
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational molecular engineering of L-amino acid deaminase for production of α-ketoisovaleric acid from L-valine by Escherichia coli
Ruoxi Liab,
Hossain Gazi Sakira,
Jianghua Lia,
Hyun-dong Shinc,
Guocheng Duab,
Jian Chena and
Long Liu *ab
*ab
aKey Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China. E-mail: longliu@jiangnan.edu.cn; Fax: +86-510-85918309; Tel: +86-510-85918312
bKey Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China
cSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta 30332, USA
First published on 20th January 2017
Abstract
The targeted modification of enzymatic efficiency can drive an increased production of desired metabolites. α-Ketoisovaleric acid (KIV) is a candidate material for use in the pharmaceutical and food industries. In the present study, we aimed to enhance the biotransformation efficiency of L-amino acid deaminase (L-aad) from Proteus myxofaciens ATCC 19692 to improve the production of KIV from L-valine. First, L-aad was expressed in Escherichia coli BL21(DE3). We employed transformed E. coli cells as a whole-cell biocatalyst system and optimized their biochemical characteristics for the biotransformation of L-valine. Then, based on the known 3D structural model of L-aad from P. myxofaciens and the simulation results for docking with L-valine, four amino acid residues (N100, Q276, R316, and F318) were identified as potential target sites for mutagenesis. Next, we performed site-directed saturation mutagenesis to improve the biotransformation efficiency. With 11.3 g L−1 L-valine, the bioconversion efficiencies of a single-mutant strain (F318T) and a double-mutant strain (F318T and N100H) were 4.474 and 8.197 g L−1, respectively, whereas that of the wild-type strain was 2.014 g L−1 under optimal conditions. In summary, we developed a one-step process for KIV production via expressing P. myxofaciens L-aad in E. coli BL21(DE3) and enhanced the yield of KIV by site-directed saturation mutagenesis of L-aad.
Introduction
The development of microbial strains capable of efficiently producing biochemicals and biofuels is no longer limited by the technical challenge of introducing genetic variations into microorganisms. The current bottleneck is the ability to screen and develop strains as well as enzymes with enhanced productivity from the enormous genetic diversity that can now be generated. α-Ketoisovaleric acid (KIV), a branched-chain organic acid, serves as a precursor in leucine and valine synthesis. It can be used as an initial compound for vitamin B5 biosynthesis, and the conversion of KIV to α-ketopantoate constitutes the rate-limiting step in vitamin B5 production.1–3 At present, KIV is mainly synthesized by multistep chemical methods (e.g., the azlactone method and Grignard reagents with diethyloxamates).4,5 The high cost and complexity of these chemical processes restrain the industrial production of KIV. Therefore, biotechnological processes have been widely studied for the KIV production because of their high efficiency. In our previous study, whole-cell transformation was used to produce α-ketoglutarate, α-ketoisocaproate, and α-ketomethylvalerate.6–10 These studies indicated the advantages of whole-cell biocatalyst systems; for instance, it is possible to immobilize the cells and reuse them as catalysts.L-Amino acid deaminase (L-aad, E.C. 1.4.3.2) is a monomeric enzyme (49 kDa) that is located on the outer membrane of the host cell. L-aad can transform an L-amino acid to corresponding keto acids in a single-step reaction6,9,10 as shown in Fig. 1. In addition, the high catalytic efficiency, low toxicity, and broad substrate selectivity of L-aad indicate a high potential for its application to the industrial production of keto acids. Nonetheless, because L-aad is a transmembrane protein, its crystal structure remained ambiguous until recently, resulting in difficulties with confirming the active residues and the mechanism of the deamination reaction in previous studies.9
 | ||
| Fig. 1 Reaction scheme of L-amino acid deaminase from Proteus myxofaciens. | ||
Studies on α-keto acid production from amino acids by biotransformation have been mainly focused on the fermentation strategy and directed evolution based on error-prone polymerase chain reaction (ep-PCR).6–10 Recently, Motta et al. reported the crystal structure of L-aad from P. myxofaciens11 and Ju et al. reported that of L-aad from P. vulgaris.12
Site-directed mutagenesis is an effective method that is widely used for the improvement of an enzyme's catalytic characteristics.13 In this work, L-aad from P. myxofaciens was heterologously expressed in E. coli BL21(DE3). Then, four key residues were determined by means of a flexible docking experiment between L-aad and L-valine. After that, site-directed saturation mutagenesis was carried out on these four sites, and the function of the corresponding residues was demonstrated. Finally, we constructed mutant E. coli strains harboring L-aad containing multiple mutations and identified a unique mutant (F318T) that showed a 3.17-fold increase in catalytic efficiency compared with that of wild-type L-aad.
