DOI:
10.1039/C6RA26318F
(Paper)
RSC Adv., 2017,
7, 6123-6130
Hydrolytic depolymerization of corncob lignin in the view of a bio-based rigid polyurethane foam synthesis†
Received
4th November 2016
, Accepted 20th December 2016
First published on 18th January 2017
Abstract
Corncob lignin was efficiently depolymerized in an isopropanol–water mixture with NaOH as catalyst into bio-polyols with low molecular weight and suitable hydroxyl number in view of the preparation of a bio-based rigid polyurethane foam. The effects of process parameters including the reaction temperature, NaOH concentration, isopropanol–water ratio and reaction time on the product yields were investigated. The depolymerized lignin (DL) products obtained were further analyzed by GPC, FT-IR, 2D-HSQC NMR, 31P-NMR, GC-MS and thermogravimetric analysis. The results showed that 22.1% DL with a low molecular weight (860 g mol−1) and a suitable hydroxyl number (5.10 mmol g−1) together with 5.1% solid residue was obtained under an optimum condition (at 280 °C, 5% NaOH in 25% isopropanol–water ratio for 15 min). In short, this depolymerization process would effectively produce the bio-polyols, which would have great potential as a polyol substitution for partial incorporation into rigid polyurethane foams.
1. Introduction
Lignin is the second most abundant natural polymer on the earth following cellulose and is a three dimensional high molecular weight aromatic biopolymer mainly consisting of three phenyl-propanoid monomers, guaiacyl, syringyl and p-hydroxyphenyl propane connected by a variety of C–O and C–C linkages.1 Currently, the major source of lignin is produced in the pulp and paper industry as a waste-product, and mostly used as a low value fuel to provide heat and power, resulting in the massive waste of this resource as well as pollution of the environment.2
Polyurethane (PU) is usually synthesized through a polyaddition reaction between polyols (polyether or polyester polyols) and polyisocyanate to form urethane linkages. Rigid polyurethane (RPU) foams are the most important commercial products made of PU, which have been widely utilized in the appliances and construction industry because of their unique combination of excellent thermal insulation and mechanical properties. Nowadays, petroleum derived polyols are typically used, but rising petroleum prices and sustainability concerns have focused attention towards bio-polyols.3,4 Since lignin's phenolic and alcoholic hydroxyl groups in its structure as the reactive sites, the utilization of lignin as a good source of polyols for the PU formulations has received a lot of interest.5–8 However, due to the lower reactivity and stronger steric hindrance effect of lignin, the direct utilization of lignin in place of polyol is unable to prepare satisfactory RPU foam in industrial production.9
In view of the structural characteristic of the lignin, depolymerization of the bulky lignin would produce low molecular weight and more reactive aliphatic and phenolic–OH products for further utilization in RPU foam.10 Recently, depolymerization of lignin could be carried out through different thermochemical approaches, including pyrolysis, hydrolysis, hydrogenation, oxidation reaction and so on.11–13 However, most of the reported processes had more or less side reactions, such as char formation, oligomer products repolymerization and relatively low phenolic monomers yield. Hydrolysis of lignin is a very promising method to produce a multitude of high-value products via acid- or base-catalyzed cleavage of ether bonds, which is conducted under a pressurized liquid or supercritical state. For example, hydrolytic depolymerization of lignin was performed in presence of sulfuric acid, but the main drawback is that unavoidable repolymerization of lignin intermediates products resulted in a high yield of solid residue and waste disposal problem for sulfuric acid.14 In addition, acid-catalyzed depolymerization required a special reactor to resist corrosion concerns. Lavoie et al. (2011) showed that depolymerization of the steam-treated lignin, 10% phenolic monomers and 60% oligomer could be obtained at 300–330 °C using NaOH as a catalyst, however, more than 30% of char was generated.15 Mahmood et al. (2015) reported base-catalyzed depolymerization of hydrolysis lignin at 250 °C in water–ethanol mixture with 70.5% of oligomer and 9.8% of solid residue.16 A drawback of the alkaline depolymerization of lignin was related to its relatively high yield of solid residue due to repolymerization of the reaction intermediates.17,18 The use of a hydrogen donor agent would be a promising method expected to reduce repolymerization, which was due to that the formation of aldehyde group was suppressed during hydrodeoxygenation catalyzed by the ability of the solvent to donate hydrogen, leading to the stabilization of reactive phenolic compounds and reduction of char formation.19 Kleinert et al. (2009) demonstrated that reductive depolymerization of lignin in isopropanol could form in situ hydrogen gas above 200 °C.20 Therefore, lignin depolymerization using isopropanol as a hydrogen-transfer initiator was recently investigated, suggesting that the molecular weight of phenolic compounds decreased and did not undergo severe repolymerization.21,22 As above, the alkaline catalyst could enhance cleavage of ether bond and C–C bond in the lignin and increase the yield of lignin depolymerized products, meanwhile, the isopropanol as a hydrogen donor agent could effectively prevent generating the aldehyde group, which readily lead to condensation reaction. However, limited reports were found to apply isopropanol–water mixture in the depolymerization of lignin via alkaline hydrolysis.
In this work, the primary objective is to produce the bio-polyols from corncob lignin via utilizing the DL obtained in the hydrolytic depolymerization reaction. The effects of various process factors (reaction temperature, NaOH concentrations, isopropanol/water ratio and reaction time) on the yields and molecular weight distribution of the lignin depolymerized products were comprehensively investigated. The lignin degraded products were further analyzed and characterized using gel permeation chromatography (GPC), Fourier Transform Infrared (FT-IR), gas chromatography-mass spectrometry (GC-MS), two-dimensional hetero-nuclear single-quantum correlation nuclear magnetic resonance (2D-HSQC NMR), 31P NMR and thermogravimetric analysis. The lignin degraded products are expected to have desirable characteristics (lower molecular weights and moderately higher hydroxyl number), which could be a promising feedstock for the synthesis of bio-based RPU foam.
2. Material and methods
2.1. Materials
Corncob lignin used in this study was kindly provided by Shandong LongLi Biological Technology Co., LTD, China. The general production process of corncob lignin was shown as follows: firstly, the corncob was treated hydrothermally to degrade the hemicelluloses and obtain xylo-oligosaccharides. Secondly, the corncob residue was treated with an alkaline solution to release the lignin and a cellulose-rich residue, the solid residue was used as a feedstock for bio-ethanol production; finally, the effluent was adjusted to acidic condition to precipitate the corncob lignin after the alkaline treatment. Their characteristics have been previously reported by our group.23 Sodium hydroxide (NaOH), isopropanol and acetone were of analytical grade and used as received without further purification.
