
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switching of the π-electronic conjugations in the reduction of a dithienylethene-fused p-benzoquinone†
Eiji Saito,
Takumi Ako,
Yasuhiro Kobori * and
Akihiko Tsuda*
* and
Akihiko Tsuda*
Department of Chemistry, Graduate School of Science, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan. E-mail: tsuda@harbor.kobe-u.ac.jp; ykobori@kitty.kobe-u.ac.jp; Fax: +81-78-803-5671
First published on 12th January 2017
Abstract
The electron accepting character of a dithienylethene-fused p-benzoquinone derivative is significantly reduced upon ring-closing isomerization. Visible light unlocks the π-electronic conjugation of the quinone so it can be utilized for a light-driven oxidation reaction.
Quinones, an oxidized species of aromatic compounds, play important roles in biological systems, and are also valuable in industry. They serve as electron acceptors, oxidizing agents, dyes, and pigments in biochemistry and organic chemistry.1–5 The electrochemical properties and chemical reactivity of quinone are dependent on its substituent groups, and are also affected by solvent and external chemical stimuli.6 Many examples have been reported of controlling the reactions of quinones through weak interactions with additives such as metal ions.7 We previously reported supramolecular [4 + 2] cycloaddition and [2 + 2] photocycloaddition reactions of a p-benzoquinone derivative bearing an oligoether side-chain that can bind a metal ion through electrostatic interaction.8,9 The bound metal ion reduces or increases the π-electron density of the quinone, changing its reactivity in both thermal and photochemical reactions. However, to the best of our knowledge, physical operation of the chemical reactions of quinone with the light is an unprecedented subject. The light-driven chemical reactions, which allow for arbitrary control of specific target and time, promise new developments in photoresponsive reagents and catalysts, photoconductive materials, and focal reactions in living organisms and nanomaterials.10–12 In the present study, we report a light-driven oxidation reaction with a dithienylethene-fused p-benzoquinone, which unlocks the π-electronic conjugation of the quinone upon exposure to visible light.
Katsumura and coworkers reported synthesis of dithienylethene-fused p-benzoquinones using the Stille's type Pd(0) catalyzed coupling reaction.13 The ring-opening and -closing reactions of their derivatives were then reported by Liebeskind and coworkers.14 The ring-opening quinones cause ring closure on photolysis. However, the observed conversion in the photostationary state (PSS) is only 5–30%, and is also dependent on the substituent groups attached to the quinone and thiophene groups. The conversion is increased through an acid-promoted ring-closing reaction with AlCl3, FeCl3, or CF3SO3H in the dark, and the ring-closing isomer can be isolated in yields of up to 95% through column chromatography. Although the observed differences in absorption spectra between the ring-opening and -closing forms indicate significant changes in the π-electronic conjugations, there are no reports that focus on their electron accepting character and chemical reactivity. Knowing this, we herein studied redox properties of a dithienylethene-fused quinone DTQ by means of cyclic voltammetry and EPR spectroscopy together with theoretical calculations, and applied it to a light-driven chemical reaction (Scheme 1).
