
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of a crossed-layer-structure MoS2/g-C3N4 heterojunction with enhanced photocatalytic performance†
Youzhi Cao a,
Qiao Lia and
Wei Wang*ab
a,
Qiao Lia and
Wei Wang*ab
aSchool of Chemistry and Chemical Engineering, Shihezi University, Shihezi, Xinjiang, China 832003. E-mail: wangwei_group@sina.com
bKey Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, Shihezi University, Shihezi, Xinjiang, China 832003
First published on 18th January 2017
Abstract
A novel crossed-layer-structure MoS2/g-C3N4 (graphitic carbon nitride) was synthesized by a facile method, and was characterized by a collection of analytical techniques: X-ray diffraction patterns, FT-IR spectra, SEM, TEM, and XPS. The crossed-layer-structure morphologies of MoS2/g-C3N4 could be readily tailored by adjusting the molar ratio of MoS2 to dicyandiamide; with an increase in the molar ratio from 0.08% to 4%, a morphology transformation was observed. It can be used as a photocatalyst in degrading methyl orange under simulated solar light irradiation. These MoS2/g-C3N4 samples demonstrated 2 to 3 times higher performance than pure g-C3N4. We also found that the highest phenol degradation activity was with MoS2/g-C3N4-4 under visible light. The high performance was attributed to the unique morphology that provided low recombination losses of photogenerated charges.
1. Introduction
Due to the global energy crisis and environmental pollution,1–5 various technologies have been used to cope with the crisis. Photocatalysis is considered to be one of the most promising technologies to produce renewable fuels and remove pollutants via solar energy. Photocatalytic degradation of pollutants in water by using semiconductor catalysts has received extensive and increasing attention in the past few decades, and various kinds of catalyst suitable for use in natural light have been developed.6,7The key to achieving solar pollution degradation is to develop stable, efficient, nontoxic and inexpensive photocatalysts that are capable of working in the visible light, which occupies ca. 50% of the incoming solar irradiation on earth. In the last few years, a graphite-like organic polymeric carbon nitride (g-C3N4), a metal-free photoactive semiconductor consisting of abundant and nontoxic elements, has ignited increasing interest owing to its photocatalytic activity for degradation of pollutants in water under visible-light irradiation, and its cheap, stable, environmental friendly, metal-free features. In fact, g-C3N4, one of the oldest synthetic polymers, can be traced back to 1834, and reported for the first time by Liebig and named “melon”.8 Unlike graphite, g-C3N4 is a semiconductor material with an optical band gap of 2.7 eV.9
Recently, g-C3N4 derivates with various heteroatom dopants (e.g. C, B, F, S, P, O, I),10–17 have been fabricated by co-thermal condensation of suitable additives with the precursor of g-C3N4. In comparison, post-functionalization of g-C3N4 has 40 extra advantages of introducing much more functional groups or dopants into g-C3N4 after the lattice is formed at elevated temperatures. However, the high chemical stability and bad solubility in common solvents of g-C3N4 constitute a vast barrier to post-modify g-C3N4 material.18 Great efforts have been made to enhance the photocatalytic activity of g-C3N4, such as doping,19–23 copolymerization,18,24 texturization,25,26 supermolecular assembly,27,28 surface heterojunction design.29 Highly condensed g-C3N4 with fewer defects favors enhanced the photocatalytic performance by increasing the crystallinity and charge carrier mobility.30 Therefore, it is urgent for us to obtain highly efficient g-C3N4 with enhanced crystallinity and charge carrier separation efficiency in a novel way.31 To date, various nanoarchitectural g-C3N4, ranging from one-dimensional nanorods and nanowires to two-dimensional nanosheets and three-dimensional mesoporous structures, have been designed by many methods and strategies, such as hard/soft template synthesis32–34 solvothermal/molten-salt technology,35,36 ods,37,38 and supramolecular chemistry.27,39
Transition-metal dichalcogenides (TMDs), especially the ones with atomic thickness, have been regarding as a new class of nanomaterials for fundamental studies and promising applications because of their special properties. TMDs can be metals, semiconductors, or insulators,40–42 exhibiting diverse properties. MoS2 is one of the most promising semiconductors owing to its inherent and thickness-dependent band gap as well as its abundance in nature as molybdenite.43 Moreover, bulk MoS2 crystals can be exfoliated to single- or few-layer nanosheets, which exhibit unusual physical and electronic properties. Specifically, bulk MoS2 is a semiconductor with narrow band gap of about 1.3 eV, while the isolated MoS2 monolayer possesses a large band gap of 1.8–1.9 eV.43
It is worth noting that the MoS2 nanosheets,44 as an electron acceptor, can efficiently increase the visible-light absorbance, charge separation, specific surface area and active sites for reaction, which result in an enhanced photocatalysis ability. MoS2/g-C3N4 heterojunction photocatalyst has been synthesized, using basically layered self-assembly technology and the impregnation method.45–53 In this paper, a facile method, impregnation–calcination, was applied to synthesize crossed-layer-structure MoS2/g-C3N4. The activities of as-prepared photocatalysts were evaluated by photodegradation of methyl orange (MO) under simulated solar light illumination. The as-fabricated MoS2/g-C3N4 heterojunction photocatalyst exhibited enhanced photoreactivity compared with the pure g-C3N4.
2. Experimental section
2.1 Reagents
Dicyandiamide (C2H4N4, CP), isopropanol (IPA, AR), sodium molybdate (Na2MoO4, CP), thiourea (CN2H4S, CP), disodium ethylenediaminetetraacetate dehydrate (EDTA-2Na, AR), 1,4-benzoquinone (BQ, AR), methyl orange (MO, AR), and other chemicals involved were purchased from Sinopharm Chemical Reagent Co., Ltd. (China) and used directly for experimental without further purifications. All aqueous solutions throughout were prepared with the deionized water.2.2 Catalysts preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. Then dried in an oven at 60 °C for 24 h. 1 g bulk MoS2 powder was put into a 100 mL glass bottle and ultrasonicated for 48 h, then put the suspension 4 mg mL−1.