Methods
Materials, plasmids, and strains
The SanPrep Column Plasmid Mini-Preps Kit was purchased from Sangon (Shanghai, China). The DNA purification kit, PrimeSTAR® HS DNA polymerase premix, restriction enzymes, and T4 DNA ligase were acquired from Takara Bio (Ōtsu, Japan). The KIV standard sample was purchased from Sigma-Aldrich (St. Louis, MO, USA). Ampicillin was acquired from Sangon.P. myxofaciens ATCC 19692 was purchased from American Type Culture Collection (ATCC; Manassas, VA, USA) and used as a source for cloning of the ladF gene, which encodes L-aad. E. coli JM109 was used for cloning, and E. coli BL21 (DE3) was used for the expression of L-aad wild-type and mutants. The ladF gene sequence was confirmed by DNA sequencing. Primer synthesis and DNA sequencing were performed by BGI technology (Shanghai, China).
Recombinant plasmid construction and heterologous expression
The genomic DNA of P. myxofaciens ATCC 19692 was extracted using a Genomic DNA Purification Kit (Tiangen, Beijing, China), and the ladF gene was amplified by PCR using gene-specific primers (Table 1). The PCR product was resolved on a 1% agarose gel, purified by means of a DNA purification kit (Takara Bio, Dalian, China), digested with the restriction enzymes EcoRI and HindIII, and ligated into the plasmid pET20b(+) for subsequent amplification in E. coli JM109. After restriction analysis and DNA sequencing, the recombinant vector containing ladF was transfected into competent cells of E. coli BL21(DE3) for heterologous expression.| Primers | Nucleotide sequencea | Restriction sites/Tm |
|---|---|---|
| a Blanks with underlines are mutation sites. | ||
| LadF-S | CGAATTCATGAACATCTCTCGTCGTAAACTGC | EcoR I |
| LadF-A | TTACTTCTTAAAACGATCCAAACTATTCGAACGC | Hind III |
| MT316-S | CACTTATGCTGTAGCCCCA_ATCTTCACAAGCTCCATTG | 68 °C |
| MT316-A | CAATGGAGCTTGTGAAGAT_TGGGGCTACAGCATAAGTG | 68 °C |
| MT318-S | GCTGTAGCCCCACGTATC_ACAAGCTCCATTGTTAAAG | 69 °C |
| MT318-A | CTTTAACAATGGAGCTTGT_GATACGTGGGGCTACAGC | 69 °C |
| MT100-S | CAGGCCGCGCATACAGC_ATCATTAGCTACCAAAC | 65 °C |
| MT100-A | GTTTGGTAGCTAATGA_TGCTGTATGCGCGGCCTG | 65 °C |
| MT276-S | GTTGATCTTCCAACCTT_TGTTTACTTATCACAAC | 65 °C |
| MT276-A | GTTGTGATAAGTAAACA_AAGGTTGGAAGATCAAC | 65 °C |
| C6H-S | GATCGTTTTAAGAAGACCCTTGGAAGCTTG | 70 °C |
| C6H-A | CAAGCTTCCAAGGGTCTTCTTAAAACGATC | 70 °C |
The recombinant strain was inoculated into 30 mL of Luria–Bertani medium in a 250 mL flask supplemented with 100 μg mL−1 ampicillin and cultured for 6 h at 37 °C and 220 rpm. The seed medium was transferred into Terrific Broth to a final concentration of 1% (v/v). The incubation medium was cultivated at 37 °C until the optical density at 600 nm (OD600) reached 1.0. Next, 0.4 mM β-D-l-thiogalactopyranoside was added and the solution was incubated at a relatively low temperature (25 °C) to induce protein expression. The OD600 of the growth culture was measured every 2 h to obtain a growth curve. After 6 h of incubation, the cells were harvested by centrifugation at 9000 × g and 4 °C for 5 min. The precipitate was resuspended in potassium phosphate buffer (pH 6.5, 0.1 M) and washed twice with the same buffer, then kept at 4 °C for the biotransformation analysis. The concentration of cells in the final suspension was quantified by converting OD620 data to dry cell weight (DCW) using the following equation: DCW (g L−1) = 0.4442 × OD620 − 0.021 (ref. 7).
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) analysis
The cell suspension was diluted to 1.5 g DCW/L and then incubated in denaturing buffer at 100 °C for 10 min. Twenty microliters of sample was resolved by 10% PAGE (Bio-Rad Laboratories, Hercules, CA, USA). The prepared samples and marker were loaded into wells and separated by electrophoresis, and the protein bands were visualized by staining with Coomassie Brilliant Blue R-250.Computational analysis of 3D structure and docking calculations
The structure of L-aad from P. myxofaciens (PDB ID: 5FJM) was obtained using homology modeling on the SWISS MODEL server with the crystal structure of lopped L-aad as a template. Amino acid mutation analysis was incorporated into the model using the Swiss-Viewer tool followed by side chain reconstruction for neighboring amino acids and energy minimization.14 AutoDock 4 software was used to identify potential binding sites for L-valine. Default parameters were selected as described in the manual. Binding modes within 1 kcal mol−1 of the most favorable binding free energy conformation (−3 kcal mol−1) are shown in Fig. 3.Site-directed saturation mutagenesis
This procedure was carried out using targeted whole-plasmid mutagenesis. PrimeSTAR® HS DNA polymerase was utilized in one-step PCR to amplify the pET20b-ladF vector as well as the sites to be mutated. The primers are presented in Table 1. The amplified sample was treated with DpnI at 37 °C to digest the template DNA for 2 h. After purification with the DNA purification kit from Takara Bio, the PCR product was transfected into E. coli JM109. We chose four residues of L-aad based on their predicted involvement in the active site and performed saturation mutagenesis of each site to improve the catalytic efficiency of L-aad. The mutations were confirmed by DNA sequencing and finally the relevant vectors were transformed into E. coli BL21(DE3) for expression and biotransformation experiments.Measurement of the biocatalytic activity
The biocatalytic activity was determined through whole-cell transformation. First, 11.3 g L−1 L-valine was resolved in potassium phosphate buffer containing 10 g L−1 cells; thereafter, the whole reaction system was placed in a 250 mL shaking flask and incubated on a rotary shaker for 12 h. To identify the best volume, pH, and other reaction conditions, the reaction was conducted under a range of conditions such as different volumes and pH.The collected samples were centrifuged at 9000 × g for 10 min to stop the reaction and separate the enzyme from the substrate. The concentration of KIV in the supernatant was measured as described below.
High-performance liquid chromatography for KIV measurement
