
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a novel polyester building block from pentoses by tin-containing silicates†
S. G. Elliot ac,
C. Andersenb,
S. Tolborgac,
S. Meierc,
I. Sádabaa,
A. E. Daugaardb and
E. Taarning*a
ac,
C. Andersenb,
S. Tolborgac,
S. Meierc,
I. Sádabaa,
A. E. Daugaardb and
E. Taarning*a
aHaldor Topsøe A/S, Haldor Topsøes Allé 1, 2800-Kgs. Lyngby, Denmark. E-mail: esta@topsoe.dk
bDanish Polymer Centre, Department of Chemical and Biochemical Engineering Technical University of Denmark, Sølvtoftsplads, 2800-Kgs Lyngby, Denmark
cDepartment of Chemistry, Technical University of Denmark, Kemitorvet, 2800-Kgs. Lyngby, Denmark
First published on 4th January 2017
Abstract
We report here the direct formation of the new chemical product trans-2,5-dihydroxy-3-pentenoic acid methyl ester from pentoses using tin-containing silicates as catalysts. The product is formed under alkali-free conditions in methanol at temperatures in the range 140–180 °C. The highest yields are found using Sn-Beta as the catalyst. Under optimised conditions, a yield of 33% is achieved. Purified trans-2,5-dihydroxy-3-pentenoic acid methyl ester was used for co-polymerisation studies with ethyl 6-hydroxyhexanoate using Candida antarctica lipase B as the catalyst. The co-polymerisation yields a product containing functional groups originating from trans-2,5-dihydroxy-3-pentenoic acid methyl ester in the polyester backbone. The reactivity of the incorporated olefin and hydroxyl moieties was investigated using trifluoroacetic anhydride and thiol–ene chemistry, thus illustrating the potential for functionalising the new co-polymers.
Introduction
The emergence of bio-based chemical products from sugars has been particularly successful in the area of polyester materials. Simple polyester building blocks include hydroxy acids, diacids and dialcohols, such as lactic acid, succinic acid and 1,3-propylene glycol. These building blocks can be produced from sugars by fermentation or chemocatalysis.1–3 While fermentation has been the preferred method, research aimed at transforming sugars into polyester building blocks using chemo-catalytic processing has proliferated during the last decade.4–9 These activities are driven partly by the prospect of chemo-catalysis offering cost advantages over fermentation, but also by the prospect of gaining access to chemical products that are inaccessible by fermentation. Polyester building blocks that are accessible in few steps from sugars using chemo-catalysis include 2,5-furandicarboxylic acid, ethylene glycol, adipic acid, isosorbide, lactic acid and 2-hydroxy-3-butenoic acid.2,5-Furandicarboxylic acid is accessible via acid catalysed dehydration of fructose, followed by oxidation.10–12 When 2,5-furandicarboxylic acid is polymerised with ethylene glycol, polyethylene furanoate is formed.13 This polyester has been shown to have favourable barrier properties compared to PET plastics, indicating that it may be developed as a useful material in the packaging materials segment.6 Ethylene glycol itself can be produced in various ways for instance by catalytic hydrogenation of sorbitol in yields of up to 37%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 or directly from glucose in combination with retro-aldol co-catalysts such as tungsten salts in yields of 35–75%.15,16 In a different catalytic approach using acid catalysis, sorbitol can be dehydrated to give isosorbide. This diol has been successfully polymerised with different dicarboxylic acids resulting in new types of polyester materials, some of which have already become commercial products.6,7 Adipic acid is accessible from glucose in two steps by catalytic hydrogenation of glucaric acid.17 Racemic methyl lactate can be made directly from C6 sugars in yields up to 75% using tin-containing silicates as catalysts.18 Lactic acid is an established polyester building block used in the production of poly(lactic acid) which has become widely used as a bio-based polyester material within the packaging materials segment.19 The newest and least known member of this group is methyl vinyl glycolate (MVG), which can be made directly from C6 sugars in yields up to 20% as a co-product to methyl lactate and from C4 sugars in yields up to 60%.8,20–22 Due to its chemical similarity to lactic acid, MVG can be co-polymerised with lactic acid to produce a co-polyester having pendant vinyl groups that allow for post functionalisation.23 Here, we report that pentose sugars can be converted into a new activated polyester building block, trans-2,5-dihydroxy-3-pentenoic acid methyl ester (DPM), in one step using tin-containing silicates as catalysts. We have furthermore isolated gram quantities of DPM and we show the successful co-polymerisation of DPM and ethyl 6-hydroxyhexanoate (E6-HH) using enzymatic polymerisation. The co-polymers can be functionalised by thiolation or acetylation of the olefin and hydroxyl moiety, respectively.
14 or directly from glucose in combination with retro-aldol co-catalysts such as tungsten salts in yields of 35–75%.15,16 In a different catalytic approach using acid catalysis, sorbitol can be dehydrated to give isosorbide. This diol has been successfully polymerised with different dicarboxylic acids resulting in new types of polyester materials, some of which have already become commercial products.6,7 Adipic acid is accessible from glucose in two steps by catalytic hydrogenation of glucaric acid.17 Racemic methyl lactate can be made directly from C6 sugars in yields up to 75% using tin-containing silicates as catalysts.18 Lactic acid is an established polyester building block used in the production of poly(lactic acid) which has become widely used as a bio-based polyester material within the packaging materials segment.19 The newest and least known member of this group is methyl vinyl glycolate (MVG), which can be made directly from C6 sugars in yields up to 20% as a co-product to methyl lactate and from C4 sugars in yields up to 60%.8,20–22 Due to its chemical similarity to lactic acid, MVG can be co-polymerised with lactic acid to produce a co-polyester having pendant vinyl groups that allow for post functionalisation.23 Here, we report that pentose sugars can be converted into a new activated polyester building block, trans-2,5-dihydroxy-3-pentenoic acid methyl ester (DPM), in one step using tin-containing silicates as catalysts. We have furthermore isolated gram quantities of DPM and we show the successful co-polymerisation of DPM and ethyl 6-hydroxyhexanoate (E6-HH) using enzymatic polymerisation. The co-polymers can be functionalised by thiolation or acetylation of the olefin and hydroxyl moiety, respectively.
The catalytic conversion of sugars to lactic acid derivatives using tin-containing silicates has been reported for all simple monosaccharides.20,21,24,25 The first report was related to triose sugars in water and methanol and later reports described similar chemistry occurring for hexoses, tetroses and pentoses. The product selectivity varies, depending on the length of the sugar molecule. Trioses form exclusively lactates while tetroses lead to high yields of MVG. Pentoses and hexoses form multiple products, depending amongst other factors on the presence of co-solutes in the reaction medium. Recently we have shown that trans-2,5,6-trihydroxy-3-hexenoic acid methyl ester (THM) is formed in yields of 15–18% from hexoses using tin-containing silicates as catalysts and in the absence of added co-solutes. The reaction pathway was elucidated and 3-deoxyglucosone was identified as the intermediate responsible for the formation of this product and of related lactones and 5-hydroxymethyl furfural derivatives.26
We here continue our exploration of chemo-catalytic sugar processing, taking these reports into consideration. A product pattern emerges based on the prior reports consisting of the homologous series of α-hydroxy esters: THM from hexoses, MVG from tetroses and methyl lactate from trioses. From this trend, a similar C5 product can be predicted for pentoses (Scheme 1). This product, DPM, is the activated and biocompatible ester version of trans-2,5-dihydroxy-3-pentenoic acid recently identified among trihydroxypentanoic acid derivatives formed from xylose in water.29 Here, we verify that this product, DPM, is indeed formed in yields of up to 33% from the pentoses xylose, lyxose and ribose. This finding shows that tin-containing silicates display a remarkable ability to catalyse consecutive dehydrations of sugars to afford intermediary β,γ-unsaturated α-keto-aldehydes which are converted into the β,γ-unsaturated α-hydroxy ester end products (Scheme 1).