000 rpm. Then dried in an oven at 60 °C for 24 h. 1 g bulk MoS2 powder was put into a 100 mL glass bottle and ultrasonicated for 48 h, then put the suspension 4 mg mL−1.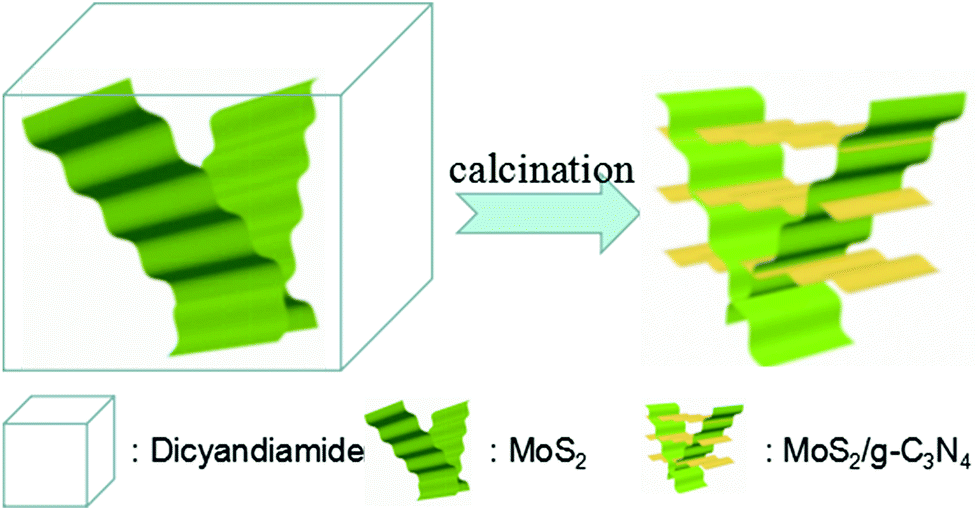 | ||
| Scheme 1 Schematic illustration for the preparation of crossed-layer-structure MoS2/g-C3N4 heterojunction photocatalyst. | ||
2.3 Catalysts characterization
X-ray diffraction (XRD) of the samples was carried out on a Bruker AXS D8 X-ray diffractometer with a Cu-Ka X-ray source operating at 40 kV and 100 mA. The morphologies of the samples were observed using a scanning electron microscope (SEM, JEOL JSM-6490LV) and a transmission electron microscope (TEM, FEI Tecnai G2). X-ray photoelectron spectroscopic (XPS) measurements were made on an Escalab 250Xi system. UV-vis diffuse reflection spectra of the as-prepared products were carried out using an Evolution 220 UV-vis spectrophotometer (Thermo Fisher) from 200 to 800 nm. Fourier transform infrared (FT-IR) spectra of the samples were measured by a Nicolet 5700 Fourier transform infrared spectrometer. The photoluminescence spectra (PL) of the samples were obtained using a fluorescence spectrometer (Hitachi F-4500) at 293 K. The photocurrent curves and electrochemical impedance spectroscopy (EIS) plots were collected by a CHI660 electrochemical analyzer.2.4 Photocatalytic experiments
The activity of the photocatalysts was evaluated via the photocatalytic degradation of MO under simulated solar light irradiation. In a typical experiment, 0.05 g photocatalyst was suspended in 100 mL 10 mg L−1 MO solution. The suspension was first dispersed by sonication for 30 min and stirred for 30 min in the dark to obtain adsorption–desorption equilibrium between the MO and the photocatalyst. The suspension was then stirred and irradiated under a 500 W Xe lamp. During the irradiation process, about 3 mL of suspension was taken from the reaction cell every 30 min and centrifuged to remove the photocatalyst. The absorbance of the MO solution in degradation was detected on TU-1900 UV-vis spectrophotometer.2.5 Electrochemical measurements
Photoelectrochemical measurements were performed in a three-electrode, single compartment quartz cell on an electrochemical station (CHI 660). Samples on ITO glass with an active area of ca. 1.0 cm2 (1 mg of photocatalyst) were prepared as the working electrode. The platinum sheet and saturated calomel electrode (SCE) were served as the counter electrode and reference electrode, respectively. Besides, a bias voltage of 0.5 V was used for driving the photo-generated electrons transfer from the working electrode to the platinum electrode. The light source (300 W xenon lamp) with ultraviolet filter (λ > 400 nm) was mounted 10.0 cm away from the photoelectrochemical cell. A 0.50 M Na2SO4 aqueous solution worked as the electrolyte.3. Results and discussion
The XRD patterns of the as-prepared pure g-C3N4, MoS2, and MoS2/g-C3N4 nanocomposites were shown in Fig. 1. These two peaks at 13.057° and 27.76° matched well with the (100) and (002) crystal planes of graphitic carbon nitride. This result demonstrate that the synthesized samples were graphitic carbon nitride. The small angle peak at 13.057°, which corresponded to a d-spacing of 0.677 nm, was due to the in-plane structural packing motif. The strong peak at 27.76° was a characteristic interlayer stacking peak and corresponded to an interlayer distance of d = 0.321 nm, as shown in Fig. 1a. Existing two diffraction peaks at 12.91° and 14.17° corresponded to g-C3N4 (100) peak and MoS2 (002) peak in MoS2/g-C3N4-50 and MoS2/g-C3N4-100, however no distinct diffraction peaks corresponding to other MoS2 co-catalysts could be observed. This might be due to the small amount of MoS2 contents, having small amount of MoS2 contents in the surface of g-C3N4 and highly dispersion in the interior of polymeric g-C3N4 photocatalysts. The presence of MoS2 in the MoS2/g-C3N4 composite samples could be confirmed by TEM, SEM and XPS analyses, as discussed later. From the inset, the (002) peak of MoS2 co-catalysts shifted to left comparing with pure g-C3N4.51 Moreover, the diffraction peaks at 14.0°, 33.4° and 58.7° were in good agreement with the (002), (100) and (110) planes of the hexagonal phase of MoS2 in Fig. 1b.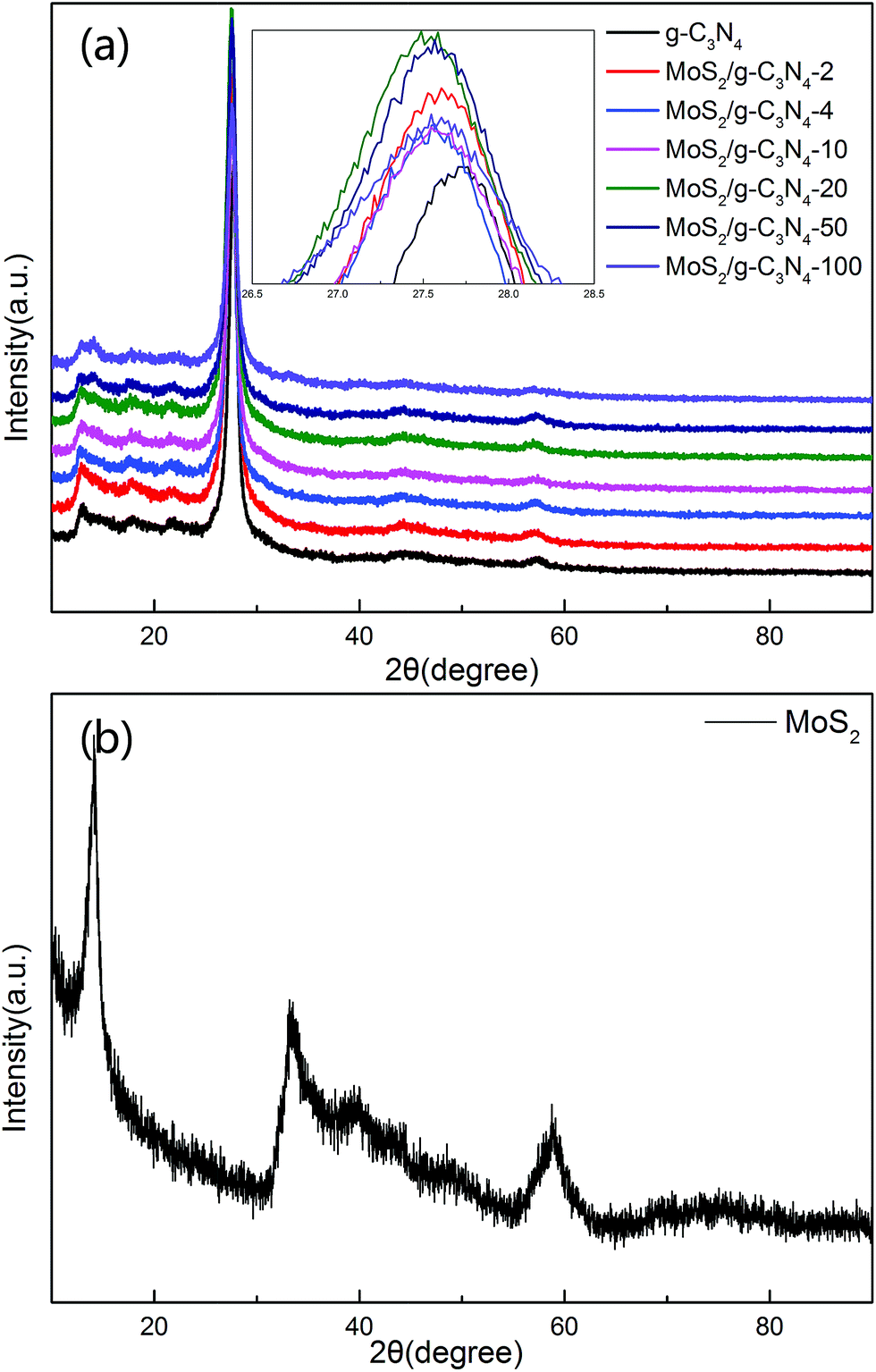 | ||
| Fig. 1 XRD patterns of different photocatalyst: (a) pure g-C3N4 and a series of MoS2/g-C3N4 (b) MoS2. | ||
The structural information of the as-developed g-C3N4 and MoS2/g-C3N4 samples were confirmed by the FT-IR analysis (Fig. 2). FT-IR spectrum showed the presence of polar oxygen-containing functionalities at 1062 cm−1 (C–O–C stretching) and 1205 cm−1 (phenolic C–OH stretching). Further observation, the characteristic peaks of MoS2/g-C3N4 were found to be almost identical to the pure g-C3N4, inferring that the impregnation of MoS2 did not destroy the in-plane tri-s-triazine units. The MoS2/g-C3N4 sample revealed almost similar characteristic features to the pure g-C3N4, verifying that the structural integrity of g-C3N4 remained intact after the incorporation with MoS2. Several strong absorption bands in the range of 1200–1650 cm−1 was originated from the skeletal stretching of C–N heterocycles with peaks positioned at 1632, 1573, 1423, 1329 and 1245 cm−1, comprising both trigonal N–(C)3 (full condensation) and bridging C–NH–C units (partial condensation). This exemplified the successful development of the extended C–N–C network. The broad peak ranging from 3000 to 3700 cm−1 was assigned to the N–H and O–H stretches due to the free amino groups and adsorbed hydroxyl species, respectively, whereas the sharp band at 812 cm−1 was derived from the breathing vibration of tri-s-triazine units.54 The virtually identical X-ray diffraction patterns (Fig. 1) and FT-IR spectra (Fig. 2) of g-C3N4 and MoS2/g-C3N4 reveal that loading with MoS2 does not change the bulk structure of g-C3N4.
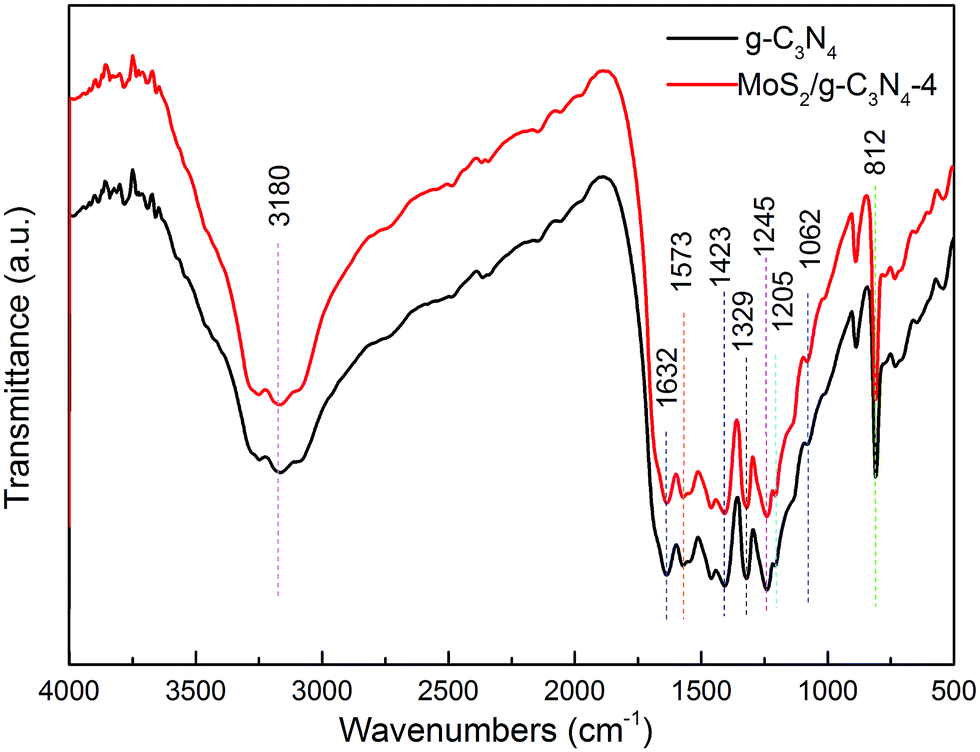 | ||
| Fig. 2 FT-IR spectra of pure g-C3N4 and MoS2/g-C3N4-4 heterojunction photocatalyst. | ||
XPS of MoS2/g-C3N4 samples were measured on an Escalab 250Xi system and shown in Fig. 3. However, the C/N atomic ratio increased from 0.77 for g-C3N4 to 0.84 for MoS2/g-C3N4-4 (Fig. 3a). There were mainly two carbon species presented in the samples (Fig. 3b): one (284.6 eV) was sp2 C–C bonds, and the other one (288.0 eV) was sp2-hybridized carbon in N-containing aromatic ring (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). The latter was considered as the major carbon species in C3N4 layer. The high resolution N1s spectra could be also deconvoluted into three different peaks at binding energies of 398.7, 400.2, 401.3 eV, respectively (Fig. 3c). These peaks could be ascribed to the sp2-hybridized nitrogen involved in triazine rings (C–NC), the tertiary nitrogen N–(C)3 groups, and the free amino groups (C–N–H), respectively. A weak O1s peak at 532.1 eV in the MoS2/g-C3N4-4 was attributed to the adsorbed H2O or CO2, which was a common phenomenon found in literatures (Fig. 3d).55 For Mo3d spectra, two peaks, accredited to the doublet Mo3d5/2 and Mo3d3/2, were located at 229.4 and 232.5 eV (Fig. 3e). The other peak, corresponding to the S2p3/2 orbital of divalent sulfide ions (S2−), was observed at 162.3 eV (Fig. 3f). Those results indicate the existence of Mo4+ and S2−, with an atomic composition ratio for Mo and S of 1:2. Of course, existing two peaks in the Fig. 5C corresponding to S2p3/2 and S2p1/2 orbital of tetravalent sulphide ions (S4−), were observed at 167.5 and 168.6 eV.56 This is contributed to high-temperature calcination, which make divalent sulfide ions (S2−) be reduced to tetravalent sulfide ions (S4−), and produce a lot sulfide vacancy, which lead to great improvement of catalyst activity.
N). The latter was considered as the major carbon species in C3N4 layer. The high resolution N1s spectra could be also deconvoluted into three different peaks at binding energies of 398.7, 400.2, 401.3 eV, respectively (Fig. 3c). These peaks could be ascribed to the sp2-hybridized nitrogen involved in triazine rings (C–NC), the tertiary nitrogen N–(C)3 groups, and the free amino groups (C–N–H), respectively. A weak O1s peak at 532.1 eV in the MoS2/g-C3N4-4 was attributed to the adsorbed H2O or CO2, which was a common phenomenon found in literatures (Fig. 3d).55 For Mo3d spectra, two peaks, accredited to the doublet Mo3d5/2 and Mo3d3/2, were located at 229.4 and 232.5 eV (Fig. 3e). The other peak, corresponding to the S2p3/2 orbital of divalent sulfide ions (S2−), was observed at 162.3 eV (Fig. 3f). Those results indicate the existence of Mo4+ and S2−, with an atomic composition ratio for Mo and S of 1:2. Of course, existing two peaks in the Fig. 5C corresponding to S2p3/2 and S2p1/2 orbital of tetravalent sulphide ions (S4−), were observed at 167.5 and 168.6 eV.56 This is contributed to high-temperature calcination, which make divalent sulfide ions (S2−) be reduced to tetravalent sulfide ions (S4−), and produce a lot sulfide vacancy, which lead to great improvement of catalyst activity.
