DOI:
10.1039/C6RA25319A
(Paper)
RSC Adv., 2017,
7, 975-984
A series of coordination polymers based on terphenyl tetracarboxylates and bis-pyridyl ligands with water vapor sorption properties†
Received
16th October 2016
, Accepted 21st November 2016
First published on 4th January 2017
Abstract
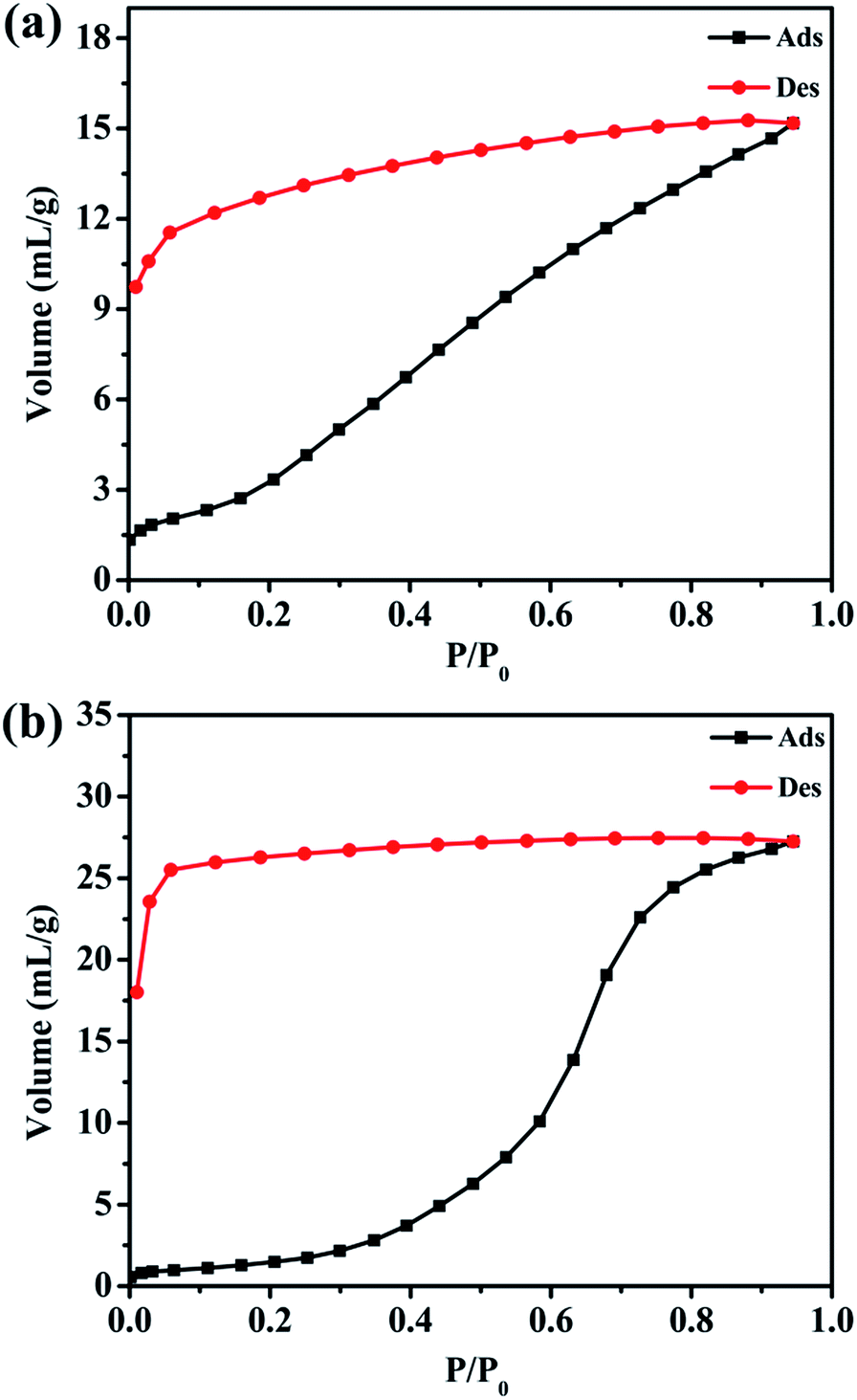
Six new coordination polymers, namely, {[Mn2(L1)(bpfp)(H2O)2]·6H2O·(CH3OH)}n (1), {[Mn4(μ3-O)2(L1)2(bpe)2(H2O)2]·2H2O}n (2), [Ni(HL1)(Hbpmp)(H2O)2]n (3), [Co2(μ2-OH)(HL1)(bpmp)(H2O)4]n (4), {[Co2(L2)(bpfp)(H2O)2]·4H2O}n (5), {[Co2(L2)2(bpe)2(H2O)2·2H2O]·(NH4)4}n (6) (H4L1 = [1,1′:4′,1′′-terphenyl]-2,2′′,5,5′′-tetracarboxylic acid, H4L2 = [1,1′:4′,1′′-terphenyl]-3,3′′,5,5′′-tetracarboxylic acid, bpfp = bis(4-pyridylformyl)piperazine, bpe = 1,2-bis(pyridin-4-yl)ethane and bpmp = N,N′-bis(4-pyridyl)piperazine) were synthesized under hydrothermal conditions by terphenyl tetracarboxylates, bis-pyridyl ligands and transition metal salts. These six complexes were characterized by elemental analysis, infrared (IR) spectroscopy, thermogravimetric analysis (TGA), single-crystal X-ray diffraction, powder X-ray diffraction (PXRD) and fluorescence spectroscopy. Complexes 1 and 5 possess three-dimensional (3D) (4,6)-connected networks with {43·63}2{46·66·83} and {44·610·8}{44·62} topologies, respectively. Complexes 2–4 and 6 feature two-dimensional (2D) networks, which are expanded into 3D supramolecular frameworks via hydrogen bonding interactions. Desorption of lattice water molecules in complexes 1 and 2 was analyzed, and the values of water vapor uptake are 15.17 mL g−1 and 27.24 mL g−1, respectively.
Introduction
In recent years, the design and synthesis of hybrid metal–organic coordination polymers have attracted intense interest due to their various architectures, interesting topologies and desirable applications such as sorption,1–3 separation,4 luminence,5,6 catalysis7 and magnetism.8 To date, a large number of metal–organic coordination polymers with diverse structures have been synthesized.9,10 In the process of synthesis, many factors influence the final structure, such as molar ratio of reactants, temperature, pH value, solvent system and auxiliary ligands, etc.11,12 Organic ligands, as one of the most important factors, are crucial to constructing novel architectures.13,14 The O-donor polycarboxylate ligands are excellent organic ligands in forming various structures such as 0D cages, 1D chains, 2D grids and 3D frameworks.15 Among these, rigid tetracarboxylate ligands based on terphenyl moieties have been used to generate a large number of coordination polymers with remarkable properties.16 Additionally, N-donor ligands can offer nitrogen coordination donors to the metal centers, acting as hydrogen bond donors and acceptors. Therefore, N-donor ligands as auxiliary ligands are also wonderful candidates in producing novel coordination polymers.17
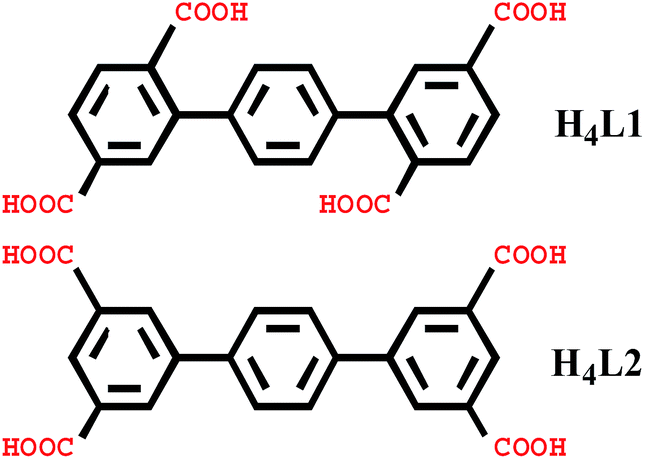
In this paper, we selected two types of rigid tetracarboxylate ligands based on terphenyl moieties [1,1′:4′,1′′-terphenyl]-2,2′′,5,5′′-tetracarboxylic acid (H4L1) and [1,1′:4′,1′′-terphenyl]-3,3′′,5,5′′-tetracarboxylic acid (H4L2) as the main ligands, three N-donor ligands bis(4-pyridylformyl)piperazine (bpfp), 1,2-bis(pyridin-4-yl)ethane (bpe), and N,N′-bis(4-pyridyl)piperazine (bpmp) as the auxiliary ligands, to react with different transition metal salts under hydrothermal conditions. Six complexes, {[Mn2(L1)(bpfp)(H2O)2]·6H2O·(CH3OH)}n (1), {[Mn4(μ3-O)2(L1)2(bpe)2(H2O)2]·2H2O}n (2), [Ni(HL1)(Hbpmp)(H2O)2]n (3), [Co2(μ2-OH)(HL1)(bpmp)(H2O)4]n (4), {[Co2(L2)(bpfp)(H2O)2]·4H2O}n (5), {[Co2(L2)2(bpe)2(H2O)2·2H2O]·(NH4)4}n (6) were synthesized. These six complexes were characterized by elemental analysis, infrared (IR) spectroscopy, thermogravimetric analysis (TGA), single-crystal X-ray diffraction, powder X-ray diffraction (PXRD) and fluorescence spectroscopy. Desorption of lattice water molecules in complexes 1 and 2 was investigated and the values of water vapor uptake are 15.17 mL g−1 and 27.24 mL g−1, respectively (Scheme 1).