2.2. Depolymerization of the lignin
The lignin depolymerization experiment was carried out in a batch reactor (100 mL internal volume, Senlong Instruments Company, Beijing, China) with a magnetic stirrer and a temperature controller. Typically in each run, the reactor was loaded with 1 g lignin and NaOH catalyst (2.5–10 wt% of the solvent) in 20 mL isopropanol–water mixture. Then the reactor was sealed and purged with N2 for three times before each experiment to remove air from the reactor. When the desired temperature achieved, the reaction time was initiated and temperature kept constant. The reactor was continuously stirred at 1000 rpm. After the reaction, the reactor was rapidly cooled to room temperature by ice water. Reaction products were completely transferred into a beaker after releasing pressure, and then the reactor was thoroughly rinsed with distilled water. The detailed flow diagram of the separation process for the reaction products is shown in Fig. S1.† The reaction mixtures were adjusted to pH 1–2 using 1.0 M H2SO4 solution to precipitate the DL products, and were separated from the liquid by filtration. The filtrate was evaporated under reduced pressure at 60 °C to remove water and volatile organic compounds (VOC). Then water-soluble oil (WSO) was obtained by oven-dried at 50 °C to constant weight. Meanwhile, the liquid phase obtained was extracted using dichloromethane (DCM), which was further dried with anhydrous sodium sulfate, filtered and finally vacuum-dried to obtain DCM-soluble fraction. The solid phase was washed with acetone and stirred for 3 h in a beaker. Then, the mixtures were filtrated, the acetone insoluble fraction was dried at 105 °C, and were weighed to obtain the yield of solid residue (SR). The acetone soluble filtrate was transferred to a pre-weighed erlenmeyer flask to remove acetone with a rotary evaporator under reduced pressure to obtain the DL. Each experiment was repeated at three times to ensure that the experimental errors were within ±5%. The yields of DL, SR and WSO fraction were calculated with formulas as following:| | |
Yield of DL (%) = Wd/WF × 100%
| (1) |
| | |
Yield of SR (%) = WS/WF × 100%
| (2) |
| | |
Yield of WSO (%) = WWSO/WF × 100%
| (3) |
WF: the weight of raw lignin; Wd: the weight of DL; WS: the weight of SR; WWSO, the weight of WSO.
2.3. Characterization of the degraded products from lignin
FT-IR spectra were recorded using a Thermo Scientific Nicolet iN10 FT-IR Microscope (Thermo Nicolet Corporation, Madison, WI) equipped with a liquid nitrogen cooled mercury cadmium telluride (MCT) detector. Samples were ground and pelletized using BaF2 and their spectra were recorded in the range from 4000 to 700 cm−1 at 4 cm−1 resolution and 128 scans per sample. The molecular weight distributions were determined by gel permeation chromatography (GPC, Agilent 1200, USA) with a refraction index detector (RID) on a PL-gel 10 μm mixed-B 7.5 mm ID column, calibrated with PL polystyrene standards. 4 mg sample was dissolved in 2 mL tetrahydrofuran (THF), and 20 μL solutions were injected. The column was operated at ambient temperature and eluted with THF at a flow rate of 1.0 mL min−1. NMR spectra were recorded on a Bruker AVIII 400 MHz spectrometer (Germany) at 25 °C according to the method described previously.24,25 For the 2D-HSQC NMR spectra analysis, 50 mg sample was dissolved in 0.5 mL DMSO-d6, and then the dissolved samples were transferred to NMR sample tubes. 31P NMR spectra were acquired after reacting lignin with 2-chloro-4,4′,5,5′-tetramethyl-1,3,2-dioxaphospholane (TMDP).26 The sample (20 mg) was dissolved in 500 μL anhydrous pyridine and deuterated chloroform (1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) under stirring. This was followed by the addition of 100 μL cyclohexanol (10.85 mg mL−1) as an internal standard (IS), and 100 μL chromium(III) acetylacetonate solution (5 mg mL−1 in anhydrous pyridine and deuterated chloroform 1.6:1, v/v) as relaxation reagent. Finally, the mixture was treated with 100 μL phosphorylating reagent (TMDP) and was transferred into a 5 mm NMR tube for analysis. The parameters used in quantitative 31P NMR experiment were listed as follows: 30° pulse angle; 2 s relaxation delay; 64 K data points, and 1024 scans. Qualitative analysis of the DCM-soluble fraction was performed by GC-MS (Agilent 7890A/5975, USA) using a 30 m × 250 μm × 0.25 μm capillary column (HP-5ms). The oven temperature was programmed as 40 °C hold 2 min, before increased to 280 °C with 5 °C min−1. The injector was at 300 °C with a spit ratio of 5:1 and injection volume was 1 μL. The flow rate of the carrier gas (helium) was 1 mL min−1. The compounds were identified by comparison with library spectra supplied by the NIST database. Thermogravimetric analysis (TGA) experiments were performed using a simultaneous thermal analyzer (SDT Q600 TGA/DSC, TA Instrument, USA). The samples weighed around 6 mg were heated from room temperature to 600 °C at a heating rate of 10 °C min−1 under an inert atmosphere of N2.
:1, v/v) under stirring. This was followed by the addition of 100 μL cyclohexanol (10.85 mg mL−1) as an internal standard (IS), and 100 μL chromium(III) acetylacetonate solution (5 mg mL−1 in anhydrous pyridine and deuterated chloroform 1.6:1, v/v) as relaxation reagent. Finally, the mixture was treated with 100 μL phosphorylating reagent (TMDP) and was transferred into a 5 mm NMR tube for analysis. The parameters used in quantitative 31P NMR experiment were listed as follows: 30° pulse angle; 2 s relaxation delay; 64 K data points, and 1024 scans. Qualitative analysis of the DCM-soluble fraction was performed by GC-MS (Agilent 7890A/5975, USA) using a 30 m × 250 μm × 0.25 μm capillary column (HP-5ms). The oven temperature was programmed as 40 °C hold 2 min, before increased to 280 °C with 5 °C min−1. The injector was at 300 °C with a spit ratio of 5:1 and injection volume was 1 μL. The flow rate of the carrier gas (helium) was 1 mL min−1. The compounds were identified by comparison with library spectra supplied by the NIST database. Thermogravimetric analysis (TGA) experiments were performed using a simultaneous thermal analyzer (SDT Q600 TGA/DSC, TA Instrument, USA). The samples weighed around 6 mg were heated from room temperature to 600 °C at a heating rate of 10 °C min−1 under an inert atmosphere of N2.