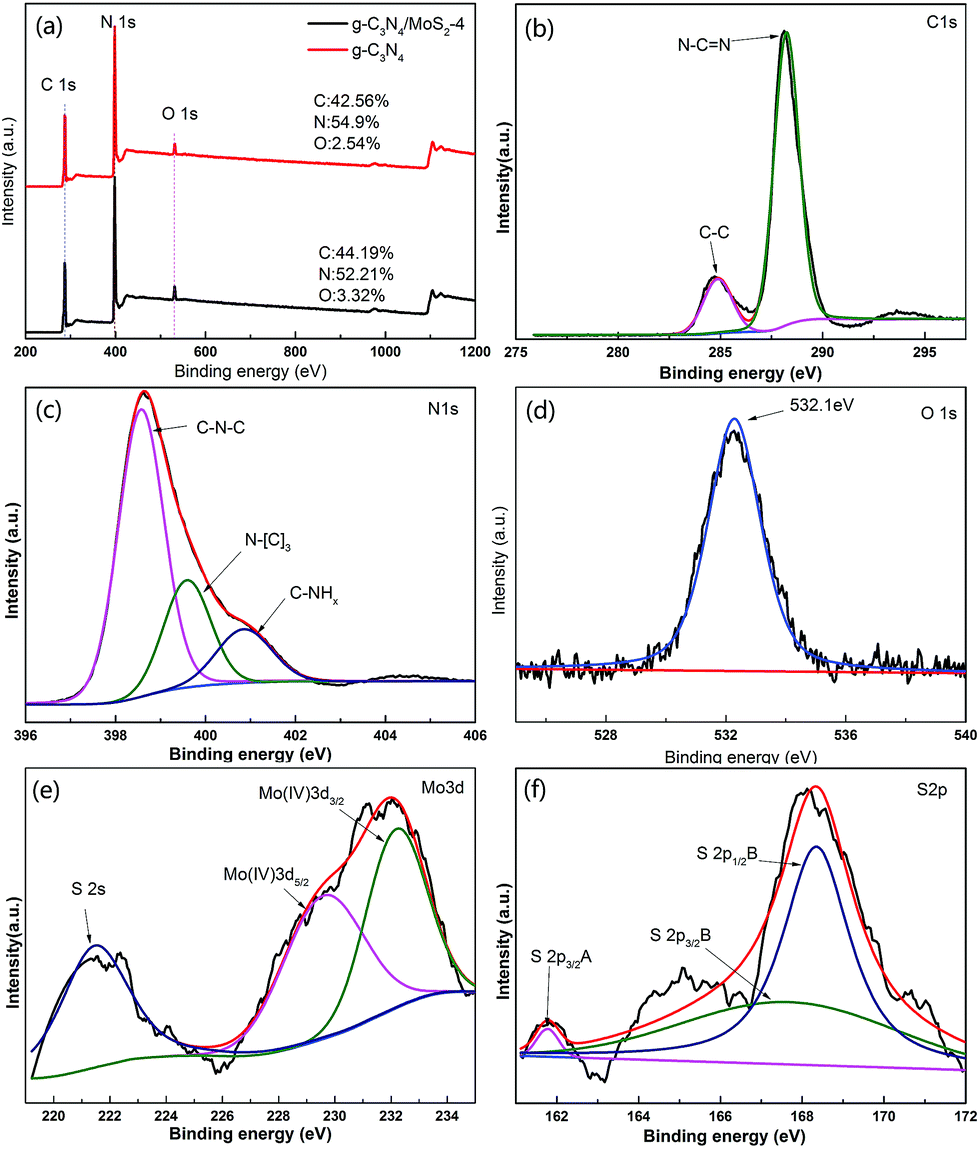 | ||
| Fig. 3 XPS spectra of as-fabricated photocatalysts: (a) survey of g-C3N4 and MoS2/g-C3N4-4 and (b) C1s; (c) N1s; (d) O1s; (e) S2p; (f) Mo3d of MoS2/g-C3N4-4. | ||
Transmission electron microscopy (TEM) image were showed in Fig. 4. Compared with pure g-C3N4 (Fig. 4c), the as-prepared MoS2/g-C3N4 samples with laminar morphology exhibited nearly transparent feature with black lines and black cycles, indicating its crossed-layer-structure (Fig. 4a and b). Transparent layer structure of MoS2 was observed (Fig. 4d), indicating that the black lines and black cycles in MoS2/g-C3N4 samples (Fig. 4a and b) were MoS2. The morphology of crossed-layer-structure MoS2/g-C3N4 was also investigated via scanning electron microscopy (SEM). As shown in Fig. 5a and b, crossed-layer-structure MoS2/g-C3N4 samples still maintained loose and irregular cross-like 3D morphology, although a great portion of g-C3N4 nanosheets was stacked. Its morphology was quite different from that of the pure g-C3N4 with a typical layer structure stacked layer by layer (Fig. 5c) and MoS2 with sheet structure (Fig. 5d). To further verify the crossed-layer-structure, EDX elements mappings were carried out and the results were shown in Fig. 6. Elements mapping of MoS2/g-C3N4-4 reveals that the sample mainly contains five elements (C, N, O, S and Mo), which is coincident with the results from XPS (Fig. 3). Of course, the results also confirmed further the existence of the crossed-layer-structure.
 | ||
| Fig. 4 Typical TEM images of (a) MoS2/g-C3N4-4, (b) MoS2/g-C3N4-100, (c) g-C3N4, (d) MoS2 samples. | ||
 | ||
| Fig. 5 Typical SEM images of (a) MoS2/g-C3N4-4, (b) MoS2/g-C3N4-100, (c) pure g-C3N4 and (d) MoS2 samples. | ||
 | ||
| Fig. 6 SEM images and corresponding mapping images of MoS2/g-C3N4-4. | ||
The BET surface areas and porous structures of the photocatalysts were investigated by nitrogen adsorption–desorption. Fig. 7a and b showed the nitrogen adsorption–desorption isotherms and the corresponding pore-size distribution curves of pure g-C3N4 and MoS2/g-C3N4 heterojunction catalysts (MoS2/g-C3N4-2, MoS2/g-C3N4-4 and MoS2/g-C3N4-50). It was obvious that the nitrogen adsorption–desorption isotherms for pure g-C3N4 and MoS2/g-C3N4 heterojunction catalysts belonged to type IV, indicating the presence of macropores. The increase of the surface areas was accompanied by the increase of the pore volumes, resulting from the increased size of the generated pores as suggested by the slight shift of the hysteresis onset in the corresponding nitrogen sorption isotherms to higher P/P0 as well as by the results of pore size analysis (Fig. 7a and b). Pure g-C3N4 catalyst had a Brunauer–Emmett–Teller (BET) surface area of 34.38 m2 g−1 and a pore volume of 0.077 cm3 g−1. While the BET surface areas and pore volumes of MoS2/g-C3N4 samples were less than pure g-C3N4. For MoS2/g-C3N4 samples, with the mass of MoS2 increasing, the BET surface areas and pore volume were increasing (Table 1).