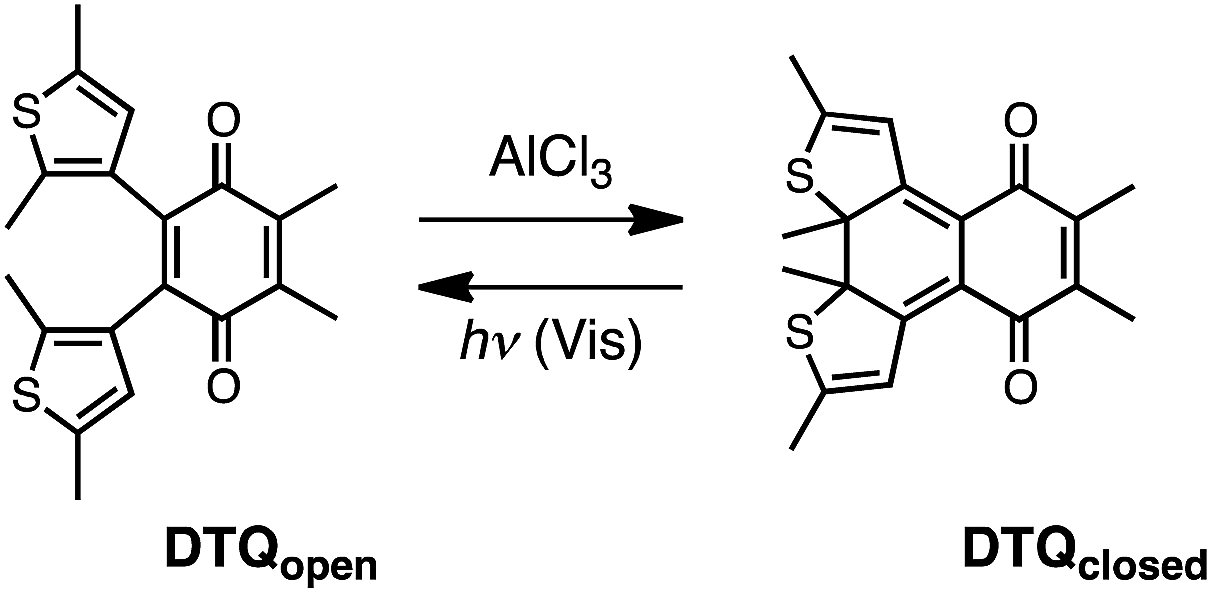 | ||
| Scheme 1 Ring-closing and -opening reactions of DTQ with AlCl3 and visible light, respectively. | ||
We synthesized DTQ in line with the literature.13,14 The molecular structure of ring-opening DTQopen was characterized by means of 1H and 13C NMR and electronic absorption spectroscopies, mass spectrometry, and X-ray crystallography (Fig. S1 and Table S1†).15 When DTQopen was mixed with 3 equiv. of AlCl3 in CH2Cl2, it was converted immediately to the ring-closing isomer DTQclosed. Through chromatographic separation, it was isolated in 71% yield (Fig. S2†). The sample solutions of DTQopen and DTQclosed showed orange and reddish violet with λmax in the lowest energy absorption band at 446 and 520 nm with the molar absorption coefficient of 1750 and 5941 M−1 cm−1, respectively (Fig. 1). These spectral data then allowed estimation of the ratio of DTQopen and DTQclosed in the solution. When a cyclohexane solution of DTQopen was exposed to 320 nm UV-light with a 500 W xenon lamp for 20 min, the spectral change due to photoisomerization occurred, and 26% conversion to DTQclosed was evaluated in the photostationary state (PSS) (Fig. S3†). On the other hand, DTQclosed showed quantitative conversion to DTQopen, when exposed to visible light (>380 nm) (Fig. S4†). The quantum yield (Φ) of this ring-opening reaction upon excitation at 420 nm was determined to be 0.78,16 which is larger than that of diarylethene derivatives reported previously.17 However, without photo-irradiation, no isomerization occurred when a sample solution of DTQclosed was left for 14 days in the dark at room temperature (Fig. S5†). This is the characteristic P-type feature of diarylethene. The sample solution, thus, reversibly changes the absorption spectrum upon photo-irradiation with visible and UV light (Fig. S6†).