3. Result and discussion
3.1. Effect of reaction temperature on the lignin depolymerization
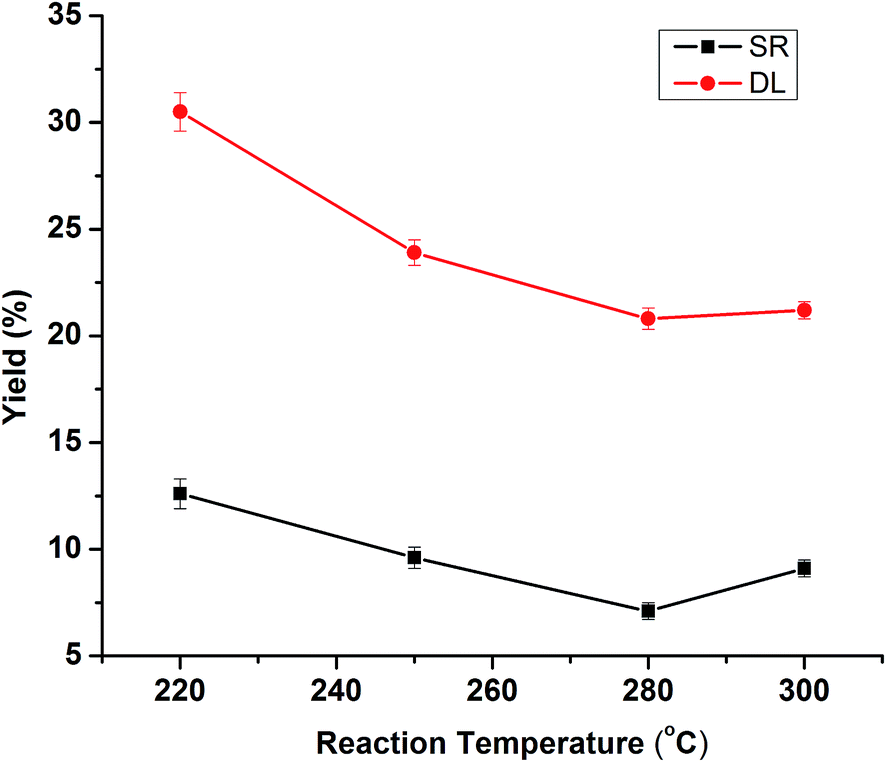
Temperature is the critical parameter for lignin depolymerization in isopropanol–water mixture. The influence of reaction temperature on the yield of DL and SR is shown in Fig. 1. The initial reaction condition at selected temperature was fixed at 50% isopropanol/water ratio with the catalyst of 5% NaOH in 30 min. The DL yields decreased sharply from 32.5% to 23.9% when the reaction temperature increased from 220 °C to 250 °C, while the DL yields decreased continually to 20.8% as the reaction temperature further increased to 280 °C. Lignin depolymerization is endothermic reaction and hence thermodynamically favorable at a higher temperature.27 The high yield of DL at low temperature of 220 °C was 32.5%, which was probably due to lower degree of lignin depolymerization. A large amount of lignin may be degraded into small fragments and dissolved in dichloromethane at higher temperature (250–280 °C), thus probably in turn led to reduced DL yields. The SR yield decreased from 12.6% to 7.1% with corresponding temperature from 220 °C to 280 °C, likely due to the formation of more aqueous products (monomers, aldehydes, alcohols, etc.) with increasing temperature. However, the SR yield increased to 9.1% as the temperature further increased to 300 °C. The relatively high yield of solid/char at higher temperature probably resulted from repolymerization/condensation of the lignin-derived intermediates to form tar/char, which was insoluble in water/acetone.14 Therefore, with respect to both SR and DL yield, the most appropriate reaction temperature was selected at 280 °C.
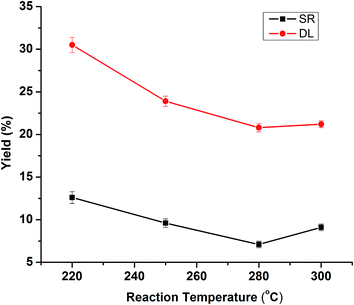 |
| | Fig. 1 Effects of reaction temperatures on the product yields of DLs and SR. (Reaction condition: 5% NaOH, 20 mL isopropanol/water (50%) and 30 min). | |
3.2. Effects of NaOH dosage on the lignin depolymerization
The effect of NaOH dosage on the lignin depolymerization was determined by various loading rates (2.5%, 5% and 10%), while keeping all other variables constant. The DL and SR yield at 280 °C, 30 min and 50% isopropanol/water ratio with various NaOH concentration are shown in Fig. 2. The high yield of DL was 24.9% at lower NaOH concentration (2.5%), accompanied with high solid yield (9.6%), the possible reason was that this reaction condition was not enough for depolymerizing lignin adequately.11 Another possibility was that the input alkali has been neutralized by the carboxylic acids produced during this depolymerization reaction, thus retarding the depolymerization process. The DL yield dropped to 20.8% with increasing loading of NaOH from 2.5% to 5%, and subsequently remained almost constant (20.8–19.7%) with NaOH concentration further increased from 7.5% to 10%. These results suggested that the effect of NaOH on the DL yield was minimal after the NaOH/lignin mass ratio was above 5%. It was worthy that the SR yield increased from 7.1% to 8.8% as the NaOH concentration increased from 5% to 10%, likely due to the repolymerization of lignin-derived intermediate products. Thus, concerning both SR and DL yield, NaOH concentration was fixed at 5%.
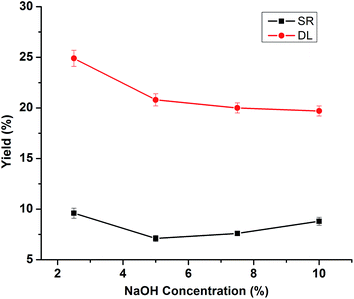 |
| | Fig. 2 Effects of NaOH dosage on the product yields of DLs and SR. (Reaction condition: 280 °C, 20 mL isopropanol/water (50%) and 30 min). | |
3.3. Effects of isopropanol/water ratio on the lignin depolymerization
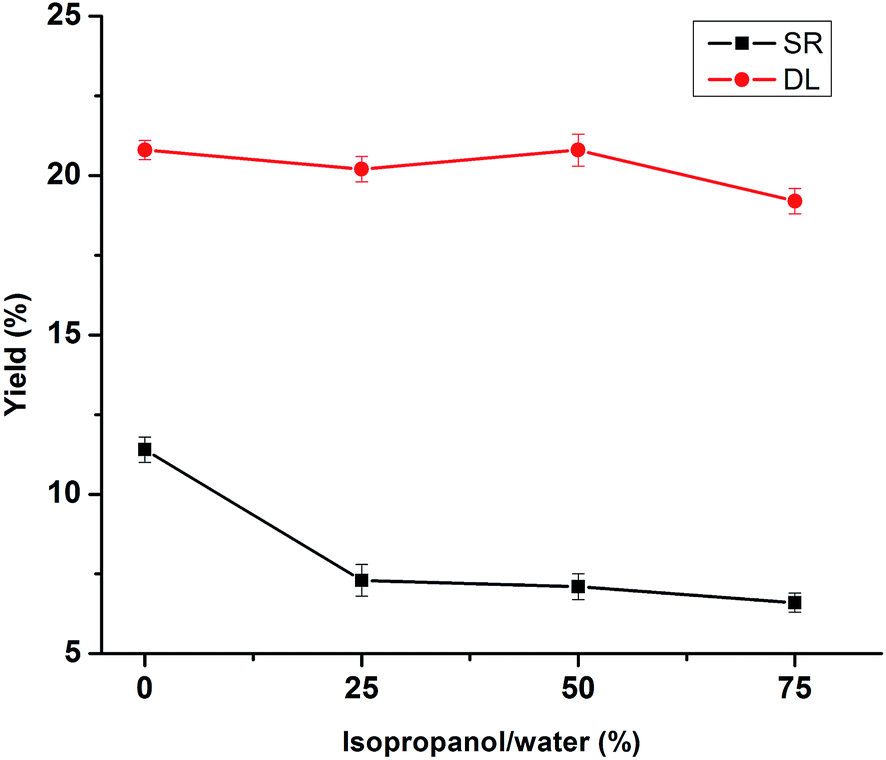
The influences of solvent medium (isopropanol/water ratio) on the depolymerization of lignin under 280 °C, 30 min at 5% NaOH are shown in Fig. 3. The solvent medium using water alone in lignin depolymerization resulted in 11.4% yield of SR. While the isopropanol–water mixture (25%, 50% and 75%) were used for lignin depolymerization, it showed a decreasing tendency for SR yield from 7.3% to 6.6% with the increase of isopropanol from 25% to 75% in this medium. This phenomenon was probably due to that the isopropanol dissolved the depolymerized lignin products and stabilize the intermediates generated during the reaction. In particular, it was noted that the DL yield remained basically unchanged (approximately 20%) regardless of isopropanol content in this medium, which was probably due to the hydrogen-donor capability of isopropanol to stabilize the intermediates generated.21 Although the SR yield dropped to 6.6% in the isopropanol–water mixture (75%), considering that the isopropanol usage, DL and SR yield, and the isopropanol–water mixture (25%) were selected for the lignin depolymerization.
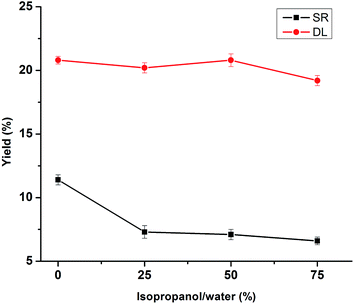 |
| | Fig. 3 Effects of isopropanol/water ratios on the product yields of DLs and SR. (Reaction condition: 280 °C, 5% NaOH and 30 min). | |
3.4. Effects of reaction time on the lignin depolymerization
The further experiments were conducted at 280 °C, 5% NaOH and isopropanol–water mixture (25%) in reaction times ranging from 5 min to 45 min. As shown in Fig. 4, the yield of SR decreased from 7.1% to 5.1% with reaction time increased from 5 min to 15 min, the corresponding DL yield decreased from 25.1% to 22.1%, likely due to the initial depolymerization effect was not sufficient to break the ether linkages of lignin. The yield of SR increased from 5.1% to 8.4% when prolong the reaction time from 15 min to 45 min, which was probably owing to the repolymerization reaction between lignin-derived intermediate products. However, further increasing the reaction time (30–45 min) had a minimal effect on the DL yield (approximately 20%), suggesting that this system might have already reached an equilibrium state. Therefore, 15 min under this condition was selected as an optimal reaction time for lignin depolymerization. Accordingly, in order to minimize the formation of SR, the best operation condition appeared at 280 °C for 15 min in the isopropanol–water mixture (25%) with 5% NaOH as a catalyst, leading to 5.1% yield of SR and 22.1% yield of DL, respectively. Additionally, the yield of WSO measured was 61.3% under this most appropriate condition.
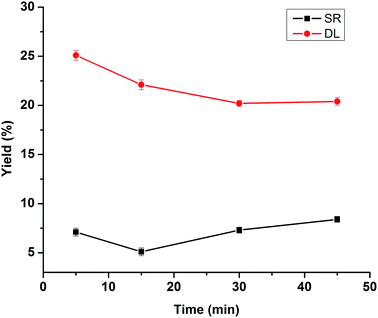 |
| | Fig. 4 Effects of reaction time on the product yields of DLs and SR. (Reaction condition: 280 °C, 5% NaOH, 20 mL isopropanol/water (25%)). | |
3.5. Characterization of original and depolymerized lignin
3.5.1 GPC analysis. Weight-average (Mw), number average (Mn) molecular weight and polydispersity (Mw/Mn) of all the DLs at different conditions are listed in Table 1. As compared with the Mw of original lignin (1970 g mol−1),23 Mw of all the DLs was obviously decreased. Mw of the DLs decreased from 1130 g mol−1 to 850 g mol−1 as the reaction temperature was increased from 220 °C to 280 °C. This was due to the sufficient energy available to break the ether bond of lignin at higher temperature. However, the increment of the DL's Mw at 300 °C was probably attributed to the repolymerization of lignin intermediate products at higher temperature. The depolymerization and repolymerization of lignin occurred simultaneously, when the concentration of NaOH increased from 2.5% to 10% at 280 °C, 0.5 h and 50% isopropanol/water ratio, the value of Mw of the lignin was firstly decreased and then kept almost constant. The possible reason was that the depolymerization was the main reaction with increasing the NaOH concentration (2.5–5.0%), thus leading to the decreased Mw of depolymerized lignin; whereas the repolymerization reaction became more serious with further increasing the NaOH concentration (5–10%). However, the repolymerization reaction was limited to a certain extent, resulting in an equilibrium state of the Mw value of repolymerized lignin. Their PDI were gradually decreased with increasing the NaOH concentration, which was mainly due to that the molecular weights distribution of depolymerized/repolymerized lignin became more narrower. As compared with the Mw of DL in water solvent (1020 g mol−1), Mw of the DLs were decreased in isopropanol–water mixtures. Moreover, the Mw of DLs was slightly decreased with increasing the isopropanol–water ratio (25–75%). The PDI kept constant in the different isopropanol–water ratios increased from 0 to 25%, which was probably due to the hydrogen-donor capability and good solubility of isopropanol to stabilize the depolymerized lignin generated, and making their PDI stable. When the isopropanol/water ratio was further increased to 50%, the depolymerization reaction was also main reaction, resulting in that more depolymerized lignin products generated and their PDI of DLs became broader. However, their PDI were obviously decreased in 75% isopropanol–water ratio, which was probably due to the change of DLs' molecular weight stable, and leading to their PDI became narrower. Furthermore, Mw of the DLs decreased from 950 g mol−1 to 860 g mol−1 as the reaction time was increased from 5 min to 15 min, which was probably due to the initial depolymerization reaction was not sufficient. Whereas the DL's Mw increased to 970 g mol−1 at 45 min, attributed to the repolymerization reaction between lignin-derived intermediates products at longer reaction time. In addition, it should also be pointed out that all the DLs exhibited relatively low PDI. These results indicated that the hydrolytic degradation in the isopropanol–water system could effectively break the bulky structure of the lignin into oligomer units. These lower molecular weight of DL products would be more suitable for producing the lignin based RPU foam. With respect to both yield and Mw of the DL products, the optimal condition was selected at 280 °C for 15 min in isopropanol–water mixture (25%) with 5% NaOH as a catalyst, producing lower Mw of DL (860 g mol−1).