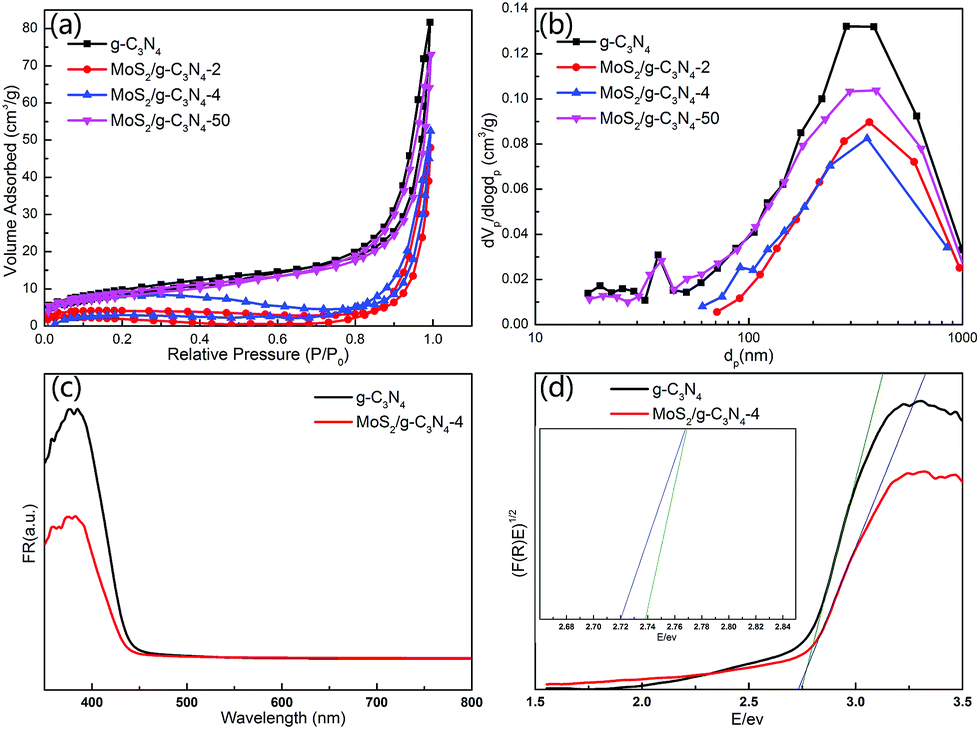 | ||
| Fig. 7 (a) N2 adsorption isotherms and (b) pore size distribution of the g-C3N4 and MoS2/g-C3N4, (c) UV-vis spectra of g-C3N4 and MoS2/g-C3N4-4 composites. (d) The plot of (F(R)E)1/2 versus energy (E) for the band gap energy of g-C3N4 and MoS2/g-C3N4-4. | ||
| Samples | g-C3N4 | MoS2/g-C3N4-2 | MoS2/g-C3N4-4 | MoS2/g-C3N4-50 |
|---|---|---|---|---|
| BET (m2 g−1) | 34.3807 | 11.3649 | 24.527 | 31.109 |
| Pore volume (cm3 g−1) | 0.0778 | 0.0368 | 0.0465 | 0.0717 |
The UV-Vis diffuse reflectance spectra (DRS) and the band gap values calculated by plots of (F(R)E)1/2 versus photo energy of g-C3N4 and MoS2/g-C3N4 were shown in Fig. 7c and d. The band gap energy of g-C3N4 in the onset is about 2.75 eV, corresponding to an absorption edge of 450 nm. After hybridization with MoS2, the band gap of MoS2/g-C3N4-4 was about 2.72 eV, with an absorption edge of 455 nm. The composite photocatalyst can improve the utilization of the visible light.
The charge carrier separation and recombination rates of the photoexcited carriers were investigated using room temperature photoluminescence spectra with an excitation wavelength of 431 nm. As shown in Fig. 8, g-C3N4 exhibited a broad emission peak that was centered at 450 nm due to the band–band PL phenomenon with the energy of light, which was approximately equal to the bandgap energy of g-C3N4. The emission peak of MoS2/g-C3N4-4 shifted to ca. 455 nm, which was consistent with DRS results. The PL intensity for MoS2/g-C3N4-4 decreased significantly compared to that of g-C3N4. In general, a decrease in the PL intensity indicates a suppressed electron–hole pair recombination, which makes MoS2/g-C3N4-4 to generate more photoelectrons and holes to participate in the photocatalytic reaction. This may be because the heterojunction formed at the interface and interior between g-C3N4 and MoS2 with a high rate of carrier mobility can restrain the recombination of photo-generated charge effectively and accelerate charge transport.
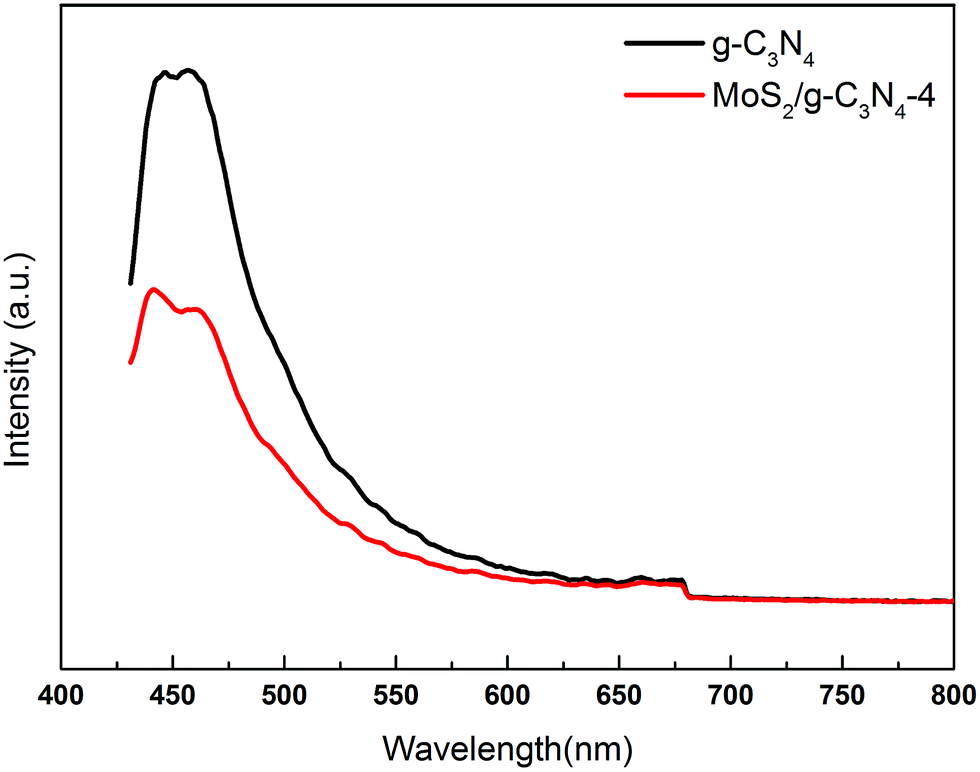 | ||
| Fig. 8 PL spectra of pure g-C3N4 and MoS2/g-C3N4-4 composites under 431 nm excitation at room temperature. | ||
To gain deeper insights into the charge transport behavior in the MoS2/g-C3N4 system, we conducted electrochemical impedance spectroscopy (EIS) measurements (Fig. 9). In the Nyquist diagram, the radius of each arc is associated with the charge-transfer process at the corresponding electrode/electrolyte interface with a smaller radius corresponding with a lower charge-transfer resistance.49,56 The MoS2/g-C3N4-4 exhibited the smallest charge transfer resistance among all g-C3N4-based electrodes under visible light irradiation, suggesting that effective shuttling of charges between the electrode and the electrolyte, and faster interfacial charge transfer occurred at the composite interface and interior owing to the formation of the crossed-layer-structure.
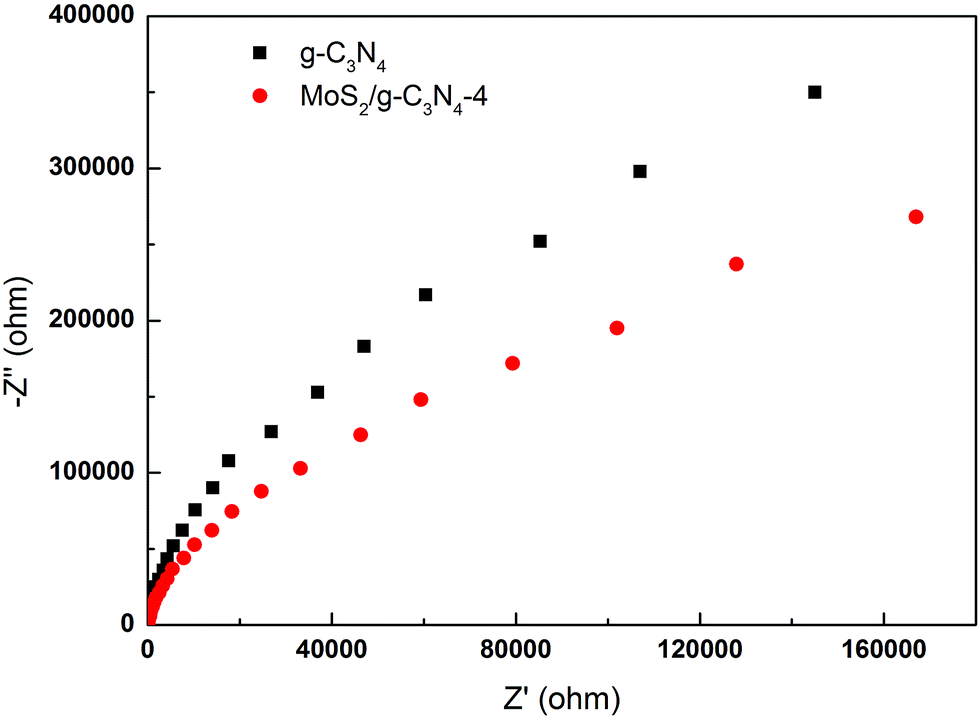 | ||
| Fig. 9 Electrochemical impedance spectroscopy of the g-C3N4 and MoS2/g-C3N4-4 samples. | ||
Semiconductors exhibit unique photoelectric properties owing to their essential band-gap structure, which provides a simple and economical light-to-electric conversion approach for various energy-related applications. Separation of electrons and holes plays a significant role in the photocatalytic reaction, and the photocurrent derives from electrons stimulated from the valence band to the conduction band under visible-light irradiation. Therefore, a higher photocurrent response indicates better separation efficiency of electrons and holes, which boosts the photocatalytic activity of the photocatalysts.57 Fig. 10 displayed the transient photocurrent response of as-fabricated product printed on indium tin oxide (ITO) glass plates in a 0.5 M Na2SO4 aqueous solution under 300 W Xe lamp light irradiation (λ > 400 nm). The transient photocurrents of the samples were measured during repeated ON/OFF illumination cycles at 0.5 V. All of the samples exhibit prompt and reproducible photocurrent responses on each illumination. When the irradiation was interrupted, the photocurrent rapidly dropped to almost zero (steady-state value), and the photocurrent reverted to the original value once light was switched back on again. The photocurrent density of the MoS2/g-C3N4-4 is about 1.93 times higher than that of the pure g-C3N4. In addition, the transient photocurrent is widely regarded as the most efficient evidence to characterize charge separation.58 The unique crossed-layer-structure can further shorten the photoelectron transport distance, thus facilitating fast electron transfer across the crossed-layer-structure. These results suggest that the enhanced light harvesting, efficient separation of photogenerated electron–hole pairs and the crossed-layer-structure of the MoS2/g-C3N4 have a significant role in improving the overall photoelectrochemical performance.
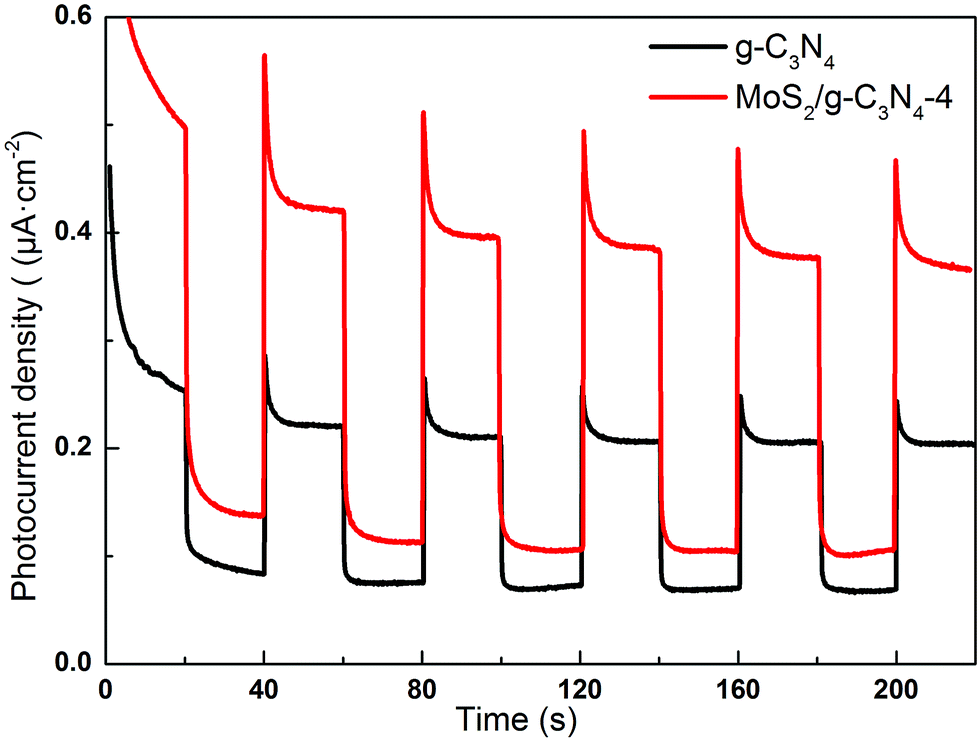 | ||
| Fig. 10 Transient photocurrent response for the g-C3N4 and MoS2/g-C3N4-4 sample. | ||
The photocatalytic activities of the as-prepared g-C3N4, and MoS2/g-C3N4 were evaluated by the degradation of MO in aqueous solution under visible light irradiation. We studied the influence of the MoS2 amount in MoS2/g-C3N4 on the photocatalytic activity under the same conditions. As shown in Fig. 11, the removal rate of MO using MoS2/g-C3N4 as the photocatalyst was higher than that of pure g-C3N4, and it improved gradually with the increase of the MoS2 amount. When the percent of MoS2 reached 0.16% (MoS2/g-C3N4-4), it exhibited the highest photocatalytic activity. However, photocatalytic activity of MoS2/g-C3N4-20 (0.8%) was significantly lower than that of MoS2/g-C3N4-4 (0.16%). The reason might be that the too many active sites of g-C3N4 were covered by MoS2, leading to weaken the absorption of visible light by g-C3N4, and causing the decrease of photocatalytic activity. Yet according to the results of BET analysis, surface areas and pore volumes were not main reason for enhancing photocatalytic degradation activity. We inferred that the enhanced activities were due to the synergistic effect between g-C3N4 and MoS2. The optimum photocatalytic degradation efficiency of MO was 68% for MoS2/g-C3N4-4 (0.16%), which is much higher than that of g-C3N4. In other words, it demonstrated 3.4 times higher performance than g-C3N4. Of course, we also tested the visible light activity for degradation of colorless pollutant (phenol) in Fig. S1.† Compared with degradation MO under stimulated solar light in Fig. 11, we observed that the tendency was consistent and MoS2/g-C3N4-4 showed optimal activity among the MoS2/g-C3N4 heterojunction catalysts. The result of photocatalytic degradation of phenol by thermal treatment g-C3N4 was added in the Fig. S2,† there were no obvious change in degradation phenol under visible light, so in the photocatalytic degradation phenol process, the thermal treatment could not promote the photocatalytic activity of g-C3N4. In summary, the high performance was attributed to the enhanced light harvesting efficient separation of photogenerated electron–hole pairs, and the crossed-layer-structure of the MoS2/g-C3N4.
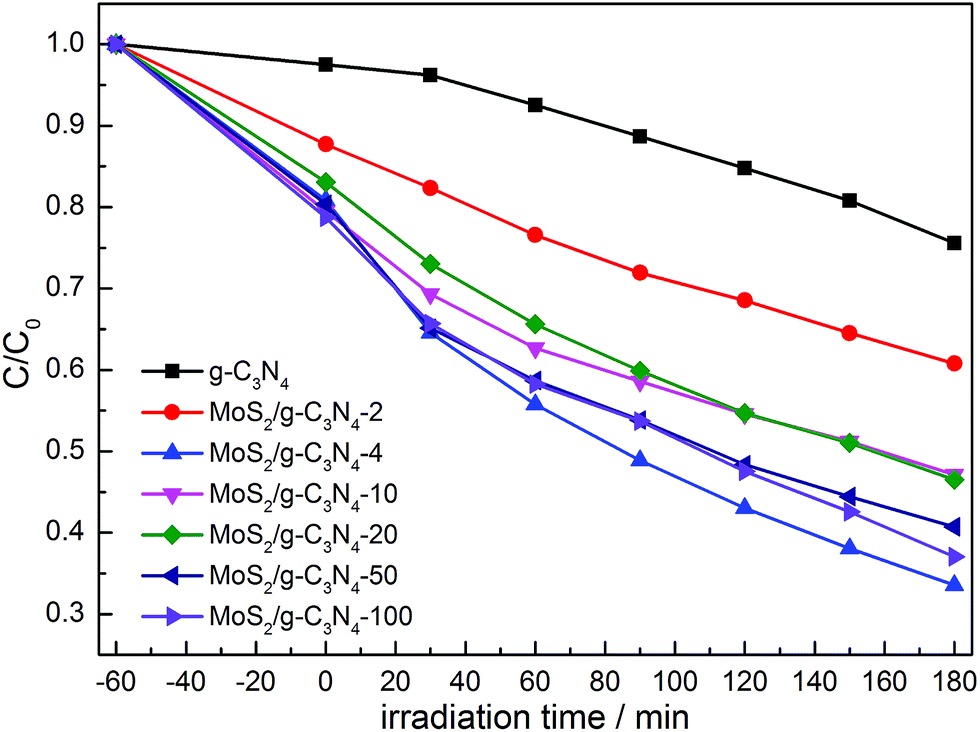 | ||
| Fig. 11 Photocatalytic degradation of MO by g-C3N4 and various MoS2/g-C3N4 samples under simulated solar light. | ||
In order to confirm the main active species for the photocatalytic degradation of MO over MoS2/g-C3N4, the scavengers BQ, disodium EDTA and IPA were added to quench the possible active species ˙O2−, hole (h+) and ˙OH, respectively.41–44 As shown in Fig. 12, when disodium EDTA was added, the removal rate of MO had no obvious decrease, implying that h+ did not dominate the photocatalytic reaction with MoS2/g-C3N4. However, the dramatic decline of MO removal was achieved in the presence of BQ and IPA, suggesting that ˙O2− and ˙OH were the main active species. According to the above results, we inferred that the enhanced activities stemmed from the synergistic effect of ˙O2− and ˙OH active species between g-C3N4 and MoS2.
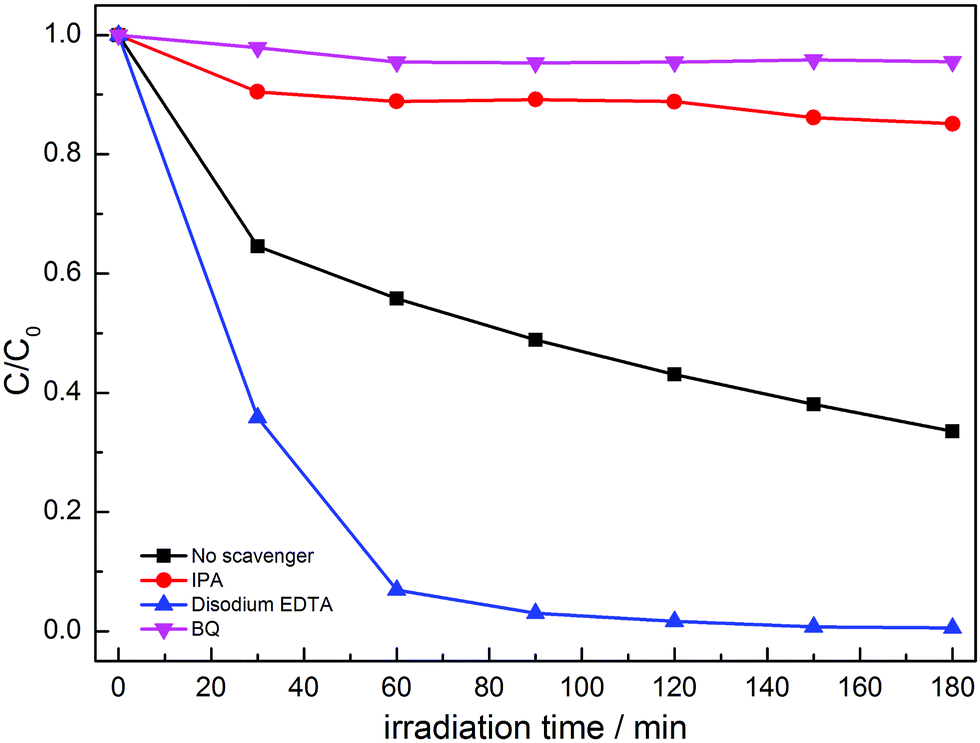 | ||
| Fig. 12 Photocatalytic performances for the degradation of MO over MoS2/g-C3N4-4 composite with different sacrificial agents under simulated solar light irradiation. | ||
The photocatalytic mechanisms are illustrated in Fig. 13. The photoexcited electrons would transfer from g-C3N4 to MoS2 due to the lower CB positions,53,58 and the separated electrons on the surface of MoS2 would combine with adsorbed O2 to produce ˙O2− radicals (the presence of ˙O2− was confirmed by previous report59). Subsequently, it will combine with H+ to produce H2O2 which finally decomposes into hydroxyl radicals.60,61 At the same time, the accumulated holes on the surface of g-C3N4 would oxidize H2O to ˙OH, then ˙OH would oxidize dye molecules to H2O/CO2. In Fig. 11, disodium EDTA, as a sacrificial agent of hole, was added to the reaction system, with the holes decreasing, meanwhile, the balance moves to the direction which can produce holes and electrons. The mentioned factor would produce a large amount of electrons and improve the separation efficiency of photogenerated electron–hole pairs. In the reaction of dye degradation, the addition of disodium EDTA made its reaction rate increasing greatly. The observed MO degradation was more than 90% in an hour. When IPA and BQ were added into the reaction system, the ˙OH and ˙O2− were captured, which led to active component of dye degradation sharply reducing and we could observe the degradation ability had fallen dramatically. Compared with pure g-C3N4, the MoS2/g-C3N4, the MoS2/g-C3N4 heterostructure had extended the lifetime of separated electron–hole pairs, resulting in the improvement of photocatalytic degradation efficiency.