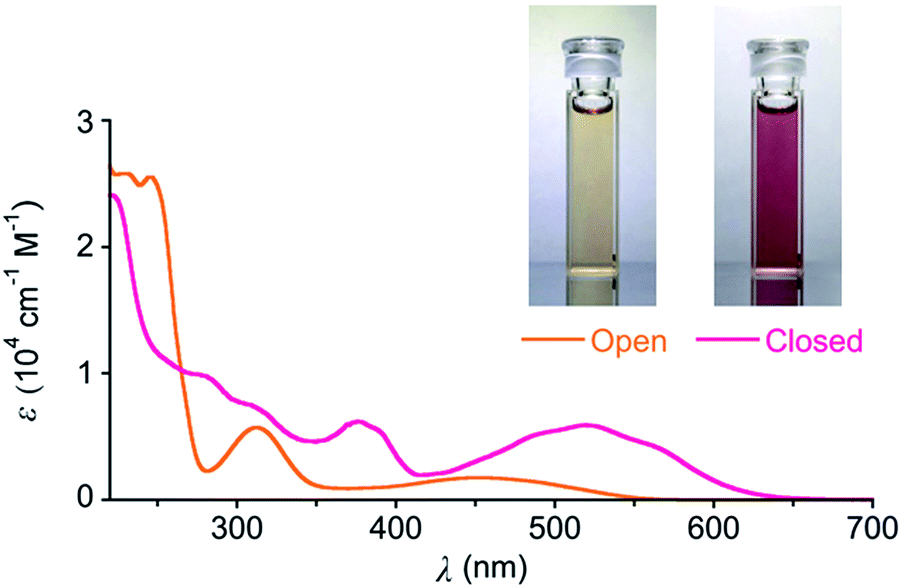 | ||
| Fig. 1 Photos and UV-Vis absorption spectra of cyclohexane solutions of DTQopen and DTQclosed at 20 °C. The spectra were taken on the concentration at 4.0 × 10−5 M. | ||
The reduction potentials of DTQopen and DTQclosed in CH2Cl2 containing 0.1 M Bu4NClO4 as a supporting electrolyte were measured by cyclic voltammetry (Fig. 2). The ring-opening DTQopen showed two redox couples at E1/2 = −1.24 V and −2.01 V (vs. Fc/Fc+), which may correspond to DTQopen/DTQopen˙− and DTQopen˙−/DTQopen2−, respectively. This is a characteristic redox profile observed in quinone compounds. On the other hand, the ring-closing DTQclosed showed a negatively shifted one redox peak at E1/2 = −1.80 V. Plots of Epa and Epc values with respect to the square root of the scan rate (ν1/2) in the range of 50–500 mV s−1 provided a potential difference ΔEp = 57.9 mV (Fig. S7†). Since ΔEp should be 59/n mV, where n is the number of electrons, at 25 °C in an ideal Nernstian process,18 the observed redox couple was assigned as a 1e− transition. The observed large negative shift of the reduction potential in DTQclosed may originate in the fact that its radical anion, including 1,4-semidion, does not undergo aromatic stabilization upon a one-electron reduction.19
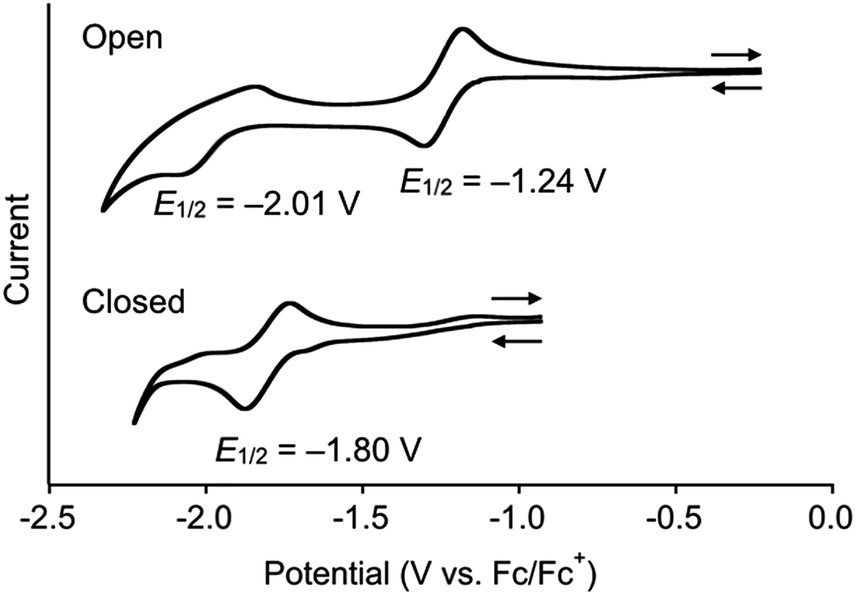 | ||
| Fig. 2 Redox profiles in cyclic voltammetry (V vs. Fc/Fc+) of mixtures of DTQopen and DTQclosed in CH2Cl2. Scan rate, 100 mV s−1; working electrode, Pt; supporting electrolyte, 0.1 M Bu4NClO4. | ||
We then demonstrated EPR spectroscopy for the reduced forms of DTQopen and DTQclosed. With a sample solution prepared upon mixing DTQopen and metallic sodium in CH2Cl2 at room temperature, the EPR signal was observed at g = 2.005 with hyperfine splitting most likely due to methyl protons attached at 5,6-positions (Fig. 3a). The hyperfine coupling constant (hfc) was estimated to be a(6H) = 1.59 G by a computer-simulated spectrum of DTQopen˙− with the DFT calculation at UB3LYP/6-311+G(d,p) level.20 The sample solution prepared upon mixing DTQclosed and metallic sodium also provided the EPR signal at g = 2.005 (Fig. 3b). However, the observed hyperfine structure was entirely different from that of DTQopen˙−, and was simulated by applying to the three hfcs a(6H) = 1.20 G, a(6H) = 