The KIV concentration in the supernatant was determined using an HPLC system (Agilent 1260 Infinity, Santa Clara, CA, USA) with a Bio-Rad Aminex HPX-87H column (300 × 7.8 mm, 9 μm). The eluent was 5 mM sulfuric acid in deionized water after filtering and ultrasonic treatment. We maintained the flow rate at 0.6 mL min−1 and column temperature at 40 °C, and injected 10 μL of the mixture into the detector. KIV was quantified at the ultraviolet wavelength of 210 nm. The pure substances served as standards.Results and discussion
Expression of LadF from P. myxofaciens in E. coli BL21(DE3)
The wild-type ladF gene from P. myxofaciens ATCC 19692 was cloned into the pET20b(+) expression vector and transfected into E. coli BL21(DE3) for expression. As predicted using the TMPred server (http://embnet.vital-it.ch/software/TMPRED_form.html), L-aad is a type II transmembrane protein; therefore, a low temperature (25 °C) and sufficient time for refolding were needed in the overexpression of L-aad.15 To verify the expression of L-aad protein, lysates from the transformed cells were analyzed by SDS-PAGE (Fig. 2). However, it is difficult to obtain the purified protein in an active form, because for membrane proteins, the experimental conditions16,17 or changes to protein structure during purification6 would cause the enzyme efficiency to be altered. The optimization of detergent selection and protein dissolution are vital, because an improper detergent will cause the protein to be inactivated. For example, purification using n-dodecyl-β-D-maltoside significantly inhibited the binding between L-aad and L-phenylalanine.9 For the maximization of bioconversion, L-aad activity was measured in a bacterial whole-cell biocatalyst system containing the recombinant strain. Next, the biochemical characteristics (temperature, reaction volume, and pH) for the whole-cell transformation were optimized. It was found that under the optimized conditions (32 °C, pH 6.5 phosphate-buffered saline, and 20 mL working volume in a 250 mL flask), the yield of α-ketoisovalerate from the reaction catalyzed by the wild-type L-aad reached 13.991 mg L−1 min−1 g−1 DCW after 12 h. | ||
| Fig. 2 Whole cell SDS-PAGE analysis. The band of L-aad protein was highlighted by an arrow. Lane 1: whole cell protein after transfection of pET20b-LadF; lane 2: the controlled whole cell protein after transfection of empty pET20b plasmid. | ||
Selection of residues involved in L-valine binding and catalysis