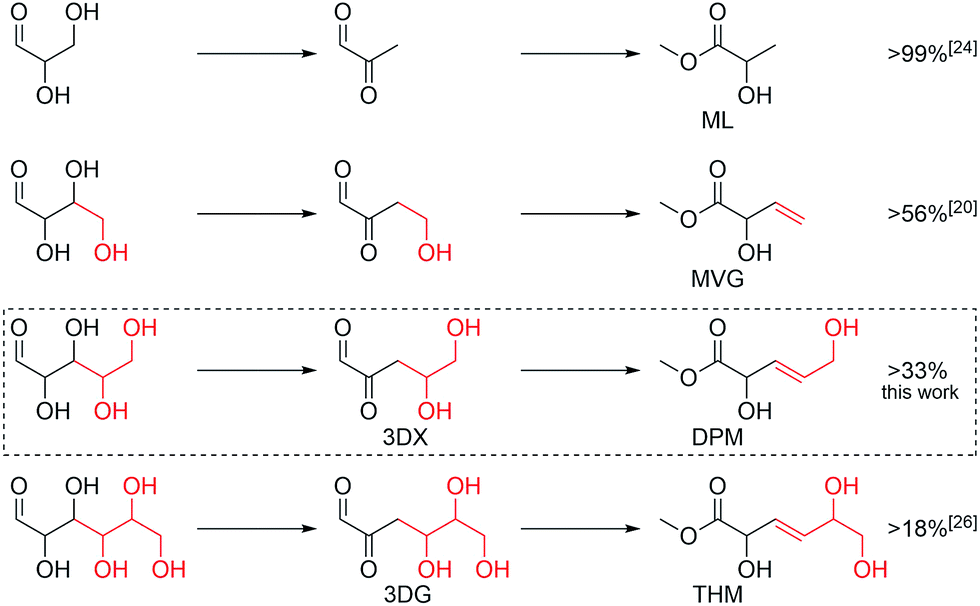 | ||
| Scheme 1 Formation of homologous α-hydroxy ester products from C3–C6 sugars catalysed by tin-containing silicates. The reaction is applicable both to aldoses and ketoses. | ||
Tin-containing silicates are solid Lewis acid materials that are capable of activating carbonyl groups in small molecules and catalyse simple transformations. Examples include Baeyer–Villiger oxidation, Meerwein–Ponndorf–Verley–Oppenauer redox reactions, monosaccharide isomerisation, aldol- and retro-aldol reactions and certain dehydration reactions.30–36 The most studied tin-containing silicate is Sn-Beta, in which tin is incorporated into a framework of silica having the *BEA topology. Sn-Beta is often reported as being a superior catalyst compared to other tin-containing silicates for these reactions.18,30,31,37,38 Currently, this supposed superiority of Sn-Beta is not well understood, especially under operando conditions. Recent work aimed at elucidating the catalytic functioning of Sn-Beta includes DFT calculations, FT-IR, TPR and 119Sn-NMR spectroscopy.39–44 Despite it being a crystalline material, the preparation method often greatly influences the catalytic performance. Two principally different preparation methods are normally used, direct synthesis under hydrothermal conditions using hydrofluoric acid as mineralising agent [Sn-Beta (HT)] and synthesis by post treatment of a dealuminated Beta zeolite with a tin source [Sn-Beta (PT)]. The Sn-Beta (HT) typically consists of large hydrophobic crystals of 3–5 μm with few defects. Tin loadings up to 2% are typically employed, as it is difficult to incorporate higher loadings of tin.45 Sn-Beta (PT) is made from a parent Al-Beta zeolite which has been dealuminated. The Sn-Beta (PT) crystal size is inherited from the parent material and is often in the range of 0.2–1 μm in diameter. The material contains many defects, causing it to be more hydrophilic than the Sn-Beta (HT). Several different methods of incorporating tin into the dealuminated zeolite are being employed such as vapour-phase deposition,46 solid state ion exchange47 and reflux in isopropyl alcohol with a tin salt.48
We synthesised Sn-Beta (HT) and Sn-Beta (PT) with a tin content of 1.25–1.5% alongside other tin-containing silicates and other Lewis acidic Beta zeolites. These materials were tested in the production of DPM from xylose in methanol with the aim of identifying activity patterns and optimising the DPM yield.
Experimental
Catalyst synthesis
Sn-, Zr-, Ti- and Al-Beta (Si/M = 150) via hydrothermal synthesis were prepared by modifying the route described by Valencia et al.45,49 In a typical Sn-Beta (Si/Sn = 150) synthesis procedure, 30.6 g of tetraethyl orthosilicate (Aldrich, 98%) was added to 33.1 g of tetraethylammonium hydroxide (Sigma-Aldrich, 35% in water) under careful stirring (∼60 min), and tin(IV) chloride pentahydrate (Aldrich, 98%) dissolved in 2.0 mL of demineralised water was added drop wise. The mixture was then left to stir for several hours. Finally, 3.1 g hydrofluoric acid (Fluka, 47–51%) in 1.6 g of demineralised water was added. All samples were then homogenised and transferred to a Teflon®-container placed in a stainless steel autoclave. The samples were then incubated at 140 °C for 14 days. The solid was recovered by filtration and washed with demineralised water, followed by drying overnight at 80 °C in air. The organic template contained within the material was removed by heating the sample at 2 °C min−1 to 550 °C in static air and maintaining this temperature for 6 h.Zr-Beta (Si/Zr = 150) and Al-Beta (Si/Al = 150) zeolites were prepared by the aforementioned procedure, exchanging the tin source with zirconyl chloride octahydrate (Sigma-Aldrich, 98%) or aluminium chloride hexahydrate (Fluka, ≥99%), respectively. Furthermore, Al-Beta was incubated for only 5 days at 140 °C. For Ti-Beta (Si/Ti = 150), tetraethyl orthotitanate (Aldrich) was first dissolved in a mixture of hydrogen peroxide and water and then used in a similar fashion as the metal source. SnO2-Beta (Si/Sn = 200) was synthesised using tin oxide (Aldrich, <100 nm) as the tin source. Purely siliceous Beta (Si-Beta) was prepared by omitting the addition of a metal source.
Sn-Beta (Si/Sn = 125) via post-treatment was prepared according to a modification of the procedure described by Hammond et al.47 Commercial zeolite Beta (Zeolyst, Si/Al = 12.5, NH4+-form) was calcined at 550 °C for 6 h and dealuminated by treatment with 10 g of concentrated nitric acid (Sigma-Aldrich, ≥65%) per gram of zeolite Beta powder for 12 h at 80 °C. The solid was recovered by filtration, washed and calcined (550 °C for 6 h). The dealuminated zeolite was then impregnated by incipient wetness methodology with a Si/Sn ratio of 125. For this purpose, tin(II) chloride (0.128 g, Sigma-Aldrich, 98%) was dissolved in water (5.75 mL) and added to 5 g of the solid. After the impregnation process, the sample was dried at 110 °C for 12 h and calcined again.
Sn-MFI (Si/Sn = 100) was prepared following a procedure described by Mal et al.50 In a typical synthesis, tin(IV) chloride pentahydrate (Aldrich, 98%) was dissolved in 5 g of demineralised water and added to 15.6 g of tetraethyl orthosilicate (98%, Aldrich). This mixture was then stirred for 30 min. Afterwards, 13.4 g of tetrapropylammonium hydroxide (40%, AppliChem) in 13.4 g of demineralised water was added and stirred for 1 h, and subsequently an additional 60 g of demineralised water was added. The solution was then left stirring for 20 h. The gel was transferred to a Teflon®-lined autoclave and synthesised at 160 °C for 2 days under static conditions. The solid was recovered by filtration, washed with ample water and dried overnight at 80 °C followed by calcination (2 °C min−1, 550 °C, 6 h dwell time) to obtain the finished material.
Sn-MCM-41 (Si/Sn = 200) was prepared according to the method described by Li et al.51 In a typical synthesis, 26.4 g of tetraethylammonium silicate (Aldrich, 15–20 wt% in water, ≥99.99%) was slowly added to a solution of 13.0 g of hexadecyltrimethylammonium bromide (Sigma, ≥99.0%) dissolved in 38.0 g of water and allowed to stir for 1 h. At this point, tin(IV) chloride pentahydrate (Aldrich, 98%) and hydrochloric acid (Sigma-Aldrich, min. 37%) in 2.1 g of water was added drop wise to the solution and allowed to stir for 1.5 h. To this solution, 12.2 g of tetraethylorthosilicate (98%, Aldrich) was added and stirred for 3 h. The sample was then transferred to a Teflon®-lined container in a stainless steel autoclave and placed in a pre-heated oven at 140 °C for 15 h. The solid was recovered by filtration, washed with ample water and dried overnight at 80 °C. The material was finalised by calcination, heating the sample to 550 °C at 2 °C min−1 in static air and maintaining this temperature for 6 h.
Sn-SBA-15 (Si/Sn = 200) was prepared following the synthesis route described by Ramaswamy et al.52 In a typical synthesis, 1.0 g of hydrochloric acid (37 wt%, Fluka) in 140 g of demineralised water was added to a solution of 8.0 g of Pluronic® P-123 (PEG–PPG–PEG polymer, Aldrich, Mw = ∼5800 g mol−1) in 60 g of demineralised water. The solution was then stirred for 2 h. To the synthesis mixture 18.0 g of tetraethyl orthosilicate (98%, Aldrich) was added followed by tin(IV) chloride pentahydrate (Aldrich, 98%) dissolved in 2.0 g of demineralised water. The mixture was then stirred for 24 h at 40 °C and then transferred to a Teflon®-lined autoclave and heated to 100 °C for 24 h. The solid was recovered by filtration, washed with ample water and then calcined at 550 °C for 6 hours.
Catalyst characterisation
Catalyst characterisation was performed by XRD, BET, N2-sorption and ICP-OES. Powder X-ray diffraction (XRD) patterns of the prepared and calcined samples were measured on an X'Pert diffractometer (Philips) using Cu-Kα radiation. Surface area and pore volume measurements were performed using multipoint N2 adsorption/desorption on an Autosorb automatic surface area and pore size analyser (Quantachrome Instruments). The total surface area (SBET) of the samples was obtained using the BET method and the micropore volume (Vmicro) was calculated by the t-plot method using the Autosorb3 software. The characterisation results are shown in the ESI, Fig. S2, S3 and Table S1.† The elemental composition of the prepared materials was measured using inductively coupled plasma-atomic emission spectroscopy (ICP-OES) on a PerkinElmer model Optima 3000 (Varian Vista).Catalytic tests
In a typical experiment, 4.0 g methanol (Sigma-Aldrich, >99.8%), 360 mg (2.4 mmol) xylose (Sigma-Aldrich, >99%) and 180 mg of catalyst were added to a 5 mL glass microwave vial (Biotage). No special precautions were taken to avoid moisture or oxygen. The reaction vessel was heated under stirring (600 rpm) for 2 h in a Biotage Initiator+ microwave synthesiser. After cooling, samples were retrieved and analysed. In some experiments, an alkali salt co-solute was added by replacing the appropriate portion of the methanol solvent with a 1.0 mM standard solution of potassium carbonate (Sigma-Aldrich, ≥99.0%) in methanol to obtain the desired concentration.Product analysis for catalyst testing
Analysis of the reaction mixtures was performed using a combination of GC-FID, HPLC and 1D and 2D NMR. A Bruker (Fällanden, Switzerland) Avance II 800 MHz spectrometer equipped with a TCI Z-gradient CryoProbe and an 18.7 T magnet (Oxford Magnet Technology, Oxford, U.K.) was used for the identification of unknown products. A 7890A Series GC system (Agilent Technologies) with a Phenomenex Zebron ZB-5 column and a FID detector was used for the quantification of glycolaldehyde dimethylacetal (GA-DMA), methyl lactate (ML), methyl vinyl glycolate (MVG), methyl 4-methoxy-2-hydroxybutanoate (MMHB) and 2,5-dihydroxy-3-pentenoic acid methyl ester (DPM). With the exception of DPM, commercial standards were used for the calibration. DPM was synthesised and purified as described under “Isolation of trans-2,5-dihydroxy-3-pentenoic acid methyl ester (DPM)” and the pure material was used in calibration standards.An Agilent 1200 series HPLC equipped with an Aminex HPX-87H (BioRad) column (0.004 M H2SO4, 0.6 mL min−1, 65 °C) using a refractive index and diode array detector was used for detection and quantification of furfural, furfural dimethylacetal and other analogues. The aqueous acidic eluent hydrolyses all furfural analogues back to furfural, resulting in a collective quantification of all furanics (FUR).