Table 1 Weight-average (Mw) and number-average (Mn) molecular weights and polydispersity (Mw/Mn) of the DL samples
| Lignin (g) |
Temp (°C) |
NaOHa (g) |
Isopropanol/water (v/v) |
Time (min) |
Mw (g mol−1) |
Mn (g mol−1) |
PDIb |
| NaOH is based on the amount of the solvent substrate. Polydispersity index = (Mw/Mn). |
| 1 |
220 |
5% |
50% |
30 |
1130 |
640 |
1.78 |
| 1 |
250 |
5% |
50% |
30 |
1060 |
530 |
2.00 |
| 1 |
280 |
5% |
50% |
30 |
850 |
390 |
2.15 |
| 1 |
300 |
5% |
50% |
30 |
970 |
570 |
1.70 |
| 1 |
280 |
2.5% |
50% |
30 |
1090 |
480 |
2.27 |
| 1 |
280 |
7.5% |
50% |
30 |
910 |
560 |
1.63 |
| 1 |
280 |
10% |
50% |
30 |
900 |
650 |
1.40 |
| 1 |
280 |
5% |
0% |
30 |
1020 |
520 |
1.97 |
| 1 |
280 |
5% |
25% |
30 |
880 |
450 |
1.95 |
| 1 |
280 |
5% |
75% |
30 |
820 |
490 |
1.69 |
| 1 |
280 |
5% |
25% |
5 |
950 |
500 |
1.90 |
| 1 |
280 |
5% |
25% |
15 |
860 |
480 |
1.81 |
| 1 |
280 |
5% |
25% |
45 |
970 |
480 |
2.00 |
3.5.2 FT-IR analysis. FT-IR spectra of the original lignin and DL obtained under the optimal condition were qualitatively analyzed to investigate the structural changes during this depolymerization process. As shown in Fig. S2,† the spectra showed some common features, but also small differences that were specific to the lignin and DL. They exhibited a broad band around 3500–3200 cm−1, which is attributed to the stretching of the phenolic and aliphatic hydroxyl groups. The bands around 2952 cm−1 and 2848 cm−1 arise from CH stretching in the aromatic methoxyl groups and the methyl and methylene groups of the side chains. Additionally, the absorption of carbonyl groups is observed at 1694 cm−1.28 The intensities in 1400–1700 cm−1 attributed to aromatics of lignin and DL basically keep same, suggesting that the basic aromatic structures of lignin were not significantly damaged during this process. The ether linkage at 1037 cm−1 was observable for lignin while the spectra for DL almost showed no absorption, furthermore, the signal of the lignin at 1262 cm−1 is related to the stretching vibrations of C–O connecting to aromatic ring, which was disappeared in the DL. These results preliminarily showed that the ether bonds were cleaved to form more hydroxyl groups during this process. Accordingly, the relative intensities of hydroxyl group would be higher in the DL as compared to that of the original lignin, which would be confirmed accurately by the following 31P NMR.
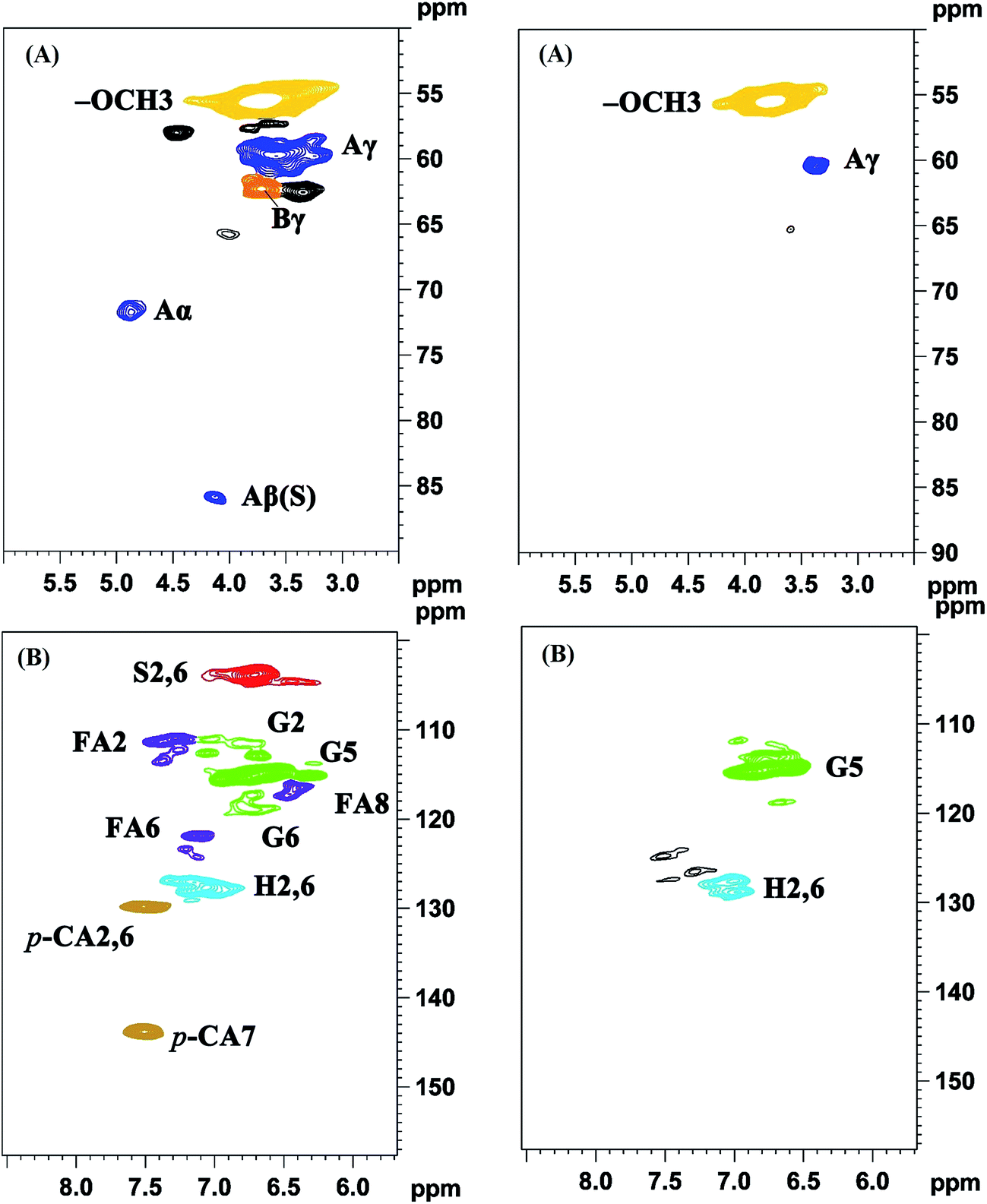
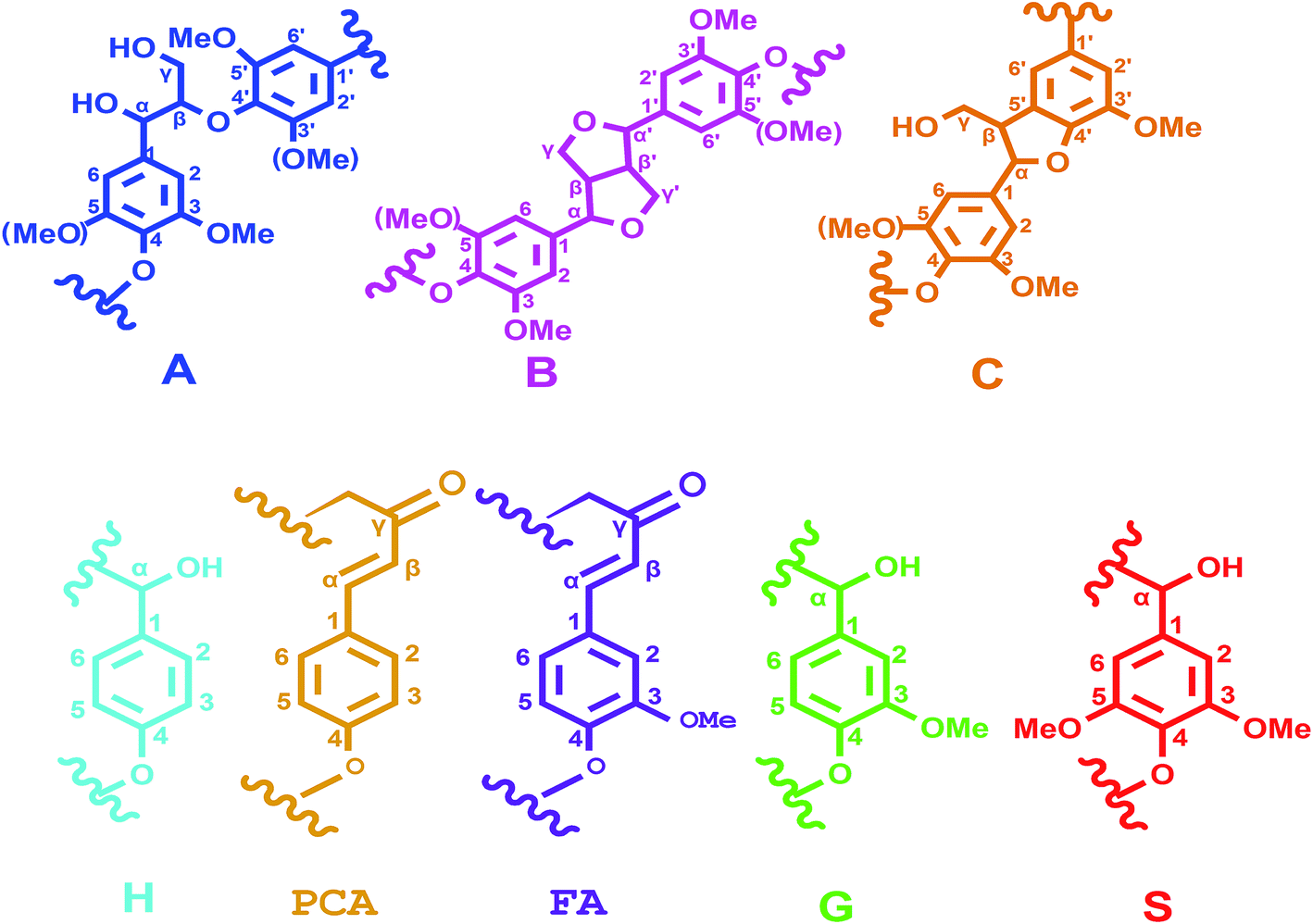
3.5.3 2D-HSQC spectra analysis. 2D-HSQC is an efficient tool to give important chemical structural information of lignin macromolecule. The structural changes of the lignin during this depolymerization process could be obtained by the comparison between original lignin and DL obtained at the optimal condition. Their side-chain region (δC/δH 50.0–90.0/2.5–6.0) and aromatic region (δC/δH 100.0–160.0/5.50–8.50) are shown in Fig. 5. The main substructures are depicted in Fig. 6. The assignments of 13C–1H correlation signals in 2D-HSQC were identified according to the previous literatures in Table S1.†24,25
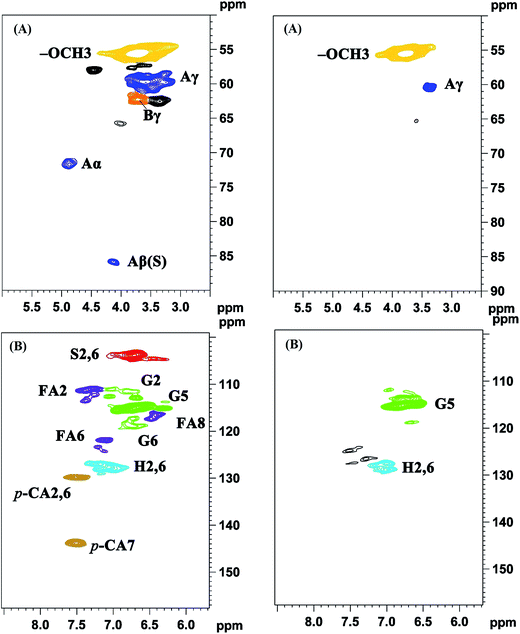 |
| | Fig. 5 2D-HSQC NMR spectra of original lignin (left) and DL sample obtained under the optimum condition (right) (A) side-chain region; (B) aromatic region. Lignin correlations are labeled with color-coded structures as given in Fig. 6. | |
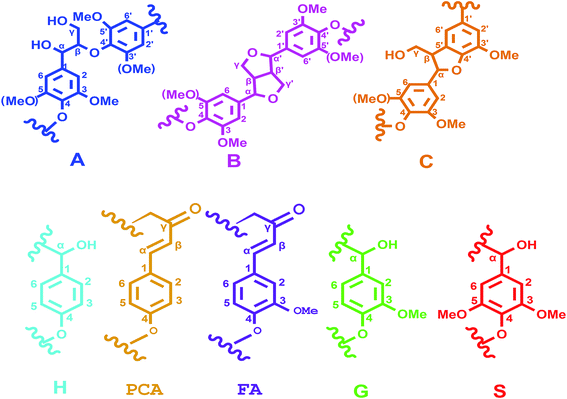 |
| | Fig. 6 Main classical substructures, involving different side-chain linkages, and aromatic units identified by 2D-HSQC NMR: (A) β-O-4′ aryl ether linkages; (B) phenylcoumarane substructures (β-5 and α-O-4 linkages); (p-CA) p-coumarates; (FA) ferulates; (H) p-hydroxyphenyl units; (G) guaiacyl units; (S) syringyl units. | |
In the side-chain region of original lignin (Fig. 5A), the prominent cross-peaks in the spectra were methoxy groups (–OCH3, δC/δH 55.7/3.70) and β-O-4′ substructures (A). The Cα–Hα correlations in β-O-4′ substructures were observed at δC/δH 71.8/4.86, whereas the cross-peaks of Cβ–Hβ in β-O-4′ substructures linked to S units were at δC/δH 85.8/4.12. Meanwhile, the obvious signal at δC/δH 59.5/3.68 is attributed to the Cγ–Hγ correlations in β-O-4′ substructures. Besides β-O-4′ substructure, the Cγ–Hγ correlations for phenylcoumaran (α-O-4′/β-5′) substructures (B) were found at δC/δH 62.3/3.70. As compared with the spectra of the original lignin, the main signals corresponding to the β-O-4′ substructures were less remained in the spectra of DL fraction. These results were agreement with previously published literatures that the β-O-4′ aryl ether linkages in lignin are readily broken at temperatures from 200 °C to 300 °C under different solvents and catalysts.14,29 Thus, the cleavage of aryl ether linkages in the original lignin was the main reaction during this depolymerization process, which could produce more free phenolic hydroxyl groups from the lignin in favor of synthesizing the bio-based RPU foam.
In the aromatic region of the original lignin (Fig. 5B), the cross-signals from guaiacyl (G), syringyl (S) and p-hydroxyphenyl (H) were obviously observed. S type lignin units showed a prominent signal for C2,6–H2,6 correlations at δC/δH 103.8/6.70. By contrast, G type lignin units exhibited different correlations for C2–H2, C5–H5 and C6–H6 at δC/δH 110.8/6.97, 114.5/6.70 and 119.0/6.78, respectively. The C2,6–H2,6 correlations of the H type lignin units were observed at δC/δH 127.2/7.12. Furthermore, the C2,6–H2,6 and C7–H7 correlations of p-coumarates (p-CA) substructure were located at δC/δH 130.2/7.48 and δC/δH 144.8/7.51, respectively. In addition, the correlations of ferulates (FA) substructure were at δC/δH 110.8/7.35 (C2–H2), 122.1/7.20 (C6–H6) and 116.5/6.36 (C8–H8). As compared with the original lignin, the signal of C2,6–H2,6 in the S unit was disappeared in the DL fraction. These results suggested that the S type units of the original lignin were probably transformed into G and H units through demethoxylation reaction and some G unit also may be changed to H units.23 The capacity of lignin or lignin fragments as a substitute for polyols in synthesizing lignin based RPU foam is related to their number of reactive sites. More G and H units revealed more active sites (unsubstituted C-3 and C-5 positions in aromatic rings) in the DL fraction, implying more suitability for the preparation of bio-based RPU foam.
3.5.4 31P NMR spectra analysis. Hydroxyl value of the lignin is a critical factor for lignin based RPU foam applications. Quantitative 31P NMR analysis was conducted to measure the aliphatic/phenolic hydroxyl groups. 31P NMR spectra of the DL obtained at the best operating condition are shown in Fig. S3.† The hydroxyl groups of lignin can be categorized in three groups: aliphatic hydroxyl units (149.0–146.0 ppm), phenolic hydroxyl units (144.5–137.1 ppm) and carboxylic groups (135.5–134.2 ppm).26 The calculated hydroxyl values for the DL sample are summarized in Table S2.† As compared with our previous studies, the raw lignin has aliphatic hydroxyl (1.58 mmol g−1), phenolic hydroxyl (1.79 mmol g−1) and carboxylic groups (0.81 mmol g−1),23 the DL obtained in this study has an aliphatic hydroxyl number of 0.12 mmol g−1, phenolic hydroxyl number of 4.38 mmol g−1, carboxylic groups 0.59 mmol g−1. As compared with the original lignin, the increase of phenolic hydroxyl groups probably caused by hydrolysis reaction of methoxy group in lignin30 and the cleavage of β-O-4′ bonds as shown in the aforementioned 2D-HSQC analysis. While the decrease of aliphatic hydroxyl groups was probably due to the cleavage of aliphatic propane chains, leading to produce more water soluble products as shown in the following analysis of GC-MS. Beauchet et al. (2012) reported that the oligomer-rich residuals obtained from the base-catalyzed depolymerization of Kraft lignin at 315 °C exhibited a similar hydroxyl value (5.25 mmol g−1).31 Xin et al. (2014) showed that partial depolymerization of enzymolysis lignin via mild hydrogenolysis under the catalysis of RANEY® Ni, and the depolymerized lignin displayed a hydroxyl value of 4.38 mmol g−1.32 Hence, the DL (total hydroxyl number = 5.10 mmol g−1) obtained in this study would be suitable as a substitution of polyols in the synthesis of bio-based RPU foam.
3.5.5 GC-MS analysis. Considering that the yield of gases usually formed was relatively low during this process due to the cleavage of aliphatic propane chains and removal of ring substituents (≤1 wt%).3 The main volatile products in DCM-soluble fraction could be attributed to the phenolic monomers, aldehydes, esters, ethers, and alcohols. The DCM-soluble fraction of WSO obtained under the optimal condition were identified by GC-MS. Each peak was assigned and its relative abundance is summarized in Table S3.† Due to each peak presented herein showed only relatively abundance of the compounds that could be detected by GC column among the lignin depolymerized compounds. Thus, the detectable compounds are mainly various types of esters, ethers, acids and phenolic compounds. These results were in consistent with the previous research in water–ethanol cosolvent.16
3.5.6 Thermal stability analysis. Thermo gravimetric analysis (TGA) and differential thermal analysis (DTA) were used to investigate comparatively the thermal stability of the lignin and DL obtained at the optimal reaction condition. As shown in Fig. S4,† the two samples began to decompose at 160 °C and 200 °C, respectively. With respect to the char residues in the TG curves, it was found that the char residues of the lignin and DL at 650 °C were 42% and 48%, respectively, indicating that the thermal stability of the DL was stronger than those of original lignin. Furthermore, the rate of weight loss of lignin fraction was faster than those of DL, and thermal decomposition temperature of their largest rate of weight loss in the original lignin and DL were at 343 °C and 379 °C, respectively. Therefore, these above results indicated that the DL was more thermally stable than original lignin, making it suitable for utilization as an insulation material in bio-based RPU foam.
4. Conclusion
Corncob lignin was efficiently depolymerized in isopropanol–water mixture with NaOH as catalyst into bio-polyols. The best operating condition appeared at 280 °C, 15 min and 25% isopropanol–water ratio with 5% NaOH in terms of the lignin depolymerized products (DL and SR) yield and their Mw. The DL obtained under the optimal condition displayed suitable molecular weight and hydroxyl value, making it potential to be used as polyols in view of the preparation of bio-based RPU. Further work will focus on the effect of the resulting DL content on the physical, mechanical, morphology and thermal properties of the bio-based RPU foam.
Acknowledgements
The authors wish to express their gratitude for the grants from the Doctoral Scientific Research Fund by Shaanxi University of Science & Technology (grant no. BJ15-26), Special research Fund by Shaanxi Provincial Department of Education (16JK1102), and State Key Laboratory of Pulp and Paper Engineering (201619).
References
- W. G. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef PubMed.
- X. Ouyang, X. Qiu and P. Chen, Colloids Surf., 2006, 282–283, 489–497 CrossRef.
- E. A. Borges da Silva, M. Zabkova, J. D. Araújo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef CAS.
- C. Bueno-Ferrer, E. Hablot, M. D. C. Garrigós, S. Bocchini, L. Averous and A. Jiménez, Polym. Degrad. Stab., 2012, 97, 1964–1969 CrossRef CAS.
- M. N. Belgacem and A. Gandini, Monomers, polymers and composites from renewable resources, Oxford, UK, 2008 Search PubMed.
- Y. Li and A. J. Ragauskas, J. Wood Chem. Technol., 2012, 32, 210–224 CrossRef CAS.
- B. L. Xue, J. L. Wen and R. C. Sun, ACS Sustainable Chem. Eng., 2014, 2, 1474–1480 CrossRef CAS.
- B. L. Xue, J. L. Wen, M. Q. Zhu and R. C. Sun, RSC Adv., 2014, 4, 36089–36096 RSC.
- N. Mahmood, Z. S. Yuan, J. Schmidt and C. Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- A. Arshanitsa, L. Krumina, G. Telysheva and T. Dizhbite, Ind. Crops Prod., 2016, 92, 1–12 CrossRef CAS.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Z. Yuan, S. Cheng, M. Leitch and C. Xu, Bioresour. Technol., 2010, 101, 9308–9313 CrossRef CAS PubMed.
- J. M. Lavoie, W. Bare and M. Bilodeau, Bioresour. Technol., 2011, 102, 4917–4920 CrossRef CAS PubMed.
- N. Mahmood, Z. S. Yuan, J. Schmidt and C. Xu, Bioresour. Technol., 2015, 190, 416–419 CrossRef CAS PubMed.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. B. Li and J. A. Lercher, Chem.–Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- A. Toledano, L. Serrano and J. Labidi, J. Chem. Technol. Biotechnol., 2012, 87, 1593–1599 CrossRef CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- M. Kleinert, J. R. Gasson, I. Eide, A. M. Hilmen and T. Barth, Cellul. Chem. Technol., 2009, 45, 3–12 Search PubMed.
- X. Y. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 1499–11503 CrossRef PubMed.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 1–7 CrossRef.
- S. Yang, T. Q. Yuan, M. F. Li and R. C. Sun, Int. J. Biol. Macromol., 2015, 72, 54–62 CrossRef CAS PubMed.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef CAS.
- J. L. Wen, B. L. Xue, F. Xu, R. C. Sun and A. Pinkert, Ind. Crops Prod., 2013, 42, 332–343 CrossRef CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- S. Cheng, C. Wilks, Z. Yuan, M. Leitch and C. Xu, Polym. Degrad. Stab., 2012, 97, 839–848 CrossRef CAS.
- O. Faix, Holzforschung, 1991, 45, 21–27 CrossRef CAS.
- X. M. Huang, T. I. Koranyi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- J. Barbier, N. Charon, N. Dupassieux, A. Loppinet-Serani, L. Mahé, J. Ponthus, M. Courtiade, A. Ducrozet, A. A. Quoineaud and F. Cansell, Biomass Bioenergy, 2012, 46, 479–491 CrossRef CAS.
- R. Beauchet, F. Monteil-Rivera and J. M. Lavoie, Bioresour. Technol., 2012, 121, 328–334 CrossRef CAS PubMed.
- J. N. Xin, P. Zhang, M. P. Wolcott, X. Zhang and J. W. Zhang, Bioresour. Technol., 2014, 155, 422–426 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26318f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Pan-Li Huangc,
Yong-Chang Sund,
Xin-Ping Lia and
Run-Cang Sunc
*ab,
Pan-Li Huangc,
Yong-Chang Sund,
Xin-Ping Lia and
Run-Cang Sunc