Rational evolution (e.g., site-directed mutagenesis) by means of computational biotechnology has been used to enhance enzymatic properties. For example, Chen et al. utilized site-directed mutagenesis to explain the inhibition mechanism of a threonine deaminase; this experiment also helped to raise ketobutyrate accumulation by 100%.18 In addition, Hossain et al. recently performed saturation mutagenesis to enhance α-ketoglutarate acid production by carrying out error-prone PCR for 6 sites in the corresponding enzyme, and the yield of α-ketoglutarate acid was increased by 1.68-fold relative to that of the control.7Since then, Motta et al. found that the activity of L-aad could be affected by its interaction with the cell membrane.11 Therefore, in the present work, the transmembrane part of L-aad was added to modify the crystal structure model by homology modeling (Fig. 3). As a result, it was possible to accurately identify the binding sites and analyze them to improve the catalytic performance of L-aad.
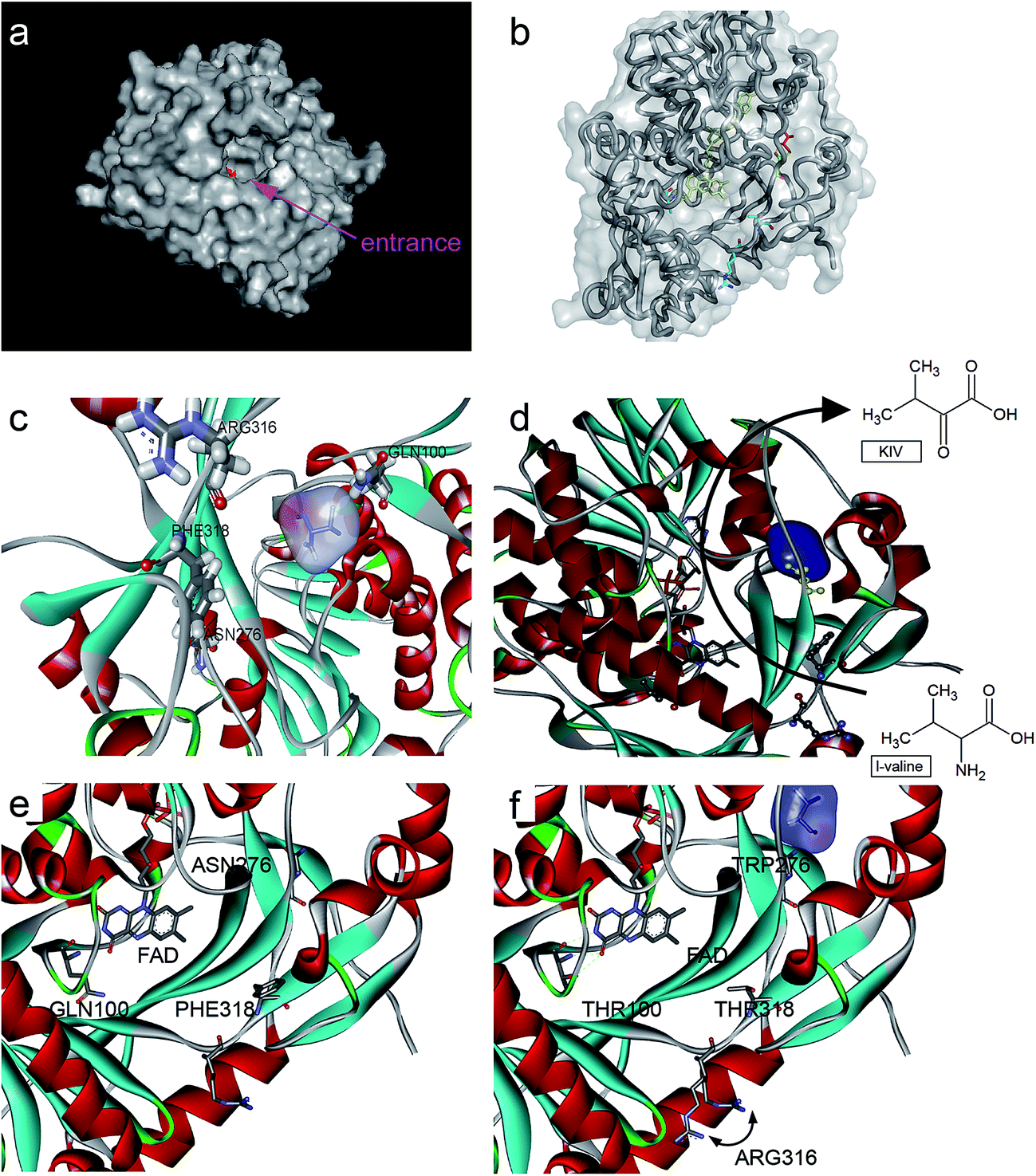 | ||
| Fig. 3 Protein structure and ligand binding to L-aad and its mutants. (a) The surface of L-aad protein and its uncommonly broad entrance, the inside active pocket was colored red; (b and c) the residues was reengineered in this work, (d) the potential process path of L-amino deaminase deamination, (e) a possible reaction path in L-aad, (e and f) the comparison of the chosen residues. | ||
In addition, to enhance the biotransformation efficiency of L-aad, the selection of the L-valine-binding sites was essential. Hence, protein molecular docking experiments between L-aad and L-valine were carried out. As indicated in the 3D structure of L-aad, the entrance of the active pocket is wide; therefore, the substrate-binding region is unusually broad and can distinguish among different amino acids. By comparing the hydrogen-bonding capability of residues in the 3D structure, we estimated that F318 and R316 are the key sites for binding to L-valine.
The hydrogen bond network around the catalytic domain usually contributes to determining the enzyme's biotransformation efficiency.19–21 In addition, L-aad is a monomer that is assisted by farnesyl adenine dinucleotide as a cofactor for the deamination reaction. It was reported that the binding of L-aad with this cofactor could influence its catalytic efficiency.22 Therefore, it will form an H-bond via N100 and Q276 when L-valine is bound to the active site of the enzyme (several binding sites). Thus, we selected all four sites (R316, F318, N100, and Q276) for saturation mutagenesis to enhance the enzyme's biotransformation capability.
Enhancing KIV production by rational molecular engineering
To enhance the biotransformation efficiency of L-aad, the amino acid residues contributing to substrate binding (R316, F318, N100, and Q276) were identified and then mutated. Fig. 4 shows the biotransformation efficiency of wild-type L-aad and mutants. The biotransformation capability was shut down by saturation mutagenesis of R316. This result revealed that when the substrate comes close to the binding pocket, the flexibility of R316 dramatically changes during the binding process.12 As shown in Fig. 3, R316 controls the “door” of the binding pocket of the substrate. The amino acid side chain of arginine consists of a 3-carbon aliphatic straight chain and the distal carbon is usually capped by a complex guanidinium group. Due to the double bond and the nitrogen long pair at the distal end, arginine residues tend to form multiple H-bonds, which interact with the nearby ligand. Additionally, arginine can retain its charge in dehydrated environments, which likely contributes to the function of an enzyme when a charge is required in the interior position of the enzyme.23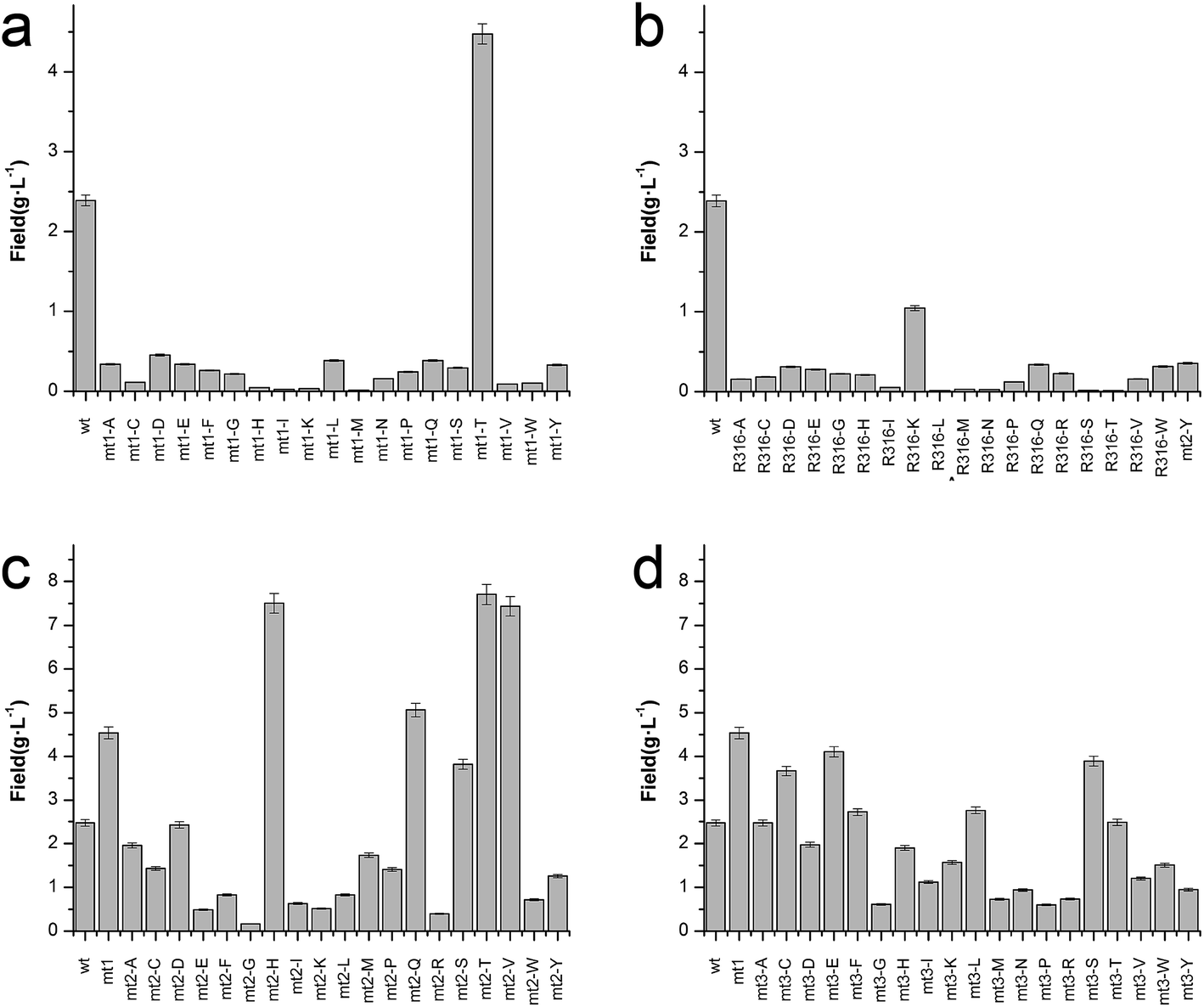 | ||
| Fig. 4 Catalytic efficiency of L-aad mutants. (a) Time-space yield of R316 saturation mutants; (b) time-space yield of F318 saturation mutants; (c) time-space yield of N100 saturation mutants based on the positive F318T mutant; (d) time-space yield of Q276 saturation mutants based on the positive F318T mutant. | ||
Moreover, the substitution of N100, Q276, and F318 had a positive effect on the biotransformation efficiency of L-aad. The bioconversion efficiency of F318T was increased by 187.38% relative to that of wild-type L-aad, but the other 18 tested substitutions completely inhibited its biotransformation efficiency. In wild-type L-aad, this phenylalanine residue (F318) is responsible for the inert and hydrophobic characteristics of the benzyl side chain. This result revealed a preference for the binding of residue 318 to aromatic groups. In addition, threonine contains one more nonhydrogen substituent than do other amino acids except valine and isoleucine; therefore, more bulkiness near the enzyme backbone is crucial for its activity. The substitution of phenylalanine with threonine at the key binding site led to an altered substrate specificity. In a recent study, it was reported that L-aad from P. myxofaciens is specific for aromatic, neutral L-amino acids but shows lower activity toward small, charged, or polar L-amino acids.11 In this work, however, mutation of the key residue F318 to T318 increased the catalytic efficiency of L-aad by 31.075 mg L−1 min−1 g−1 DCW in comparison with that of the control. This figure is 11.4-fold higher than that presented in a previous report.11 This result revealed that reaction condition optimization and the positive substitution of key amino acid residues improved the catalytic efficiency of L-aad.
Based on data from the L-aad mutant F318T, residue N100 was replaced by 19 other amino acids (one by one). As shown in Fig. 4c, the resulting L-aad mutants showed different levels of activities compared to the wild-type. Specifically, in the cases of N100H, N100Q, N100T, and N100V, the biotransformation efficiency was increased by 26.24%, 4.62%, 28.03%, and 25.62%, respectively, while in N100I, N100L, N100R, and N100Y, the biotransformation efficiency of L-aad was lost completely. The other mutants showed lower biotransformation efficiencies than that of wild-type L-aad. Among them, the N100H mutant showed a better biotransformation efficiency, leading to an increase of the α-ketoisovalerate titer to 8.197 g L−1. Saturation mutagenesis of Q276, however, did not increase the biotransformation efficiency, but shortened the reaction duration in a 250 mL flask from 12 h to 10 h.
After combining the results of saturation mutagenesis of N100, Q276, R316, and F318, it was determined that L-valine and other L-amino acid residues enter the active site of L-aad by following an interesting path from the broad entrance of L-aad, resulting in the export of KIV. Based on these results, it was interpreted that R316 controls the switch of the entrance, then the residues distributed on the sides of the entry path in addition to N100, Q276, and F318 perform flexible functions in the deamination reaction.
Previously, a His-tag was fused with membrane enzymes to improve their biotransformation efficiency.24,25 In the present work, the C-terminus of L-aad was deleted and fused with the inherent His-tag from plasmid pET20b(+). After fusion with this His tag, the yield of α-ketoisovalerate from the reaction catalyzed by L-aad was increased to 65.673 mg L−1 min−1 g−1 DCW (Fig. 5d), which is 3.17-fold higher than that of wild-type L-aad.26,27 In summary, through rational molecular engineering, we constructed an efficient biocatalyst for the production of KIV.
 | ||
| Fig. 5 Effect of altered temperature, aeration and pH on whole-cell transformation. (a) Transformation curve of L-aad from P. myxofaciens in E. coli BL21(DE3) under 37 °C, 50 mL, pH 8.0 PBS; (a) effect of temperature on enzyme activity, (b) effect of reaction volume in 250 mL flask on enzyme activity, (c) effect of pH on enzyme activity. (d) The whole cell bioconversion of L-aad from P. myxofaciens and its mutants. ■: L-aad wild type; ●: L-aad-mt1: L-aad mutant F318T; ▲: L-aad-mt2: L-aad complex mutant of F318T and N100H;▼: L-aad-mt3: L-aad complex mutant of F318T, N100H and Q276E; ◆: L-aad-mt3-C6H: complex mutant of F318T, N100H and Q276E with 6 His-tag at the C-terminus. All the optimization was performed under certain reaction condition (37 °C, 25 mL, pH 7.0) with a single variable. | ||
Acknowledgements
This work is financially supported by the Fundamental Research Funds for the Central Universities (JUSRP51612A), 863 project (2015AA021004), and 111 project (111-2-06).References
- C. Chassagnole, A. Diano, F. Létisse and N. D. Lindley, J. Biotechnol., 2003, 104, 261–272 CrossRef CAS PubMed.
- M. E. Webb, A. G. Smith and C. Abell, Nat. Prod. Rep., 2004, 21, 695–721 RSC.
- H. J. V. Werner and K. Maas, J. Bacteriol., 1953, 6, 388–393 Search PubMed.
- A. J. Cooper, J. Z. Ginos and A. Meister, Chem. Rev., 1983, 83, 321–358 CrossRef CAS.
- D. Balducci, Org. Biomol. Chem., 2004, 2, 2684–2691 Search PubMed.