One-dimensional 1H NMR spectra were used to quantify 3-deoxy-γ-pentonolactones (DPL), 2,4,5-trihydroxy-3-pentanoic acid methyl ester (TPM) and 2,5-dihydroxy-4-methoxy-pentanoic acid methyl ester (DMPM) using the CH3 signal at 1.39 ppm from ML as a reference. Spectra were recorded directly on reaction mixtures in methanol after removal of catalyst and upon addition of 10% (v/v) D4-methanol (Cambridge Isotopes). Spectra were recorded on a Bruker Avance III spectrometer equipped with a 9.4 T magnet and a BBO probe. Methanol proton resonances were suppressed by presaturation at frequencies of 3.36 ppm, 4.786 ppm using the two logical channels of the spectrometer. Spectra were recorded at 30 °C by sampling 8096 complex data points during an acquisition time of 1.02 seconds, employing an inter-scan delay of 10 seconds and accumulating 16 scans.
Two-dimensional 1H–13C HSQC spectra were used to quantify methyl glycosides (MG) and residual substrate relative to DPM at natural 13C isotopic abundance. The 1H–13C HSQC spectra had a 13C carrier offset of 101 ppm and employed a spectral width of 22 ppm to sample the anomeric region of xylose and its methyl glycosides at high resolution and sensitivity. Samples were prepared by condensing 1 mL of the filtered reaction mixture using a SpeedVac vacuum concentrator and redissolving the resultant residue in D4-methanol. These spectra were recorded on a Bruker Avance III HD spectrometer equipped with a 9.4 T magnet and a Bruker CryoProbe Prodigy, sampling 1024 and 128 complex data points in the 1H and 13C spectral dimensions for acquisition times of 292 and 58 milliseconds, respectively. Spectra were processed with extensive zero filling in all dimensions and integrated in Topspin 3.5.
In samples where DPM or ML were present in less than 10%, estimations of DPL, TPM, DMPM, MG and residual substrate were quantified by a combination of an Agilent 1200 series HPLC equipped with a Carbohydrate (Zorbax) column (60 wt% acetonitrile/water, 0.5 mL min−1, 30 °C) and two-dimensional 1H–13C HSQC employing standard addition of xylose. 50 μL of a 100 mM stock solution in D4-methanol was added to the sample and spectra were re-recorded by the aforementioned two-dimensional 1H–13C HSQC procedure.
Isolation of trans-2,5-dihydroxy-3-pentenoic acid methyl ester
Up-scaled reactions were performed in a 1 liter autoclave (Autoclave Engineers) with mechanical stirring. The autoclave was loaded with 300 g of methanol, 18 g of xylose and 4.5 g of Sn-Beta (PT) and heated under stirring to 160 °C for 16 h. The autoclave was allowed to cool, the catalyst was then removed by filtration and the reaction mixture was dried overnight with 50 g molecular sieves. The molecular sieves were filtered from the product mixture and methanol was removed in vacuo affording a crude brown residue. trans-2,5-Dihydroxy-3-pentenoic acid methyl ester was isolated from the crude by dry column vacuum chromatography using ethyl acetate/heptane as the eluent, typically yielding 2.6 g trans-2,5-dihydroxy-3-pentenoic acid methyl ester of >94% purity (GC-MS, Fig. S4,† 1H-NMR, Fig. S5†).Enzymatic polymerisation of ethyl 6-hydroxyhexanoate and trans-2,5-dihydroxy-3-pentenoic acid methyl ester to poly(E6-HH-co-DPM)
For the co-polymerisation, 0.40 g (3 mmol) of trans-2,5-dihydroxy-3-pentenoic acid methyl ester, 2.0 g (13 mmol) of ethyl 6-hydroxyhexanoate (Sigma-Aldrich, 97%) and 0.24 g of N435 (Novozymes) was added to a Schlenk tube. The mixture was placed in an oil bath under magnetic stirring at 60 °C and 200 mbar pressure for 2 h, before being reduced to 5 mbar for another 70 h. The product was dissolved in tetrahydrofuran (Sigma-Aldrich, ≥99.9%) and immediately filtered by suction. The filtrate was evaporated in vacuo, re-dissolved in tetrahydrofuran and precipitated in cold methanol (Sigma-Aldrich, ≥99%). Refrigeration at 5 °C for 30 min followed by filtration, yielded poly(E6-HH-co-DPM) as an orange sticky solid (1.088 g, 62% yield).Trifluoroacetic anhydride esterification of poly(E6-HH-co-DPM)
In a typical esterification, 63 mg poly(E6-HH-co-DPM) was dissolved in 0.7 mL D-chloroform (Sigma-Aldrich, 99.8%) and transferred to a NMR tube and 4 drops of trifluoroacetic anhydride (Sigma-Aldrich, >99%) were added. The mixture was precipitated in cold methanol (Sigma-Aldrich, ≥99%) and refrigerated at 5 °C for 30 min. The mixture was transferred to a syringe and filtered using a Teflon® syringe filter. The filter was washed with chloroform and the liquid was collected. The solvent was evaporated under ambient conditions and the product was dried overnight in a vacuum oven at 30 mbar at room temperature, yielding the functionalised polyester (polymer II). Coupling yield was determined by 1H-NMR, based on the change in chemical shift of the protons vicinal to the alcohol group of DPM.Thiol–ene functionalisation of poly(E6-HH-co-DPM)
For the thiol–ene functionalisation, 15 mg poly(E6-HH-co-DPM), 4 drops of mercaptoacetic acid (Sigma-Aldrich, ≥98%) and 2.9 mg (0.02 mmol) 2,2-dimethoxy-2-phenylacetophenone (Sigma-Aldrich, 99%) was dissolved in 0.6 mL D-chloroform (Sigma-Aldrich, 99.8%). The mixture was transferred to a NMR tube and irradiated with UV light (λ = 365 nm) for 30 min, yielding the thiol functionalised polyester (polymer IIIc). The conversion of the double bond was determined by 1H-NMR from the signals at 5.97 and 5.79 ppm corresponding to the substitution of the double bond in poly(E6-HH-co-DPM). This functionalisation was further confirmed by detection of increased presence of carboxylic acid functionality in FT-IR at 3461 cm−1. Polymers IIIa, b, d and e were prepared by the aforementioned procedure, exchanging the derivatising agent with 1-mercaptohexane, mercaptoethanol, 2-ethylhexanethiol or thiophenol respectively, and coupling yields are given in Scheme 3.Polymer characterisation
NMR characterisation of polymers was performed on a 7 Tesla Spectrospin-Bruker AC 300 MHz spectrometer at room temperature using CDCl3 as solvent. Assignments of NMR spectra were based on homonuclear and heteronuclear correlation spectroscopy (1H–1H COSY and 1H–13C HSQC spectra). Glass transition temperatures (Tg) and melting temperatures (Tm) were obtained using a Discovery DSC from TA Instruments. Thermal analyses were performed at a heating and cooling rate of 10 °C min−1 from −100 to 150 °C. Tg and Tm were measured at the inflection point and at the peak temperature, respectively. Fourier transform infrared (FT-IR) spectroscopy was conducted on an attenuated total reflection (ATR)-FT-IR (Thermo iS50 with a built-in diamond ATR with a resolution of 4 cm−1) in order to confirm the presence of functional groups. Molecular weight and polydispersities were determined using Size Exclusion Chromatography (SEC). SEC was performed in THF on a Viscotek GPCmax autosampler equipped with a Viscotek TriSEC model 302 triple detector array (RI detector, viscometer detector and light scattering detectors measuring at 90° and 7°) and a Knauer K-2501 UV detector on two Polymer Laboratories PLgel MIXED-D columns. The samples were run at a flow rate of 1 mL min−1 at 35 °C and molecular weights were determined from a PS calibration.Film casting
In a typical procedure 0.27 g of ε-polycaprolactone (Perstop, Mn = 50000 g mol−1) and 0.030 g of poly(E6-HH-co-DPM) was dissolved in 6 mL of tetrahydrofuran. The mixture was transferred to a Teflon® mold and the solvent was allowed to evaporate in a closed vessel at room temperature to ensure a slow evaporation of the solvent. The resulting film was hotpressed at 70 °C for 10 min to ensure a uniform, flat film.
Water contact angle measurements
A water droplet of 15 μL was placed on the surface with the needle inside the drop, and the drop was expanded and contracted on the surface of the polymer film at a rate of 2.0 μL s−1. All water contact angle measurements were determined as an average of three measurements of three different drops at different positions on the polymer surface.Results and discussion
Identification of trans-2,5-dihydroxy-3-pentenoic acid methyl ester and co-products
Thorough analyses of product mixtures from reactions using xylose as the substrate and Sn-Beta (HT) as the catalyst were initially performed by GC-MS. Here it was found that in addition to the previously reported retro-aldol and dehydration products, additional products were present.9,25,26 Analysis of the product mixtures using 2D heteronuclear (1H–13C) spectroscopy, confirmed the presence of DPM alongside 3-deoxy-γ-pentonolactones (DPL), 2,4,5-trihydroxypentanoic acid methyl ester (TPM) and 2,5-dihydroxy-4-methoxy-pentanoic acid methyl ester (DMPM). These products are thus homologues of those reported for the conversion of hexoses to THM and 3-deoxygluconolactones. Following this initial work, a comprehensive analysis procedure was established, combining GC, HPLC and NMR spectroscopy to quantify the different reaction products for the conversion of pentoses (Scheme 2). This analysis procedure was used for comparing different catalysts, conditions and co-solutes and for optimising the yield of DPM.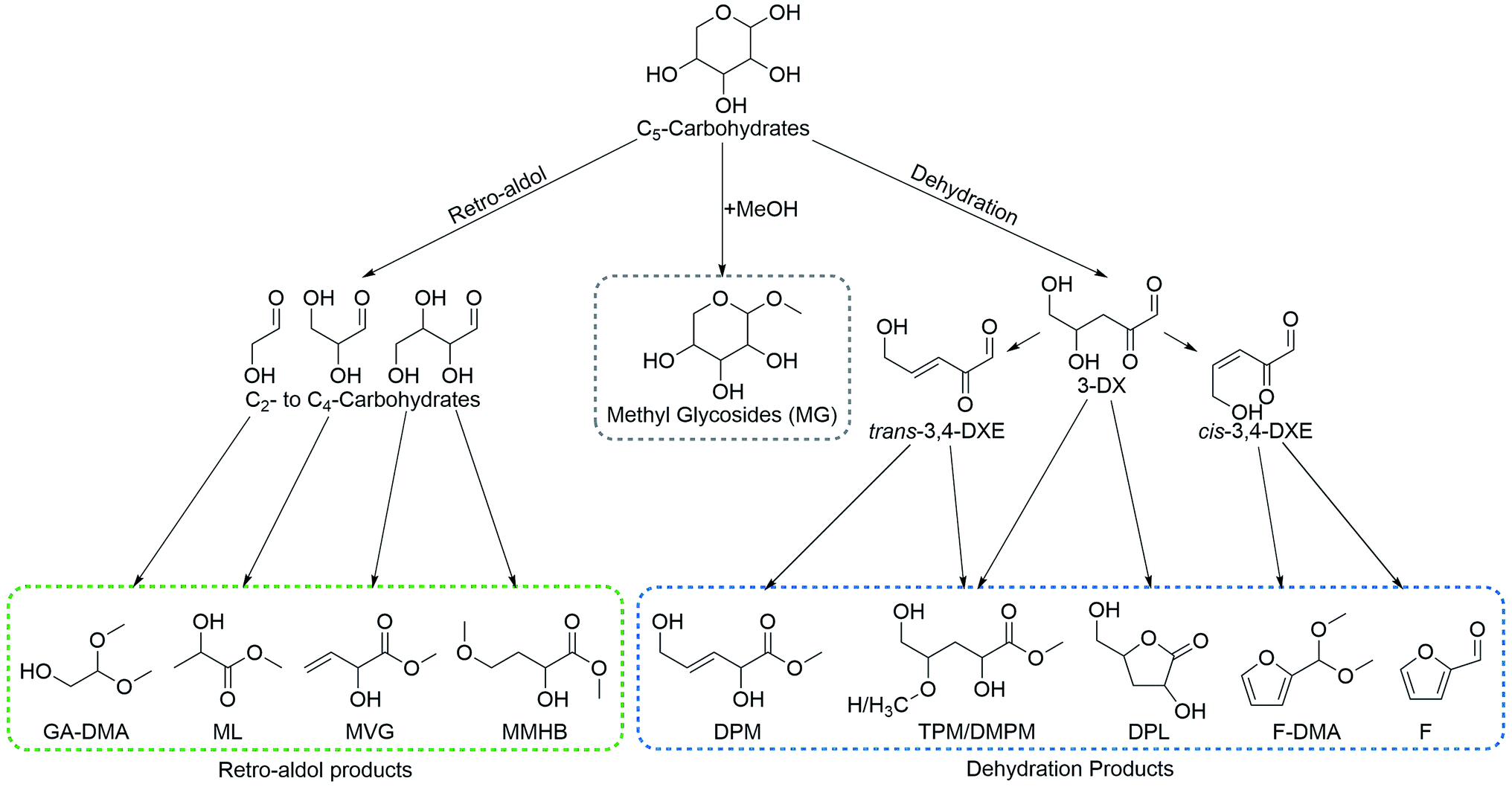 | ||
| Scheme 2 Major pathways, intermediates and products in the conversion of pentoses by Sn-Beta. Compound abbreviations: 3-deoxyxylosone (3-DX), cis-3,4-dideoxyxylos-3-enone (cis-3,4-DXE), trans-3,4-dideoxyxylos-3-enone (trans-3,4-DXE), 2,5-dihydroxy-4-methoxy-pentanoic acid methyl ester (DMPM), 3-deoxypentonolactone (DPL), trans-2,5-dihydroxy-3-pentenoic acid methyl ester (DPM), furfural (F), furfural dimethylacetal (F-DMA), glycolaldehyde dimethyl acetal (GA-DMA), methyl glycosides (MG), methyl lactate (ML), methyl 4-methoxy-2-hydroxybutanoate (MMHB), methyl vinyl glycolate, (MVG) and 2,4,5-trihydroxy-3-pentanoic acid methyl ester (TPM). | ||
Reaction pathways for pentoses
In the presence of Lewis acidic silicates, pentose sugars react along three primary reaction pathways leading to a variety of products (Scheme 2). These are grouped accordingly: (1) formation of methyl glycosides (MG) catalysed by Brønsted acids in the absence of alkali co-solutes; (2) retro-aldol reactions leading to C3–C4 α-hydroxy esters and glycolaldehyde dimethylacetal which are catalysed by Lewis acidic silicates in the presence of alkali co-solutes; (3) dehydration via 3-deoxyxylosone (3-DX) to give DPM, DPL, TPM, DMPM and furfural derivatives (F and F-DMA), catalysed by tin-containing silicates in the absence of alkali co-solutes.The central intermediate 3-DX can be transformed into DPL via a 1,2-hydride shift of its intramolecular hemiacetal. Xylose give rise to two different DPL diastereomers that can be discerned by NMR spectroscopy. We speculate these to be 3-deoxy-γ-D-xylonolactone and 3-deoxy-γ-D-lyxonolactone, formed by racemisation on C2 in the 1,2-hydride shift step. In an analogous open chain form, 3-DX can react with methanol to form a hemiacetal which can undergo a 1,2-hydride shift leading to TPM. From 3-DX a subsequent dehydration can also occur, leading to cis/trans-3,4-dideoxyxylos-3-enone (cis/trans-3,4-DXE). The trans-3,4-DXE isomer can undergo reactive esterification leading to DPM while cis-3,4-DXE is a likely precursor for furfural via a third dehydration step. Interestingly, 3-deoxy xylosone has previously been reported as an intermediate in the formation of furfural from pentoses.27,28 In the current study, we often find that the yields of furfural derivatives and DPM are correlated, supporting that this is indeed an important intermediate in the formation of furfural species using Lewis acidic catalysts. This interpretation is furthermore supported by the use of the Brønsted acidic Al-Beta as the catalyst, resulting in the formation of only trace amounts of furfural products (1%) and no detectable DPM. In contrast, Sn-Beta forms 11–17% furfural products and 23–33% DPM under comparable conditions. A small amount of DMPM was also observed as a product. This is likely formed via Michael addition of methanol to 3,4-DXE, prior to a 1,2-hydride shift, thereby being analogous to the formation of MMHB from tetroses.20,21
Testing of different catalysts
Significant yields of DPM (10–33%) were obtained only for catalysts containing tin incorporated into a siliceous matrix (Table 1, entries 1–5). A comparison of the yields of products obtained from xylose using the different tin-containing silicates is shown in Fig. 1. The products are grouped into the three categories mentioned above, according to the primary reaction pathways responsible for their formation. The highest DPM yield of 33% was obtained using Sn-Beta (HT) (Table 1, entry 1) which also resulted in the highest ratio of DPM to total dehydration products (3:5), while Sn-Beta (PT) resulted in a lower DPM yield of 23% (Table 1, entry 2) and showed a lower ratio (1:2). This difference is likely attributable to the balance of Brønsted and Lewis acidity in the two materials. Sn-Beta (PT) contains more defects due to being synthesised by a post treatment method, likely resulting in a more Brønsted acidic character.39,53 Additionally, the different preparation methods may lead to a different incorporation of tin into the *BEA siliceous matrix.39,53,54 An indication of the different acidity balance for Sn-Beta (HT) and Sn-Beta (PT) is seen when comparing their yields of MG being 6% and 23%, respectively. Of all the tin-containing silicates tested, the small pore Sn-MFI material displays the lowest selectivity towards DPM and dehydration products. The main products formed after two hours are methyl glycosides, indicating that the pentoses are not converted preferentially via Lewis acid catalysed pathways but instead have time to undergo acetalisation reactions which are typically catalysed by Brønsted acids.55 Other tin-containing silicates such as the ordered mesoporous materials Sn-SBA-15 and Sn-MCM-41 were also active for the formation of DPM, resulting in yields of 12% and 16% yield, respectively. These catalysts all belong to the group of tin-containing silicates and are all able to form DPM in significant yields. In contrast, materials having tin outside of the siliceous matrix, such as dispersed SnO2 nanoparticles on Si-Beta, did not catalyse the formation of DPM under these reaction conditions (Table 1, entry 6). This finding highlights the importance of successfully incorporating tin in the silicate matrix to obtain catalytically active heterogeneous catalysts.