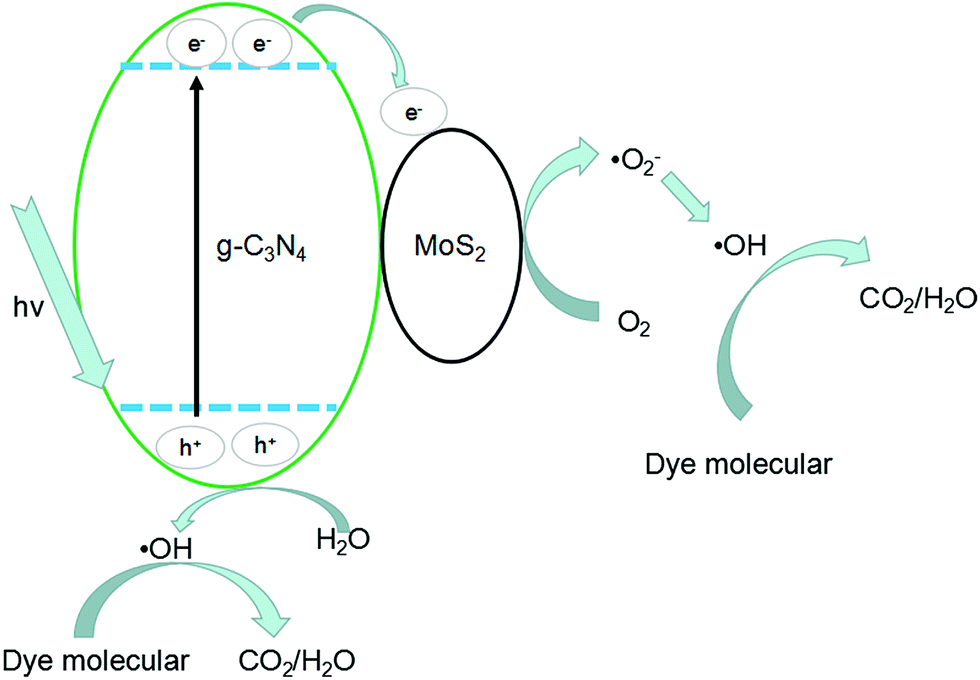 | ||
| Fig. 13 Photocatalytic mechanism of MoS2/g-C3N4 heterostructures. | ||
4. Conclusions
We have demonstrated a novel method to prepare crossed-layer-structure MoS2/g-C3N4 heterojunction photocatalyst by impregnation-calcination, which have been evidenced by TEM, SEM, XRD, XPS, IR and UV-vis DRS analysis. Under simulated solar light irradiation for 3 h, the optimum photocatalytic degradation efficiency of MO was 68% for MoS2/g-C3N4-4 (0.16%), which is much higher than that of g-C3N4. In other words, it demonstrated 3.4 times higher performance than g-C3N4. Of course, MoS2/g-C3N4-4 also showed optimal activity in phenol degradation under visible light. The high performance was attributed to the enhanced light harvesting efficient separation of photogenerated electron–hole pairs and the crossed-layer-structure of the MoS2/g-C3N4, which play a significant role in improving the overall photocatalytic performance.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant No. 21267020).Notes and references
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- P. Wang, S. Sun, X. Zhang, X. Ge and W. Lü, RSC Adv., 2016, 6, 33589–33598 RSC.
- B. Choudhury and P. K. Giri, RSC Adv., 2016, 6, 24976–24984 RSC.
- S. Le, T. Jiang, Q. Zhao, X. Liu, Y. Li, B. Fang and M. Gong, RSC Adv., 2016, 6, 38811–38819 RSC.
- J. Liebig, Ann. Pharm., 1834, 10, 1–47 CrossRef.
- J. Wen, X. Li, H. Li, S. Ma, K. He, Y. Xu, Y. Fang, W. Liu and Q. Gao, Appl. Surf. Sci., 2015, 358, 204–212 CrossRef CAS.
- H. Zhang, L. Zhao, F. Geng, L.-H. Guo, B. Wan and Y. Yang, Appl. Catal., B, 2016, 180, 656–662 CrossRef CAS.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178 RSC.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017 RSC.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- X. Du, G. Zou, Z. Wang and X. Wang, Nanoscale, 2015, 7, 8701–8706 RSC.
- Z.-F. Huang, J. Song, L. Pan, Z. Wang, X. Zhang, J.-J. Zou, W. Mi, X. Zhang and L. Wang, Nano Energy, 2015, 12, 646–656 CrossRef CAS.
- X. Ding, W. Ho, J. Shang and L. Zhang, Appl. Catal., B, 2016, 182, 316–325 CrossRef CAS.
- Z. Ni, F. Dong, H. Huang and Y. Zhang, Catal. Sci. Technol., 2016, 6, 6448–6458 CAS.
- F. Dong, Z. Zhao, Y. Sun, Y. Zhang, S. Yan and Z. Wu, Environ. Sci. Technol., 2015, 49, 12432–12440 CrossRef CAS PubMed.
- T. Xiong, W. Cen, Y. Zhang and F. Dong, ACS Catal., 2016, 6, 2462–2472 CrossRef CAS.
- D. Zheng, C. Pang, Y. Liu and X. Wang, Chem. Commun., 2015, 51, 9706–9709 RSC.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. M. Lu, Adv. Mater., 2013, 25, 998–1003 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- J. Zhu, P. Xiao, H. Li and S. A. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 1139, DOI:10.1038/ncomms2152.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., 2012, 51, 11814–11818 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Z. Lin and X. Wang, Angew. Chem., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- Y. S. Jun, J. Park, S. U. Lee, A. Thomas, W. H. Hong and G. D. Stucky, Angew. Chem., 2013, 52, 11083–11087 CrossRef CAS PubMed.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano, V. R. Cooper, L. Liang, S. G. Louie, E. Ringe, W. Zhou, S. S. Kim, R. R. Naik, B. G. Sumpter, H. Terrones, F. Xia, Y. Wang, J. Zhu, D. Akinwande, N. Alem, J. A. Schuller, R. E. Schaak, M. Terrones and J. A. Robinson, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- X. Zhang, Z. Lai, C. Tan and H. Zhang, Angew. Chem., 2016, 55, 8816–8838 CrossRef CAS PubMed.
- S. S. Chou, B. Kaehr, J. Kim, B. M. Foley, M. De, P. E. Hopkins, J. Huang, C. J. Brinker and V. P. Dravid, Angew. Chem., Int. Ed., 2013, 52, 4160–4164 CrossRef CAS PubMed.
- Q. Li, N. Zhang, Y. Yang, G. Wang and D. H. L. Ng, Langmuir, 2014, 30, 8965–8972 CrossRef CAS PubMed.
- M. Li, L. Zhang, X. Fan, M. Wu, Y. Du, M. Wang, Q. Kong, L. Zhang and J. Shi, Appl. Catal., B, 2016, 190, 36–43 CrossRef CAS.
- D. Zheng, G. Zhang, Y. Hou and X. Wang, Appl. Catal., A, 2016, 521, 2–8 CrossRef CAS.
- X. Lu, Y. Jin, X. Zhang, G. Xu, D. Wang, J. Lv, Z. Zheng and Y. Wu, Dalton Trans., 2016, 45, 15406–15414 RSC.
- Y. Hou, A. B. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., 2013, 52, 3621–3625 CrossRef CAS PubMed.
- H. Yu, P. Xiao, P. Wang and J. Yu, Appl. Catal., B, 2016, 193, 217–225 CrossRef CAS.
- J. Li, E. Liu, Y. Ma, X. Hu, J. Wan, L. Sun and J. Fan, Appl. Surf. Sci., 2016, 364, 694–702 CrossRef CAS.
- W.-C. Peng and X.-Y. Li, Catal. Commun., 2014, 49, 63–67 CrossRef CAS.
- L. Ge, C. Han, X. Xiao and L. Guo, Int. J. Hydrogen Energy, 2013, 38, 6960–6969 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nano Energy, 2015, 13, 757–770 CrossRef CAS.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- L. Ma, H. Fan, J. Wang, Y. Zhao, H. Tian and G. Dong, Appl. Catal., B, 2016, 190, 93–102 CrossRef CAS.
- J. Yan, Z. Chen, H. Ji, Z. Liu, X. Wang, Y. Xu, X. She, L. Huang, L. Xu, H. Xu and H. Li, Chemistry, 2016, 22, 4764–4773 CrossRef CAS PubMed.
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117, 9952–9961 CAS.
- Q. Li, C. Zhang, J. Ma, G. Wang and D. H. L. Ng, ChemCatChem, 2014, 6, 1392–1400 CAS.
- Q. Li, J. Bian, L. Zhang, R. Zhang, G. Wang and D. H. L. Ng, ChemPlusChem, 2014, 79, 454–461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26925g |
| This journal is © The Royal Society of Chemistry 2017 |