
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fe3O4@SiO2–MoO3H nanoparticles: a magnetically recyclable nanocatalyst system for the synthesis of 1,8-dioxo-decahydroacridine derivatives†
Mahtab Kiani*a and
Mohammad Mohammadipourb
aYoung Researchers and Elite Club, Karaj Branch, Islamic Azad University, Karaj, Iran. E-mail: mahtabkiani47@yahoo.com
bDepartment of Chemistry, Semnan University, Semnan 35131-19111, Iran
First published on 4th January 2017
Abstract
Molybdic acid-functionalized silica-coated nano-Fe3O4 particles (Fe3O4@SiO2–MoO3H) have been prepared as a novel heterogeneous acid catalyst using a facile process. The material was subsequently identified as an efficient catalyst for the synthesis of 1,8-dioxo-decahydroacridine derivatives under solvent free conditions. The catalyst could be readily recovered using a simple external magnet and reused several times without any significant loss in activity. Short reaction time, excellent yields and simple work-up are the advantages of this procedure.
Introduction
Nanocatalysis is a rapidly growing field which involves the use of nanomaterials as catalysts for a variety of homogeneous and heterogeneous catalysis applications.1–3Magnetic nanoparticles (MNPs) have attracted growing interest in biomedical applications such as targeted drug delivery, magnetic resonance contrast, therapeutic agents, labeling and sorting of cells and bioseparation.4,5
Fe3O4 is a very popular magnetic media type absorber due to its unique magnetic features, low-cost and strong absorption characteristics.6 However, pure Fe3O4 materials usually suffer from ease of oxidation, fast heat transfer and poor flame-retardant effect, which limit their applications. To solve these problems, various inorganic and polymeric materials have been reported as carriers of magnetic materials.7 Among them, the silica coating is a very good surface modifier, because of its excellent stability, biocompatibility, nontoxicity and easily furthered conjugation with various functional groups, thus enabling the coupling and labeling of biotargets with selectivity and specificity.8
Magnetic hollow silica materials with different morphology and structure have been synthesized by the different methods.9 The protective silica layer can minimize the aggregation of MNPs by reducing the magnetic dipolar attraction and provides a chemically inert surface to MNPs in biological environments. Furthermore, silica-coated MNPs can be easily activated to anchor various functional groups owing to abundant Si–OH groups on the silica surface. The fabrication of silica-coated MNPs with controlled magnetism require the simultaneous control of surface morphology and the thickness of nonmagnetic silica layer relative to the size of magnetic core.10
Acridine-1,8-dione derivatives as a class of interesting heterocyclic compounds exhibit numerous publications in organic, pharmaceutical and medicinal chemistry fields because of their potential biological activities and presence in a variety of significant natural products and synthetic dye-stuffs.11 Acridine derivatives have been used as anti-tumor,12 cytotoxic,13 anti-fungal properties,14 antimicrobial,15 anti-cancer16 and anti-multidrug-resistant.17
There are several methods reported in the literature for the synthesis of 1,8-dioxo-decahydroacridines via three component coupling of aldehydes, dimedone and amines in the presence of diverse catalysts including proline,18 benzyltriethyl ammonium chloride (TEBAC),19 fluorotailed acidic imidazolium salts,20 1-methylimidazolium trifluoroacetate [Hmim]TFA,21 ceric ammonium nitrate (CAN),22 silica-bonded N-propyl sulfamic acid (SBNPSA),23 amberlyst-15,24 carbon-based solid acid (SBSA),25 and 4-dodecylbenzenesulfonic acid (DBSA).26 However, many of these methods suffer from disadvantages such as low yields, toxic organic solvents, long reaction times, expensive catalysts, the requirement of special apparatus, laborious work-up procedures, and harsh reaction conditions. Thus, the development of simple, efficient, high-yielding and eco-friendly methods using new catalysts for the synthesis of these compounds would be highly desirable.
During the course of our recent research program on the development of new condition for organic transformations,27–29 recently, molybdic acid-functionalized silica-coated nano-Fe3O4 particles have been prepared in our laboratory. It displayed a high stability and an impressive catalytic activity in the synthesis of 1,8-dioxo-decahydroacridine derivatives under solvent free conditions. The performance and recyclability behavior of this nano magnetic catalyst in the preparation of 1,8-dioxo-decahydroacridine is reported in this study to extend possibilities of this new catalyst.
Experimental
Methods and materials
The chemicals were purchased from Merck and Aldrich chemical companies. The reactions were monitored by TLC (silica-gel 60 F 254, hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc). Fourier transform infrared (FT-IR) spectroscopy spectra were recorded on a Shimadzu-470 spectrometer, using KBr pellets and the melting points were determined on a KRUSS model instrument. 1H NMR spectra were recorded on a Bruker Avance II 400 NMR spectrometer at 400 MHz, in which DMSO-d6 was used as solvent and TMS as the internal standard. X-ray diffraction (XRD) pattern was obtained by Philips X Pert Pro X diffractometer operated with a Ni filtered Cu Kα radiation source. Transmission electron microscopy (TEM) images of the electrocatalyst were recorded using a Philips CM-10 TEM microscope operated at 100 kV. Field emission scanning electron microscopy (SEM) and X-ray energy dispersive spectroscopy (EDS) analyses were carried out on a PHILIPS XL30, operated at a 20 kV accelerating voltage. Thermo gravimetric analyses (TGA) were conducted on a Rheometric Scientific Inc. 1998 thermal analysis apparatus under a N2 atmosphere at a heating rate of 10 °C min−1. The magnetic measurement was carried out in a vibrating sample magnetometer (Model 7407 VSM system, Lake Shore Cryotronic, Inc., Westerville, OH, USA) at room temperature. The inductively coupled plasma (ICP-OES) spectra were recorded using a PerkinElmer Optima 7300 DV series.
:EtOAc). Fourier transform infrared (FT-IR) spectroscopy spectra were recorded on a Shimadzu-470 spectrometer, using KBr pellets and the melting points were determined on a KRUSS model instrument. 1H NMR spectra were recorded on a Bruker Avance II 400 NMR spectrometer at 400 MHz, in which DMSO-d6 was used as solvent and TMS as the internal standard. X-ray diffraction (XRD) pattern was obtained by Philips X Pert Pro X diffractometer operated with a Ni filtered Cu Kα radiation source. Transmission electron microscopy (TEM) images of the electrocatalyst were recorded using a Philips CM-10 TEM microscope operated at 100 kV. Field emission scanning electron microscopy (SEM) and X-ray energy dispersive spectroscopy (EDS) analyses were carried out on a PHILIPS XL30, operated at a 20 kV accelerating voltage. Thermo gravimetric analyses (TGA) were conducted on a Rheometric Scientific Inc. 1998 thermal analysis apparatus under a N2 atmosphere at a heating rate of 10 °C min−1. The magnetic measurement was carried out in a vibrating sample magnetometer (Model 7407 VSM system, Lake Shore Cryotronic, Inc., Westerville, OH, USA) at room temperature. The inductively coupled plasma (ICP-OES) spectra were recorded using a PerkinElmer Optima 7300 DV series.
General procedure for the preparation of nano-Fe3O4 1
FeCl3·6H2O (20 mmol) and FeCl2·4H2O (10 mmol) were dissolved in distilled water (100 ml) in a three-necked round-bottom flask (250 ml). The resulting transparent solution was heated at 90 °C with rapid mechanical stirring under N2 atmosphere for 1 h. A solution of concentrated aqueous ammonia (10 ml, 25 wt%) was then added to the solution in a drop-wise manner over a 30 min period using a dropping funnel. The reaction mixture was then cooled to room temperature and the resulting magnetic particles collected with a magnet and rinsed thoroughly with distilled water.General procedure for the preparation of nano-Fe3O4@SiO2 2
Nano-Fe3O4@SiO2 was synthesized according to a previously published literature method. Magnetic nano particles (1.0 g) were initially diluted via the sequential addition of water (20 ml), ethanol (60 ml) and concentrated aqueous ammonia (1.5 ml, 28 wt%). The resulting dispersion was then homogenized by ultrasonic vibration in a water bath. A solution of TEOS (0.45 ml) in ethanol (10 ml) was then added to the dispersion in a drop-wise manner under continuous mechanical stirring. Following a 12 h period of stirring, the resulting product was collected by magnetic separation and washed three times with ethanol.General procedure for the preparation of nano-Fe3O4@SiO2–OMoO3H 3
To an oven-dried (125 °C, vacuum) sample of nano-Fe3O4@SiO2 (2 g) in a round bottomed flask (50 ml) equipped with a condenser and a drying tube, thionyl chloride (8 ml) was added and the mixture in the presence of CaCl2 as a drying agent was refluxed for 48 h. The resulting dark powder was filtered and stored in a tightly capped bottle. To a mixture of Fe3O4@SiO2–Cl (1 g) and sodium molybdate (0.84 g) n-hexane (5 ml) was added. The reaction mixture was stirred under refluxing conditions (70 °C) for 4 h. After completion of the reaction, the reaction mixture was filtered and washed with distilled water, and dried and then stirred in the presence of 0.1 N HCl (20 ml) for an hour. Finally, the mixture was filtered, washed with distilled water, and dried to afford nano-Fe3O4@SiO2–OMoO3H.General procedure for the preparation of 1,8-dioxo-decahydroacridine derivatives 8
A mixture of 1,3-cyclohexanedione (2 mmol), aromatic aldehyde (1 mmol), ammonium acetate (1 mmol) and nano-Fe3O4@SiO2–OMoO3H (0.02 g) in a round bottom flask was heated with stirring in the oil bath at 100 °C for appropriate times. During the procedure, the reaction was monitored by Thin Layer Chromatography (TLC). The reaction mixture was cooled, eluted with hot ethanol (5 ml), and centrifuged to collect the catalyst. The solvent was evaporated with reduced pressure to collect the formed precipitate. The crude product was recrystallized from ethanol to yield pure 1,8-dioxo-decahydroacridines.Spectral data
Results and discussion
The MNPs 1 were prepared via a chemical co-precipitation30 and were subsequently coated with a layer of silica using the sol–gel method31 to provide reaction sites for further functionalization and thermal stability. The Fe3O4@SiO2 nanoparticles 2 were then transformed to Fe3O4@SiO2–Cl with thionyl chloride and Fe3O4@SiO2–Cl were then transformed to Fe3O4@SiO2–OMoO3H 3 via a nucleophilic substitution with anhydrous sodium molybdate in n-hexane to afford a dark gray powder (Scheme 1). We used acid–base potentiometric titration to measure the acid capacities of this catalyst. To this account, 100 mg of the catalyst was added to 10 ml of a 1 M NaCl solution and stirred continuously for 24 h at room temperature. The catalyst was separated using an external magnet, and the NaCl solution was decanted and saved. According to the obtained results, loading of acid sites was about 3.80 mmol H+ per g of catalyst. The resulting MNP acid catalyst was characterized by XRD, FT-IR, ICP-OES, TEM, SEM and EDX. The percentage of molybdenum (Mo) in catalyst was also determined by ICP-OES at 8000–10000 K which showed 1.68 (% w/w) molybdenum.
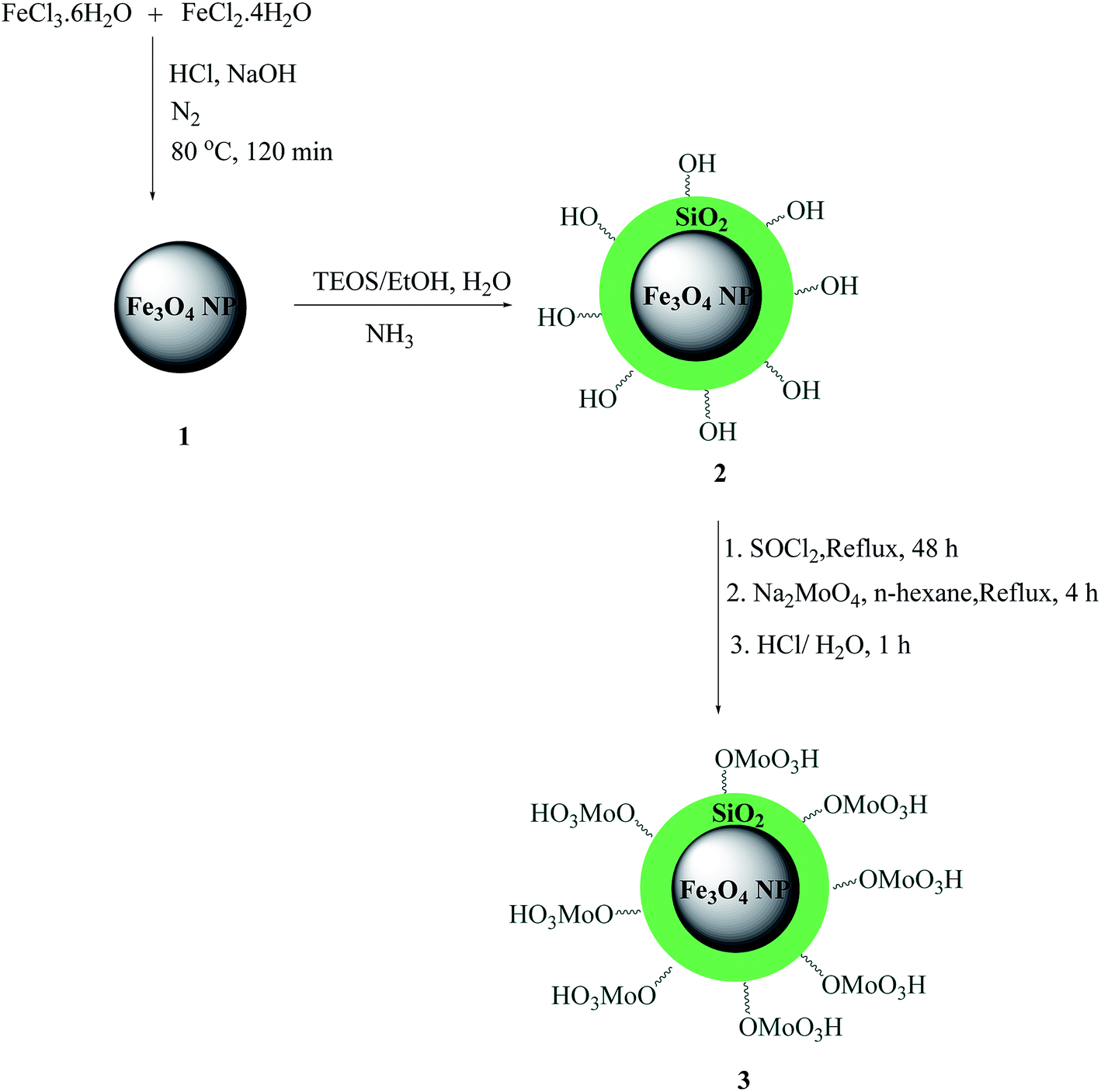 | ||
| Scheme 1 Schematic of the procedure for the preparation of Fe3O4@SiO2–MoO3H. | ||
FT-IR spectra of catalyst are presented the band in the region of 560 cm−1 is attributed to the stretching vibrations of the (Fe–O) bond and the band at about 1090 cm−1 belongs to (Si–O) stretching vibrations. FT-IR analysis was used to characterize the presence of the –MoO3H groups on the surface of the MNPs (Fig. 1). As shown in Fig. 1c, the FT-IR spectra of Fe3O4@SiO2–MoO3H was clearly different from those of Fe3O4 (Fig. 1a) and Fe3O4@SiO2 (Fig. 1b). The broad peak in range of 1080–1200 cm−1 (attributed to the overlapping of peaks corresponding to Si–O–Si, Fe–O–Si, and symmetric MoO2 stretching) indicated that the MNPs possessed MoO3H groups.
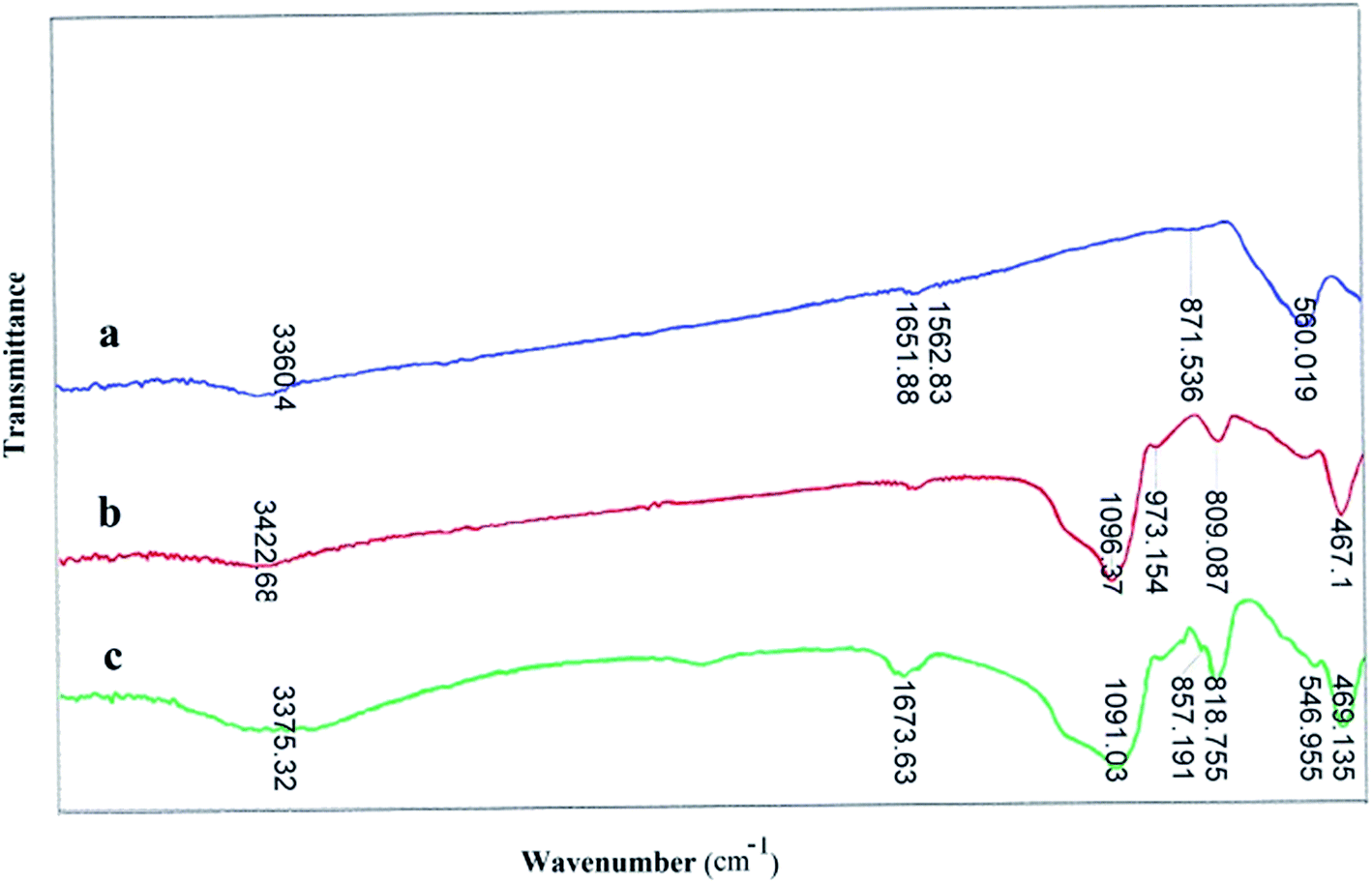 | ||
| Fig. 1 The comparative FT-IR spectra of (a) Fe3O4 NPs (b) Fe3O4@SiO2 and (c) Fe3O4@SiO2–OMoO3H. | ||
The crystalline structure of the different synthesized materials was investigated by XRD. For the Fe3O4 particles (Fig. 2a), the diffraction peaks at 2θ = 30.1°, 35.4°, 43.1°, 53.6°, 57°, and 62.8° corresponded to the spinel structure of Fe3O4, which can be assigned to the diffraction of the (220), (311), (400), (422), (511), and (440) planes of the crystals, respectively.32 For the Fe3O4@SiO2–OMoO3H (Fig. 2b), the XRD pattern indicated that the crystalline structure of the Fe3O4 particles was retained after the deposition of SiO2 layers. The broad peak at around 2θ = 20° to 27° indicated the presence of amorphous silica in Fe3O4@SiO2–OMoO3H. The intensity of this peak increased with the introducing of molybdate on the silica-coated magnetic nanoparticles, which can be attributed to the amorphous molybdate supported on the composite. The XRD results showed that the Fe3O4@SiO2 particles have been successfully coated with molybdate.
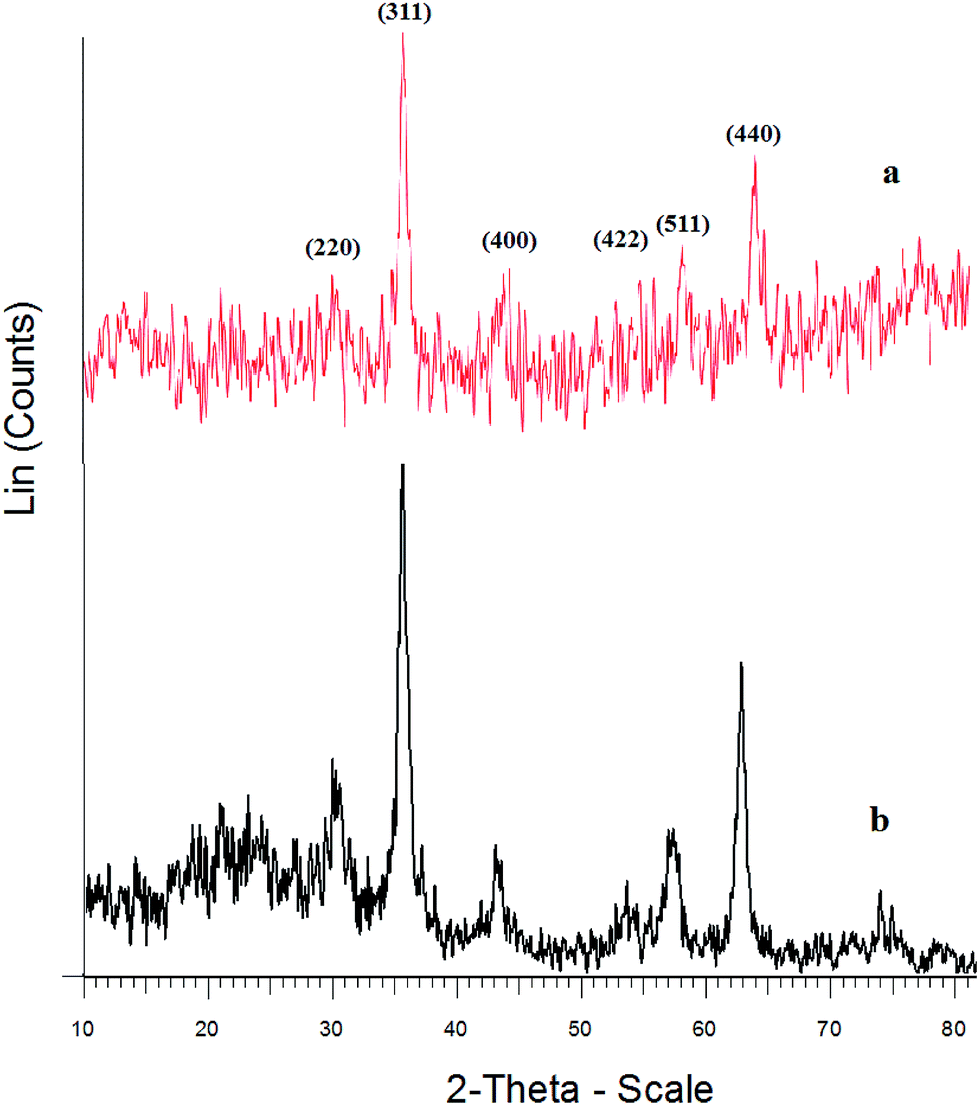 | ||
| Fig. 2 X-ray powder diffraction patterns of (a) Fe3O4 NPs, (b) Fe3O4@SiO2–OMoO3H. | ||
The TEM image revealed that the Fe3O4@SiO2–MoO3H nanoparticles were uniform with an average size in the range of 10–30 nm (Fig. 3a).
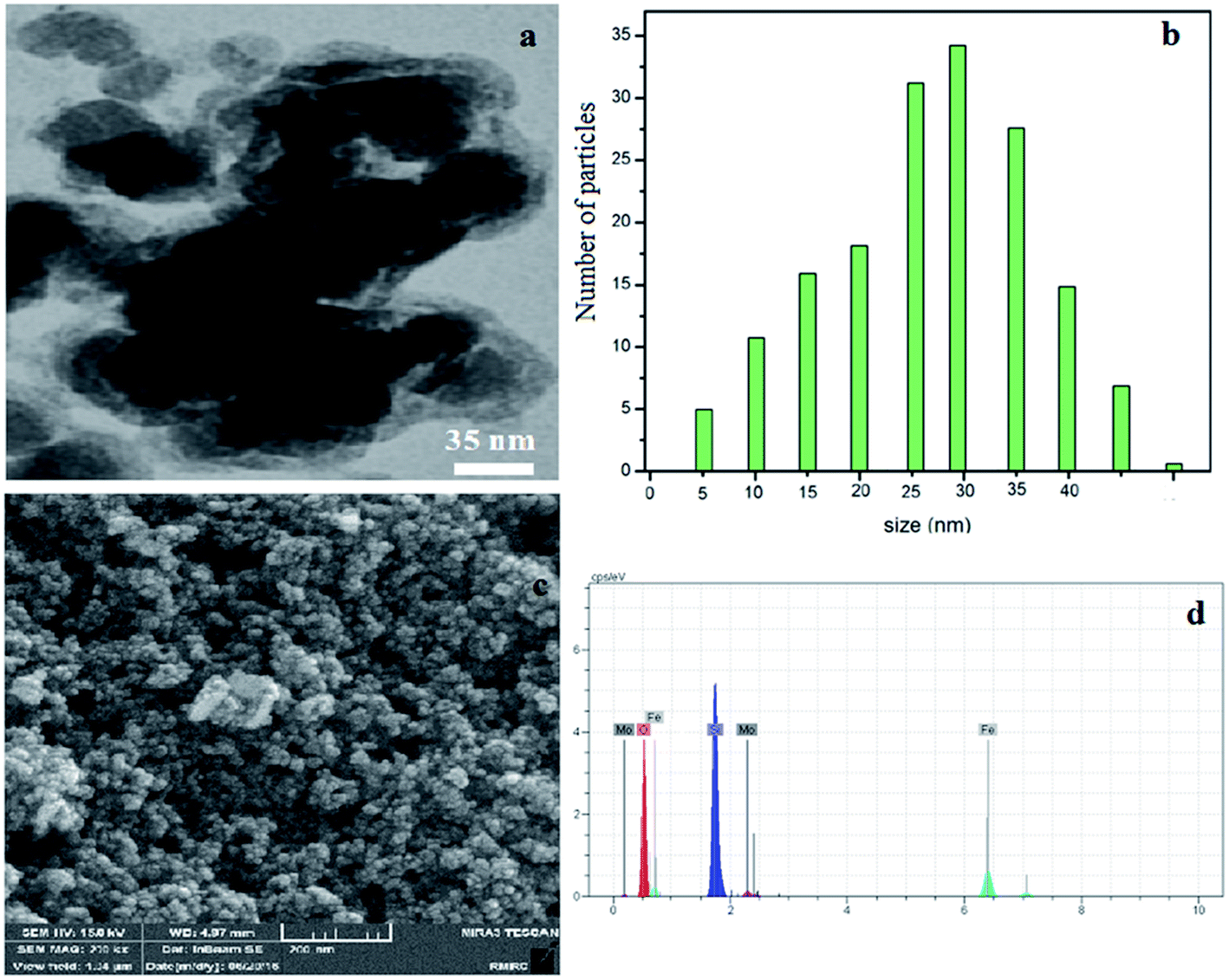 | ||
| Fig. 3 TEM image of Fe3O4@SiO2–MoO3H (a), histogram of particle size distribution (b), SEM image of Fe3O4@SiO2–MoO3H (c) and EDAX spectrum of Fe3O4@SiO2–MoO3H (d). | ||
The SEM image shown in (Fig. 3c) demonstrates that Fe3O4@SiO2–MoO3H nanoparticles are nearly spherical with more than 20 nm in size.
The successful incorporation of molybdate groups was also confirmed by EDAX analysis (Fig. 3d), which showed the presence of to Fe, Si, Mo and O elements.
The thermogravimetric analysis (TGA) was used to study the thermal stability of the acid catalyst (Fig. 4). The first weight loss which occurred below 150 °C, displayed a mass loss that was attributable to the loss of adsorbed solvent or trapped water from the catalyst. A weight loss of approximately 5% weight occurred between 300 and 500 °C which can be attributed to the loss of molybdate groups covalently bound to silica surface. Thus, it can be concluded that the catalyst is stable up to 300 °C.
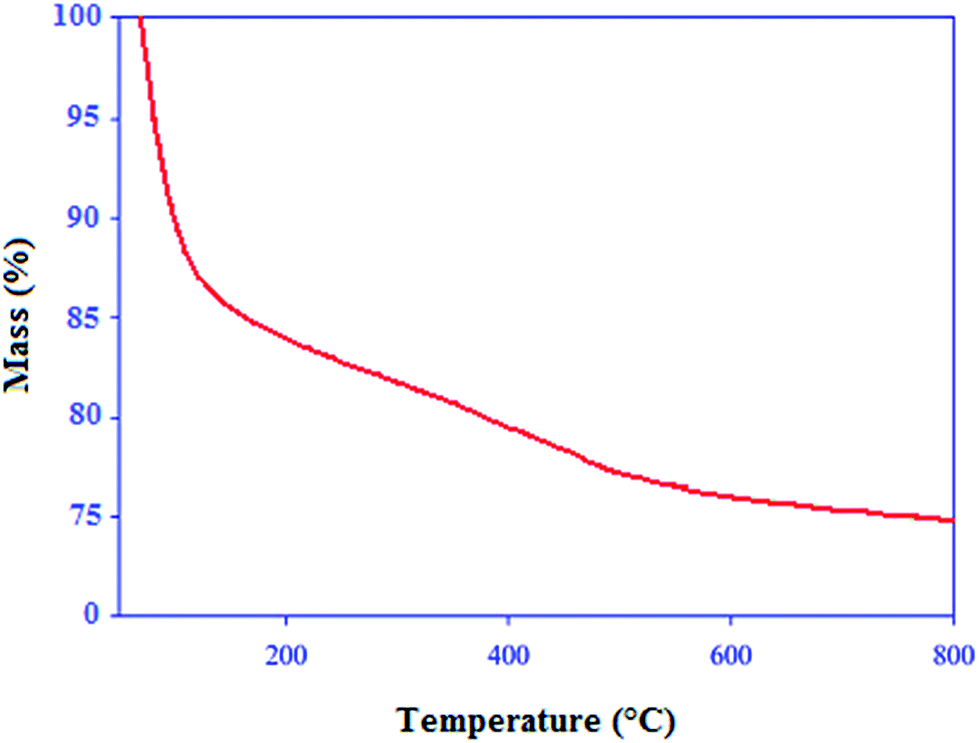 | ||
| Fig. 4 TGA curve of Fe3O4@SiO2–MoO3H. | ||
Typical magnetization curve for Fe3O4 nanoparticles and Fe3O4@SiO2–MoO3H are shown in Fig. 5. Room temperature specific magnetization (M) versus applied magnetic field (H) curve measurements of the sample indicate a saturation magnetization value (Ms) of 20.30 emu g−1, lower than that of bare MNPs (59.14 emu g−1) due to the coated shell.
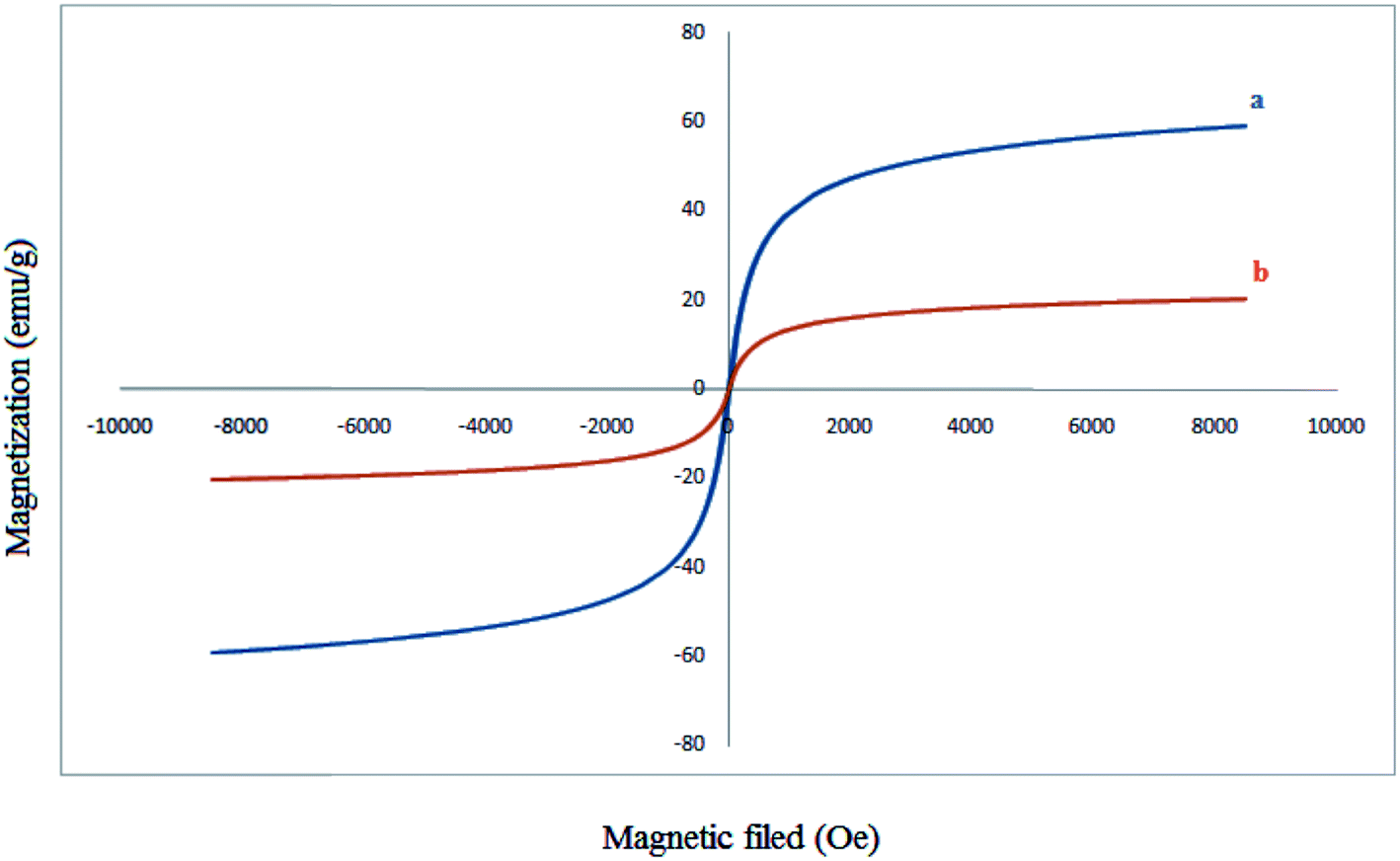 | ||
| Fig. 5 Magnetization curves for the prepared Fe3O4 MNPs (a) and Fe3O4@SiO2–MoO3H (b). | ||
Fig. 6a shows the photograph of Fe3O4@SiO2–MoO3H microspheres that dispersed in water. After a magnet was placed aside, the black microspheres can be magnetized in 3 min, leaving a clear solution (Fig. 6b). That is to say, the Fe3O4@SiO2–MoO3H nanoparticles were shown good magnetic responsibility even if the SiO2–MoO3H layer was increased to 20 nm.
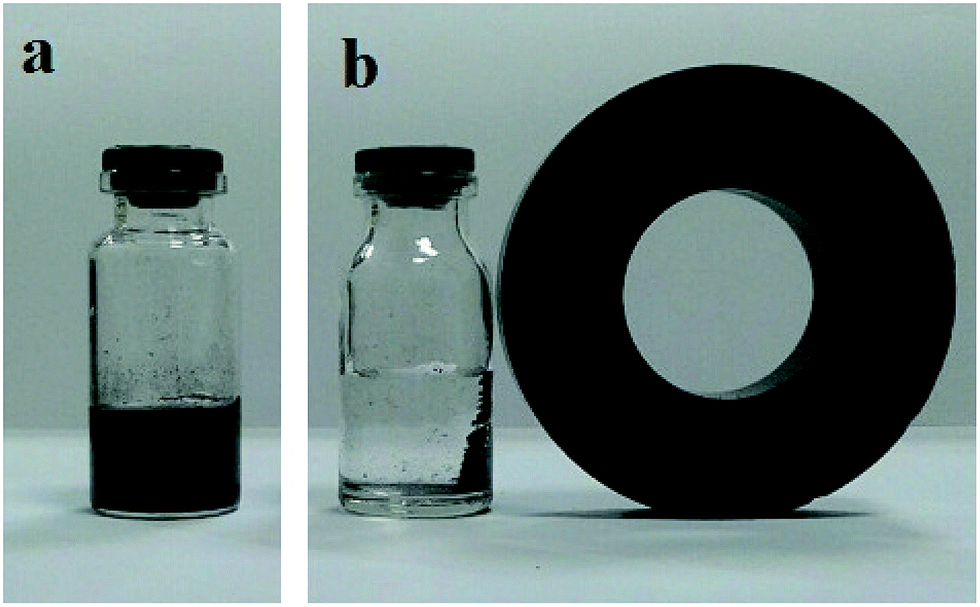 | ||
| Fig. 6 Digital camera images of the aqueous solution with dispersed magnetic Fe3O4@SiO2–MoO3H composite particles (a) and after applied magnetic field (b). | ||
In continuation of our interest in the development of novel, efficient, and green procedures for the synthesis of organic compounds using safe catalysts,33–35 we decided to prepare 1,8-dioxo-decahydroacridines 7,8 was obtained was synthesized by condensation between dimedone (4) (2 mmol), benzaldehyde (5) (1 mmol) and ammonium acetate (6) (1 mmol) under solvent-free conditions as a model reaction (Scheme 2).
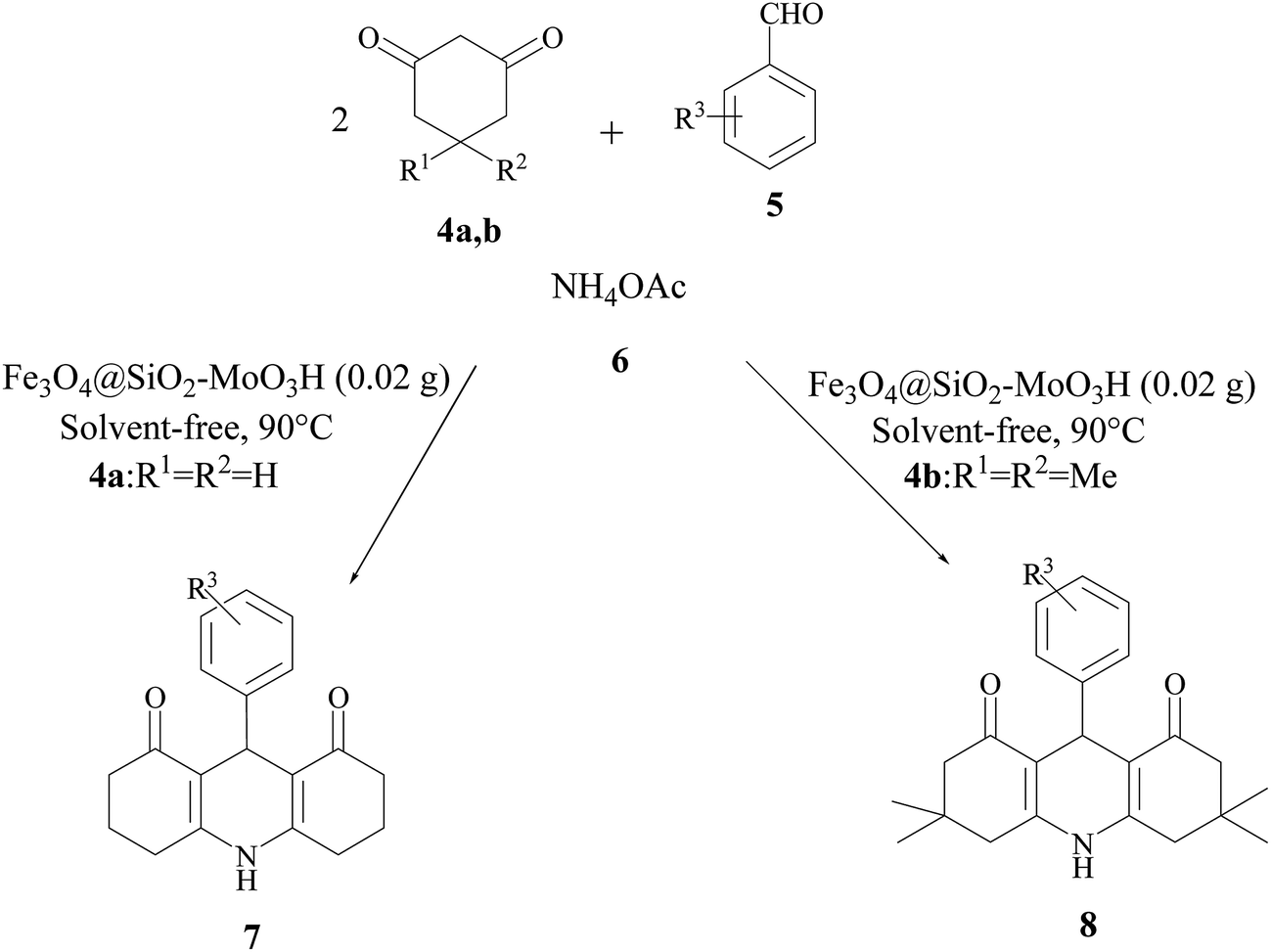 | ||
| Scheme 2 Synthesis of 1,8-dioxo-decahydroacridine derivatives by Fe3O4@SiO2–MoO3H. | ||
The reaction conditions were optimized on the basis of the catalyst, solvent and different temperature for the synthesis of 1,8-dioxo-decahydroacridines. This reaction was firstly examined in the absence of catalyst which did not show any appreciable progress even after 360 min. Upon screening, the results well showed that the reaction proceeds efficiently by adding 1 mol% of Fe3O4@SiO2–MoO3H. Also, increasing the catalyst amount did not improve the results (Fig. 7).
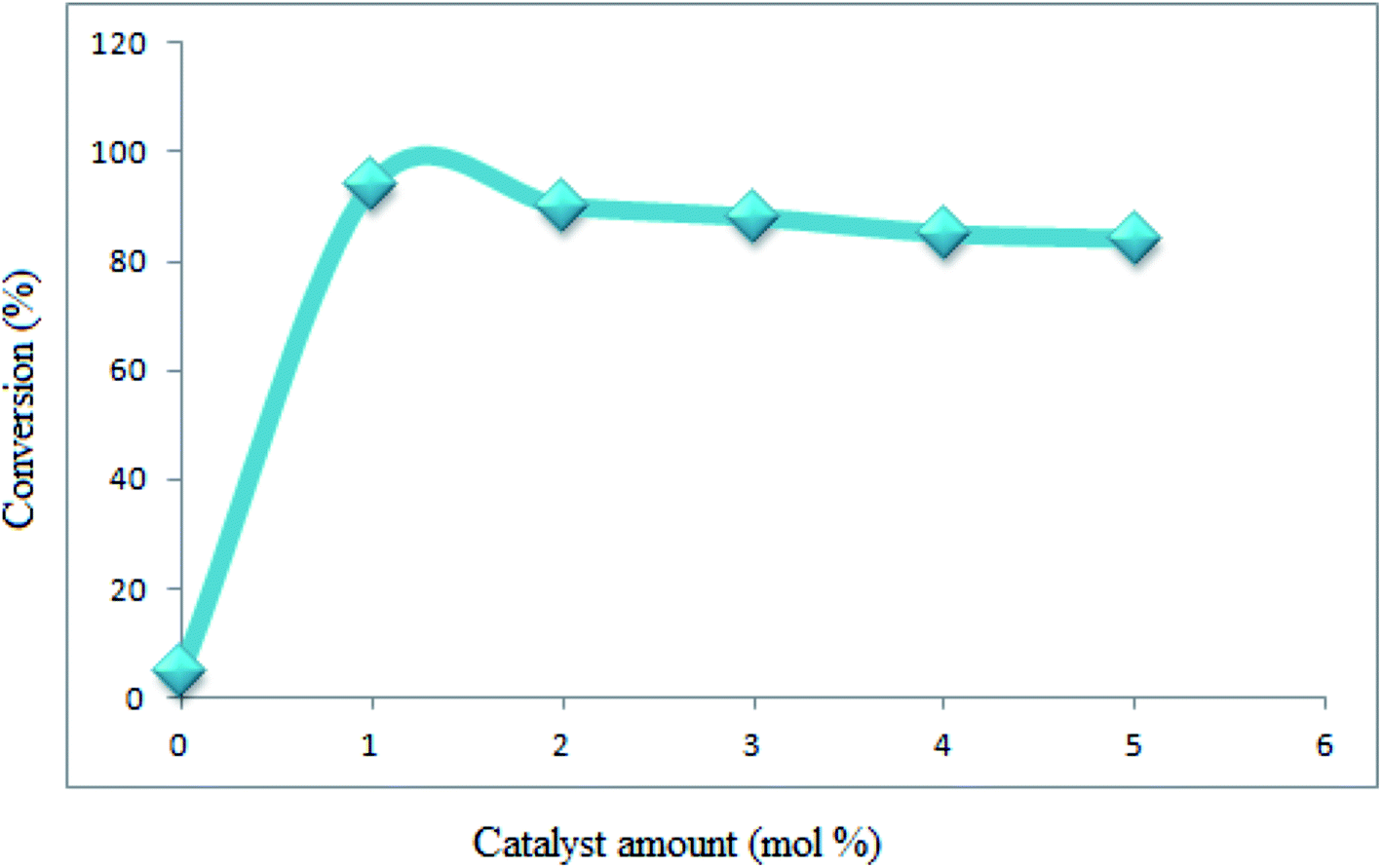 | ||
| Fig. 7 Optimization of the catalyst amount. | ||
Eventually to making sure that the solvent free condition is appropriate, it has been decided to investigate the effect of different classical solvents such as EtOH, MeOH, CHCl3, toluene, and DMF (Table 1, entries 1–7). We found that polar and protic solvents, such as EtOH or MeOH, afford better yields than aprotic ones. As it is shown in Table 1, using these solvents gave significantly lower yields and longer reaction times. Increasing the reaction times did not improve the yields. So the best yield of product was provided in solvent free conditions (Table 1, entry 8).
| Entry | Catalyst (mol%) | Solvent | Time (min) | Isolated yield (%) |
|---|---|---|---|---|
| 1 | 1 | EtOH | 80 | 50 |
| 2 | 2 | EtOH | 80 | 45 |
| 3 | 3 | EtOH | 80 | 35 |
| 4 | 1 | MeOH | 80 | 55 |
| 5 | 1 | CHCl3 | 100 | 30 |
| 6 | 1 | Toluene | 140 | 20 |
| 7 | 1 | DMF | 220 | 25 |
| 8 | 1 | Solvent-free | 35 | 93 |
| 9 | 2 | Solvent-free | 35 | 90 |
| 10 | 3 | Solvent-free | 35 | 88 |
After optimization of the reaction conditions, in order to extend the scope of this reaction, a wide range of aromatic aldehydes were used with 4 and 6 (Table 2). All the products were characterized by comparison of their spectra and physical data with those reported in the literature.36,37
| Entry | R1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) R2 R2 |
R3 | Product | Time (min) | Isolated yield (%) | Melting point (°C) |
|---|---|---|---|---|---|---|
| 1 | H | H | 7a | 35 | 93 | 278–279 |
| 2 | H | 4-Cl | 7b | 20 | 89 | 295–297 |
| 3 | H | 4-Br | 7c | 25 | 91 | 311–312 |
| 4 | H | 4-CH3 | 7d | 45 | 88 | 254–255 |
| 5 | H | 4-OH | 7e | 55 | 86 | 303–305 |
| 6 | H | 4-OCH3 | 7f | 50 | 86 | 302–304 |
| 7 | H | 3-NO2 | 7g | 25 | 90 | 280–282 |
| 8 | CH3 | H | 8a | 25 | 92 | 291–293 |
| 9 | CH3 | 4-OCH3 | 8b | 40 | 90 | 275–277 |
| 10 | CH3 | 4-OH | 8c | 45 | 88 | 302–304 |
| 11 | CH3 | 4-CH3 | 8d | 55 | 90 | 267–269 |
| 12 | CH3 | 4-F | 8e | 30 | 94 | 289–291 |
| 13 | CH3 | 4-Cl | 8f | 20 | 92 | 295–297 |
| 14 | CH3 | 2-Cl | 8g | 20 | 90 | 222–224 |
| 15 | CH3 | 4-Br | 8h | 25 | 91 | 310–312 |
| 16 | CH3 | 4-NO2 | 8i | 35 | 93 | 287–289 |
| 17 | CH3 | 3-NO2 | 8j | 30 | 91 | 290–292 |
| 18 | CH3 | 2-Naphthaldehyde | 8k | 45 | 86 | 264–266 |
| 19 | CH3 | 4-(CH3)2–N | 8l | 50 | 88 | 217–219 |
As indicated in Table 2, the new conditions are very suitable for a vast variety of aromatic aldehydes with both electron-donating and electron-withdrawing groups to give corresponding 1,8-dioxo-decahydroacridine derivatives in good to excellent yields. In all cases, the reactions proceeded within 20–55 minute. However, it is notable that substituted aromatic aldehydes with electron-withdrawing groups increase the rate of reaction (Table 2, entries 2, 3, 7, 12–17) probably by activating the carbonyl group as electrophile center. Contrarily in the case of electron-donating groups, the reaction was more slowly (Table 2, entries 4–6 and 9–11).
The reaction mechanism is shown in Scheme 3. At first, the acid catalyst changes the aldehyde into convenient electrophile via protonation of the carbonyl group and then one molecule of dimedone condenses with the aromatic aldehyde to produce intermediate 9. Then the active methylene group of the second molecule of dimedone reacts with 9 to give intermediate 10. Nucleophilic attack of amine group of ammonium acetate to carbonyl group creates intermediate 11. In the next step, cyclization will occur by the nucleophilic attack of amine group to carbonyl group to obtain intermediate 12. Finally, by the removal of one water molecule, the acridine derivatives 7 or 8 will be generated.
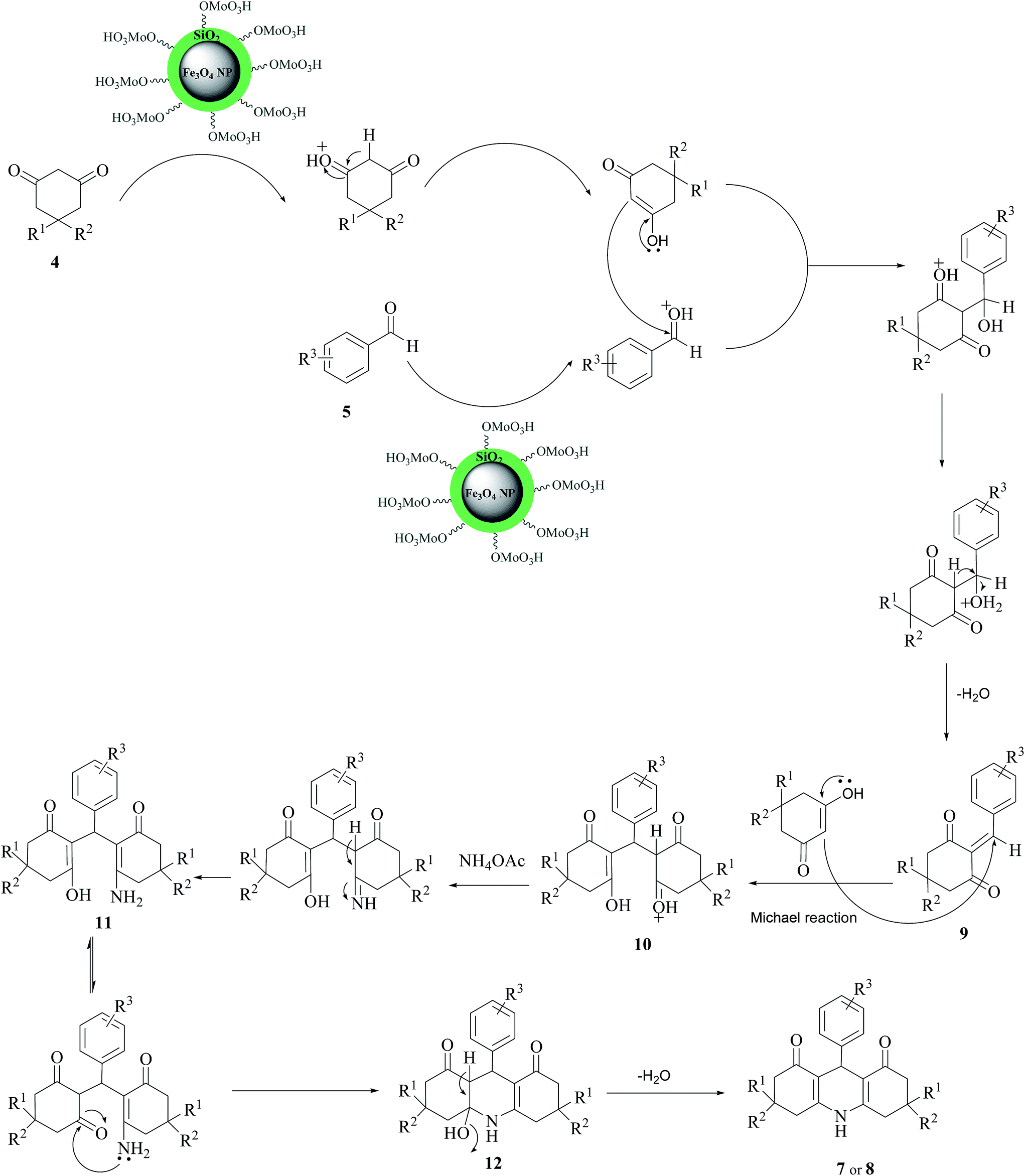 | ||
| Scheme 3 Reasonable mechanism for the formation of decahydroacridine-1,8-diones. | ||
The design and synthesis of recoverable catalysts is a highly challenging interdisciplinary field, which combines chemistry, materials science, and engineering from economic and environmental perspective. The main disadvantage for many of the reported methods is that the catalysts are destroyed in the work-up procedure and cannot be recovered or reused. In this process, as outlined in Fig. 8, the recycled catalyst can be used in up to eight cycles, during which there are negligible losses in the catalytic activity. The percentage of molybdenum (Mo) in recovered catalyst was also determined by ICP-OES at 8000–10000 K which showed 1.67 (% w/w) molybdenum. Additionally the acid capacity of catalyst was 3.7 mmol H+ per g after eighth recovery which confirms excellent efficiency of the immobilization of acidic group on the surface of the catalyst.
 | ||
| Fig. 8 Reusability of Fe3O4@SiO2–MoO3H for synthesis of 7a. | ||
After the eight recycles, the recovered catalyst had similar morphology as the fresh one (Fig. 9) and no noticeable change in structure was observed, by reference to the EDAX analysis as compared to the fresh catalyst (Fig. 10).
 | ||
| Fig. 9 FE-SEM images of (a) fresh Fe3O4@SiO2–MoO3H, (b) after reused eight times. | ||
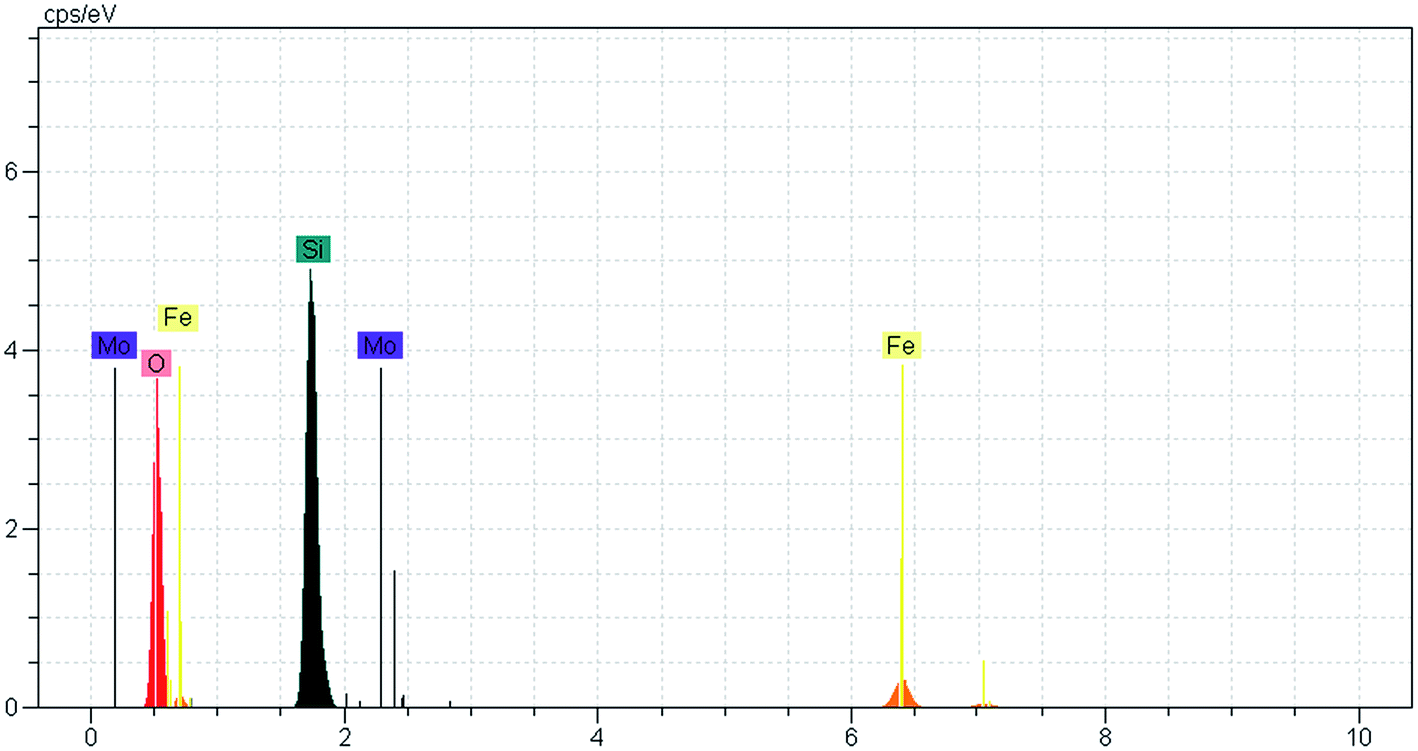 | ||
| Fig. 10 EDAX spectrum of Fe3O4@SiO2–MoO3H after reused eight times. | ||
Conclusions
In summary, we found Fe3O4@SiO2–OMoO3H as an effective and environmentally safe acidic magnetic catalyst which successfully catalyzed to produce 1,8-dioxo-decahydroacridines from aromatic aldehydes, an amine and a dimedone under solvent free conditions. By this development, the scope of heterocyclic compounds was increased. High catalytic activity under solvent free conditions, high yields, a clean process, reusable several times without loss of activity or selectivity simple catalyst preparation, easy separation after the reaction by a magnet and green conditions are advantages of these protocols.Acknowledgements
The authors gratefully acknowledge partial support of this work by Semnan University and Karaj Islamic Azad University, Iran.Notes and references
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- E. L. S. Ngee, Y. Gao, X. Chen, T. M. Lee, Z. Hu, D. Zhao and N. Yan, Ind. Eng. Chem. Res., 2014, 53, 14225–14233 CrossRef CAS.
- Y. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210–6224 RSC.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- J. Zou, Y. G. Peng and Y. Y. Tang, RSC Adv., 2014, 4, 9693–9700 RSC.
- R. Zhao, K. Jia, J. J. Wei, J. X. Pu and X. B. Liu, Mater. Lett., 2010, 64, 457–459 CrossRef CAS.
- J. Liu, R. Che, H. Chen, F. Zhang, F. Xia, Q. Wu and M. Wang, Small, 2012, 8, 1214–1221 CrossRef CAS PubMed.
- Y. Piao, A. Burns, J. Kim, U. Wiesner and T. Hyeon, Adv. Funct. Mater., 2008, 18, 3745–3758 CrossRef CAS.
- C. Wang, J. Yan, X. Cui and H. Wang, J. Colloid Interface Sci., 2011, 354, 94–99 CrossRef CAS PubMed.
- H. D. Oh and S. W. Lee, Mater. Res. Bull., 2013, 48, 2191–2193 CrossRef CAS.
- M. A. Ghasemzadeh, J. Safaei-Ghomi and H. Molaei, C. R. Chim., 2012, 15, 969–974 CrossRef CAS.
- Y. Mikata, M. Yokoyama, K. Mogami, M. Kato, I. Okura, M. Chikira and S. Yano, Inorg. Chim. Acta, 1998, 279, 51–57 CrossRef CAS.
- I. Antonini, P. Polucci, L. R. Kelland, E. Menta, N. Pescalli and S. Martelli, J. Med. Chem., 1999, 42, 2535–2541 CrossRef CAS PubMed.
- M. J. Wainwright, J. Antimicrob. Chemother., 2001, 47, 1–13 CrossRef CAS PubMed.
- L. Ngadi, A. M. Galy, J. P. Galy, J. Barbe, A. Cremieux, J. Chevalier and D. Sharples, Eur. J. Med. Chem., 1990, 25, 67–70 CrossRef CAS.
- S. A. Gamega, J. A. Spicer, G. J. Atwell, G. J. Finlay, B. C. Baguley and W. A. Deny, J. Med. Chem., 1999, 42, 2383–2393 CrossRef PubMed.
- S. Gallo, S. Atifi, A. Mohamoud, C. Santelli-Rouvier, K. Wolfart, J. Molnar and J. Barbe, Eur. J. Med. Chem., 2003, 38, 19–26 CrossRef CAS PubMed.
- K. Venkatesan, S. S. Pujari and K. V. Srinivasan, Synth. Commun., 2009, 39, 228–241 CrossRef CAS.
- X. S. Wang, D. Q. Shi, D. Q. Zhang, Y. F. Wang and S. J. Tu, Chin. J. Org. Chem., 2004, 24, 430–432 CAS.
- W. Shen, L. M. Wang, H. Tian, J. Tang and J. J. Yu, J. Fluorine Chem., 2009, 130, 522–527 CrossRef CAS.
- M. Dabiri, M. Baghbanzadeh and E. Arzroomchilar, Catal. Commun., 2008, 9, 939–942 CrossRef CAS.
- M. Kidwai and D. Bhatnagar, Tetrahedron Lett., 2010, 51, 2700–2703 CrossRef CAS.
- F. Rashedian, D. Saberib and K. Niknam, J. Chin. Chem. Soc., 2010, 57, 998–1006 CrossRef CAS.
- B. Das, P. Thirupathi, I. Mahender, V. S. Reddy and Y. Koteswara, J. Mol. Catal. A: Chem., 2006, 247, 233 CrossRef CAS.
- A. Davoodnia, A. Khojastehnezhad and N. Tavakoli-Hoseini, Bull. Korean Chem. Soc., 2011, 32, 2243 CrossRef CAS.
- T. S. Jin, J. S. Zhang, T. T. Guo, A. Q. Wang and T. S. Li, Synthesis, 2004, 12, 2001–2005 CrossRef.
- B. Karami and M. Kiani, Catal. Commun., 2011, 14, 62–67 CrossRef CAS.
- B. Karami, M. Kiani, S. J. Hosseini and M. Bahrami, New J. Chem., 2015, 39, 8576–8581 RSC.
- B. Karami and M. Kiani, Res. Chem. Intermed., 2016, 42, 3373–3383 CrossRef CAS.
- A. Bee, R. Massart and S. J. Neveu, J. Magn. Magn. Mater., 1995, 149, 6–9 CrossRef CAS.
- R. V. Ferreira, I. L. S. Pereira, L. C. D. Cavalcante, L. F. Gamarra, S. M. Carneiro, E. J. Amaro, J. D. Fabris, R. Z. Domingues and A. L. Andrade, Hyperfine Interact., 2010, 195, 265–274 CrossRef CAS.
- L. Cabrera, S. Gutierrez, M. P. Morales, N. Menendez and P. Herrasti, J. Magn. Magn. Mater., 2009, 321, 2115–2120 CrossRef CAS.
- B. Karami and M. Kiani, J. Chin. Chem. Soc., 2015, 62, 756–765 CrossRef.
- B. Karami and M. Kiani, J. Iran. Chem. Soc., 2016, 13, 111–116 CrossRef CAS.
- B. Karami, M. Kiani and M. A. Hoseini, Chin. J. Catal., 2014, 35, 1206–1211 CrossRef CAS.
- G. M. Ziarani, A. Badiei, M. Hassanzadeh and S. Mousavi, Arabian J. Chem., 2014, 7, 335–339 CrossRef CAS.
- M. Nasr-Esfahani and T. Abdizadeh, C. R. Chim., 2015, 18, 547–553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25571j |
| This journal is © The Royal Society of Chemistry 2017 |