1.11 G, and a(2H) = 0.39 G (Fig. 3b). The result indicates contributions of β-protons on the thiophene rings and methyl protons attached to the quinone and thiophene rings in the spectrum. The spin density distributions of DTQopen˙− and DTQclosed˙− were then calculated by DFT program (Fig. S8†). It showed that spin density of DTQopen˙− is distributed mainly on the quinone moiety, especially at the carbonyl oxygens. This result would explain the observed hfc in EPR spectrum. On the other hand, the calculated spin density of DTQclosed˙− is only slightly distributed on the thiophene rings, but is concentrated at unsaturated 1,4-diketone moiety. This does not agree with the fact that its EPR spectrum, as described above, indicates that there should be a reduced contribution of the quinone moiety with the adding of contributions from thiophene groups to the observed hfcs in Fig. 3b. However, we found that this disagreement can be reasonably explained by the frontier MOs and the orbital energies calculated for DTQclosed (Fig. 4). The calculation showed that DTQclosed has two nearly degenerated LUMO and LUMO+1 levels with a small energy gap of 0.07 eV. The LUMO has larger coefficients of the atomic orbitals in the quinone moiety, while LUMO+1 has them in both the quinone and the thiophene rings. The observed three different hfcs of DTQclosed˙− is well characterized with the orbital character of LUMO+1 of DTQclosed. Hence, the contribution of the degenerated LUMO+1, which provides larger distribution of the spin onto the thiophene rings, in the formation of its anion radical, may allow delocalization of the radical over the components. Conclusively, in DTQopen, where the attached thiophene groups adopt almost perpendicular conformation, the quinone moiety undergoes an aromatic stabilization by forming a semiquinone radical anion, as in the case of usual quinones. However, such an aromatic stabilization is not expected in DTQclosed˙−, because its planar structure allows orbital delocalizations over the entire molecule with an antibonding π* character between the quinone and thiophene moieties in LUMO and in LUMO+1 (Fig. 4). This is coincident with the above-mentioned negatively shifted redox peak at E1/2 = −1.80 V and with the three hfcs in the EPR spectrum (Fig. 2 and 3b, respectively).
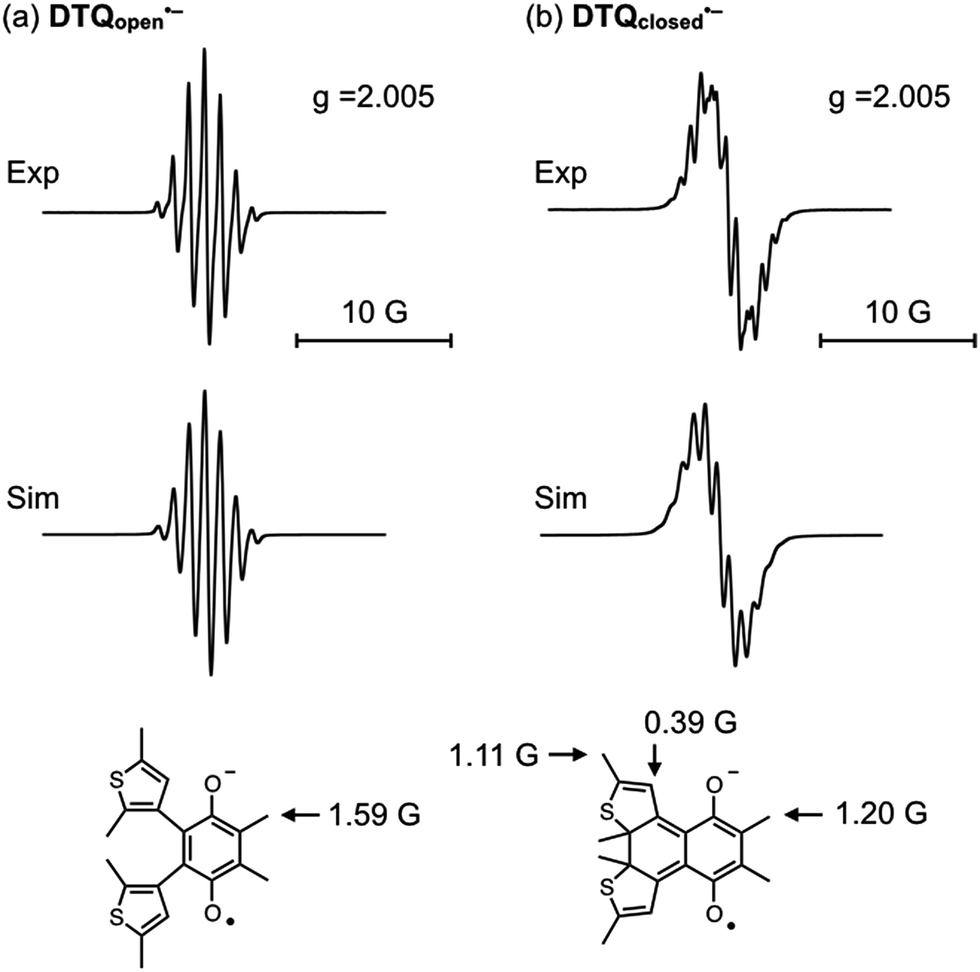 | ||
| Fig. 3 EPR spectra of (a) DTQopen˙− and (b) DTQclosed˙− generated upon mixing DTQ with metallic sodium in deaerated CH2Cl2 with the computer simulation spectra and the hyperfine coupling constants. The spectral measurements were demonstrated at room temperature. | ||
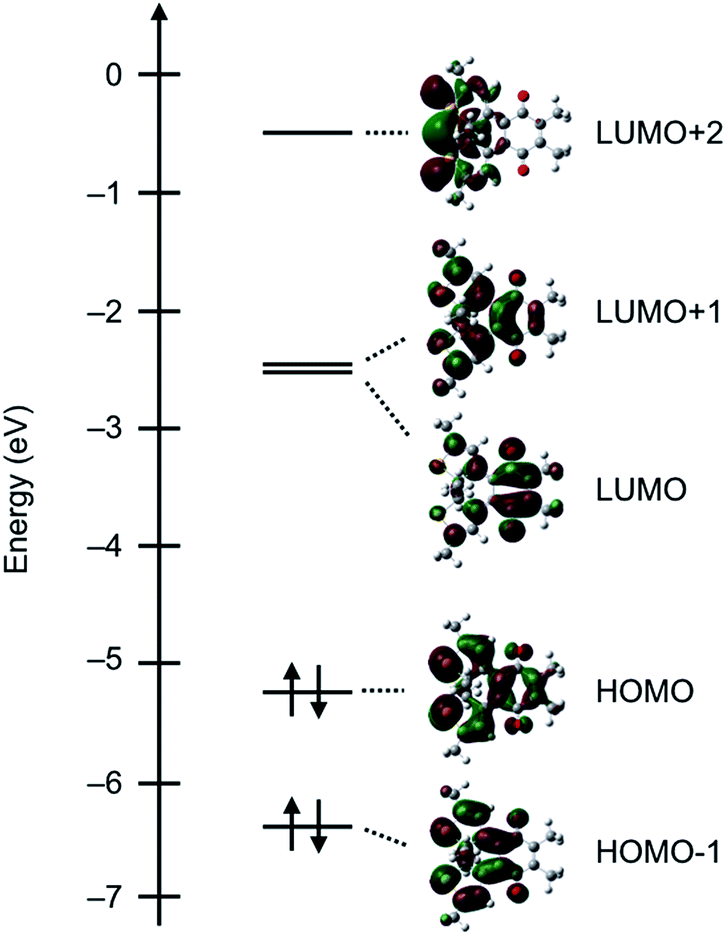 | ||
| Fig. 4 Calculated frontier MOs and the orbital energies of DTQclosed in DFT with B3LYP/6-311G(d,p) level. Red and green colors indicate the positive and negative orbitals, respectively. | ||
The electrochemical properties of the ring-opening and -closing DTQ revealed by the above experiments prompted us to expect different reactivity in their chemical reductions. Considering their one-electron reduction potentials described above, we investigated a possible hydrogen exchange reaction between DTQ and tetramethylhydroquinone Me4HQ, a reduced form of tetramethyl-p-benzoquinone Me4Q (Fig. 5).21 Judging from the reduction potential of Me4Q [E1/2 = −1.40 V (vs. Fc/Fc+)], which is lower than that of DTQopen, but is higher than that of DTQclosed, we expected the reaction (Fig. S9†). The reaction was demonstrated in CDCl3 at room temperature, and monitored by 1H NMR spectroscopy. When the ring-opening DTQopen was mixed with Me4HQ, its 1H NMR peaks disappeared, and two sets of new peaks appeared with other peaks corresponding to tetramethyl quinone Me4Q (Fig. S10†). Since 2D-NMR spectroscopies of the isolated product actually provided two sets of the correlation profiles (Fig. S11†), and further, its elemental analysis and mass spectral profile were consistent with those calculated for the reduced DTQ (Fig. S12†), the peaks may be originated from the mixture of anti- and parallel conformational isomers of the reduced DTQopen (DTHQ).22 The product was thus reconverted quantitatively to DTQopen upon oxidation with Ag2O (Fig. S13†). In sharp contrast to the result with DTQopen, interestingly, no reaction was observed with DTQclosed even upon mixing with 10 equiv. amounts of Me4HQ (Fig. 5). With these results in mind, we then demonstrated a possible light-driven oxidation reaction of Me4HQ with DTQclosed. When the sample solution, which showed no reaction in the dark, was exposed to visible light (>380 nm) for 10 min, a hydrogen-exchange reaction occurred to give DTHQ in quantitative yield (Fig. 5). Here, the photo-conversion from DTQclosed to DTQopen most likely triggered this reaction. The results can be explained by the order of first reduction potentials in cyclic voltammetry (DTQopen > Me4Q > DTQclosed), which may correlate with the thermal stability of the semiquinone radical, a possible intermediate in the formation of the corresponding hydroquinone.
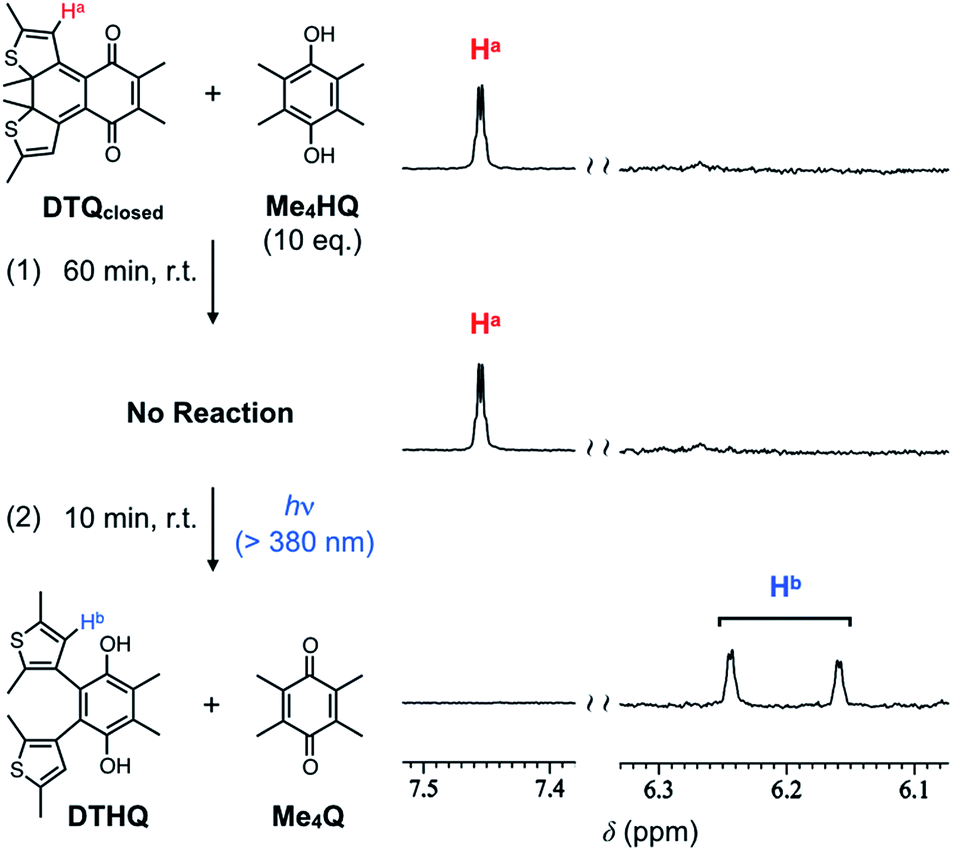 | ||
Fig. 5 1H NMR spectral profiles for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mixture of DTQclosed and Me4HQ after (1) stirring of the sample solution for 60 min in the dark and (2) photoirradiation with visible light for 10 min at room temperature. :10 mixture of DTQclosed and Me4HQ after (1) stirring of the sample solution for 60 min in the dark and (2) photoirradiation with visible light for 10 min at room temperature. | ||
Conclusions
In this study, we found that a dithienylethene-fused p-benzoquinone derivative DTQ changes its electronic properties dramatically through ring-opening and -closing isomerizations. Both isomers underwent one-electron reduction to give the corresponding anion radicals. The spectral studies together with DFT calculations indicate that DTQopen˙− adopts semiquinone form, while DTQclosed˙− allows spin delocalization between quinone and thiophene groups. The electron accepting character as well as oxidizability of DTQclosed is reduced significantly in comparison with that of DTQopen. Visible light unlocks the π-electronic conjugation of the quinone in DTQ utilizing it for a light-driven oxidation reaction.Acknowledgements
We thank Prof. T. Osakai for his helpful discussion on CV measurements. The present work was sponsored by Nakanishi Scholarship Foundation, Tokyo Ohka Foundation for The Promotion of Science and Technology, and Tokyo Kasei Chemical Promotion Foundation.Notes and references
- M. Di Donato, A. Peluso, G. Villani, M. Di Donato, D. Chimica and V. Uni, J. Phys. Chem. B, 2004, 108, 3068 CrossRef CAS; Z. Zhu and M. R. Gunner, Biochemistry, 2005, 44, 82 CrossRef PubMed; J. Madeo, M. Mihajlovic, T. Lazaridis and M. R. Gunner, J. Am. Chem. Soc., 2011, 133, 17375 CrossRef PubMed.
- Z. Song, H. Zhan and Y. Zhou, Chem. Commun., 2009, 448 RSC; B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195 CrossRef CAS PubMed; K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374 CrossRef PubMed.
- Y. Zhang and C. J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; S. Bhunia, S. Ghosh, D. Dey and A. Bisai, Org. Lett., 2013, 15, 2426 CrossRef PubMed.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS PubMed; B. Koszarna and D. T. Gryko, Chem. Commun., 2007, 2994 RSC; N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945 CrossRef PubMed; Y. Li and Z. Wang, Org. Lett., 2009, 11, 1385 CrossRef PubMed; Y. Li, J. Gao, S. Di Motta, F. Negri and Z. Wang, J. Am. Chem. Soc., 2010, 132, 4208 CrossRef PubMed.
- T. Prestera, H. J. Prochaska and P. Talalay, Biochemistry, 1992, 31, 824 CrossRef CAS PubMed; S. Kacprzak and M. Kaupp, J. Phys. Chem. B, 2006, 110, 8158 CrossRef PubMed; H. Takezawa, S. Akiba, T. Murase and M. Fujita, J. Am. Chem. Soc., 2015, 137, 7043 CrossRef PubMed.
- N. Gupta and H. Linschitz, J. Am. Chem. Soc., 1997, 119, 6384 CrossRef CAS; M. Gómez, F. J. González and I. González, J. Electroanal. Chem., 2005, 578, 193 CrossRef; T. Kawashima, K. Ohkubo and S. Fukuzumi, Phys. Chem. Chem. Phys., 2011, 13, 3344 RSC; Y. R. Kim, R. S. Kim, S. K. Kang, M. G. Choi, H. Y. Kim, D. Cho, J. Y. Lee, S. K. Chang and T. D. Chung, J. Am. Chem. Soc., 2013, 135, 18957 CrossRef PubMed.
- S. Fukuzumi, K. Ohkubo and T. Okamoto, J. Am. Chem. Soc., 2002, 124, 14147 CrossRef CAS PubMed; H. Wu, D. Zhang, L. Su, K. Ohkubo, C. Zhang, S. Yin, L. Mao, Z. Shuai, S. Fukuzumi and D. Zhu, J. Am. Chem. Soc., 2007, 129, 6839 CrossRef PubMed; J. Yuasa, S. Yamada and S. Fukuzumi, Chem.–Eur. J., 2008, 14, 1866 CrossRef PubMed.
- A. Tsuda and T. Oshima, New J. Chem., 1998, 1027 RSC; A. Tsuda and T. Oshima, J. Org. Chem., 2002, 67, 1282 CrossRef CAS PubMed; A. Tsuda, C. Fukumoto and T. Oshima, J. Am. Chem. Soc., 2003, 125, 5811 CrossRef PubMed.
- H. Yamamoto, K. Ohkubo, S. Akimoto, S. Fukuzumi and A. Tsuda, Org. Biomol. Chem., 2014, 12, 7004 CAS.
- I. Akiko, S. Yatsuda, S. Akiko, T. Takeshi and A. Munetaka, Inorg. Chem., 2007, 46, 2432 CrossRef CAS PubMed; D. A. Nicewicz and D. W. C. Macmillan, Science, 2008, 322, 77 CrossRef PubMed; J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef PubMed; C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef PubMed; E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064 CrossRef PubMed.
- A. Balamurugan and H. J. Lee, Macromolecules, 2016, 49, 2568 CrossRef CAS.
- M. Kurihara and H. Nishihara, Coord. Chem. Rev., 2002, 226, 125 CrossRef CAS; H. Nishihara, Coord. Chem. Rev., 2005, 249, 1468 CrossRef; L. Sun, S. Wang and H. Tian, Chem. Lett., 2007, 36, 250 CrossRef; M. M. Paquette, R. A. Kopelman, E. Beitlera and N. L. Frank, Chem. Commun., 2009, 5424 RSC.
- S. Yoshida, H. Kubo, T. Saika and S. Katsumura, Chem. Lett., 1996, 139 CrossRef CAS.
- X. Deng and L. S. Liebeskind, J. Am. Chem. Soc., 2001, 123, 7703 CrossRef CAS PubMed.
- Crystallographic data for the structure of DTQopen was deposited with the Cambridge Crystallographic Data Center as supplementary publication numbers CCDC 1508336.
- Determined by ferrioxalate actinometry.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174 CrossRef CAS PubMed.
- A. J. Bard and L. Y. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2004 Search PubMed.
- G. A. Russell, R. L. Blankespoor, J. Mattox, P. R. Whittle, D. Symalla and J. R. Dodd, J. Am. Chem. Soc., 1974, 96, 7250 Search PubMed.
- DFT calculations were carried out using Gaussian 09. Geometry optimizations were performed for a single molecule in a gas phase.
- L. Vigderman and E. R. Zubarev, Chem. Mater., 2013, 25, 1450 CrossRef CAS.
- M. Irie, O. Miyatake and K. Uchida, J. Am. Chem. Soc., 1992, 114, 8715 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization, and other materials. CCDC 1508336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27001h |
| This journal is © The Royal Society of Chemistry 2017 |