- L. Liu, G. S. Hossain, H. D. Shin, J. Li, G. Du and J. Chen, J. Biotechnol., 2013, 164, 97–104 CrossRef CAS PubMed.
- G. S. Hossain, J. Li, H. D. Shin, R. R. Chen, G. Du, L. Liu and J. Chen, J. Biotechnol., 2014, 169, 112–120 CrossRef CAS PubMed.
- G. S. Hossain, J. Li, H. D. Shin, L. Liu, M. Wang, G. Du and J. Chen, J. Biotechnol., 2014, 187, 71–77 CrossRef CAS PubMed.
- Y. Hou, G. S. Hossain, J. Li, H. D. Shin, L. Liu and G. Du, Appl. Microbiol. Biotechnol., 2015, 99, 8391–8402 CrossRef CAS PubMed.
- Y. Song, J. Li, H. D. Shin, G. Du, L. Liu and J. Chen, Sci. Rep., 2015, 5, 12614 CrossRef CAS PubMed.
- P. Motta, G. Molla, L. Pollegioni and M. Nardini, J. Biol. Chem., 2016 DOI:10.1074/jbc.M115.703819.
- Y. Ju, S. Tong, Y. Gao, W. Zhao, Q. Liu, Q. Gu, J. Xu, L. Niu, M. Teng and H. Zhou, J. Struct. Biol., 2016, 195, 306–315 CrossRef CAS PubMed.
- J. M. Blair, V. N. Bavro, V. Ricci, N. Modi, P. Cacciotto, U. Kleinekathӧfer, P. Ruggerone, A. V. Vargiu, A. J. Baylay and H. E. Smith, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3511–3516 CrossRef CAS PubMed.
- C. Bessler, J. Schmitt, K. H. Maurer and R. D. Schmid, Protein Sci., 2003, 12, 2141–2149 CrossRef CAS PubMed.
- R. M. Bill, P. J. Henderson, S. Iwata, E. R. Kunji, H. Michel, R. Neutze, S. Newstead, B. Poolman, C. G. Tate and H. Vogel, Nat. Biotechnol., 2011, 29, 335–340 CrossRef CAS PubMed.
- S. Wagner, M. M. Klepsch, S. Schlegel, A. Appel, R. Draheim, M. Tarry, M. Högbom, K. J. Van Wijk, D. J. Slotboom and J. O. Persson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14371–14376 CrossRef CAS PubMed.
- B. L. Nannenga and F. Baneyx, Microb. Cell Fact., 2011, 10, 1 CrossRef PubMed.
- L. Chen, Z. Chen, P. Zheng, J. Sun and A. P. Zeng, Appl. Microbiol. Biotechnol., 2013, 97, 2939–2949 CrossRef CAS PubMed.
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. da Costa Sousa, D. Gao, A. M. Cheh, U. P. Agarwal, C. M. Bianchetti, G. N. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CrossRef CAS PubMed.
- P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 737–758 RSC.
- E. Denke, T. Merbitz-Zahradnik, O. M. Hatzfeld, C. H. Snyder, T. A. Link and B. L. Trumpower, J. Biol. Chem., 1998, 273, 9085–9093 CrossRef CAS PubMed.
- A. Ciulli, G. Williams, A. G. Smith, T. L. Blundell and C. Abell, J. Med. Chem., 2006, 49, 4992–5000 CrossRef CAS PubMed.
- M. J. Harms, J. L. Schlessman and G. R. Sue, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18954–18959 CrossRef CAS PubMed.
- P. Jin, Z. Kang, N. Zhang, G. Du and J. Chen, Sci. Rep., 2014, 4 CAS.
- J. M. Dickson, W.-J. Lee, P. R. Shepherd and C. M. Buchanan, Biosci. Rep., 2013, 33, e00079 CrossRef PubMed.
- P. Ledent, C. Duez, M. Vanhove, A. Lejeune, E. Fonzé, P. Charlier, F. Rhazi-Filali, I. Thamm, G. Guillaume, B. Samyn, B. Devreese, J. Van Beeumen, J. Lamotte-Brasseur and J.-M. Frère, FEBS Lett., 1997, 413, 194–196 CrossRef CAS PubMed.
- D. Quaglia, J. A. Irwin and F. Paradisi, Mol. Biotechnol., 2012, 52, 244–250 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |