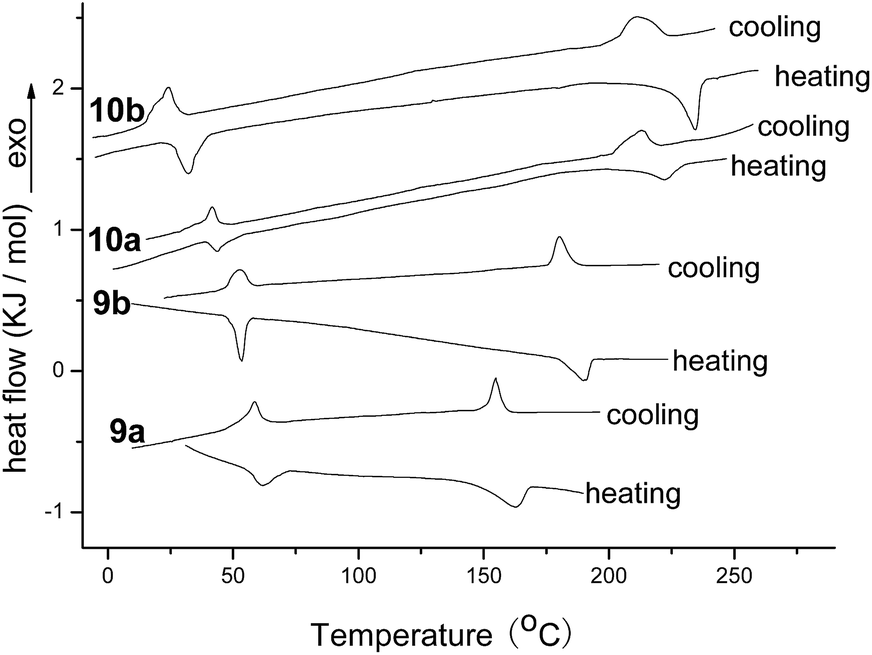
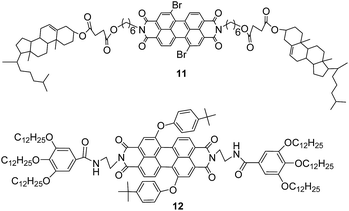
Compounds 9a, 9b, 10a and 10b were studied preliminarily by differential scanning calorimeter (DSC) to investigate their phase transition behaviours. Compound 11 with two cholesterol units on imides position and compound 12 with similar perylene core were used as reference compounds for the comparison (Fig. 1).28,34 The DSC curves of compounds 9a, 9b, 10a and 10b for second heating and cooling were illustrated in Fig. 2. The phase transition temperatures and enthalpy changes for these compounds were summarized in Table 1. It can be seen that all of new perylene derivatives possessed two phase transition peaks on second heating at 32–61 °C and 162–234 °C, respectively. Upon cooling, two corresponding exothermic peaks appeared at 28–58 °C and 154–215 °C, respectively. These data indicated they had good reversible phase transition behaviours at similar temperatures on heating and cooling. Based on these data, it was reasonable to deduce the transition of crystal phase-mesophase-isotropic phase for the phase transition process. The slightly hysteresis phenomena could be attributed to the super-cooling behaviours for such viscous materials. Thus, these DSC curves suggested that compounds 9a, 9b, 10a and 10b were perylene liquid crystals with low mesomorphic temperatures and wide scopes of mesophase.
 |
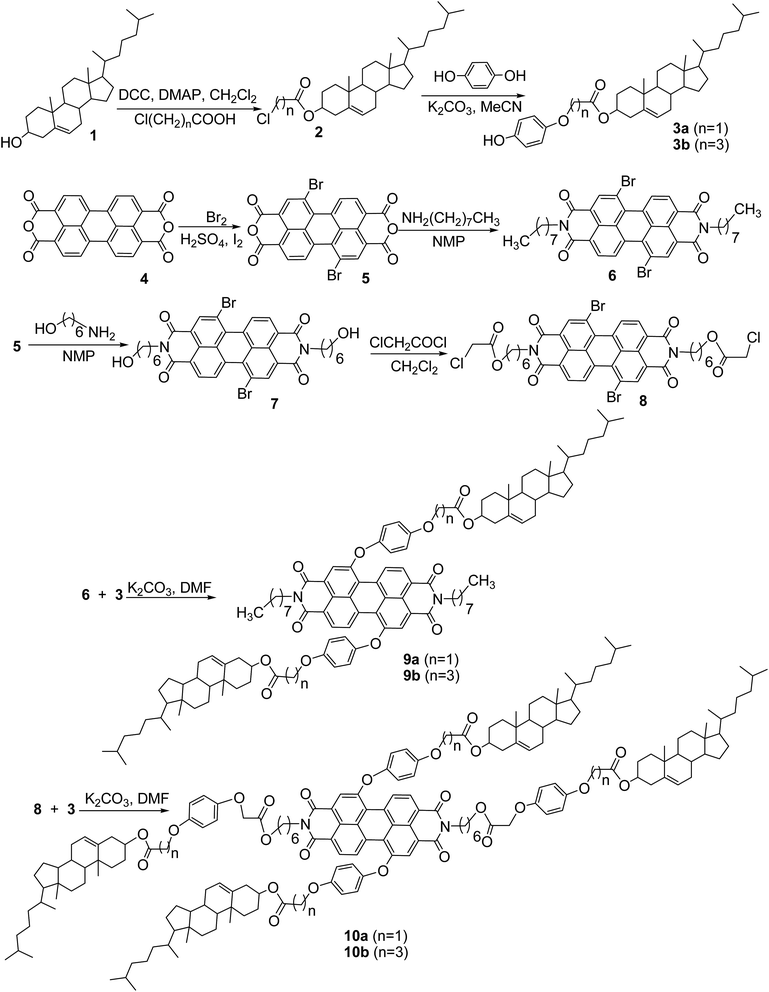
| | Fig. 1 The reference compounds 11 and 12 for comparison. | |
 |
| | Fig. 2 The DSC traces of compounds 9a, 9b, 10a and 10b on second heating and cooling (scan rate 10 °C min−1). | |
Table 1 Transition temperatures (°C) and enthalpy changes (kJ mol−1) of compounds 9a, 9b, 10a and 10b
| Compd |
Phase transitiona |
Heating scan T(ΔH) |
Cooling scan T(ΔH) |
| Cr = crystalline, Col = columnar mesophase, Iso = isotropic. These data were cited from ref. 34. These data were cited from ref. 28. |
| 9a |
Cr-Col |
61.8(10.6) |
58.5(8.5) |
| Col-Iso |
162.9(13.8) |
154.8(9.9) |
| 9b |
Cr-Col |
53.3(12.2) |
52.1(8.9) |
| Col-Iso |
190.5(11.1) |
180.1(10.4) |
| 10a |
Cr-Col |
43.6(12.4) |
41.5(10.6) |
| LC-Iso |
222.1(19.3) |
212.9(22.3) |
| 10b |
Cr-Col |
32.4(16.6) |
23.9(18.1) |
| Col-Iso |
234.3(23.8) |
210.8(21.9) |
| 11b |
Cr-Col |
34.9(20.1) |
32.6(13.9) |
| Col-Iso |
197.7(16.3) |
180.4(8.8) |
| 12c |
Cr-Col |
61.7(14.89) |
59.3(13.02) |
| Col-Iso |
247.9(19.34) |
239.6(18.68) |
Moreover, in comparison with compound 11 containing two cholesterol units on imides position, compounds 9a and 9b with two cholesterol units on bay-positions, in which two phenyls on bay-positions enlarged the areas of π–π stacking of rigid perylene core, still showed the low mesomorphic temperatures, although these temperatures were higher little than that of compound 11. Also, by comparing with compound 12 possessing similar perylene core, compounds 9a and 9b exhibited similar mesomorphic temperatures at about 60 °C. These results suggested that the perylene liquid crystals with cholesterol units at the bay-position maintained the excellent mesomorphic properties as the similar perylene liquid crystal with alkyl units at the imide positions. On the other hand, compounds 10a and 10b with four cholesterol units on both bay-position and imides position showed lower mesomorphic temperatures and wider scopes of mesophase than that of compounds 9a, 9b, 11, and 12 with two alkyl units on imides or bay-position. Especially, the mesomorphic temperature of compound 10b was as low as 32 °C and the scope of mesophase was as wide as 202 °C after calculation, which was seldom observed for perylene liquid crystals. As to the influences of different spacers between perylene and cholesterol moieties, compounds 9b and 10b derived from chlorobutyric acid exhibited lower mesomorphic temperatures and wider scopes of mesophase than compounds 9a and 10a derived from chloroacetic acid, indicating that the long spacer was favorable for good mesomorphic property. Based on these DSC analyses, it could be concluded that triads 9a and 9b, and pentads 10a and 10b were excellent perylene liquid crystals with low mesomorphic temperatures and wide scopes of mesophase. The perylene liquid crystals with alkyl units on bay-position possessed good mesomorphic properties as the analogous perylene liquid crystals with alkyl units at the imides position. The more cholesterol units in perylene liquid crystals leaded to the lower mesophase temperature and the wider scopes of phase transfer temperatures. The longer spacer also resulted in the lower mesophase temperature and the wider scopes of mesophase.
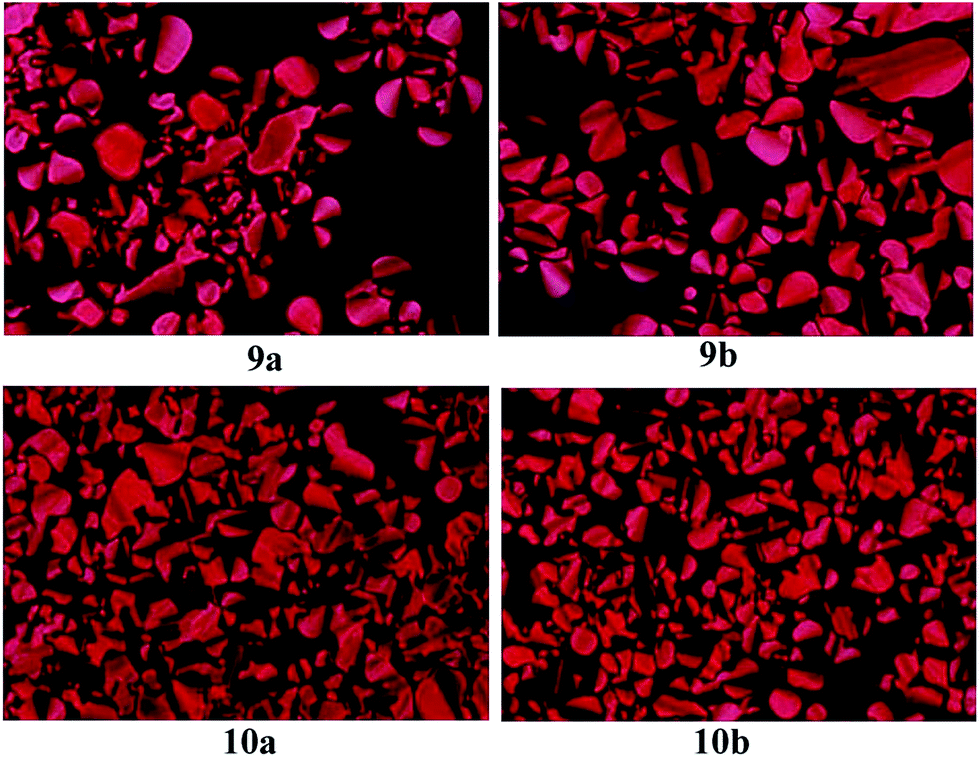
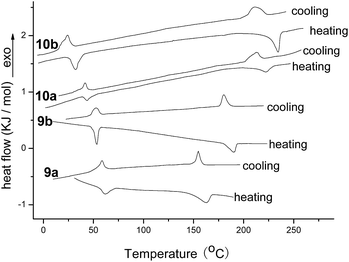
Polarizing optical microscopy (POM) was then employed to study the mesophase of compounds 9a, 9b, 10a and 10b. At the approximate temperatures of phase transition in DSC curves, the phase transfer behaviours of Cr-Col and Col-Iso phase were observed for compounds 9a, 9b, 10a and 10b. After being heated to isotropic phase, they were cooled slowly to mesophase and the columnar textures appeared gradually. Fig. 3 illustrated their mesomorphic textures at 130 °C. All of them exhibited the pseudo-confocal conic textures which were typical columnar textures, indicating they were columnar liquid crystals.
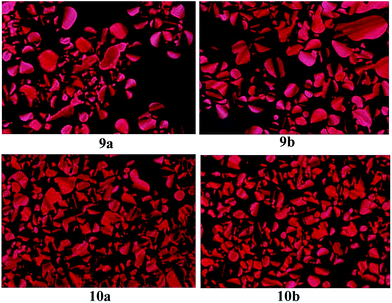 |
| | Fig. 3 The textures of compounds 9a, 9b, 10a and 10b obtained with POM on cooling at 130 °C (×400). | |
As the liquid crystalline phase were confirmed by DSC and POM, compounds 9a, 9b, 10a and 10b were further examined by X-ray diffraction (XRD) to study their molecular stacking behaviors in mesophase. Fig. 4 illustrated their XRD traces at 130 °C for the mesophase. In the small angle region, three peaks at 2.55°, 4.43° and 5.15° for 9a, 2.42°, 4.23° and 4.86° for 9b, 2.15°, 3.73° and 4.36° for 10a, 2.05°, 3.58° and 4.12° for 10b were observed obviously, respectively. Based on Bragg equation, the d-spacing could be calculated by formula d = λ/(2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ). The calculating results for these reflections were 34.61 Å, 19.93 Å and 17.15 Å for 9a, 36.47 Å, 20.87 Å and 18.18 Å for 9b, 41.05 Å, 23.67 Å and 20.25 Å for 10a, 43.06 Å, 24.66 Å and 21.48 Å for 10b, respectively. These distances were in agreement with the ratios of 1:1/√3:1/√4 for (100), (110) and (200) reflections, which were the typical reflection mode for the hexagonal columnar liquid crystals. Furthermore, in the wide angle region, the broad halos between 15° and 30°, indicating the mean distance of 4.5 Å approximately, could be assigned for the reflections of very short correlation length of the molten alkyl chains. Moreover, a small reflection at 22.43°, 23.19°, 22.10°, and 23.35° could be distinguished for compounds 9a, 9b, 10a and 10b, respectively. These peaks implied the spacing of 3.96 Å, 3.83 Å, 4.01, and 3.97 Å, respectively. These spacings were in accordance with the typical characteristic of π–π interaction for the intracolumnar distance of ordered hexagonal columnar liquid crystals. Moreover, based on the above analysis, the calculated lattice parameter α for compounds 9a, 9b, 10a and 10b was 39.86 Å, 41.74 Å, 47.34 Å and 49.32 Å, respectively. These data were 30% less than the diameters of compounds 9 (∼60 Å) and 10 (∼80 Å) simulated by CPK molecular model. These results suggested that the flexible alkyl chains of these perylene derivatives might be curled and fold or interdigitated in liquid crystalline phase. On the other hand, the average number (n) of molecules in one single disk of the column could be calculated according to the following formula.35,36
sinθ). The calculating results for these reflections were 34.61 Å, 19.93 Å and 17.15 Å for 9a, 36.47 Å, 20.87 Å and 18.18 Å for 9b, 41.05 Å, 23.67 Å and 20.25 Å for 10a, 43.06 Å, 24.66 Å and 21.48 Å for 10b, respectively. These distances were in agreement with the ratios of 1:1/√3:1/√4 for (100), (110) and (200) reflections, which were the typical reflection mode for the hexagonal columnar liquid crystals. Furthermore, in the wide angle region, the broad halos between 15° and 30°, indicating the mean distance of 4.5 Å approximately, could be assigned for the reflections of very short correlation length of the molten alkyl chains. Moreover, a small reflection at 22.43°, 23.19°, 22.10°, and 23.35° could be distinguished for compounds 9a, 9b, 10a and 10b, respectively. These peaks implied the spacing of 3.96 Å, 3.83 Å, 4.01, and 3.97 Å, respectively. These spacings were in accordance with the typical characteristic of π–π interaction for the intracolumnar distance of ordered hexagonal columnar liquid crystals. Moreover, based on the above analysis, the calculated lattice parameter α for compounds 9a, 9b, 10a and 10b was 39.86 Å, 41.74 Å, 47.34 Å and 49.32 Å, respectively. These data were 30% less than the diameters of compounds 9 (∼60 Å) and 10 (∼80 Å) simulated by CPK molecular model. These results suggested that the flexible alkyl chains of these perylene derivatives might be curled and fold or interdigitated in liquid crystalline phase. On the other hand, the average number (n) of molecules in one single disk of the column could be calculated according to the following formula.35,36
where the notation “
α” is the hexagonal lattice parameter,
NA is Avogadro's number,
M is the molecular mass of the compound and the density (
ρ) of these compounds is assumed as 1 g cm
−3.
35,36 After calculation, the number of molecules (
n) per slice was approximately 2 for compounds
9a,
9b,
10a and
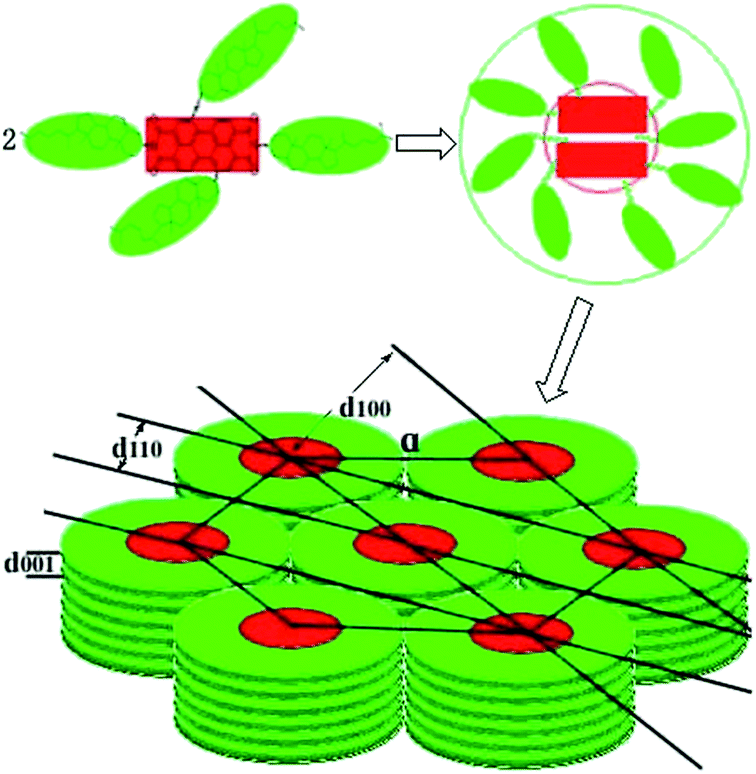
10b. Thus, the possible arrangement for the molecular stacking of hexagonal columns of compounds
9a,
9b,
10a and
10b were proposed as shown
Fig. 5. All these XRD analyses suggested that triads
9a and
9b, and pentads
10a and
10b were hexagonal columnar liquid crystals, indicating the alkyl units on bay-position or on both bay-position and imides position of perylene derivatives were favorable for the formation of hexagonal columnar liquid crystals.
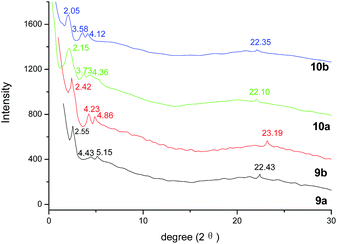 |
| | Fig. 4 XRD traces of compounds 9a, 9b, 10a and 10b measured at 130 °C. | |
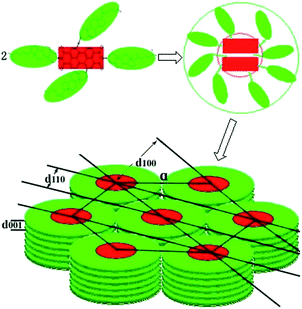 |
| | Fig. 5 The proposed molecular stacking of hexagonal columns of compounds 9a, 9b, 10a and 10b. | |
Photophysical properties
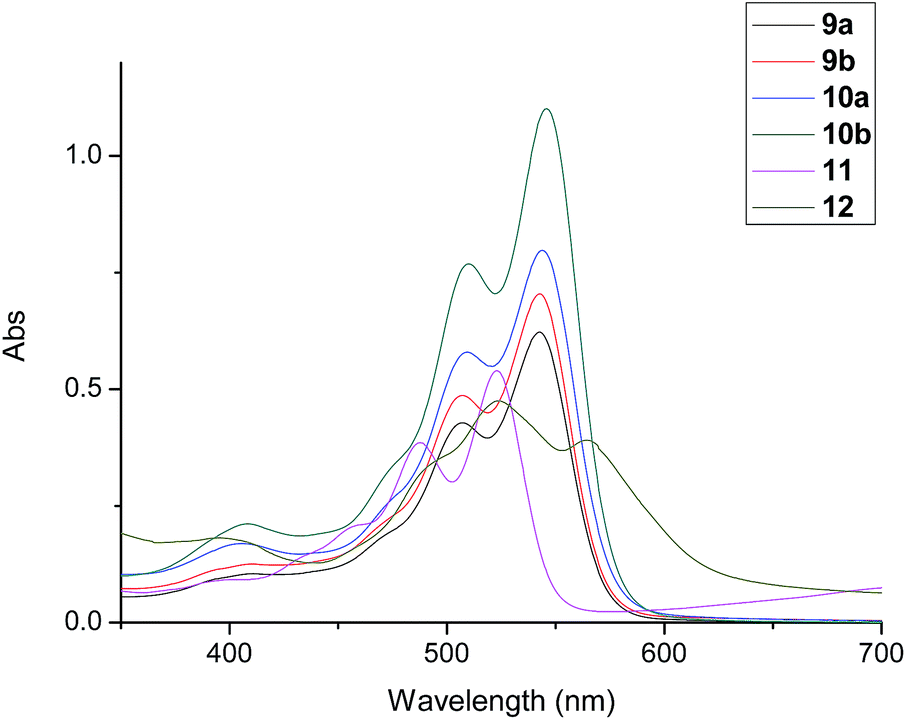
The excellent photophysical property is the important characteristic of perylene derivatives. In order to study the influence of the different number and position of alkyl substituents on photophysical properties, the absorption spectra and fluorescence spectra of compounds 9a, 9b, 10a and 10b were investigated and compared with that of reference compounds 11 and 12. Firstly, the solvent effect for absorption spectrum and fluorescence spectrum were studied in different solvents. They showed similar change in the tested solvents. The representative spectra of compound 12 were illustrated in Fig. S25 and S26.† The maximal absorption wavelength changed obviously between 450 nm and 600 nm with different absorbance. The particularly red-shifted spectra of 12 in polar solvents could be attributed to the lower excited state energy of π–π* transition in polar solvents. The emission wavelengths of fluorescence spectra in different solvents were similar but the strongest fluorescence emission appeared in toluene solution. Therefore, toluene was used as solvent for the investigation of the absorption spectra and fluorescence spectra of compounds 9a, 9b, 10a and 10b. The results were shown in Fig. 6 and 7. Their corresponding spectral data were listed in Table 2. Comparing with compound 11, compounds 9a, 9b, 10a and 10b showed red shifts for the wavelength of absorption and fluorescence spectra. These phenomena could be attributed to the electronic effect of phenolic groups on bay-position, which enlarged the aromatic conjugated system of perylene core. However, although compounds 9a, 9b, 10a and 10b possessed the similar aromatic conjugated system of perylene core with compound 12, the wavelength of absorption and fluorescence spectra of compounds 9a, 9b, 10a and 10b presented obviously blue shifts. These results could be explained by that the bulky cholesterol units on bay-position of compounds 9a, 9b, 10a and 10b produced the strong steric hindrance and restrained the conjugates between phenolic group and perylene core.
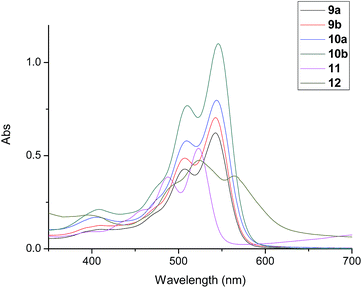 |
| | Fig. 6 Absorption spectra of compounds 9a, 9b, 10a and 10d, and reference compounds 11 and 12 (10−5 M). | |
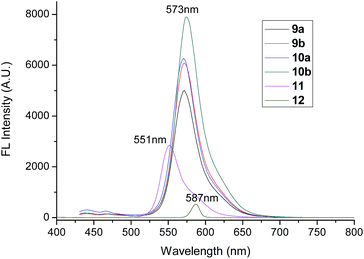 |
| | Fig. 7 Fluorescence emission spectra of compounds 9a, 9b, 10a and 10b in toluene (10−5 M). The excitation wavelength was 518 nm. The excitation wavelength of reference compounds 11 and 12 were 520 nm and 523 nm, respectively. | |
Table 2 Absorption and fluorescence data for the compounds 9a, 9b, 10a and 10b in toluene at 298 K
| Comp. |
λabs/nm |
λem/nm |
Stokes shift/nm |
ΦF |
| The data of compounds 11 and 12 were cited from ref. 28 and 34, respectively. |
| 9a |
507, 543 |
571 |
28 |
0.94 |
| 9b |
508, 543 |
572 |
29 |
0.95 |
| 10a |
508, 543 |
570 |
27 |
0.93 |
| 10b |
509, 544 |
572 |
28 |
0.96 |
| 11a |
487, 523 |
551 |
28 |
0.58 |
| 12a |
523, 564 |
587 |
17 |
0.37 |
Moreover, it can be seen that the fluorescence intensities of compounds 9a, 9b, 10a and 10b were far higher than that of compounds 11 and 12. The fluorescence quantum yields increased dramatically from 0.37 and 0.58 for compounds 11 and 12 to 0.93–0.96 for compounds 9a, 9b, 10a and 10b. These phenomena might be ascribed to two influencing factors. One was that the big alkyl units on bay-position enhanced the rotation limitation of phenolic group, resulting in the less energy-consuming and stronger fluorescence. These restricted intramolecular rotation mechanism was popular for explanation the fluorescence-enhancing in the field of aggregation-induced luminescence.37–39 Another factor was that the more alkyl units produced the stronger steric hindrances, which leaded to the reduction of the orderly aggregates in solution and then resulted in the enhancement of fluorescence intensity. On the other hand, compounds 9b and 10b derived from chlorobutyric acid showed stronger fluorescence than compounds 9a and 10a derived from chloroacetic acid, indicating the long soft spacer was favorable for the fluorescence. Especially, the fluorescence intensity of pentad 10b with four cholesterol units on both bay-position and imides position of perylene enhanced greatly and the fluorescence quantum yield reached as high as 0.96, which were very outstanding among all kinds of perylene liquid crystals.14–28 The fluorescence of solid films for these perylene derivatives were also studied but little fluorescence was detected, suggesting that the orderly aggregates in solid state resulted in strong aggregation-caused quenching. These phenomena were similar with reference compounds 11 and 12, and commonly observed for perylene derivatives. Based on the above spectral analysis, it could be concluded that the perylene triads 9a and 9b with two cholesterol units at the bay-position, pentads 10a and 10b with four cholesterol units on both bay-position and imides position possessed more excellent fluorescence than the similar perylene derivatives with alkyl units on imides position. The alkyl units at the bay-positions enhanced greatly the fluorescence. The more alkyl units leaded to the higher fluorescence in solution.
Conclusions
In summary, the design and synthesis of the triads 9a and 9b, pentads 10a and 10b based on perylene with two cholesterol units at the bay-position or four cholesterol units on both bay-position and imides position were reported in yields of 55–65%. Their mesomorphic properties and photophysical properties were studied by DSC, POM, XRD, absorption spectra and fluorescence spectra. The results suggested that the different number and position of cholesterol substituents influenced remarkably on mesomorphic properties and photophysical properties. In comparison with the similar perylene derivatives with alkyl units on imides position, the perylene liquid crystals with alkyl units on bay-position not only maintained excellent mesomorphic properties but also enhanced greatly the fluorescence. The more cholesterol units resulted in the lower mesophase temperature, the wider scopes of phase transfer temperatures, and higher fluorescence. The long soft spacer between perylene and cholesterol units was favorable for excellent mesomorphic properties and high fluorescence.
Acknowledgements
Financial support from the National Natural Science Foundation of China (No: 21406036), Fujian Natural Science Foundation of China (No. 2014J01034) and the Program for Innovative Research Team in Science and Technology in Fujian Province Universit were greatly acknowledged.
Notes and references
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudholter, J. Am. Chem. Soc., 2000, 122, 11057–11066 CrossRef CAS.
- Y. V. Zatsikha, V. P. Yakubovskyi, M. P. Shandura and Y. P. Kovtun, Dyes Pigm., 2015, 114, 215–221 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed.
- Y. P. Yu, F. Yang, Y. J. Ji, Y. G. Wu, A. D. Zhang, C. Li and W. W. Li, J. Mater. Chem. C, 2016, 4, 4134–4137 RSC.
- I. Meazzini, N. Willis-Fox, C. Blayo, J. Arlt, S. Clément and R. C. Evans, J. Mater. Chem. C, 2016, 4, 4049–4059 RSC.
- F. Galeotti, W. Mróz, M. Catellani, B. Kutrzeba-Kotowska and E. Kozma, J. Mater. Chem. C, 2016, 4, 5407–5415 RSC.
- Y. K. Che, A. Datar, K. Balakrishnan and L. Zang, J. Am. Chem. Soc., 2007, 129, 7234–7235 CrossRef CAS PubMed.
- K. Sugiyasu, N. Fujita and S. Shinkai, Angew. Chem., 2004, 116, 1249–1253 CrossRef.
- M. R. Islam and P. R. Sundararajan, Phys. Chem. Chem. Phys., 2013, 15, 21058–21069 RSC.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS PubMed.
- T. J. Zhang, D. M. Sun, X. K. Ren, L. L. Liu, G. Y. Wen, Z. J. Ren, H. H. Li and S. K. Yan, Soft Matter, 2013, 9, 10739–10745 RSC.
- M. Funahashi and A. Sonoda, Dalton Trans., 2013, 42, 15987–15992 RSC.
- R. A. Cormier and B. A. Gregg, Chem. Mater., 1998, 10, 1309–1315 CrossRef CAS.
- E. Wolarz, E. Mykowska, T. Martynski and R. Stolarski, J. Mol. Struct., 2009, 929, 79–84 CrossRef CAS.
- L. Wang, Q. Cui, X. F. Chen, Y. Li, Z. Q. Li, D. Wang and H. Yang, Aust. J. Chem., 2013, 66, 692–700 CrossRef CAS.
- N. Mizoshita, T. Tani and S. Inagaki, Adv. Funct. Mater., 2011, 21, 3291–3298 CrossRef CAS.
- B. A. Gregg and R. A. Cormier, J. Am. Chem. Soc., 2001, 123, 7959–7960 CrossRef CAS PubMed.
- Y. Xu, S. Leng, C. Xue, R. Sun, J. Pan, J. Ford and S. Jin, Angew. Chem., Int. Ed., 2007, 46, 3896–3899 CrossRef CAS PubMed.
- B. X. Gao, D. F. Xia, L. C. Zhang, Q. Q. Bai, L. B. Bai, T. Yang and X. W. Ba, J. Mater. Chem., 2011, 21, 15975–15982 RSC.
- Z. An, J. Yu, S. C. Jones, S. Barlow, S. Yoo, B. Domercq, P. Prins, L. D. A. Siebbeles, B. Kippelen and S. R. Marder, Adv. Mater., 2005, 17, 2580–2581 CrossRef CAS.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- L. Meng, Q. M. Wu, F. F. Yang and H. Y. Guo, New J. Chem., 2015, 39, 72–76 RSC.
- G. A. Bhavsar and S. K. Asha, Chem.–Eur. J., 2011, 17, 12646–12658 CrossRef CAS PubMed.
- M. Bagui, T. Dutta, H. Z. Zhong, S. H. Li, S. Chakraborty, A. Keightley and Z. H. Peng, Tetrahedron, 2012, 68, 2806–2818 CrossRef CAS.
- S. K. Gupta, S. Setia, S. Sidiq, M. Gupta, S. Kumar and S. K. Pal, RSC Adv., 2013, 3, 12060–12065 RSC.
- X. F. Kong, Z. Q. He, Y. N. Zhang, L. P. Mu, C. J. Liang, B. Chen, X. P. Jing and A. N. Cammidge, Org. Lett., 2011, 13, 764–767 CrossRef CAS PubMed.
- M. G. Zhu, H. Y. Guo, F. F. Yang and Z. S. Wang, Dyes Pigm., 2016, 133, 387–394 CrossRef CAS.
- H. Y. Guo, F. F. Yang, W. W. Liu and J. B. Lai, Tetrahedron Lett., 2015, 56, 866–870 CrossRef CAS.
- F. Würthner, V. Stepanenko, Z. Chen, C. R. Saha-Möller, N. Kocher and D. Stalke, J. Org. Chem., 2004, 69, 7933–7939 CrossRef PubMed.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- F. Fennel, S. Wolter, A. Q. Xie, P. A. Plötz, O. Kühn, F. Würthner and S. Lochbrunner, J. Am. Chem. Soc., 2013, 135, 18722–18725 CrossRef CAS PubMed.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem.–Eur. J., 2001, 7, 224522–224553 Search PubMed.
- M. G. Zhu, H. Y. Guo, F. F. Yang and Z. S. Wang, Liq. Cryst., 2016, 43, 1875–1883 CrossRef CAS.
- R. B. Hou, K. L. Zhong, Z. G. Huang, L. Y. Jin and B. Z. Yin, Tetrahedron, 2001, 67, 1238–1244 CrossRef.
- S. Dattaa and S. Bhattacharya, Soft Matter, 2015, 11, 1945–1963 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 28, 4332–4335 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthesis and characterisation of new compounds. See DOI: 10.1039/c6ra27003d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab and
Zusheng Wanga
*ab and
Zusheng Wanga