| Entry | Catalyst | XYL, % | MG, % | Retro-aldol | Dehydration | Carbon balance, % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GA-DMA, % | ML, % | MVG, % | MMHB, % | Total, % | DPL, % | TPM/DMPMb, % | FURc, % | DPM, % | Total, % | |||||
| a All reactions employed the standard reaction conditions: 360 mg xylose (8.3 wt%), 4 g methanol, 180 mg catalyst, 2 h, 160 °C and 600 rpm stirring. Reactions were performed in triplicates and standard deviations of the last digit are given in parenthesis for DPM; full values and deviations for all products are provided in Table S2. n.d. = not detected. Refer to Scheme 2 for product abbreviations.b Combined yields (carbon%) of 2,4,5-trihydroxy-3-pentanoic acid methyl ester and 2,5-dihydroxy-4-methoxy-pentanoic acid methyl ester.c Combined yields (carbon%) of furfural and furfural dimethyl acetal.d Dispersed SnO2 nanoparticles on Si-Beta. | ||||||||||||||
| 1 | Sn-Beta (HT) | n.d. | 4 | 2 | 14 | 2 | <1 | 19 | 10 | 13 | 11 | 33 (3) | 68 | 90 (4) |
| 2 | Sn-Beta (PT) | n.d. | 23 | 3 | 11 | <1 | <1 | 15 | 6 | 9 | 17 | 23 (1) | 55 | 93 (3) |
| 3 | Sn-MFI | <1 | 30 | 6 | 17 | 1 | <1 | 24 | 3 | 3 | 10 | 11 (1) | 27 | 82 (5) |
| 4 | Sn-MCM-41 | n.d. | 23 | 4 | 12 | <1 | <1 | 18 | 6 | 14 | 20 | 16 (1) | 57 | 98 (5) |
| 5 | Sn-SBA-15 | <1 | 32 | 6 | 11 | <1 | <1 | 18 | 3 | 10 | 18 | 12 (1) | 42 | 92 (6) |
| 6 | SnO2-Betad | 53 | 42 | 2 | <1 | <1 | <1 | 3 | 1 | 2 | 1 | <1 | 6 | 104 (7) |
| 7 | Ti-Beta | <1 | 48 | 7 | 11 | 3 | <1 | 22 | 4 | 7 | 5 | <1 | 16 | 86 |
| 8 | Zr-Beta | <1 | 39 | 12 | 10 | 2 | <1 | 25 | 2 | 2 | 4 | <1 | 9 | 73 |
| 9 | Al-Beta | 6 | 82 | n.d. | n.d. | n.d. | n.d. | 0 | n.d. | n.d. | 1 | n.d. | 1 | 89 (1) |
| 10 | Si-Beta | 13 | 42 | 5 | 2 | <1 | <1 | 8 | 2 | 3 | 3 | <1 | 8 | 71 |
| 11 | Blank | 88 | 6 | <1 | <1 | <1 | <1 | 1 | n.d. | n.d. | <1 | n.d. | 1 | 96 (1) |
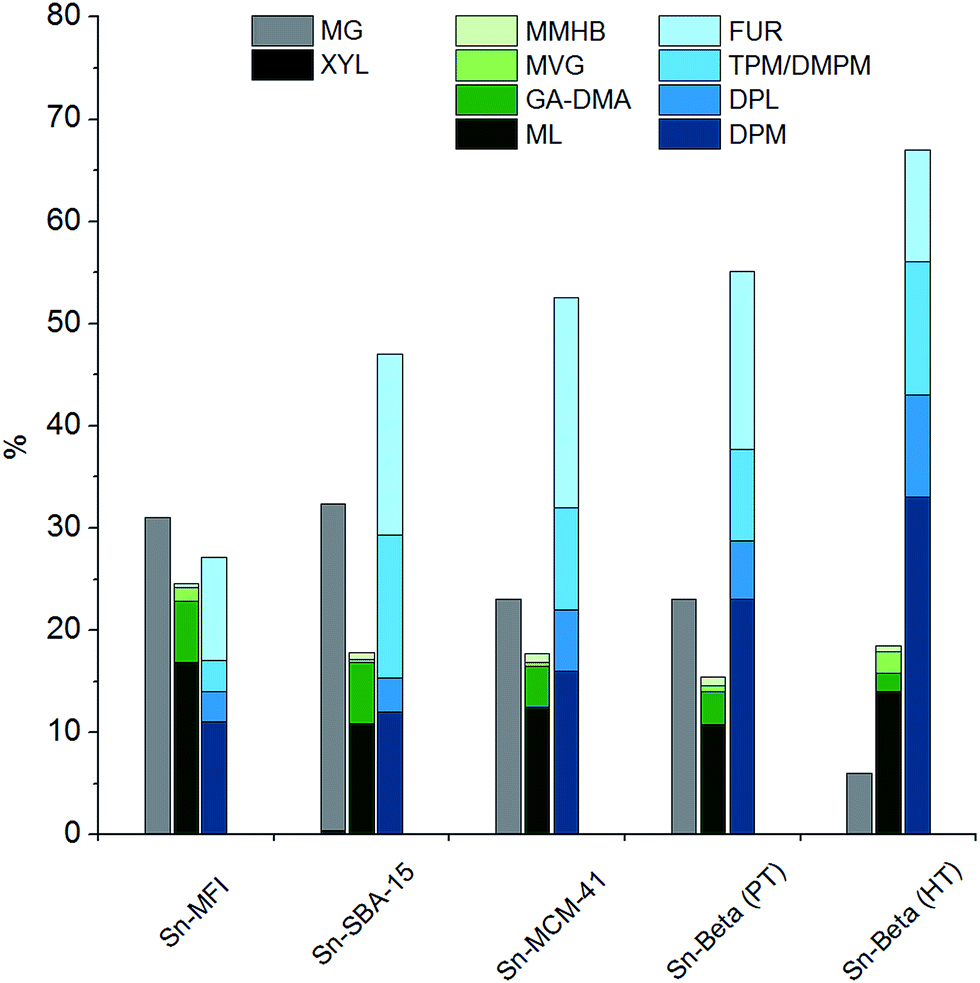 | ||
| Fig. 1 Product distribution of tin-containing silicates based on data from Table 1. Reaction conditions: 0.360 g xylose, 0.180 g catalyst, 4.0 g methanol, 160 °C, 2 hours. | ||
The purely siliceous material Si-Beta did not catalyse the formation of significant amounts of DPM, and the main product observed was MG (Table 1, entry 10). Catalysts having other metals than tin incorporated into the framework such as titanium, zirconium and aluminium did not form significant yields of DPM from xylose either (Table 1, entry 7–9). However, both Ti-Beta and Zr-Beta gave appreciable yields of retro-aldol products (22–25%), which is in accordance with previous reports of these materials being active for the formation of lactates.20 The formation of DPM, in contrast, occurs via a different reaction pathway that is seemingly only catalysed by the tin-containing silicates. The same trend was reported for the formation of THM from hexoses using the same types of materials.26 The highly Brønsted acidic Al-Beta was found to convert xylose into methyl glycosides with a high selectivity, which is in accordance with previous findings.56 No formation of DPM was observed, while small amounts of furfural were detected (ca. 1%).
We would like to draw attention to the yields of furanic products and DPM in the comparison of Zr-Beta and Ti-Beta with all the tin-containing silicates. Low yields of furanic products (4–5%) are observed for the zirconium and titanium materials while all tin-containing silicates form substantially higher yields (10–20%). This observation supports the aforementioned hypothesis, that the majority of the furanic products is formed via a 3-DX route for the tin-containing silicates. The inability of the titanium and zirconium materials to catalyse the formation of 3-DX from xylose thus also prevents a substantial co-production of furanic compounds.
Effect of reaction parameters and co-solutes for DPM formation using Sn-Beta
The catalyst screening clarified that Sn-Beta gives higher DPM yields than other tin-containing silicates. We therefore assessed the reaction parameters in detail using Sn-Beta (HT) as the catalyst. To this end, we varied the reaction temperature (140–180 °C), the catalyst to substrate ratio (0.125–1.0), the concentration of xylose in the reaction solution (8.3–23 wt%) and examined the effect of adding alkali co-solutes in the form of potassium carbonate. Furthermore, we tested if other pentoses could be used for DPM formation. From these studies, it was deduced that high catalyst to substrate ratios and the absence of alkali co-solutes are important parameters to obtain high yields of DPM.The effect of reaction temperature on the yield of DPM was insignificant and similar yields (31–34%) were obtained in the temperature range of 140–180 °C (Table S4†). The reaction temperature effect appeared to be more pronounced for yields of other reaction products, with higher temperatures favouring retro-aldol products (9% difference) and lower temperatures favouring furanics (8% difference).
It had previously been shown for hexoses that alkali co-solutes significantly diminish the yield of THM and furanic products, leading to increased yields of retro-aldol products, from 30% in the absence of alkali to 75% in the presence of 0.065 mM of added potassium carbonate.18 We find that an analogous effect exists for the conversion of pentoses (Fig. 2), illustrating that optimal yields of DPM require a strict control of alkali contaminants. We found by ICP measurements that a background alkali level of 1.3 wt ppm was present even under “alkali free” conditions, possibly originating from the borosilicate glassware.
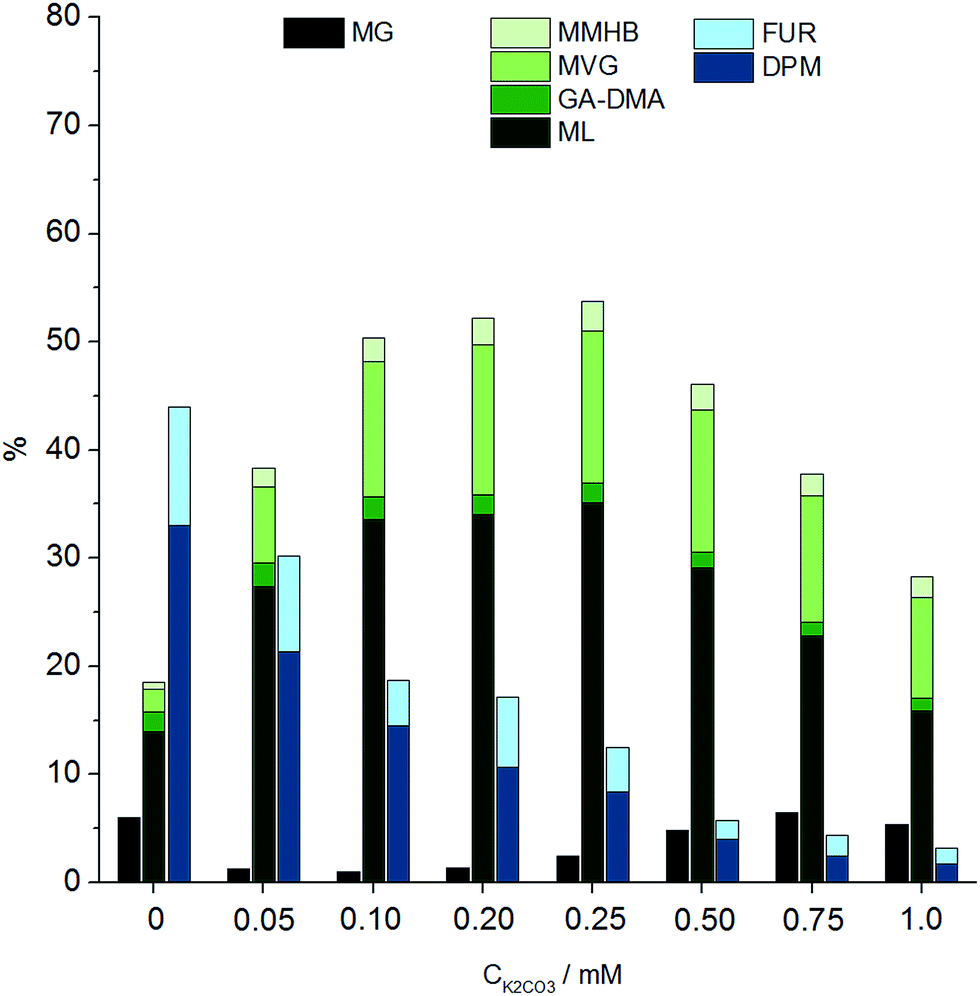 | ||
| Fig. 2 Product distribution at different alkali concentrations using Sn-Beta, based on data from Table S5.† Reaction conditions: 0.360 g xylose, 0.180 g Sn-Beta (HT, Si/Sn = 150), 4.0 g methanol, 160 °C, 2 hours. Yields of TPM and DMPM and DPL are not included in this graph. Refer to Scheme 2 for abbreviations. | ||
The catalyst to substrate ratio was varied from 0.125 to 1.0 on weight basis, while keeping the substrate concentration constant at 8.3 wt% xylose. Interestingly, a strong dependence on catalyst loading was found, as selectivity towards DPM increased significantly from a catalyst to substrate ratio of 0.125 (19% DPM) up to 0.5 (34% DPM) (Table S4†). The formation of retro-aldol products followed the opposite trend, decreasing with increased catalyst to substrate ratios from a combined yield of 32% at a catalyst to substrate ratio of 0.125 to just 15% at a ratio of 1.0.
The effect of the xylose concentration in the reaction mixture was tested, keeping the catalyst to substrate ratio constant at 0.5. Surprisingly, increasing the concentration did not lower the yield of DPM and using a 23 wt% solution of xylose in methanol gave a similar yield of 32% as an 8.3 wt% solution (Table S3†).
Finally, two other pentoses (ribose and lyxose) were tested as substrates and found to give DPM yields of 30% and 31%, respectively (Table S3†).
Polymerisation potential
In order to evaluate the usefulness of DPM as a novel polyester monomer, and having shown that DPM can be formed in acceptable yields (above 30%), the synthesis and purification of DPM was performed at larger scale (1 L). Due to the simpler synthesis route of Sn-Beta (PT), this catalyst was used at the expense of a lower overall DPM yield. From these large-scale experiments, 2.6 g of ≥94% pure (as estimated by 1H-NMR) DPM was synthesised. Having shown that DPM can be prepared in acceptable amounts, we became interested in its use as a polymer building block. It has previously been shown by Sels and co-workers that MVG can be co-polymerised with lactic acid with a 1,2-linkage to afford a polyester material having pendant vinyl groups available for post functionalisation.23 Additionally, it has recently been reported that MVG can be converted into dimethyl (E)-2,5-dihydroxyhex-3-enedioate, via a metathesis reaction.57,58 This new product has also been successfully co-polymerised with lactic acid.57 In contrast to MVG, DPM contains both a secondary and a primary alcohol, which prevents the use of classical polymerisation catalysts due to poor selectivity control. However, enzymatic polymerisation methods permit polymerisation of such highly functional monomers.59,60 Especially the enzyme selectivity between primary and secondary alcohols enables the specific synthesis of functional linear polymers that cannot be prepared using chemo-catalysis.61,62 Unfortunately, homo-polymerisation of DPM was unsuccessful and resulted only in the formation of oligomers. As an alternative to homo-polymerisation, co-polymerisations were conducted using the commercially available monomer ethyl 6-hydroxyhexanoate (E6-HH) as shown in Scheme 3. Polymerisation of pure E6-HH affords polycaprolactone (PCL) which is a linear and biodegradable polyester produced on industrial scale. Co-polymerisation with DPM could be envisaged to be a useful way to modify the properties of PCL. This approach incorporates DPM into linear polymers with a linear 1,5-linkage, leaving the secondary alcohol and olefin moieties available for post-functionalisation (Scheme 3, polymer I).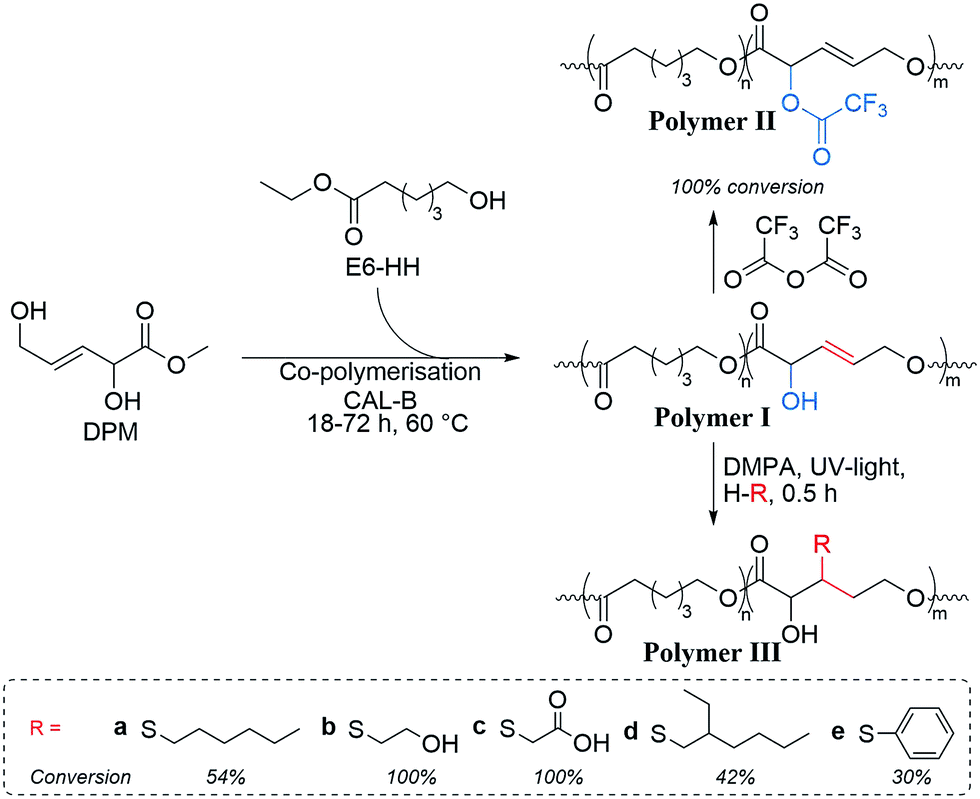 | ||
| Scheme 3 Enzymatic co-polymerisation of DPM and ethyl 6-hydroxyhexanoate (E6-HH) using Candida antarctica lipase B (CAL-B) and subsequent functionalisation. The reaction of the thiols can supposedly occur on either carbon of the olefinic moieties, but only one form is represented in the scheme for clarity. | ||
The enzymatic co-polymerisations between DPM and E6-HH were performed at 60 °C in bulk monomer in accordance with the general procedure described in the Experimental section. The NMR spectrum of the resulting polymer (Fig. 3) shows that that the vinylic hydrogen atoms from DPM are present, while the alcohol, alkene and ester functional groups were confirmed by FT-IR spectroscopy (Fig. S6†).
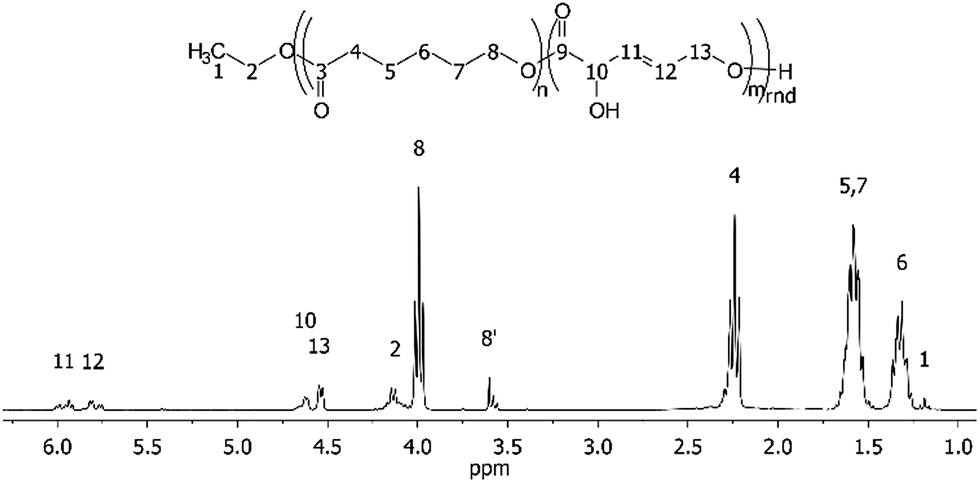 | ||
| Fig. 3 1H NMR assignment of the co-polymer product, poly(E6-HH-co-DPM), from Table 2 entry 4. | ||
The polymer synthesis procedure was varied to study the effects of feed ratio and polymerisation time on the resulting polymers. Polymerisation time was studied by conducting co-polymerisation experiments using a DPM to E6-HH molar ratio of 0.22 and varying the reaction times (Table 2, entries 2–4). The shortest polymerisation time of 18 h resulted in the incorporation of approximately 55% (0.12 molar ratio) of the DPM from the feed into the co-polymer. By increasing the polymerisation time to first 42 h and subsequently to 72 h, incorporation was increased to 77% (0.17 molar ratio). At all tested reaction times reasonable degrees of polymerisation (Mw = 10000–12350 g mol−1) and polydispersity indices (PDI = 2.1–2.5) were obtained, comparable to typical results from enzymatic polymerisation of monomers containing both secondary and primary alcohols.61,62
| Entry | Time h | Feed MRb | Prod. MRb,c | Tgd, °C | Tmd, °C | Mne, g mol−1 | Mwe, g mol−1 | PDIe |
|---|---|---|---|---|---|---|---|---|
| a All reactions were performed at 60 °C and the pressure was held at 200 mbar for 2 h whereafter it was reduced to 5 mbar in accordance with optimisations performed with E6-HH (see ESI).b Molar ratio (MR) listed as DPM/E6-HH [mol mol−1].c Determined by 1H NMR.d Determined by DSC.e Determined by SEC in THF using PS standards.f Using only E6-HH as monomer. | ||||||||
| 1f | 18 | 0 | 0 | −60.2 | 48.9 | 3600 | 4700 | 1.3 |
| 2 | 18 | 0.22 | 0.12 | −49.3 | 42.8 | 5150 | 10050 |
2.1 |
| 3 | 42 | 0.22 | 0.14 | −49.3 | 42.5 | 4490 | 10750 |
2.4 |
| 4 | 72 | 0.22 | 0.17 | −50.0 | 34.2 | 5000 | 12350 |
2.5 |
| 5 | 72 | 0.44 | 0.44 | −55.5 | 15.1 | 1900 | 4500 | 2.4 |
| 6 | 72 | 0.66 | 0.66 | −52.3 | 7.5 | 1760 | 3700 | 2.1 |
These findings clearly showed that DPM was more challenging to polymerise than E6-HH, which was also evident by DPM being unable to homo-polymerise. Nevertheless, DPM should become incorporated at longer polymerisation times and higher DPM fractions in the feed (Table 2).
A study of varying the feed ratio of E6-HH to DPM from a molar ratio of 0.22 to 0.66 showed that the content of DPM in the co-polymer increased with increased ratio. This lead to full incorporation of DPM into the co-polymers at polymerisation times of 72 h and molecular ratios above 0.44. Despite full incorporation of DPM, these polymerisations resulted in molecular weights of only 4500 and 3700 g mol−1 for 0.44 and 0.66 molar ratio, respectively. Molecular weights above 10000 g mol−1 as observed with 0.22 molar ratio DPM could not be obtained for these co-polymerisations.
The thermal properties of all the co-polymers were investigated by DSC and showed glass transition temperatures (Tg) between −49 °C to −56 °C, which is typical for aliphatic polyesters and confirms the flexible nature of the polymer chain. The melting temperature (Tm) at 0.12 molar ratio of DPM was determined to be 42.8 °C and Tm decreased with increasing content of DPM to 7.5 °C at 0.66 molar ratio DPM. The incorporation of DPM clearly prevents the regularity and close chain-to-chain packing required for the polymer to crystallise, thereby reducing the melting temperature.
These initial studies clearly show that DPM can be successfully polymerised with other similar monomers thereby providing access to functional polymers. Furthermore, the degree of incorporation may be used to manipulate the physical properties of the co-polymer.
Post-functionalisation
Post-functionalisation of polymers offers the possibility to tailor their properties. The DPM containing co-polymers were tested for their ability to undergo functionalisation of the α-hydroxyl and olefinic groups (Scheme 3). Functionalisation of the α-hydroxyl groups of DPM was performed using trifluoroacetic anhydride (TFA), a highly reactive reagent widely used in hydroxyl labelling of polymers, allowing for clear detection of functionality by NMR analysis.63,64 The structure of the TFA modified co-polymer (Scheme 3, polymer II) was verified by 1H NMR (see experimental section “Thiol–ene functionalisation of poly(E6-HH-co-DPM)”), showing that full functionalisation was achieved.Additionally, thiol–ene chemistry was used to demonstrate the reactivity of the intra-chain alkene.23 Thiol–ene chemistry is a well-established protocol for functionalisation of polymers by formation of alkyl sulphides via reaction of a thiol and an alkene using radical chemistry.65,66 Here, a photo initiator (2,2-dimethoxy-2-phenylacetophenone, DMPA) was used together with a selection of thiol compounds (Scheme 3) to illustrate the potential for post-polymerisation functionalisation.
The functionalisation of DPM sub-units in the thiol–ene experiments was determined using 1H and 1H–13C NMR following diminished double bond proton intensities within the polymer upon functionalisation. Thiol–ene reactions varied between 30 and 100% conversion, with the least sterically hindered substituents yielding the highest conversion (Scheme 3, polymers IIIa–e). Poor solubility of the functionalised co-polymer materials in DMSO and chloroform prevented conclusive studies of the regioselectivity in the thiol–ene reactions. We found that in particular mercaptoacetic acid and mercaptoethanol (Scheme 3, polymer IIIb–c) were effective grafting reagents, making it possible to fully convert the in-chain double bond, clearly demonstrating the polymers high potential for functionalisation. In-chain alkenes are estimated to have an approximately 10-fold lower reactivity compared to corresponding pendent alkenes.67 The high extent of functionalisation in the polymer observed herein is thus extraordinary and underlines the potential of in-chain alkenes for preparation of functional polymers.
Co-polymer blends with polycaprolactone in thin films
The prepared co-polymers have a high structural similarity to PCL and therefore blends between some of the co-polymers and PCL were investigated. PCL often finds uses in biomedical devices, where control of surface properties and functionality are highly desired properties and therefore simple blending could be an interesting application of the new co-polymers.Three different films consisting of pure PCL, a PCL blend with 10 wt% poly(E6-HH-co-DPM) (PECD–PCL) and a PCL blend with 10 wt% trifluroacetic acid functionalised poly(E6-HH-co-DPM) (PECD(TFA)–PCL) were prepared by solvent casting and hot-pressing. The prepared films were investigated by water contact angle (WCA) measurements to determine the impact on surface properties of the blend (Table 3).
| Film | Advancing WCA deg | Receding WCA deg |
|---|---|---|
| a All water contact angles (WCA) were determined as averages of three measurements of three different drops at different positions on the polymer surface and standard deviations of the last digit are given in parenthesis.b WCA value for PCL from literature.68c The film contained 10 wt% poly(E6-HH-co-DPM).d The film contained 10 wt% TFA functionalised poly(E6-HH-co-DPM). | ||
| PCLb | 90 (1) | 46 (1) |
| PCL | 85 (2) | 46 (2) |
| PECD–PCLc | 75 (1) | 38 (2) |
| PECD(TFA)–PCLd | 93 (2) | 36 (2) |
The pure PCL film had an advancing WCA of 85°, which was reduced to 75° by blending with the DPM co-polymer. This shows the impact of the free hydroxyl groups from the co-polymer, and could be exploited to increase the hydrophilicity of the thin film. This was corroborated with the receding WCA, which is reduced by almost 10° for the blend, indicating the presence of more polar groups in the system. Conversely, blending PCL with the trifluroacetic acid functionalised poly(E6-HH-co-DPM) resulted in an increase in the advancing WCA up to 93°, which is expected from increasing the hydrophobicity of the film due to the fluorine in the co-polymer. The receding WCA was not affected from blending in the fluorinated co-polymer, which showed that the system is still amphiphilic. Both results show that the co-polymer is able to modify the surface properties of the thin film blend, and that post-modification of the co-polymer permits exploitation of the functional groups for further modification of the surface of thin film blends.
Conclusion
In the current study, we have shown that the novel polyester building block DPM can be synthesised from pentoses using tin-containing silicates as catalysts in yields up to 32% with xylose concentrations of 23 wt%. This approach provides a simple and low cost route from a plentiful and sustainable feedstock to an intriguing chemical product. Future improvements to the DPM synthesis may encompass ways to suppress the formation of furanics or of retro-aldol products, or to better control the formation of ester products from 3-DX. DPM has been shown to be an interesting monomer for the preparation of functional polyesters in co-polymerisation with E6-HH. Enzymatic polymerisations were effective for the synthesis of the highly functional polyesters with 0.17 to 0.66 molar ratio content of DPM. The co-polymers could easily be post-functionalised using either TFA or thiol–ene chemistry to extend the potential applications of these co-polymers. As an example, simple blends with PCL were investigated and it was shown how the co-polymer could be used to affect the surface properties of the polymer film.The chemoenzymatic approach described herein thus enables future bio-refineries to better utilise pentoses from hemicellulose-containing biomass, providing the chemical industry with new types of interesting polymer building blocks.
Acknowledgements
We gratefully acknowledge funding by the Bio-Value platform (biovalue.dk) under the SPIR initiative by The Danish Council for Strategic Research and The Danish Council for Technology and Innovation (case number 0603-00522B), as well as the Innovation Fund Denmark (case number 5150-00023B). SM gratefully acknowledges funding by Grant 2013_01_0709 of the Carlsberg Foundation. Novozymes A/S is gratefully acknowledged for supplying enzymes for polymerisation. 800 MHz NMR spectra were recorded on the spectrometer of the DTU NMR center.References
- E. Castro-Aguirrea, F. Iñiguez-Francoa, H. Samsudinb, X. Fangc and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef PubMed.
- A. Carlson, B. Coggio, K. Lau, C. Mercogliano and J. Millis, in Chemicals and Fuels from Bio-Based Building Blocks, Wiley-VCH, 2016, ch. 7, pp. 173–190 Search PubMed.
- H. Liu, Y. Xu, Z. Zheng and D. Liu, Biotechnol. J., 2010, 5, 1137–1148 CrossRef CAS PubMed.
- M. Sara, S. K. Brar and J. F. Blais, in Platform Chemical Biorefinery, Elsevier Inc., 1st edn, 2016, ch. 14, pp. 249–283 Search PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- E4tech, Re-CORD and WUR, From the Sugar Platform to biofuels and biochemicals, Report ENER/C2/423–2012/SI2.673791, European Commission Directorate-General Energy, 2015 Search PubMed.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- R. De Clercq, M. Dusselier, C. Christiaens, J. Dijkmans, R. I. Iacobescu, Y. Pontikes and B. F. Sels, ACS Catal., 2015, 5, 5803–5811 CrossRef CAS.
- Y. Wang, J. D. Lewis and Y. Román-Leshkov, ACS Catal., 2016, 6, 2739–2744 CrossRef CAS.
- P. Verdeguer, N. Merat and A. Gaset, J. Mol. Catal., 1993, 85, 327–344 CrossRef CAS.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef CAS PubMed.
- J. Ma, Y. Pang, M. Wang, J. Xu, H. Ma and X. Nie, J. Mater. Chem., 2012, 22, 3457–3461 RSC.
- S. P. Chopade, D. J. Miller, J. E. Jackson, T. A. Werpy, J. G. Frye Jr and A. H. Zacher, Board of Trustees operating Michigan State University, US 6291725 B1, Battelle Memorial Institute, Pacific Northwest Laboratory, 2001.
- M.-Y. Zheng, A.-Q. Wang, N. Ji, J.-F. Pang, X.-D. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS PubMed.
- R. Ooms, M. Dusselier, J. A. Geboers, B. O. de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC.
- T. R. Boussie, E. L. Dias, Z. M. Fresco and V. J. Murphy, US 8501989 B2, Rennovia Inc., 2013.
- S. Tolborg, I. Sádaba, C. M. Osmundsen, P. Fristrup, M. S. Holm and E. Taarning, ChemSusChem, 2015, 8, 613–617 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, F. de Clippel, J. Dijkmans, D. W. Gammon and B. F. Sels, ChemCatChem, 2013, 5, 569–575 CrossRef CAS.
- I. Z. Sádaba and E. Taarning, Haldor Topsøe A/S, WO 2016083137 A1, 2016.
- M. Dusselier, P. Van Wouwe, S. De Smet, R. De Clercq, L. Verbelen, P. Van Puyvelde, F. E. Du Prez and B. F. Sels, ACS Catal., 2013, 3, 1786–1800 CrossRef CAS.
- E. Taarning, S. Saravanamurugan, M. S. Holm, J. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef CAS PubMed.
- M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- S. Tolborg, S. Meier, I. Sádaba, S. G. Elliot, S. K. Kristensen, S. Saravanamurugan, A. Riisager, P. Fristrup, T. Skrydstrup and E. Taarning, Green Chem., 2016, 18, 3360–3369 RSC.
- T. Usui, S. Yanagisawa, M. Ohguchi, M. Yoshino, R. Kawabata, J. Kishimoto, Y. Arai, K. Aida, H. Watanabe and F. Hayase, Biosci., Biotechnol., Biochem., 2007, 71, 2465–2472 CrossRef CAS PubMed.
- M. Hellwig, J. Degen and T. Henle, J. Agric. Food Chem., 2010, 58, 10752–10760 CrossRef CAS PubMed.
- H. S. Chen, A. Wang, H. Sorek, J. D. Lewis, Y. Román-Leshkov and A. Bell, ChemistrySelect, 2016, 1, 4167–4172 CrossRef CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS PubMed.
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef CAS PubMed.
- C.-C. Chang, Z. Wang, P. Dornath, H. J. Cho and W. Fan, RSC Adv., 2012, 2, 10475–10477 RSC.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottira, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- Q. Guo, F. Fan, E. A. Pidko, W. N. P. van der Graaff, Z. Feng, C. Li and E. J. M. Hensen, ChemSusChem, 2013, 6, 1352–1356 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli, J. Jagielski, R. Hauert and J. Perez-Ramirez, Catal. Sci. Technol., 2014, 4, 2302–2311 CAS.
- M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11777–11782 CrossRef CAS PubMed.
- M. Sasidharan, Y. Kiyozumi, N. K. Mal, M. Paul, P. R. Rajamohanan and A. Bhaumik, Microporous Mesoporous Mater., 2009, 126, 234–244 CrossRef CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- P. Wolf, M. Valla, F. Núñez-Zarur, A. Comas-Vives, A. J. Rossini, C. Firth, H. Kallas, A. Lesage, L. Emsley and C. Copéret, ACS Catal., 2016, 6, 4047–4063 CrossRef CAS.
- G. Yang, X. Li and L. Zhou, RSC Adv., 2016, 6, 8838–8847 RSC.
- B. D. Montejo-Valencia, J. L. Salcedo-Pérez and M. C. Curet-Arana, J. Phys. Chem. C, 2016, 120, 2176–2186 CAS.
- Y. G. Kolyagin, A. V. Yakimov, S. Tolborg, P. N. Vennestrøm and I. I. Ivanova, J. Phys. Chem. Lett., 2016, 7, 1249–1253 CrossRef CAS PubMed.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- R. Otomo, R. Kosugi, Y. Kamiya, T. Tatsumi and T. Yokoi, Catal. Sci. Technol., 2016, 6, 2787–2795 CAS.
- S. Tolborg, A. Katerinopoulou, D. D. Falcone, I. Sádaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup and M. S. Holm, J. Mater. Chem. A, 2014, 2, 20252–20262 CAS.
- P. Li, G. Liu, H. Wu, Y. Liu, J.-g. Jiang and P. Wu, J. Phys. Chem. C, 2011, 115, 3663–3670 CAS.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- J. Dijkmans, D. Gabriels, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- S. Valencia and A. Corma, UOP LLC, US 6306364 B1, 1999.
- N. K. Mal, V. Ramaswamy, P. R. Rajamohanan and A. V. Ramaswamy, Microporous Mater., 1997, 12, 331–340 CrossRef CAS.
- L. Li, C. Stroobants, K. Lin, P. A. Jacobs, B. F. Sels and P. P. Pescarmona, Green Chem., 2011, 13, 1175–1181 RSC.
- V. Ramaswamy, P. Shah, K. Lazar and A. V. Ramaswamy, Catal. Surv. Asia, 2008, 12, 283–309 CrossRef CAS.
- J. Dijkmans, M. Dusselier, W. Janssens, M. Trekels, A. Vantomme, E. Breynaert, C. Kirschhock and B. F. Sels, ACS Catal., 2015, 6, 31–46 CrossRef.
- P. Wolf, W.-C. Liao, T.-C. Ong, M. Valla, J. W. Harris, R. Gounder, W. N. P. van der Graaff, E. A. Pidko, E. J. M. Hensen, P. Ferrini, J. Dijkmans, B. Sels, I. Hermans and C. Copèret, Helv. Chim. Acta, 2016, 99, 916–927 CrossRef CAS.
- S. Saravanamurugan, A. Riisager, E. Taarning and S. Meier, ChemCatChem, 2016, 8, 3107–3111 CrossRef CAS.
- M. Paniagua, S. Saravanamurugan, M. Melian-Rodriguez, J. A. Melero and A. Riisager, ChemSusChem, 2015, 8, 1088–1094 CrossRef CAS PubMed.
- A. Dewaele, L. Meerten, L. Verbelen, S. Eyley, W. Thielemans, P. Van Puyvelde, M. Dusselier and B. Sels, ACS Sustainable Chem. Eng., 2016, 4, 5943–5952 CrossRef CAS.
- A. Sølvhøj, E. Taarning and R. Madsen, Green Chem., 2016, 18, 5448–5455 RSC.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- N. Miletić, K. Loos and R. A. Gross, in Biocatalysis in Polymer Chemistry, Wiley-VCH, 2010, ch. 4, pp. 83–129 Search PubMed.
- B. J. Kline, E. J. Beckman and A. J. Russell, J. Am. Chem. Soc., 1998, 120, 9475–9480 CrossRef CAS.
- A. S. Kulshrestha, W. Gao and R. A. Gross, Macromolecules, 2005, 38, 3193–3204 CrossRef CAS.
- A. Holländer, S. Kröpke and F. Pippig, Surf. Interface Anal., 2008, 40, 379–385 CrossRef.
- C. Hoffmann, M. C. Stuparu, A. Daugaard and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 745–749 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- N. Ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
- D. S. Jones, J. Djokic, C. P. McCoy and S. P. Gorman, Biomaterials, 2002, 23, 4449–4458 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26708d |
| This journal is © The Royal Society of Chemistry 2017 |