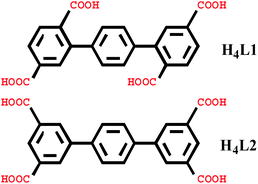 |
| | Scheme 1 Schematic structure of the organic linker H4L1 and H4L2. | |
Experimental section
Materials and methods
All reagents and solvents were obtained commercially without further purification. Mn(OAc)2·4H2O, Ni(NO3)2·6H2O and Co(NO3)2·6H2O are from Beijing Tong Guang Fine Chemicals Company, and the purity are 99%. H4L1, H4L2, bpfp, bep, bpmp ligands are from Jinan Camolai Trading Company, and the purity are 98%. Elemental analysis for C, H and N were performed on a Perkin-Elmer 2400 CHN elemental analyzer. The IR spectra were recorded (as KBr pressed pellets) on a Nicolet 170SX spectrometer in the range of 4000–400 cm−1. TGA were carried out on a Exstar SII TG/DTA 7200 thermal analyzer from room temperature to 800 °C with a heating rate of 10 °C min−1 under the protection of nitrogen. Powder PXRD patterns were obtained using a Siemens D5005 diffractometer (Cu Kα radiation, λ = 1.5410 Å) with the 2θ from 5 to 55° under the 0.03° scan step. Fluorescence spectra were recorded on a Hitachi F-4600 fluorescence/phosphorescence spectrophotometer with a xenon lamp light source. The magnetic measurements were performed on the Quantum Design SQUID PPMS60000 instruments in a magnetic field of 1000 Oe in the temperature range of 1.8–300 K. The 2D and 3D models for complexes 1–6 were derived with Topos4.0 software.
Synthesis of {[Mn2(L1)(bpfp)(H2O)2]·6H2O·(CH3OH)}n (1)
A mixture of Mn(OAc)2·4H2O (73.5 mg, 0.3 mmol), H4L1 (20.3 mg, 0.05 mmol), bpfp (29.6 mg, 0.1 mmol) and H2O (10 mL) was sealed in a 23 mL Teflon-lined autoclave and heated at 150 °C for 3 days. After cooling to room temperature at a speed of 0.1 °C min−1, the colorless crystals of 1 (90.12 mg) were obtained and washed with distilled water. 61% yield based on Mn. Elemental analysis for C39H46Mn2N4O19, calcd (%): C, 47.53; H, 4.67; N, 5.68. Found (%): C, 47.48; H, 4.73; N, 5.62. IR data (KBr, cm−1): 3406 (s), 3011 (w), 1523 (s), 1441 (s), 1353 (s), 1262 (m), 1166 (w), 1084 (m), 912 (w), 890 (m), 832 (m), 801 (m), 767 (m), 680 (m), 576 (w), 527 (w).
Synthesis of {[Mn4(μ3-O)2(L1)2(bpe)2(H2O)2]·2H2O}n (2)
A mixture of Mn(OAc)2·4H2O (73.5 mg, 0.3 mmol), H4L1 (20.3 mg, 0.05 mmol), bpe (9.1 mg, 0.05 mmol), H2O (10 mL) and 1 mol L−1 HCl aqueous solution (0.05 mL) was sealed in a 23 mL Teflon-lined autoclave and heated at 150 °C for 3 days. After cooling to room temperature at a speed of 0.1 °C min−1, the colorless crystals of 2 (23.52 mg) were obtained and washed with distilled water. 21% yield based on Mn. Elemental analysis for C68H48Mn4N4O22, calcd (%): C, 54.66; H, 3.21; N, 3.75. Found (%): C, 54.61; H, 3.27; N, 3.77. IR data (KBr, cm−1): 3439 (s), 3071 (w), 1552 (s), 1405 (s), 1352 (m), 1268 (w), 1130 (w), 1079 (w), 1026 (w), 939 (w), 851 (w), 771 (m), 686 (w), 549 (w).
Synthesis of [Ni(HL1)(Hbpmp)(H2O)2]n (3)
A mixture of Ni(NO3)2·6H2O (87.3 mg, 0.3 mmol), H4L1 (20.3 mg, 0.05 mmol), bpmp (13.4 mg, 0.05 mmol), H2O (10 mL) and 1 mol L−1 NaOH aqueous solution (0.05 mL) was sealed in a 23 mL Teflon-lined autoclave and heated at 150 °C for 3 days. After cooling to room temperature at a speed of 0.1 °C min−1, the green crystals of 3 (108.32 mg) were obtained and washed with distilled water. 47% yield based on Ni. Elemental analysis for C38H36NiN4O10, calcd (%): C, 59.42; H, 4.69; N, 7.30. Found (%): C, 59.39; H, 4.64; N, 7.27. IR data (KBr, cm−1): 3445 (s), 3012 (w), 1556 (s), 1441 (s), 1361 (s), 1242 (m), 1128 (w), 1070 (m), 914 (w), 899 (m), 839 (m), 768 (m), 672 (m), 587 (w), 524 (w).
Synthesis of [Co2(μ2-OH)(HL1)(bpmp)(H2O)4]n (4)
A mixture of Co(NO3)2·6H2O (87.1 mg, 0.3 mmol), H4L1 (20.3 mg, 0.05 mmol), bpmp (26.8 mg, 0.1 mmol) and H2O (10 mL) was sealed in a 23 mL Teflon-lined autoclave and heated at 150 °C for 3 days. After cooling to room temperature at a speed of 0.1 °C min−1, the red crystals of 4 (48.66 mg) were obtained and washed with distilled water. 37% yield based on Co. Elemental analysis for C38H40Co2N4O13, calcd (%): C, 51.90; H, 4.55; N, 6.37. Found (%): C, 51.88; H, 4.57; N, 6.33. IR data (KBr, cm−1): 3558 (s), 3231 (s), 1522 (s), 1394 (s), 1261 (m), 1155 (w), 1125 (w), 1051 (w), 966 (w), 851 (m), 778 (m), 670 (w), 570 (w), 514 (w).
Synthesis of {[Co2(L2)(bpfp)(H2O)2]·4H2O}n (5)
A mixture of Co(NO3)2·6H2O (17.5 mg, 0.06 mmol), H4L2 (8.1 mg, 0.02 mmol), bpfp (17.7 mg, 0.06 mmol), H2O (10 mL) and 1 mol L−1 NaOH aqueous solution (0.1 mL) was sealed in a 23 mL Teflon-lined autoclave and heated at 160 °C for 3 days. After cooling to room temperature at a speed of 0.1 °C min−1, the red crystals of 5 (3.06 mg) were obtained and washed with distilled water. 11% yield based on Co. Elemental analysis for C38H38Co2N4O16, calcd (%): C, 49.32; H, 4.11; N, 6.05. Found (%): C, 49.35; H, 4.14; N, 6.00. IR data (KBr, cm−1): 3440 (s), 1641 (m), 1549 (s), 1504 (s), 1442 (m), 1371 (s), 1260 (m), 1192 (s), 1138 (s), 1060 (s), 1026 (w), 951 (w), 880 (w), 798 (m), 727 (w), 630 (w), 502 (w).
Synthesis of {[Co2(L2)2(bpe)2(H2O)2·2H2O]·(NH4)4}n (6)
A mixture of Co(NO3)2·6H2O (58.2 mg, 0.2 mmol), H4L2 (40.6 mg, 0.1 mmol), bpe (36.4 mg, 0.2 mmol), H2O (10 mL) and 1 mol L−1 HCl aqueous solution (0.05 mL) was sealed in a 23 mL Teflon-lined autoclave and heated at 150 °C for 3 days. After cooling to room temperature at a speed of 0.1 °C min−1, the red crystals of 6 (10.02 mg) were obtained and washed with distilled water. 7% yield based on Co. Elemental analysis for C68H64Co2N8O20, calcd (%): C, 57.02; H, 4.47; N, 7.82. Found (%): C, 57.05; H, 4.44; N, 7.84. IR data (KBr, cm−1): 3417 (w), 3030 (m), 2772 (m), 2432 (m), 1655 (s), 1561 (s), 1481 (m), 1401 (s), 1355 (s), 1261 (m), 1108 (m), 1026 (m), 921 (w), 849 (m), 783 (m), 660 (w), 575 (w), 531 (w), 502 (w).
Single-crystal X-ray crystallography
Single-crystal X-ray diffraction data for complexes 1–6 were collected on a Bruker APEX diffractometer with Mo Kα radiation (graphite monochromator, λ = 0.71073 Å) at 298(2) K. The structures of complexes 1–6 were solved by direct methods and refined by full-matrix least-squares methods on F2 using SHELXTL crystallographic software package.18 All the non-hydrogen atoms were refined anisotropically. The hydrogen atoms of the organic ligands were generated theoretically on to the specific atoms and refined isotropically through fixed thermal factors. The crystallographic data and structure refinement parameters of complexes 1–6 are listed in Table 1. The selected bond distances (Å) and angles (°) for complexes 1–6 are summarized in Table S1 (ESI†). The hydrogen bond parameters of complexes 2, 3, 4, 6 are presented in Table S2 (ESI†). The CCDC reference numbers are 1056874–1056879 for complexes 1–6.
Table 1 Crystal data and structure refinement for complexes 1–6
| R1 = Σ(||Fo| − |Fc||)/Σ|Fo|. wR2 = [Σw(|Fo|2 − |Fc|2)2/(Σw|Fo|2)2]1/2. |
| Complex |
1 |
2 |
3 |
4 |
5 |
6 |
| Formula |
C39H46Mn2N4O19 |
C68H48Mn4N4O22 |
C38H36NiN4O10 |
C38H40Co2N4O13 |
C38H38Co2N4O16 |
C68H64Co2N8O20 |
| Formula wt |
984.68 |
1492.86 |
767.42 |
878.60 |
924.58 |
1431.13 |
| Cryst. syst. |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
P |
P |
P |
| a (Å) |
10.2909(9) |
11.4630(9) |
13.5450(12) |
12.3610(11) |
8.5810(7) |
10.7230(9) |
| b (Å) |
10.6550(10) |
11.8350(10) |
20.0472(18) |
12.7309(12) |
10.2780(8) |
11.8829(11) |
| c (Å) |
11.4221(11) |
11.8930(11) |
12.8501(11) |
12.7571(14) |
12.2140(11) |
13.6481(12) |
| α (°) |
79.594(2) |
74.1490(10) |
90 |
83.027(2) |
79.344(2) |
88.316(2) |
| β (°) |
75.334(2) |
88.610(2) |
91.3810(10) |
82.9210(10) |
71.7490(10) |
70.2590(10) |
| γ (°) |
61.9060(10) |
87.414(2) |
90 |
70.9820 |
68.8610(10) |
78.075(2) |
| V (Å3) |
1066.09(17) |
1550.4(2) |
3488.3(5) |
1876.4(3) |
951.03(14) |
1600.0(2) |
| Z |
1 |
1 |
4 |
2 |
1 |
1 |
| Dcalc (g cm−3) |
1.534 |
1.599 |
1.461 |
1.555 |
1.614 |
1.485 |
| μ (mm−1) |
0.677 |
0.883 |
0.623 |
0.957 |
0.955 |
0.603 |
| F(000) |
510 |
760 |
1600 |
908 |
476 |
742 |
| Rint |
0.0384 |
0.1129 |
0.0598 |
0.0000 |
0.0725 |
0.0886 |
| R1a [I > 2σ(I)] |
0.0621 |
0.1324 |
0.0460 |
0.1307 |
0.1007 |
0.1471 |
| wR2b (all data) |
0.1514 |
0.3214 |
0.0818 |
0.3409 |
0.2669 |
0.2957 |
| GOF |
1.030 |
1.038 |
1.056 |
1.256 |
1.016 |
1.017 |
| Δρmax (e Å−3) |
0.603 |
1.108 |
0.544 |
1.520 |
1.584 |
0.862 |
| Δρmin (e Å−3) |
−0.519 |
−0.971 |
−0.397 |
−0.942 |
−0.968 |
−0.499 |
Results and discussion
Description of crystal structures
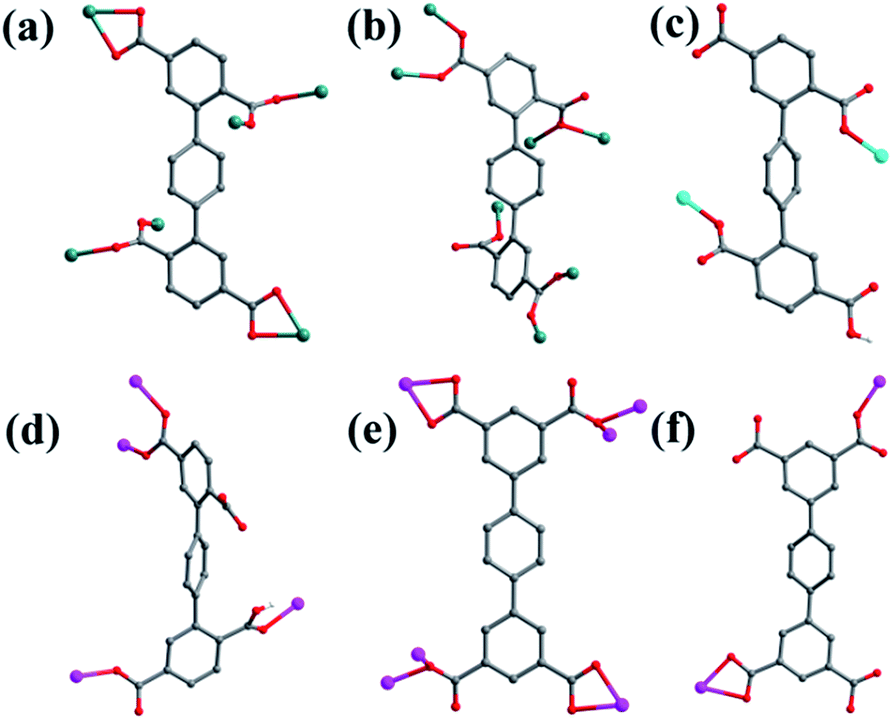
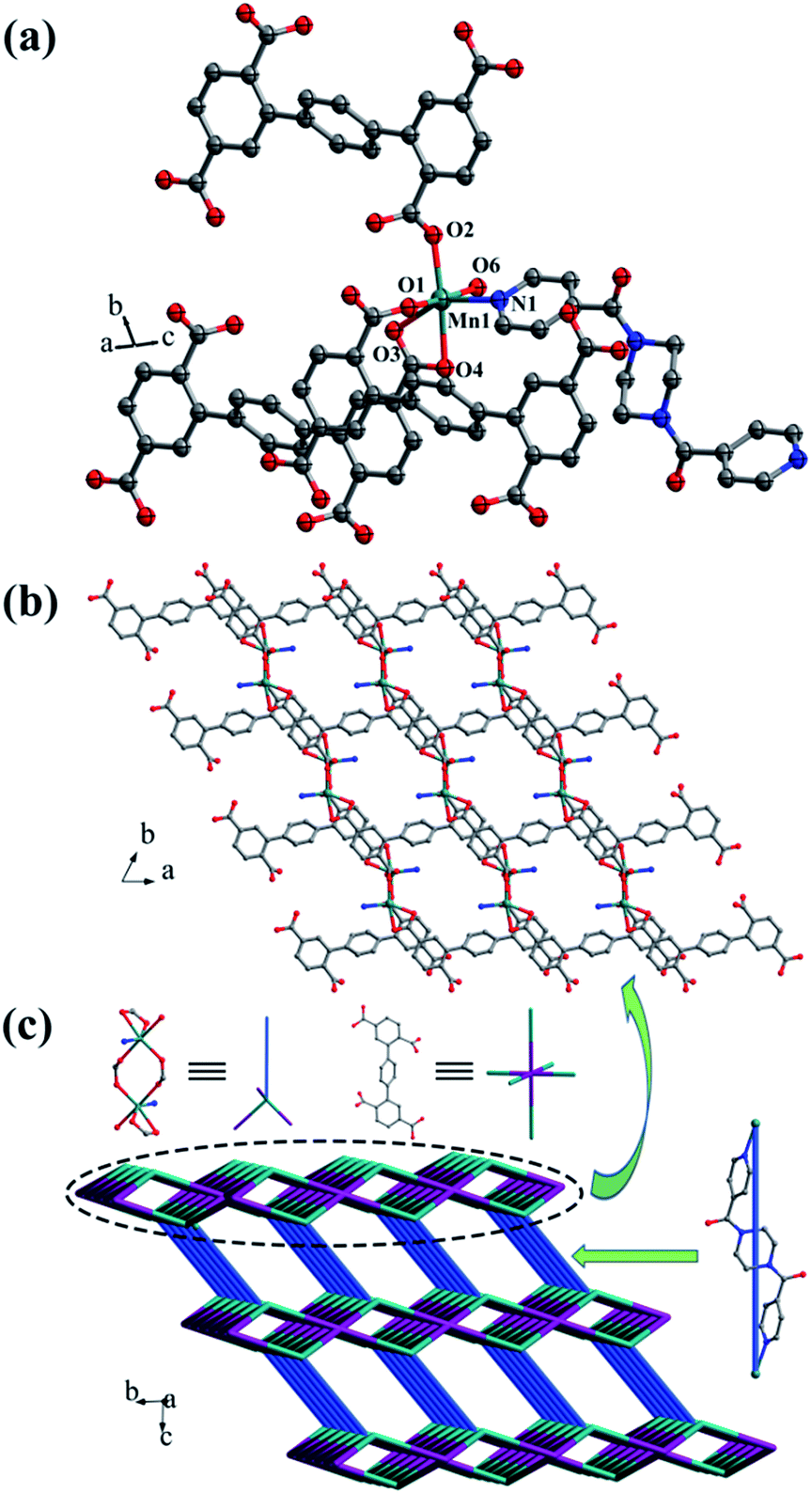
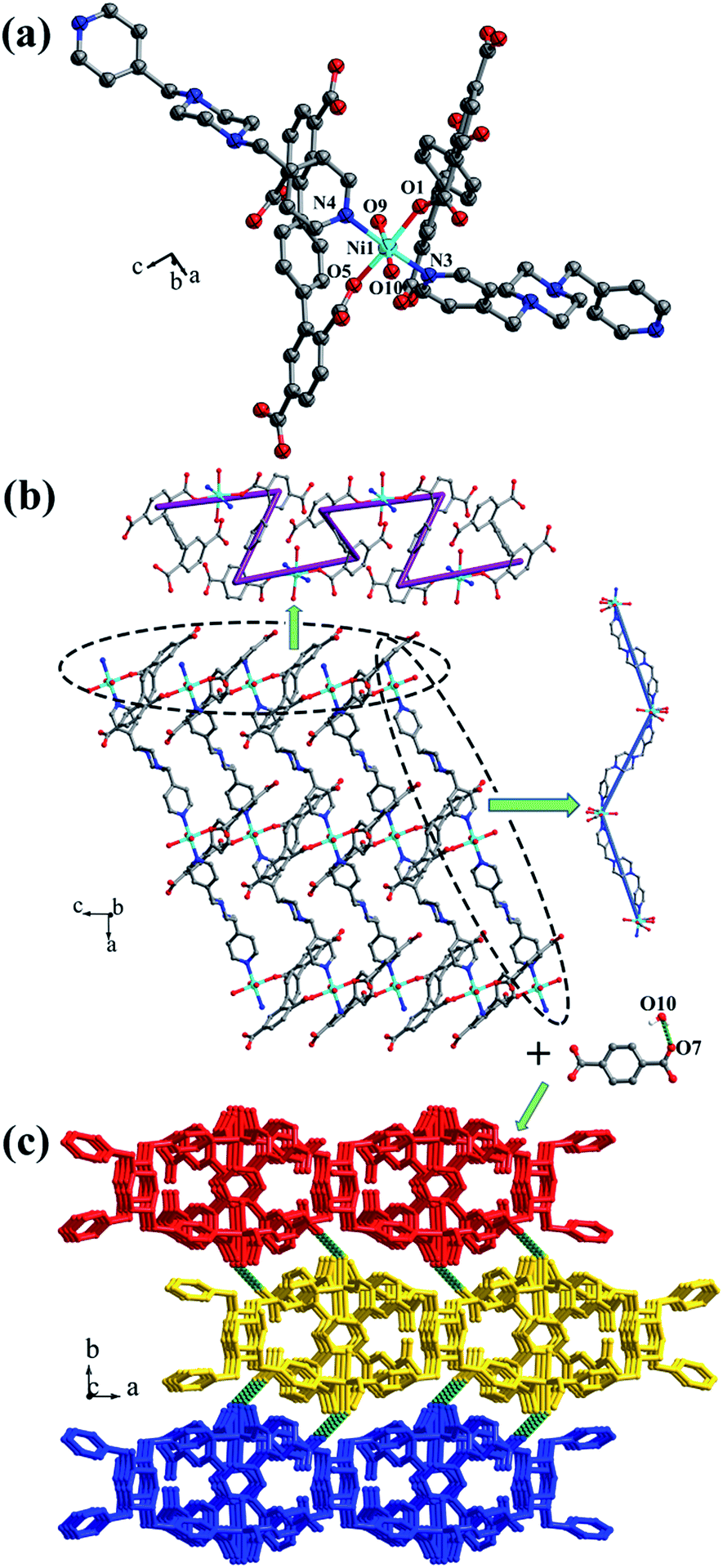
Crystal structure of {[Mn2(L1)(bpfp)(H2O)2]·6H2O·(CH3OH)}n (1). Structural analysis indicates that complex 1 is a 3D framework containing 2D Mn–L1 coordination layers. Complex 1 crystallizes in the triclinic P space group, and the asymmetric unit is composed of one MnII ion, one half of L1 ligand, one half of bpfp ligand, one coordinated water molecule, three free water molecules and one half of free methanol molecule. Mn1 is coordinated by four carboxylic oxygen atoms from three separate L1 ligands, one pyridine nitrogen atom from bpfp ligand and one coordinated water molecule, adopting {MnO5N} coordination geometry (Fig. 2a). The distances of Mn–O are in the range of 2.093(3)–2.265(3) Å and the distance of Mn1–N1 is 2.208(4) Å. The completely deprotonated L1 ligand exhibits μ1-η1:η1 and μ2-η1:η1 coordination modes (Fig. 1a). The lateral phenyl rings are parallel. The dihedral angles between lateral phenyl rings and the central phenyl ring are both 44.03°. The μ6-bridging L1 ligands are linked by MnII ions to generate a 2D coordination layer structure (Fig. 2b), and the μ2-bridging N-donor bpfp ligands with the parallel pyridine rings link the neighbouring 2D layer structures to produce a 3D framework (Fig. S2†).
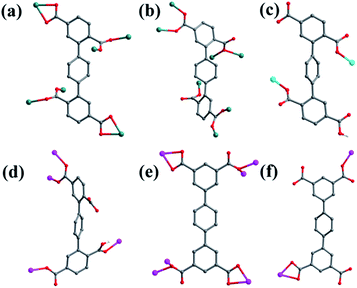 |
| | Fig. 1 Coordination modes of H4L1 and H4L2 ligands in complexes 1–6. (a) Complex 1; (b) complex 2; (c) complex 3; (d) complex 4; (e) complex 5; (f) complex 6. Color legend: teal, Mn; turquoise, Ni; pink, Co; gray, C; red, O; white, H. | |
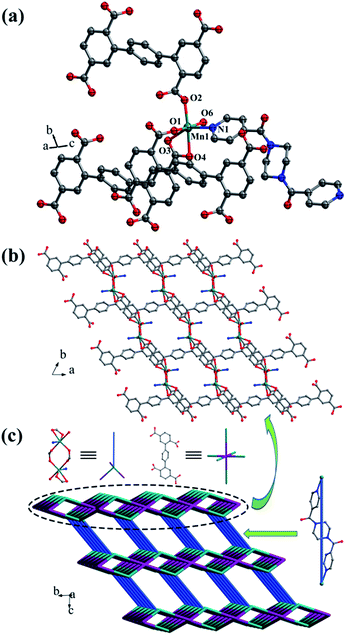 |
| | Fig. 2 (a) Coordination environment of the MnII ion in complex 1; (b) 2D layer structure in 1; (c) schematic of the (4,6)-connected 3D framework with {43·63}2{46·66·83} topology. Color legend: teal, Mn; gray, C; blue, N; red, O. All hydrogen atoms, the lattice H2O and CH3OH are omitted for clarity. | |
From a topological viewpoint, Mn1 is coordinated by three L1 ligands and one bpfp ligand, which is considered as 4-connected node. The L1 ligand linking six Mn1 is considered as 6-connected node, and the bpfp ligand acts as a linker connecting two MnII metal ions. Therefore, the 3D structure can be generalized as a (4,6)-connected net with the Schläfli symbol of {43·63}2{46·66·83} (Fig. 2c). The topology type has been reported by our group.19
Crystal structure of {[Mn4(μ3-O)2(L1)2(bpe)2(H2O)2]·2H2O}n (2). Compared with complex 1, bpfp ligand was replaced by bpe ligand, and 2D layer with (4,6)-connected net was obtained. The X-ray crystal data reveal that complex 2 also crystallizes in the triclinic P space group. The asymmetric unit is made up of two crystallographically independent MnII ions, one L1 ligand, one bpe ligand, one coordinated water molecule, one μ3-O and one free water molecule (Fig. 3a). Mn1 is located in a distorted {MnO6} square-pyramidal coordination configuration, which is surrounded by five oxygen atoms from three carboxylic oxygen atoms (O1, O3, O7) of three separate L1 ligands, one oxygen atom (O10) of one coordinated water molecule and one oxygen atom (O9) of μ3-O. The distances of Mn1–O are in the range of 2.095(9)–2.501(9) Å. Mn2 is located in a distorted {MnO5N} octahedral coordination geometry, which is surrounded with five oxygen atoms and one nitrogen atom from three carboxylic oxygen atoms (O4A, O5, O8A) of three separate L1 ligands, two oxygen atoms (O9, O9A) of two separate μ3-O and one nitrogen atom (N1) of one bpe ligand. The distances of Mn2–O are in the range of 2.149(10)–2.323(9) Å and the distance of Mn2–N1 is 2.342(12) Å. The completely deprotonated L1 ligand displays μ1-η1:η0, μ2-η2:η0 and μ2-η1:η1 coordination patterns (Fig. 1b). The bpe acts as terminal ligand linking with Mn2. The dihedral angle between the lateral phenyl rings is 82.81°, while the dihedral angles between lateral phenyl rings and the central phenyl ring are 37.52° and 45.49°, respectively. Each μ6-bridging L1 ligand is linked by dinuclear MnII units to form a (3,6)-connected 2D layer with the Schläfli symbol of {43}2{46·66·83} (Fig. 3b and S3†). The topology type has been reported by Zhao' group.20
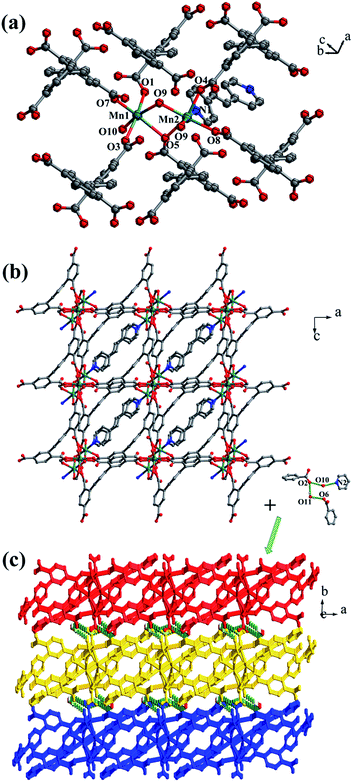 |
| | Fig. 3 (a) Coordination environment of the MnII ion in complex 2; (b) 2D layer structure in 2; (c) 3D supramolecular framework formed by hydrogen-bonding interactions in 2. Color legend: teal, Mn; gray, C; blue, N; red, O. All hydrogen atoms from ligands are omitted for clarity. | |
The 2D layer is further extended to generate a 3D supramolecular framework by three types of hydrogen-bonding interactions (Fig. 3c). The distance of O10–H10D⋯O2 formed between the coordinated water molecule and the carboxylic oxygen atom is 2.767 Å. O10–H10E⋯N2 is formed between the coordinated water molecule and the bpe ligand with the distance of 2.797 Å. O11–H11C⋯O2 and O11–H11D⋯O6 are formed between the lattice water molecules and the carboxylic oxygen atoms with the distances of 3.029 Å and 2.906 Å, respectively.
Crystal structure of [Ni(HL1)(Hbpmp)(H2O)2]n (3). Single-crystal X-ray structural analysis shows that complex 3 belongs to the monoclinic P21/c space group. The asymmetric unit has one NiII ion, one HL1 ligand, one protonated Hbpmp ligand and two coordinated water molecules. The NiII metal ion is surrounded by two oxygen atoms (O1, O5A) from two separate HL1 ligands, two nitrogen atoms (N3, N4) from Hbpmp ligands and two oxygen atoms (O9, O10) from two coordinated water molecules, displaying a distorted {NiO4N2} octahedral coordination configuration (Fig. 4a). The coordinated water molecules are in trans configuration. The distances of Ni–O are in the range of 2.023(2)–2.146(2) Å, and Ni–N are 2.111(2) Å, 2.118(2) Å. The partially deprotonated HL1 ligands adopt μ1-η1:η0 patterns linking with NiII ion (Fig. 1c). The dihedral angle between the lateral phenyl rings is 4.78°. The dihedral angles between lateral phenyl rings and the central phenyl ring are 59.34° and 63.83°, respectively. Each μ2-bridging HL1 ligand is connected by NiII metal ions to form a “Z”-type 1D chain, and two nitrogen atoms from bis-pyridine Hbpmp ligands link NiII metal ions to form a 1D chair-like chain (Fig. 4b), displaying a 4-connected 2D layer with the Schläfli symbol of {44·62} (Fig. S4†).
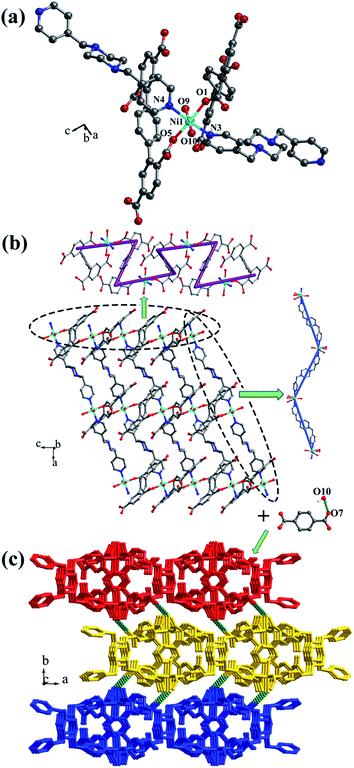 |
| | Fig. 4 (a) Coordination environment of the NiII ion in complex 3; (b) 2D layer structure with two types of 1D chain in 3; (c) 3D supramolecular framework formed by hydrogen-bonding interactions in 3. Color legend: turquoise, Ni; gray, C; blue, N; red, O. All hydrogen atoms from ligands are omitted for clarity. | |
The 3D supramolecular framework is generated by hydrogen-bonding interactions between the coordinated water molecules (O10) and carboxylic oxygen atoms (O7). The distance of the O10–H10D⋯O7 hydrogen bond is 2.741 Å (Fig. 4c).
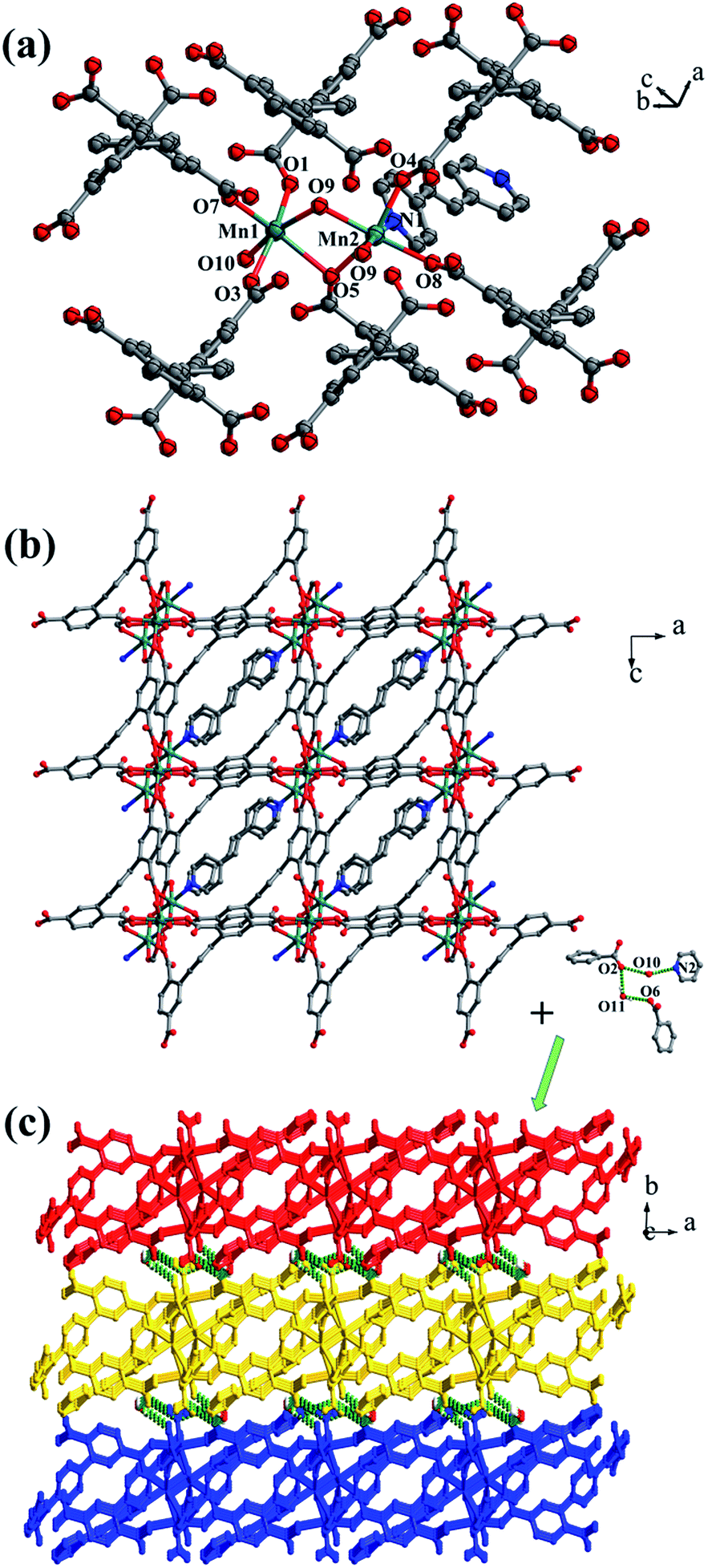
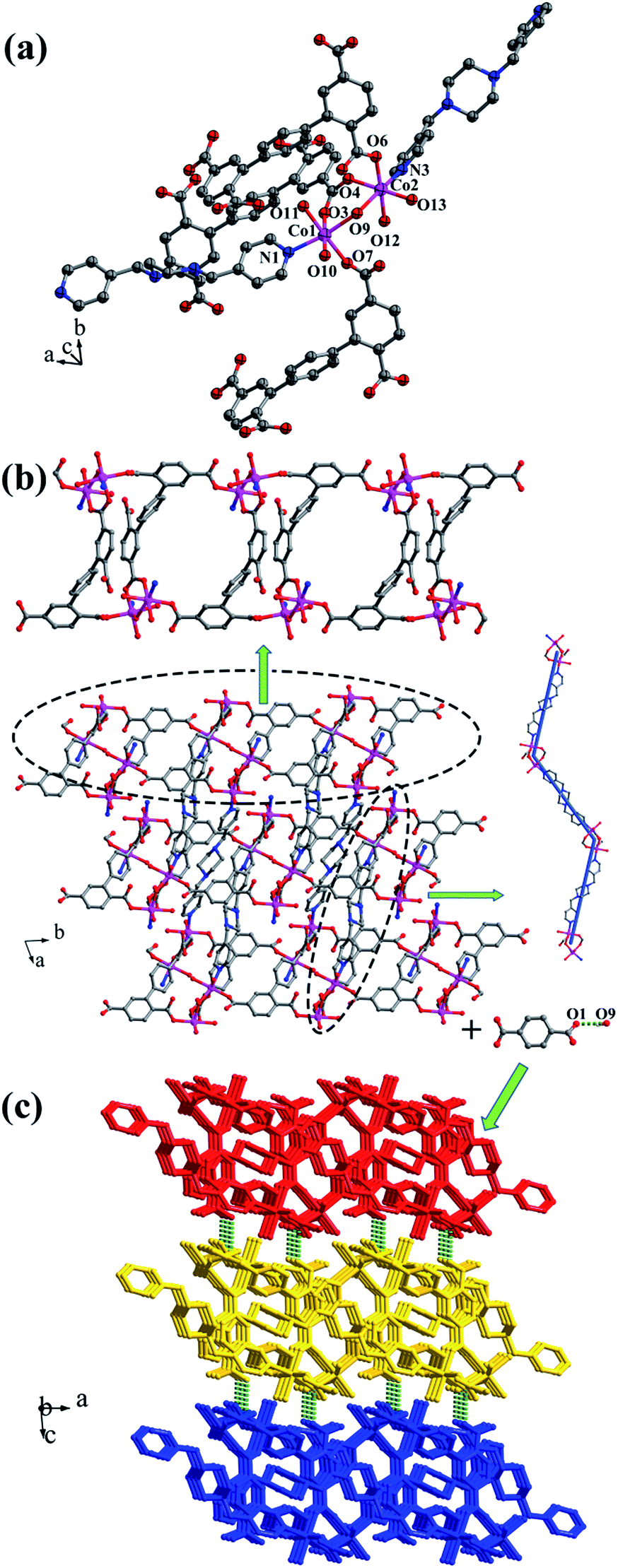
Crystal structure of [Co2(μ2-OH)(HL1)(bpmp)(H2O)4]n (4). The space group of complex 4 is triclinic P, and complex 4 is a 3D supramolecular framework based on 4-connected 2D layer. The asymmetric unit is composed of two crystallographically independent CoII ions, one HL1 ligand, one bpmp ligand, four coordinated water molecules and one μ2-OH (Fig. 5a). Co1 and Co2 are all located in distorted octahedral configuration, which are bonded by one hydroxyl oxygen atom (O9). Co1 and Co2 are coordinated with two oxygen atoms (O3 and O7 for Co1, O4 and O6A for Co2) from carboxyl groups of two separated HL1 ligands, one nitrogen atom (N1 for Co1, N3 for Co2) of two separated bpmp ligands, two oxygen atoms (O10 and O11 for Co1, O12 and O13 for Co2) from the coordinated water molecules and one hydroxyl oxygen atom (O9). The distances are in the range of 2.068(8)–2.298(8) Å for Co1, 2.056(8)–2.201(7) Å for Co2, and the distances of Co1–N1 and Co2–N3 are 2.221(10) Å, 2.153(9) Å, respectively. The partially deprotonated HL1 ligand possesses two kinds of coordination patterns: monodentate μ1-η1:η0 and bidentate μ2-η1:η1 (Fig. 1d). The dihedral angle between lateral phenyl rings is 75.93°, and the dihedral angles are 50.58° and 53.56° between the lateral phenyl rings and the central phenyl ring, respectively. Each μ4-bridging HL1 ligand is linked with CoII metal ions to form a 1D chain, and the μ2-bridging N-donor bpmp ligands connect the neighbouring 1D chains to give a 4-connected 2D layer (Fig. 5b) with the Schläfli symbol of {4·9·10}{4·92}{42·9 × 103} (Fig. S5†).
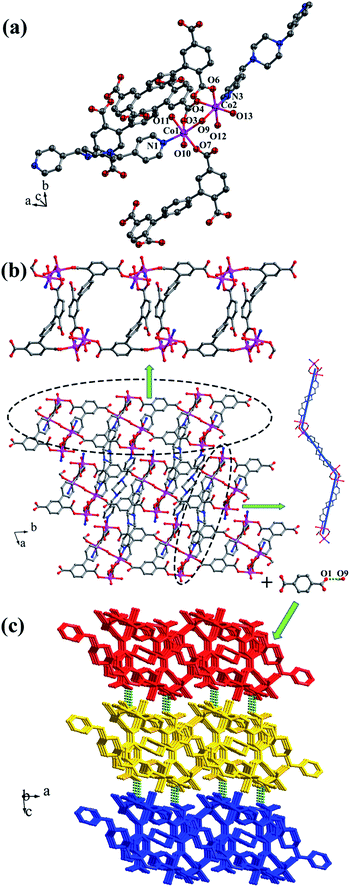 |
| | Fig. 5 (a) Coordination environment of the CoII ion in complex 4; (b) 2D layer structure with two types of 1D chain in 4; (c) 3D supramolecular framework formed by hydrogen-bonding interactions in 4. Color legend: pink, Co; gray, C; blue, N; red, O. All hydrogen atoms from ligands are omitted for clarity. | |
The 3D supramolecular framework is formed by O–H⋯O hydrogen-bonding interactions between the coordinated water molecules (O9) and carboxylic oxygen atoms (O1). The distance of the O9–H9A⋯O1 hydrogen bond is 2.751 Å (Fig. 5c).
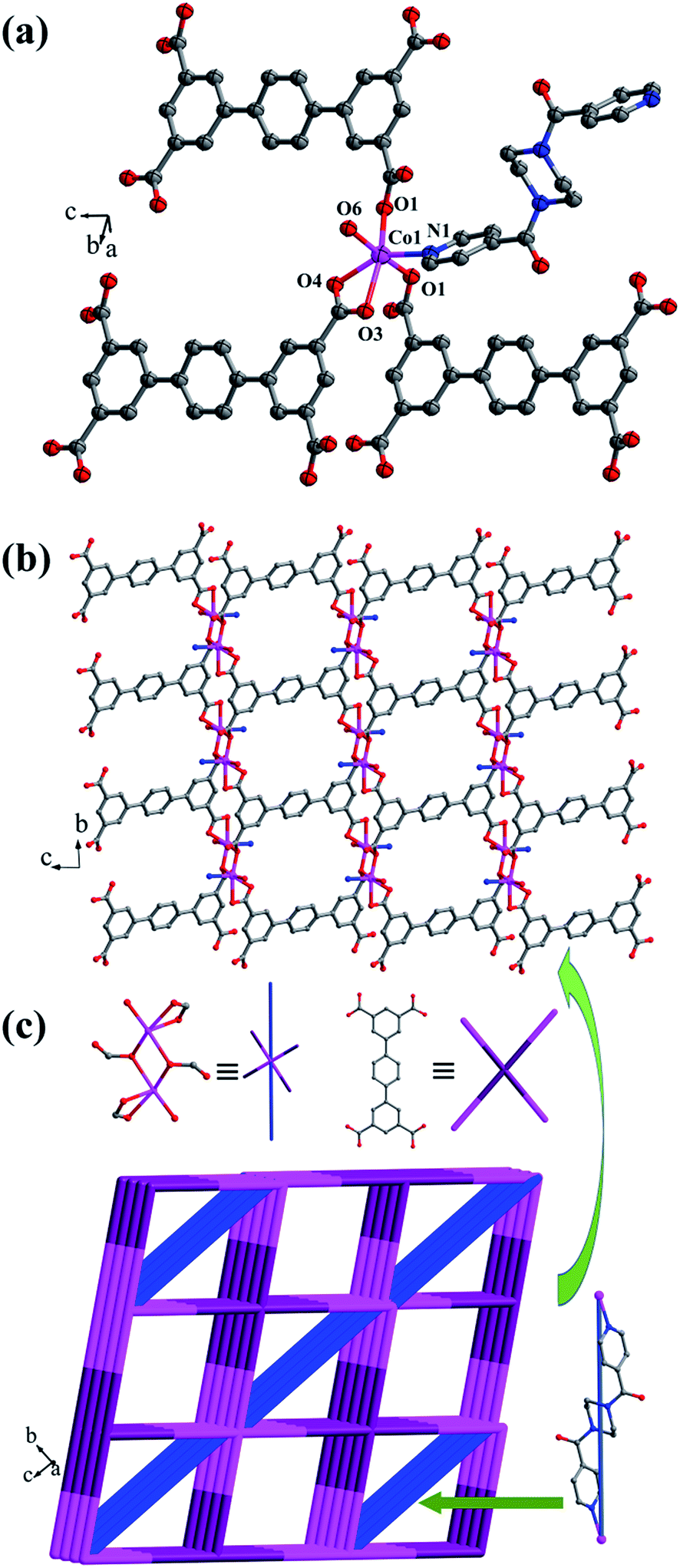
Crystal structure of {[Co2(L2)(bpfp)(H2O)2]·4H2O}n (5). X-ray crystal data reveal that complex 5 crystallizes triclinic P space group, and it shows a 3D framework based on the Co–L2 coordination layers. The asymmetric unit is made up of one CoII ion, a half L2 ligand, a half bpfp ligand, one coordinated water molecule and two free water molecules. Each CoII ion is in a distorted {CoO5N} coordination environment and is surrounded by four oxygen atoms (O1, O1B, O3A, O4A) of three separate L2 ligands, one oxygen atom (O6) of coordinated water molecule and one pyridine nitrogen atom (N1) of bpfp ligand (Fig. 6a). The distances of Co–O are in the range of 2.071(5)–2.306(5) Å, and Co–N are 2.099(6) Å. The completely deprotonated L2 ligand adopts two kinds of coordination patterns: μ1-η1:η1 and μ2-η2:η0 (Fig. 1e). The two parallel phenyl rings have a dihedral angle of 15.63° with the central phenyl ring. Each μ6-bridging L2 ligand is connected with CoII metal ions to form a 2D layer (Fig. 6b), and the μ2-bridging N-donor bpfp ligands link the neighbouring 2D layers to generate a 3D framework (Fig. S6†).
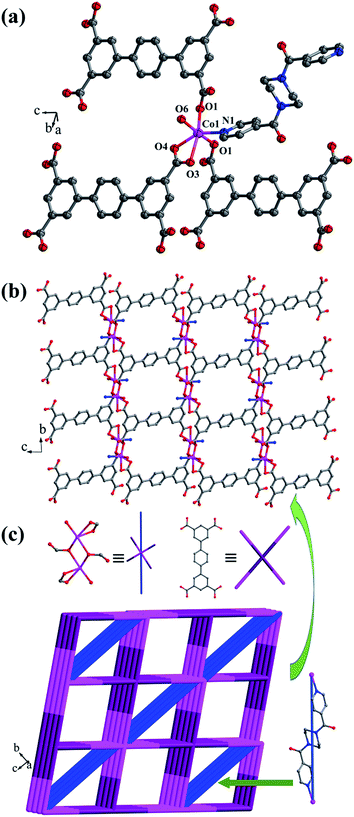 |
| | Fig. 6 (a) Coordination environment of the CoII ion in complex 5; (b) 2D layer structure in 5; (c) Schematic of the (4,6)-connected 3D framework with {44·610·8}{44·62} topology. Color legend: pink, Co; gray, C; blue, N; red, O. All hydrogen atoms and the lattice H2O are omitted for clarity. | |
From a topological viewpoint, Co1 ions are coordinated by three L2 ligands and one bpfp ligand, which are considered as 4-connected nodes. The L2 ligand connected with six Co1 ions is considered as 6-connected node, and the bpfp ligand acts as a linker. Therefore, the 3D structure can be simplified as a (4,6)-connected {44·610·8}{44·62} framework (Fig. 6c).
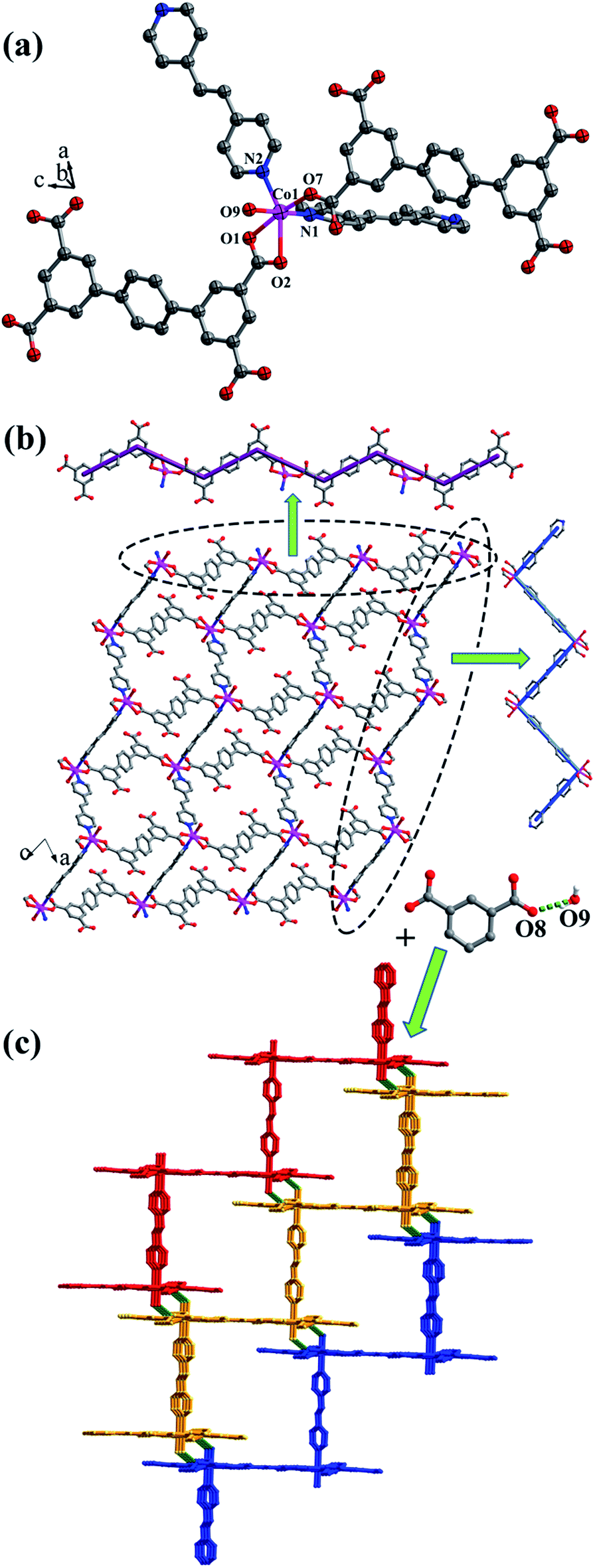
Crystal structure of {[Co2(L2)2(bpe)2(H2O)2·2H2O]·(NH4)4}n (6). When bpe ligand was used instead of bpfp ligand, a 3D supramolecular framework containing (4,4)-connected net was obtained. Single-crystal X-ray structural analysis indicates that complex 6 belongs to the triclinic P space group. The asymmetric unit is composed of one CoII ion, one L2 ligand, one bpe ligand, one coordinated water molecule, one free water molecule and two NH4+. The CoII ion is surrounded by three oxygen atoms (O1, O2, O7A) from two separate L2 ligands, two nitrogen atoms (N1, N2) from bpe ligands and one oxygen atom (O9) of coordinated water molecule, adopting a distorted {CoO4N2} octahedral coordination configuration (Fig. 7a). The distances of Co–O are in the range of 2.006(9)–2.304(9) Å, and Co–N are 2.133(11) Å, 2.146(13) Å. The fully deprotonated L2 ligands possess μ1-η1:η0 and μ1-η1:η1 patterns (Fig. 1f). The dihedral angle between the lateral phenyl rings is 2.60°. The dihedral angles between lateral phenyl rings and the central phenyl ring are 32.58°. Each μ2-bridging L2 ligand is linked with CoII metal ions to form a wave-type 1D chain, and bpe ligands connected the neighbouring CoII metal ions to form a 4-connected 2D layer (Fig. 7b) with the Schläfli symbol of {44·62} (Fig. S7†).
 |
| | Fig. 7 (a) Coordination environment of the CoII ion in complex 6; (b) 2D layer structure with two types of 1D chain in 6; (c) 3D supramolecular framework formed by hydrogen-bonding interactions in 6. Color legend: pink, Co; gray, C; blue, N; red, O. All hydrogen atoms from ligands, the lattice H2O and NH4+ are omitted for clarity. | |
The 2D layer is further extended to form a 3D supramolecular framework by O–H⋯O hydrogen-bonding interactions between the coordinated water molecules (O9) and carboxylic oxygen atoms (O8). The distance of the O9–H9D⋯O8 hydrogen bond is 2.771 Å (Fig. 7c).
Influence of the different bis-pyridyl ligands and metal ions on the architectures of complexes 1–6
In our work, [1,1′:4′,1′′-terphenyl]-2,2′′,5,5′′-tetracarboxylic acid (H4L1), [1,1′:4′,1′′-terphenyl]-3,3′′,5,5′′-tetracarboxylic acid (H4L2) as the main ligands react with the transition metal ions (MnII, NiII and CoII) and different auxiliary bis-pyridyl ligands (bpfp, bpe and bpmp) to construct six 2D and 3D coordination polymers. In the structures of complexes 1 and 2, the completely deprotonated L1 ligands exhibit multiply bidentate and monodentate coordination modes. Complex 1 is a 3D {43·63}2{46·66·83} framework based on 2D Mn–L1 layers and the bridging bpfp ligand, while complex 2 is a {43}2{46·66·83} 2D layer constructed from MnII ions, L1 ligands, and the terminal bpe ligands. The reason for the different structures may be that bpfp ligand is more flexible than bpe ligand in the presence of piperazine ring. For complexes 5 and 6, the fully deprotonated L2 ligands display multiply coordination modes. Complex 5 is a 3D {44·610·8}{44·62} net based on a 2D Co–L2 layers and the bridging bpfp ligand, while complex 6 is a {44·62} 2D layer formed with Co–L2 chain and Co–bpe chain. For complexes 3 and 4, different transition metal ions coordinate with the partially deprotonated HL1 ligands and bpmp ligands. In complex 3, a {44·62} 2D layer is constructed from Ni–HL1 chain and Ni–bpmp chain. The NiII metal ion is coordinated with four oxygen atoms and two nitrogen atoms, exhibiting a distorted octahedral coordination configuration. Compared with complex 3, the geometry of CoII ions for complex 4 is the same as that of NiII ions. However, the coordination environments of the two crystallographically independent CoII ions are different from that of NiII ions. In the final structures, Co–HL1 chain and Co–bpmp chain form a {4·9·10}{4·92}{42·9 × 103} 2D layer.
X-ray powder diffraction
In order to prove the purities of complexes 1–6, the X-ray powder diffraction (XRPD) patterns have been detected (Fig. S8†). The experimental patterns match the simulated patterns, confirming the phase purities of the samples. The slight differences of intensity between the simulated and experimental peaks may be attributed to the preferred orientation of the crystals.
Thermal stability analysis
Thermal behaviors of complexes 1–6 were performed by thermogravimetric (TG) analyses (Fig. S9†) in a flowing nitrogen atmosphere between 30 °C and 800 °C at a heating rate of 10 °C min−1. TGA reveal that complexes 1–6 exhibit two weight loss steps. For complex 1, the weight loss in the first step is 17.20% between 30 °C and 127 °C, which is corresponding to the loss of the coordinated water molecules, the lattice water molecules and methanol molecules (calc. 17.87%). The second weight loss is 67.86% between 404 °C and 563 °C should be attributed to the release of H4L1 and bpfp ligands (calc. 67.71%). The possible residual is MnO (14.94%, calc. 14.42%). For complex 2, the first weight loss is 5.15% due to the loss of the coordinated water molecules and the lattice water molecules (calc. 4.82%). The second weight loss is 75.60% between 110 °C and 530 °C, which should be ascribed to the decomposition of H4L1 and bpe ligands (calc. 76.18%). The possible residual MnO is about 19.25% (calc. 19.00%). For complex 3, the weight loss in the first step is 4.75% between 180 °C and 295 °C due to the loss of coordinated water molecules (calc. 4.69%). The second weight loss is 85.45% between 295 °C and 620 °C, which should be attributed to the release of H4L1 and bpfp ligands (calc. 85.58%). The possible residual is NiO (9.80%, calc. 9.73%). For complex 4, the first weight loss is 8.30% between 141 °C and 178 °C corresponding to the loss of coordinated water molecule (calc. 8.19%). The second weight loss is 74.74% between 178 °C and 620 °C due to the decomposition of H4L1 and bpmp ligands (calc. 74.75%). The possible residual CoO is about 16.96% (calc. 17.06%). For complex 5, the weight loss in the first step is 10.70% in the temperature range of 150–190 °C, which is attributed to the loss of the coordinated water molecules and the lattice water molecules (calc. 11.68%). The second weight loss of 73.24% between 430 °C and 650 °C should be ascribed to the release of H4L2 and bpfp ligands (calc. 72.11%). The possible residual is CoO (16.06%, calc. 16.21%). For complex 6, a weight loss of 10.35% between 125 °C and 155 °C is due to the loss of the coordinated water molecules, the lattice water molecules and NH4+ (calc. 10.07%). The second weight loss of 79.02% between 155 °C and 600 °C should be attributed to the decomposition of H4L2 and bpe ligands (calc. 79.46%). The possible residual is CoO (10.63%, calc. 10.47%).
Fluorescence properties
The solid-state fluorescence properties of complexes 1–6 and the free organic ligands (H4L1, H4L2, bpfp, bpe and bpmp) have been researched at room temperature. The H4L1 and H4L2 ligands show intense emission peaks with maxima at 436 nm and 431 nm, respectively. The N-donor ligands (bpfp, bpe and bpmp) exhibit no significant fluorescent emission (Fig. S10†). Complexes 1–6 display no obvious fluorescence emission, which may be ascribed to d–d electron transitions and vibrations of single electrons in Mn, Ni or Co complexes.21
Water vapor sorption of complexes 1 and 2
Since there are many interstitial water and methanol molecules in complexes 1 and 2, we tried to investigate the sorption properties and the reversibility of the sorption properties. The TGA studies indicate that the solvent molecules could be removed by heating the samples above 130 °C. Therefore, we measured the water vapor sorption behaviour of complexes 1 and 2. Before the measurements, the samples of complexes 1 and 2 were heated at 130 °C for 12 h under vacuum to remove the guest molecules. Water vapor sorption isotherms at 298 K are presented in Fig. 8. Complexes 1 and 2 display the increasing uptake of water, reaching a maximum value of 15.17 mL g−1 and 27.24 mL g−1 at P/P0 = 0.95 (P0 is the saturation pressure of H2O at 298 K), respectively. The reason of the obvious hysteresis for desorption isotherms may be the strong adsorption for the water molecules in the pores of these complexes due to the hydrogen bonding interactions in the structure.
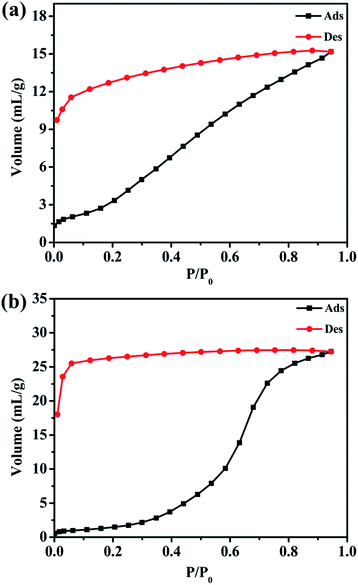 |
| | Fig. 8 Water adsorption–desorption isotherms for (a) complex 1; (b) complex 2 at 298 K. | |
Conclusions
In summary, six coordination polymers based on the main ligands [1,1′:4′,1′′-terphenyl]-2,2′′,5,5′′-tetracarboxylic acid (H4L1), [1,1′:4′,1′′-terphenyl]-3,3′′,5,5′′-tetracarboxylic acid (H4L2) and three types of auxiliary bis-pyridyl ligand have been synthesized under hydrothermal conditions. The different flexibilities of the bis-pyridyl ligands have great influences on the architectural diversity of complexes 1–6. Complexes 1 and 2 exhibit a certain water vapor uptake. This work evidently indicates that the effect of auxiliary ligands and coordination modes of H4L1 and H4L2 are conclusive in the construction of coordination polymers.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21271024 and 20971014) and the 111 Project (B07012) in China.
Notes and references
-
(a) M. Hirscher, Angew. Chem., Int. Ed., 2011, 50, 581 CrossRef CAS PubMed;
(b) J. R. Li and H. C. Zhou, Nature, 2010, 2, 893 CAS;
(c) H. L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351 RSC;
(d) R. B. Getman, Y. S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703 CrossRef CAS PubMed;
(e) Z. Y. Guo, H. Wu, G. Srinivas, Y. M. Zhou, S. C. Xiang, Z. X. Chen, Y. T. Yang, W. Zhou, M. O'Keeffe and B. L. Chen, Angew. Chem., Int. Ed., 2011, 50, 3178 CrossRef CAS PubMed.
-
(a) Z. Y. Guo, J. B. Yu, G. H. Li, Z. J. Si, H. D Guo and H. J. Zhang, CrystEngComm, 2009, 11, 2254 RSC;
(b) M. H. Zeng, Q. X. Wang, Y. X. Tan, S. Hu, H. X. Zhao, L. S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561 CrossRef CAS PubMed;
(c) X. Zhao, X. H. Bu, Q. G. Zhai, H. Tran and P. Y. Feng, J. Am. Chem. Soc., 2015, 137, 1396 CrossRef CAS PubMed;
(d) F. Sun, Z. Yin, Q. Q. Wang, D. Sun, M. H. Zeng and M. Kurmoo, Angew. Chem., Int. Ed., 2013, 52, 4538 CrossRef CAS PubMed.
-
(a) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650 CrossRef CAS PubMed;
(b) A. Z. Yin, Q. X. Wang and M. H. Zeng, J. Am. Chem. Soc., 2012, 134, 4857 CrossRef PubMed;
(c) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed.
-
(a) G. H. Chen, Z. J. Zhang, S. C. Xiang and B. L. Chen, CrystEngComm, 2013, 15, 5232 RSC;
(b) J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed;
(c) Y. J. Cui, Y. F. Yue, G. D. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(d) L. J. Dong, W. Chu, Q. L. Zhu and R. D. Huang, Cryst. Growth Des., 2011, 1, 93 CrossRef;
(e) C. A. Kent, D. Liu, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2012, 134, 3991 CrossRef CAS PubMed.
-
(a) N. B. Shustova, B. D. McCarthy and M. Dinca, J. Am. Chem. Soc., 2011, 133, 20126 CrossRef CAS PubMed;
(b) C. B. He, K. D. Lu and W. B. Lin, J. Am. Chem. Soc., 2014, 136, 12253 CrossRef CAS PubMed.
-
(a) G. L. Zhuang, L. Tan, W. L. Chen, J. Q. Bai, X. Zhong and J. G. Wang, CrystEngComm, 2014, 16, 6963 RSC;
(b) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed;
(c) Z. Y. Guo, H. Xu, S. Q. Su, J. F. Cai, S. Dang, S. C. Xiang, G. D. Qian, H. J. Zhang, M. O'Keeffe and B. L. Chen, Chem. Commun., 2011, 47, 5551 RSC;
(d) Z. C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
-
(a) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC;
(b) H. C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC;
(c) A. Karmakar, L. M. D. R. S. Martins, S. Hazra, M. F. C. G. D. Silva and A. J. L. Pombeiro, Cryst. Growth Des., 2016, 16, 1837 CrossRef CAS;
(d) W. Y. Gao, Y. Chen, Y. H. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. F. Cai, Y. S. Chen and S. Q. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615 CrossRef CAS PubMed.
-
(a) X. Y. Cao, L. Li, C. X. Li, L. Lv and R. D. Huang, CrystEngComm, 2014, 16, 5093 RSC;
(b) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC;
(c) M. H. Zeng, Z. Yin, Y. X. Tan, W. X. Zhang, Y. P. He and M. Kurmoo, J. Am. Chem. Soc., 2014, 136, 4680 CrossRef CAS PubMed;
(d) L. J. Dong, X. F. Li, J. Cao, W. Chu and R. D. Huang, Dalton Trans., 2012, 42, 1342 RSC.
-
(a) J. C. Jin, J. Wu, G. P. Yang, Y. L. Wu and Y. Y. Wang, Chem. Commun., 2016, 52, 8475 RSC;
(b) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed;
(c) J. C. Jiang, H. Furukawa, Y. B. Zhang and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 10244 CrossRef CAS PubMed.
-
(a) Z. J. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920 CrossRef CAS PubMed;
(b) D. M. Chen, J. Y. Tian and C. S. Liu, Inorg. Chem., 2016, 55, 8892 CrossRef CAS PubMed;
(c) S. Ganguly, S. Mukherjee and P. Dastidar, Cryst. Growth Des., 2016, 16, 5247 CrossRef CAS.
-
(a) D. Wu, J. J. Gassensmith, D. Gouvêa, S. Ushakov, J. F. Stoddart and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 6790 CrossRef CAS PubMed;
(b) F. Gross, E. Sherman, S. L. Mahoney and J. J. Vajo, J. Phys. Chem. A, 2013, 117, 3771 CrossRef PubMed;
(c) T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427 CrossRef CAS PubMed;
(d) V. Guillerm, L. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, L. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673 CAS.
-
(a) J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195 CrossRef CAS PubMed;
(b) X. Zhou, P. Liu, W. H. Huang, M. Kang, Y. Y. Wang and Q. Z. Shi, CrystEngComm, 2013, 15, 8125 RSC;
(c) Y. Q. Lan, H. L. Jiang, S. L. Li and Q. Xu, Inorg. Chem., 2012, 51, 7484 CrossRef CAS PubMed;
(d) H. J. Li, B. Zhao, R. Ding, Y. Y. Jia, H. W. Hou and Y. T. Fan, Cryst. Growth Des., 2012, 12, 4170 CrossRef CAS.
-
(a) N. Chen, M. X. Li, P. Yang, X. He, M. Shao and S. R. Zhu, Cryst. Growth Des., 2013, 13, 2650 CrossRef CAS;
(b) Q. G. Zhai, X. H. Bu, C. Y. Mao, X. Zhao and P. Y. Feng, J. Am. Chem. Soc., 2016, 138, 2524 CrossRef CAS PubMed.
-
(a) X. J. Gao, M. X. Zheng, L. Qin, K. Shen and H. G. Zheng, Cryst. Growth Des., 2016, 16, 4711 CrossRef CAS;
(b) N. C. Thacker, Z. K. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. B. Lin, J. Am. Chem. Soc., 2016, 138, 3501 CrossRef CAS PubMed;
(c) W. D. Fan, H. Lin, X. Yuan, F. N. Dai, Z. Y. Xiao, L. L. Zhang, L. W. Luo and R. M. Wang, Inorg. Chem., 2016, 55, 6420 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
-
(a) B. B. Kang, N. Wei and Z. B. Han, RSC Adv., 2015, 5, 1605 RSC;
(b) Y. L. Gai, F. L. Jiang, L. Chen, Y. Bu, K. Z. Su, S. A. Al-Thabaiti and M. C. Hong, Inorg. Chem., 2013, 52, 7658 CrossRef CAS PubMed;
(c) Y. L. Wu, G. P. Yang, X. Zhou, J. Li, Y. Ning and Y. Y. Wang, Dalton Trans., 2015, 44, 10385 RSC;
(d) Y. L. Wu, G. P. Yang, Y. Q. Zhao, W. P. Wu, B. Liu and Y. Y. Wang, Dalton Trans., 2015, 44, 3271 RSC.
-
(a) L. L. Zhang, F. L. Liu, Y. Guo, X. P. Wang, J. Guo, Y. H. Wei, Z. Chen and D. F. Sun, Cryst. Growth Des., 2012, 12, 6215 CrossRef CAS;
(b) H. Y. Zang, Y. Q. Lan, G. S. Yang, X. L. Wang, K. Z. Shao, G. J. Xu and Z. M. Su, CrystEngComm, 2010, 12, 434 RSC;
(c) C. Pettinari, N. Masciocchi, L. Pandolfo and D. Pucci, Chem.–Eur. J., 2010, 16, 1106 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- B. Mu and R. D. Huang, CrystEngComm, 2016, 18, 986 RSC.
- X. T. Zhang, L. M. Fan, X. Zhao, D. Sun, D. C. Li and J. M. Dou, CrystEngComm, 2012, 14, 2053 RSC.
- H. Y. Li, L. H. Cao, Y. L. Wei, H. Xu and S. Q. Zang, CrystEngComm, 2015, 17, 6297 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic cif files, check cif files, selected bond lengths and angles, PXRD curve, TGA curve, fluorescence spectra and additional figures for complexes 1–6. Complexes 1–6 have been assigned the following numbers: CCDC 1056874–1056879, respectively. CCDC 1056874–1056879. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25319a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence