
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct electrochemistry and bioelectrocatalysis of glucose oxidase in CS/CNC film and its application in glucose biosensing and biofuel cells
Zepeng Kanga,
Kailong Jiaoa,
Chao Yub,
Ji Dongb,
Ruiyun Peng*b,
Zongqian Hu*b and
Shuqiang Jiao *a
*a
aState Key Laboratory of Advanced Metallurgy, University of Science and Technology Beijing, Beijing, 100083, P. R. China. E-mail: sjiao@ustb.edu.cn
bBeijing Institute of Radiation Medicine, Beijing, 100850, P. R. China. E-mail: huzongqian@hotmail.com; pengry@bmi.ac.cn
First published on 16th January 2017
Abstract
Due to their unique physicochemical properties, carbon nanochips (CNCs) have been used for studies of the direct electrochemical and electrocatalytic properties of oxidoreductase. In this report, a glassy carbon electrode (GCE) was modified with CNCs, and glucose oxidase (GOx) was immobilized on the modified electrode surface. Chitosan (CS) was employed to fix the GOx/CNCs tightly to the surface of the GCE. Characterization of the modified electrode by SEM, TEM, and FT-IR showed that GOx remained in its native structure when immobilized in CNC film. The results of electrochemical impedance spectroscopy, cyclic voltammetry, and linear sweep voltammetry studies showed that direct electrochemical and bioelectrocatalytic activities of GOx were achieved and that the presence of CNCs enhanced the electrochemical response of GOx. Glucose had a linear current response from 0 to 1.9 mM, with a detection limit of 0.16 mM and an apparent Michaelis–Menten constant of 0.15 mM. The polarization curve of the biofuel cell revealed that the bioanode afforded an open circuit voltage of 0.59 V, maximum power density of 55 μW cm−2, and maximum current density of 434 μA cm−2. These results establish that the direct immobilization of protein onto the CNC surface can be used to achieve direct electron transfer of other redox enzymes. Furthermore, the findings provide a new perspective toward understanding the kinetics and thermodynamics of biological redox processes.
1. Introduction
Direct electrochemistry between a redox protein or enzyme and the conductive electrode surface plays a key role in the development of third-generation biosensors and high performance biofuel cells.1–7 Direct electrochemistry also provides a suitable model for understanding electron transfer mechanisms in biological systems and a more effective way for fabricating biosensors, bioreactors and biomedical devices than using mediators.1 In this context, researchers seek to achieve a high rate of electron transfer between the active site of the protein or enzyme and the immobilized matrix.7Most redox proteins show a slow rate of electron transfer on conventional electrode because the electroactive centers are deeply embedded within isolated protein shells. An unfavorable orientation of protein molecules on the electrode surface can block electron transfer between the electrode and protein electroactive centers. Moreover, the adsorption of protein molecules onto the bare electrode surface would lead to their denaturation, which also decreases the direct electron-transfer rate. Electrode passivation can happen due to the adsorption of the large three-dimensional structure of protein which makes the establishment of direct electron transfer (DET) between these macromolecules and conventional electrodes generally difficult.8–10 Liang et al.7 found that DET and enzyme catalysis can not occur on the same glucose oxidase (GOx) simultaneously. These issues remain major challenge for biosensor and BFC researchers.
Modification of the electrode surface with nanomaterials allows efficient DET between redox-active proteins and electrodes. Many immobilization attempts have been employed to improve electron transfer between the active center of the redox protein and the electrode. These attempts have incorporated various inorganic/organic matrixes (e.g., gold nanoparticles,10–12 quantum dots,3 carbon nanotubes,13,14 graphene,15–17 carbon nanofibers,18 polyaniline,19,20 polypyrrole,21 and their composites6,22–24), and immobilization methods (e.g., physical adsorption,25 covalent attachment,26,27 electrostatic interactions,28 and film entrapment29).
Carbon nanochips (CNCs)30,31 are a novel class of graphite nanofibers. CNCs are created by calcination at high temperatures (2300–3000 °C) with protection by an inert gas. These materials have enhanced performance as support media for biological catalysts. High-temperature processing improves catalytic properties and increases the average pore size of CNCs compared to conventional graphite nanofibers. In addition, the conductivity of CNCs seems to be ideal for electrochemical transduction. These characteristics, combined with the ability to control the number and type of functional groups on the outer surface of CNCs, are expected to make CNCs suitable for the selective immobilization and stabilization of functional biomolecules.31 However, the use of CNCs as substrates for the immobilization of biological catalysts has barely been reported, in contrast to reports about conventional carbon nanomaterials such as fullerenes, carbon nanotubes and graphene.
In this work, a glassy carbon electrode (GCE) was modified with CNCs and a film of GOx, as the model catalyst, was cast on the modified electrode surface. Chitosan (CS) was used to fix the CNCs and GOx tightly to the surface of the GCE surface. The paper demonstrates the use of CNCs in studies of the direct electrochemical and electrocatalytic properties of GOx and discusses the advantage of using CNCs as supports for GOx immobilization.
2. Materials and methods
2.1. Materials and chemicals
CNCs (HT3000, 99.99% carbon) and GOx (EC 1.1.3.4, from Aspergillus niger, 200 U mg−1) were purchased from Sigma-Aldrich. CS and glucose were purchased from Aladdin Ltd (Shanghai, China). Na2HPO4, NaH2PO4, HPO4, K3Fe(CN)6/K4Fe(CN)6, HNO3, H2SO4 and NaOH were supplied by Sinopharm Chemical Reagent Co., Ltd. (China). All reagents were of analytical grade and used without further purification. Phosphate buffer solutions (PBS, 0.1 M, pH 7.2) were prepared with 0.1 M NaH2PO4 and 0.1 M Na2HPO4. Different stock solution concentrations of glucose were prepared in 0.1 M PBS (pH 7.2) and mutarotated at least 24 h before use. All solutions were prepared with deionized water which was purified with a Millipore-Q purification system to a specific resistance over 18.0 MΩ.2.2. Preparation of CS/GOx/CNCs/GCE
In preparing a GOx electrode, a GCE was sequentially polished using a slurry of 0.5 μm and 0.03 μm alumina power and successively washed by ultrasonication in deionized water, 1 M HNO3, deionized water and ethanol for 3 minutes, respectively. Then the electrode was activated electrochemically in 0.1 M H2SO4 by cyclic voltammetry (CV) until the CV curve to invariant.32 Thereafter, the electrode was washed and dried in argon, then 5 μL CNCs (5 mg mL−1, prepared in deionized water) suspension was dropped on the surface of the cleaned GCE. The droplet on the GCE was carefully manipulated to just cover the entire electrode surface, and was dried in air, which resulted in a uniform film. The resultant electrode is termed herein as CNCs/GCE. A 2 μL volume of GOx solution (20 mg mL−1, prepared in 0.1 M PBS, pH 4) was evenly pipetted onto the surface of the CNCs/GCE and spread evenly over the entire surface. The electrode was left in air to dry at 4 °C for 3 h. The resultant electrode is termed herein as GOx/CNCs/GCE. Before use, 5 μL CS (0.5 wt%, prepared in 0.1 M acetic acid at 60–70 °C, pH 5) was spread onto the entire surface of GOx/CNCs/GCE as a binder to hold the film on the GCE surface stably. The electrode was left in air to dry at 4 °C for 6 h. The resultant electrode is termed herein as CS/GOx/CNCs/GCE. For comparison, CS/GOx/GCE, CS/CNCs/GCE and CS/[GOx]x/[CNCs]y/GCE (x, y is volume of GOx and CNCs, respectively, μL) were prepared using a similar procedure. The prepared CS/GOx/CNCs/GCE was stored in PBS (0.1 M, pH 7.2) at 4 °C. The schematic structure of the CS/GOx/CNCs/GCE is shown in Fig. 1.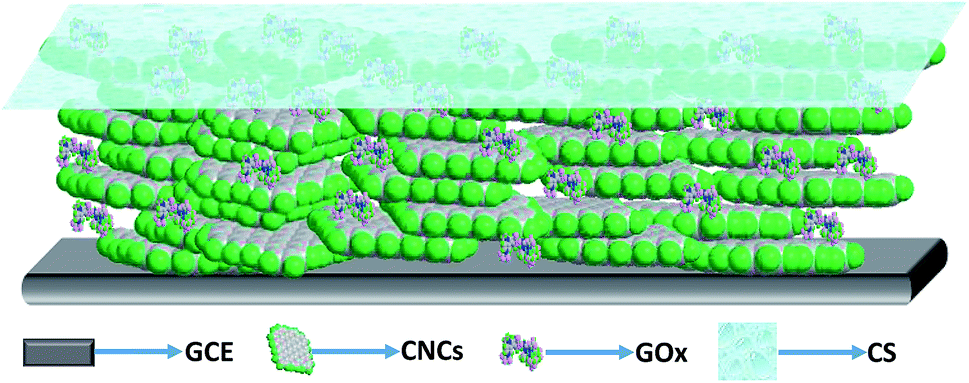 | ||
| Fig. 1 Schematic structure of CS/GOx/CNCs/GCE. | ||
2.3. Apparatus and measurements
Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) were performed using JEOL (Tokyo, Japan) instruments (JSM-6701 for FESEM and JEM-2100 for TEM). Fourier transform infrared spectroscopy (FT-IR) spectra were obtained on a Spectrum 100 FT-IR spectrophotometer from Perkin Elmer. XRD studies were performed on a PANalytical X'pert Pro X-ray diffractometer. Raman was measured by a WITec CRM200 using 532 nm laser.All electrochemical experiments were carried out by a CHI660C electrochemical workstation (CHI Instrument, Shanghai, China). The electrochemical measurements were carried out in a single-compartment, three-electrode glass cell. The working electrodes were the modified GCE (each 3 mm in diameter), the counter electrode was a Pt sheet with a surface area of 1 cm2, and the reference electrode was Ag/AgCl (3 M KCl). The electrochemical measurements were carried out in 0.1 M PBS (pH 7.2) as the supporting electrolyte which were purged with high-purity argon for at least 30 min prior to experiments and a argon environment was kept over the solution in the cell. All experiments were performed at room temperature.
For the test of the BFC, a glucose/O2 BFC consisted of the CS/GOx/CNCs/GCE anode and an Pt sheet cathode in a single-compartment. The voltages and currents of the BFC were measured by tow multimeters with an external resistance varying 100 Ω to 100 kΩ. The BFC was operated at room temperature and fed with a fuel solution of 10 mM glucose stock solution (0.1 M PBS, pH 6) with saturated air. The current density and power density of the BFC were calculated from the data of the voltages and currents.
3. Results and discussion
3.1. General characteristics
Fig. 2A shows the FESEM and TEM images of CNCs, which presented a nanofiber shape with a diameter of 20–45 nm and a length of several micrometers (Fig. 2A(a)). High-resolution images (Fig. 2A(b)) obviously exhibits that the CNCs have straight, branched, twisted, helical and coiled structures. Fig. 2A(c) showed the Raman spectrum of CNCs. The peaks at 1350 cm−1 (D-band) and 1580 cm−1 (G-band) which were attributed to vibration of sp2 and sp3 hybridize carbon atoms, respectively. The intensity ratio of D-band to G-band (ID/IG), which can be used to evaluate the degree of crystallization,17 was calculated to be 0.334. This value suggested a high degree of graphitization. Chemical structure and composition of CNCs were further studied by XRD. The strong observed peak at 26.0° (Fig. 2A(d)) can be attributed to the (002) plane of the hexagonal graphite structure, indicating a high degree of crystallization.17 The high active surface area and good electrical conductivity make CNCs extremely attractive as sensing materials,30 and the combination of CNCs and GOx would bring new opportunities for designing novel bioelectrocatalytic systems.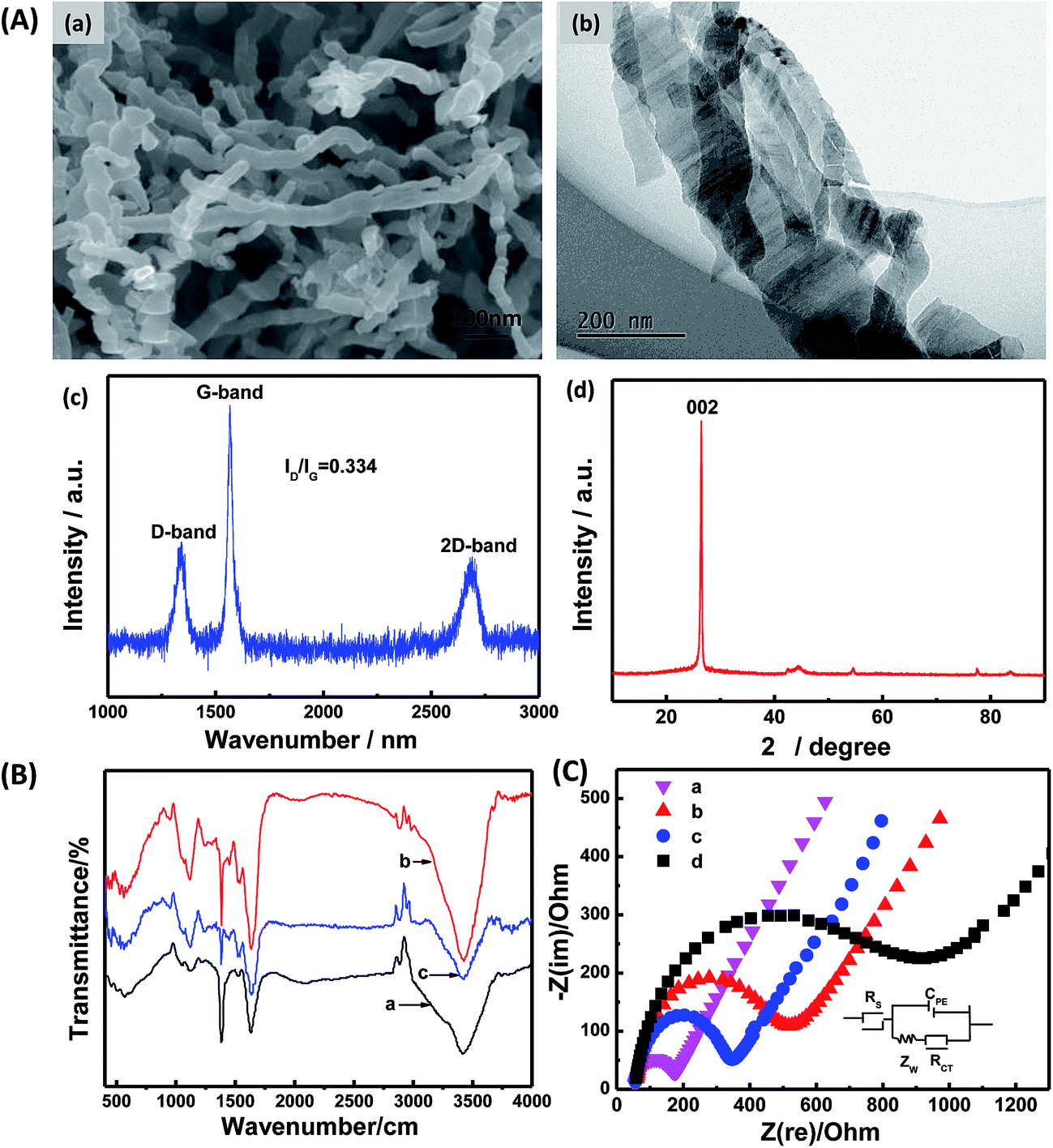 | ||
| Fig. 2 (A) SEM image (a), TEM image (b), Raman spectrum (c), and XRD pattern (d) of CNCs. (B) FT-IR spectra of (a) CNCs, (b) GOx, and (c) GOx/CNCs. (C) Nyquist plots of (a) bare GCE, (b) CS/GCE, (c) CS/CNCs/GCE, and (d) CS/GOx/CNCs/GCE. | ||
Next, the structural features of CNC-immobilized GOx were investigated. FT-IR spectroscopy is a common technology to investigate secondary-structures of polypeptide chains and conformational changes of proteins.33 FT-IR spectroscopy was used to compare structural variations of native and immobilized GOx. The amide I band at 1700–1625 cm−1 was caused by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the peptide linkage in the backbone of the protein. The amide II band at 1625–1500 cm−1 was assigned to the combination of the N–H in-plane bending and the C–N stretching vibration of the peptide groups.34 Denaturation of the enzyme molecule will result in extensive reduction or even disappearance of the amide I and II bands.26 As shown in Fig. 2B, spectra of the amide I and II bands of GOx in the GOx/CNCs film at 1635, 1538 and 1448 cm−1 were nearly the same as the corresponding bands on GOx film. This finding suggests that GOx retained the essential features of its native structure on the CNC surface.
O stretching vibration of the peptide linkage in the backbone of the protein. The amide II band at 1625–1500 cm−1 was assigned to the combination of the N–H in-plane bending and the C–N stretching vibration of the peptide groups.34 Denaturation of the enzyme molecule will result in extensive reduction or even disappearance of the amide I and II bands.26 As shown in Fig. 2B, spectra of the amide I and II bands of GOx in the GOx/CNCs film at 1635, 1538 and 1448 cm−1 were nearly the same as the corresponding bands on GOx film. This finding suggests that GOx retained the essential features of its native structure on the CNC surface.
Electrochemical impedance spectroscopy (EIS) can provide information on impedance changes of the interface of the electrode surface/electrolyte solution. This technique is a powerful tool for investigating interface properties of the modified electrode.35 EIS was used to validate the interaction between GOx and CNCs. The EIS results of bare GCE (a), CS/GCE (b), CS/CNCs/GCE (c), and CS/GOx/CNCs/GCE (d) are shown in Fig. 2C. The EIS results of electrodes modified with different films displayed a linear part and a semicircular part with different diameters. A Randles circuit (inset in Fig. 2C) was chosen to fit the impedance data obtained. The equivalent electrical network of the electrochemical interface can be represented by the electrolyte resistance between the working electrode and the reference electrode, Rs, in series with a double-layer capacitance, CPE, in parallel with the charge-transfer resistance, Rct, and the mass transport resistance, Zw. For diffusive species, the EIS data include a semicircular part at high frequencies (corresponding to the electron-transfer-limited process) and a linear part at low frequencies (corresponding to the diffusion process). The semicircle diameter observed at higher frequencies represents the electron-transfer kinetics of the redox probe at the electrode/electrolyte interface.6 The diameter of the semicircular part increased among samples, with GC < CS/CNCs/GCE < CS/GCE < CS/GOx/CNCs/GCE. These findings reflect the idea that CNCs played an important role in enhancing and promoting electron transfer, thereby decreasing resistance of the CS/GCE to Fe(CN)63− and Fe(CN)64−. On the other hand, these results may also reflect the interaction between GOx and CNCs. In PBS at pH 7.2, GOx has a negative charge (isoelectric point is 4.2), which will repel Fe(CN)63− and Fe(CN)64−. The large semicircle diameter of CS/[GOx]/[CNCs]/GCE indicates that GOx was successfully immobilized to CNCs. We believe that GOx could be immobilized to CNCs through the aid of the functional group on CNCs, adsorption, or hydrophobic interactions.
3.2. Direct electrochemistry of CS/GOx/CNCs/GCE
Direct electrochemistry of GOx on CNCs/GCE was investigated by CV. Fig. 3A shows the CVs of different kinds of modified electrodes in Ar-saturated PBS (0.1 M, pH 7.2). Obviously, no redox peaks were observed at the bare GCE (curve a), CS/GOx/GCE (curve b), and CS/CNCs/GCE (curve c) in the same potential range, however, a pair of well-defined redox peaks was observed at the CS/GOx/CNCs/GCE (curve d) at a scan rate of 100 mV s−1. The formal potential (E′0), which was calculated from the midpoint of the anodic and cathodic peak potentials, was −454 mV. The peak-to-peak potential difference (ΔEP) was 42 mV. These results indicate that immobilized GOx on the CNCs/GCE surface displayed a two-electron quasi-reversible electrochemical reaction.18 The CV peaks were located at the potential location of the cofactor flavin adenine dinucleotide (FAD) in GOx.1,36 Previous researchers36 proposed a reaction mechanism of GOx on an electrode, without the interference of oxygen, in which GOx(FAD) was reduced to GOx(FADH2), as represented by reaction (i):| GOx(FAD) + 2H+ + 2e− ↔ GOx(FADH2) | (i) |
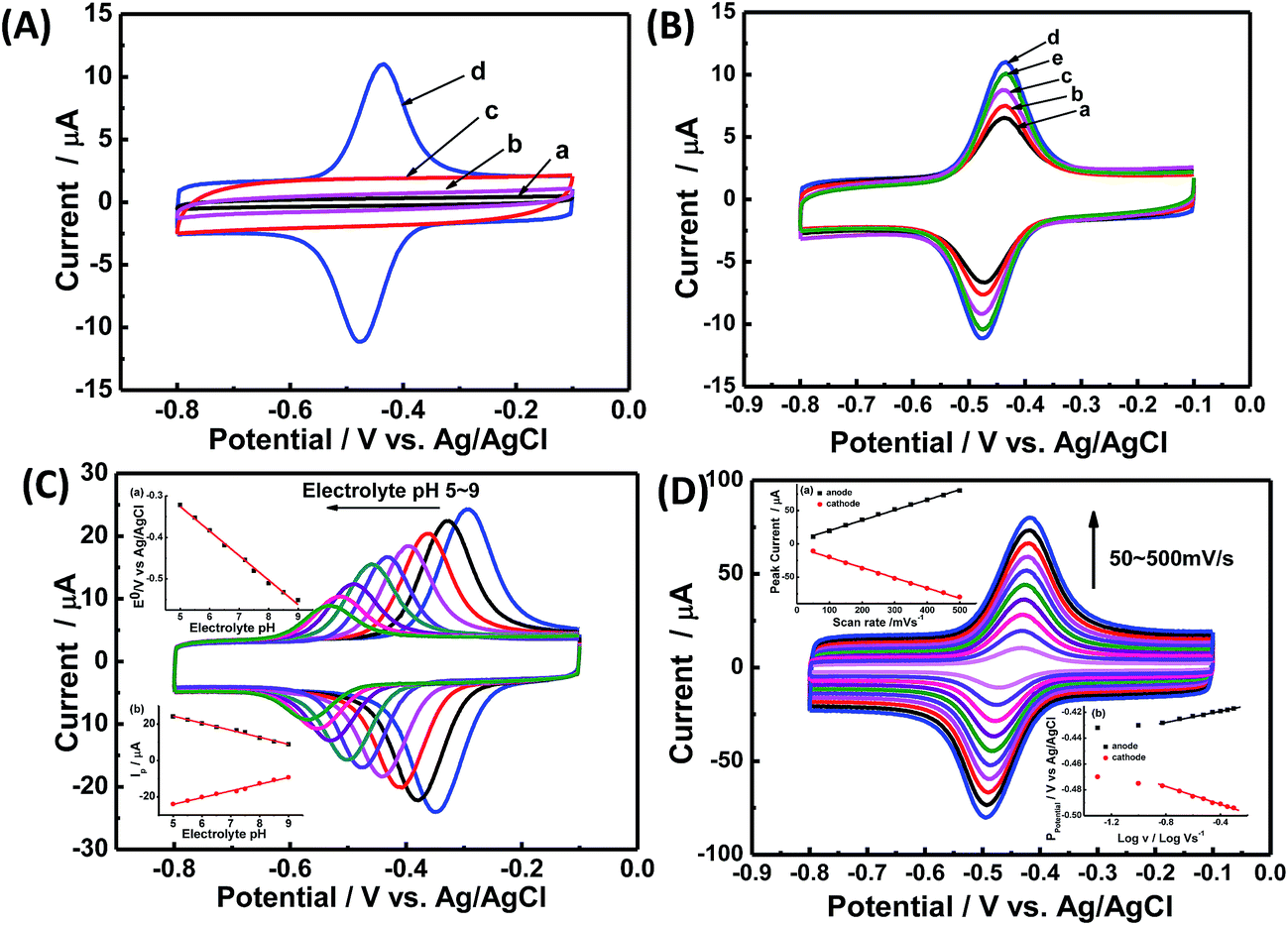 | ||
Fig. 3 (A) CV curves at 100 mV s−1 in Ar-saturated PBS (0.1 M, pH 7.2) for (a) bare GCE, (b) CS/GOx/GCE, (c) CS/CNCs/GCE, and (d) CS/GOx/CNCs/GC. (B) CV curves at 100 mV s−1 in Ar-saturated PBS (0.1 M, pH 7.2) for CS/[GOx]x/[CNCs]y/GCE with x of (a) 2, (b) 3, (c) 4, (d) 5, and (e) 6, and y of 5, where x and y are volumes of GOx and CNC solutions, respectively, in μL. (C) CV curves of CS/[GOx]5/[CNCs]5/GCE in 0.1 M Ar-saturated PBS with pH values of 5.0, 5.5, 6.0, 6.5, 7.2, 7.5, 8.0, 8.5, and 9.0 (from right to left), at a scan rate of 100 mV s−1. Insets are (a) E′0 vs. pH, and (b) IP vs. pH. (D) CV curves of CS/[GOx]5/[CNCs]5/GCE in Ar-saturated PBS (0.1 M, pH 7.2) at scan rates of 50, 100, 150, 200, 250, 300, 350, 400, 450, and 500 mV s−1 (from inner to outer). Insets are (a) IP vs. v and (b) EP vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) v. v. | ||
Taken together these results indicate that the direct electrochemistry of GOx was achieved through CNCs on the surface of GCE, and that the GOx molecules retained their electrochemical properties after they were modified on the CNCs surface.
The loading amount of GOx is important when seeking to optimize the catalytic of the modified electrode. Fig. 3B presents the CV results for the modified electrode with different volumes of GOx solution (x) under a constant volume of CNC suspension (5 μL). When the GOx volume was 5 μL, the redox current peak was at its maximum, and the GOx/CNCs produced the best catalytic activity. When the GOx volume was less than 5 μL (x = 2, 3, or 4 μL), the peak current of the modified electrode did not reach the maximum value. When the GOx volume was greater than 5 μL (x = 6 μL), the peak current of GOx for producing electrons was not sufficient to reach maximal catalytic activity. The amount of GOx for producing electrons was not sufficient to reach maximum catalytic activity when GOx volume was less than 5 μL. On the other hand, when the GOx volume was 6 μL, electrons generated at the upper layer of GOx most likely were not transferred to the CNCs, leaving the overall catalytic activity diminished. Based on these results of Fig. 3B, we considered [GOx]5/[CNCs]5 to be the optimal layer.
The pH of buffer solution affects electrochemical behavior of GOx in CS/GOx/CNCs/GCE.1 As shown in Fig. 3C, CVs curves with stable and well-defined peaks were observed from pH 5.0 to 9.0; however, the anodic and cathodic peaks shifted to the negative direction as buffer pH increased. Quantitative analysis showed that E′0 had a linear dependence on buffer pH, with a slope of −59.1 mV per pH (R2 = 0.994). This slope is close to the theoretical value of −59.2 mV per pH for the two-proton-transfer coupled and two-electron-transfer reaction.1–3 The redox peak current decreased with increasing buffer pH and showed a linear dependence on buffer pH. This finding further proves that the electrochemical reaction of GOx involved proton exchange.1,5
Fig. 3D shows CVs curve of CS/[GOx]5/[CNCs]5/GCE in Ar-saturated 0.1 M PBS (pH 7.2) with different scan rates. Redox peak currents increased gradually with increasing scan rate, accompanied by positive shifts of the anodic peaks and negative shifts of the cathodic peaks. Anodic and cathodic peak currents exhibited linear relationship with the scan rate in range from 50 to 500 mV s−1 (inset a of Fig. 3D), indicating a quasi-reversible surface-controlled electrochemical process.6,14,26 Electron transfer between immobilized GOx(FAD/FADH2) and the CNCs modified electrode was analyzed by measuring CV characteristics of the anode at different scan rates. Dependence of the anodic and cathodic peak potentials on the logarithm of scan rates from 50 to 500 mV s−1 (inset b of Fig. 3D) was used to determine the anodic electrochemical parameters according to eqn (1) and (2):37
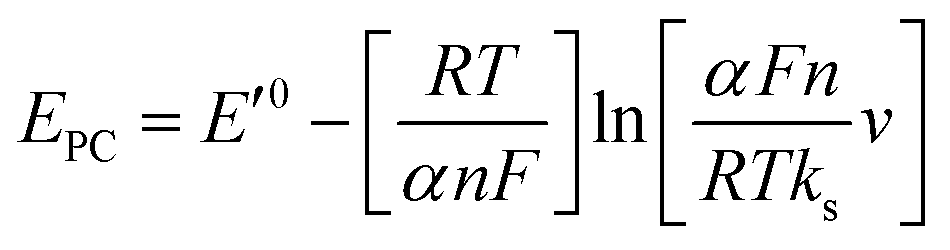 | (1) |
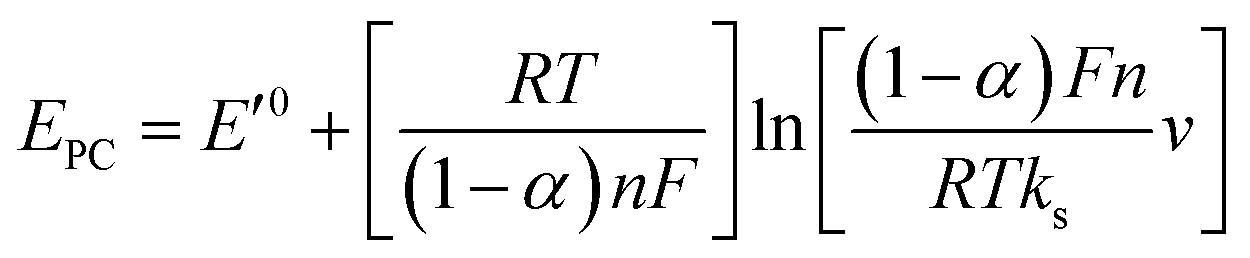 | (2) |
In these equations, E′0 is the formal potential, α is the charge transfer coefficient of the system, v is the scan rate, n is the electron transfer number, and ks is the heterogeneous electron transfer rate constant, T, R, and F are as the usual meaning. Consistent with findings in previous reports, we observed that, E′0 was linear with v above 150 mV s−1 (inset b of Fig. 3D), using the slopes of the lines, the α and n were calculated to be 0.32 and 1.53, respectively. Additionally, the ks was calculated by eqn (3):38
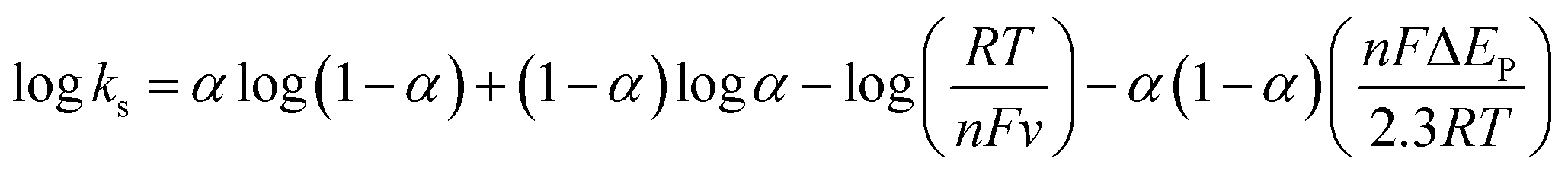 | (3) |
Thus ks of the [GOx]5/[CNCs]5 system was estimated to be 6.0 at 500 mV s−1. According to the Laviron eqn (4):38
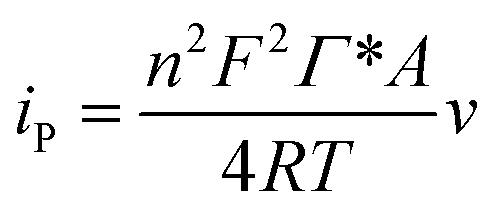 | (4) |
3.3. Electrocatalytic activity of CS/GOx/CNCs/GCE
As a fast and convenient tool for characterizing glucose biosensors,33 CV was used to characterize the electrocatalytic activity of CS/GOx/CNCs/GCE towards glucose. Fig. 4 shows the CVs of CS/[GOx]5/[CNCs]5/GCE in the absence or presence of different concentrations of glucose in air-saturated PBS at a scan rate of 100 mV s−1. After a certain volume of glucose was added to PBS, a significant cathodic peak appeared at about −485 mV. This new peak was due to glucose oxidation and was confirmed by adding different concentrations of glucose. When glucose was added to the PBS, the GOx(FAD) reduction peak of CS/[GOx]5/[CNCs]5/GCE increased, accompanied by a slight decrease of the GOx(FADH2) oxidation peak. The decrease (increase) in the oxidation peak (reduction peak) increased as increasing amounts of glucose were added. This behavior is characteristic of enzymatic catalysis. Thus, we postulated the following potential mechanism for the catalytic reaction of glucose by CS/[GOx]5/[CNCs]5/GCE:7| GOx(FAD) + glucose → GOx(FADH2) + gluconolactone | (ii) |
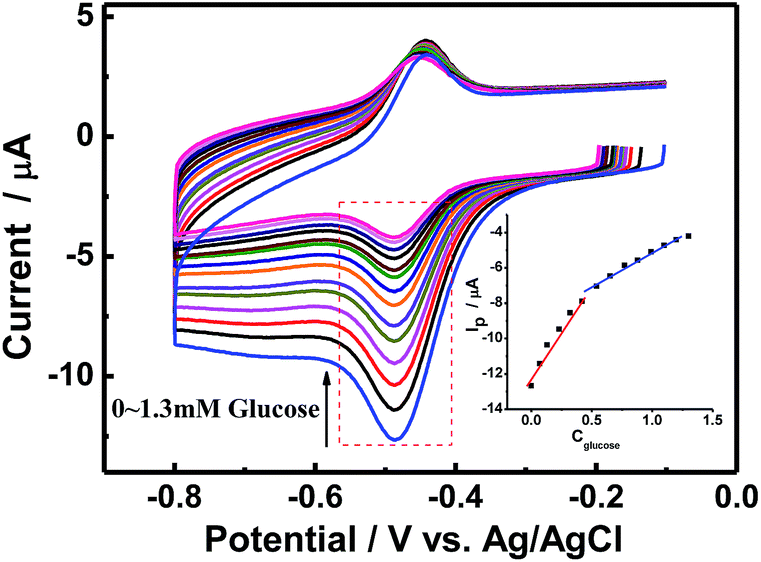 | ||
| Fig. 4 CV curves of CS/[GOx]5/[CNCs]5/GCE in air-saturated PBS (0.1 M, pH 7.2) solution with various concentrations of glucose. Scan rate: 100 m s−1. Inset: calibration curve of the linear dependence of cathodic peak current on the glucose concentration. | ||
Calibration curves (inset of Fig. 4B) showed that the CV response was linear in glucose concentration ranging 0.02–0.44 mM and 0.55–1.3 mM with correlation coefficients of 0.997 and 0.995, respectively.
Although Fig. 4 explains the DET activity of immobilized GOx on CNCs toward glucose, this figure cannot directly prove the glucose bioelectrocatalytic activity. As shown in Fig. 5A, when glucose was added to PBS, CV curves of CS/[GOx]5/[CNCs]5/GCE showed no variation, which is consistent with a previous report.7 It is well known that DET-based third-generation glucose biosensors and bioelectrodes of BFCs do not require mediators, including oxygen.7 In experiment, the hydrogen evolution reaction may occur on the surface of the Pt counter electrode:
| 2H+ + 2e− → H2 | (iii) |
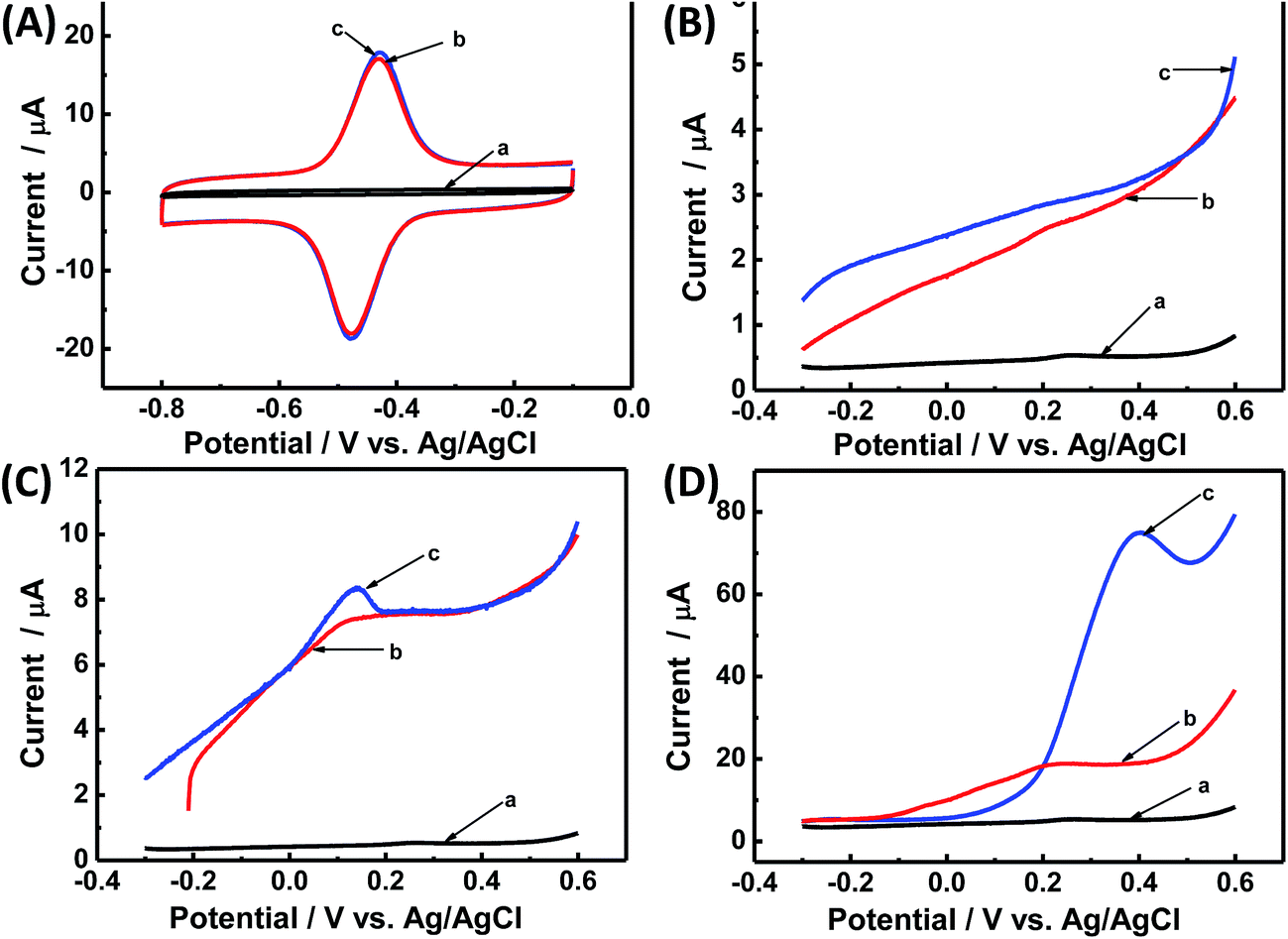 | ||
| Fig. 5 (A) CV curves of (a) CS/CNCs/GCE, and (b and c) CS/[GOx]5/[CNCs]5/GCE in Ar-saturated PBS (0.1 M, pH 7.2) in the presence of 10 mM glucose (a and c) or in the absence of glucose (b), scan rate 100 mV s−1. (B) LSV curves of (a) CS/CNCs/GCE and (b and c) CS/[GOx]5/[CNCs]5/GCE in a single compartment containing Ar-saturated PBS (0.1 M, pH 7.2) in the presence of 10 mM glucose (a and c) or in the absence of glucose (b). Scan rate: 20 mV s−1. (C) LSV curves of (a) CS/CNCs/GCE, and (b and c) CS/[GOx]5/[CNCs]5/GCE in a single compartment containing Ar-saturated PBS (0.1 M, pH 6) in the presence of 10 mM glucose (a and c) or in the absence of glucose (b). Scan rate: 20 mV s−1. (D) LSV curves of (a) CS/CNCs/GCE and (b and c) CS/[GOx]5/[CNCs]5/GCE in a double compartment separated by a Nafion membrane. Anode holder is placed in Ar-saturated PBS (0.1 M, pH 7.2) in the presence of 10 mM glucose (a and c) or in the absence of glucose (b). Cathode holder is placed in O2-saturated PBS (0.1 M, pH 3). Scan rate: 20 mV s−1. | ||
This reaction occurred very slowly in the potential window of the experiment under neutral conditions, which will be the limiting step in the three-electrode system.39
To investigate the bioelectrocatalytic activity of modified electrodes, linear sweep voltammetry (LSV) experiments were performed through improving operational conditions for Pt counter electrode in Ar-saturated PBS (0.1 M, pH 7.2) with or without glucose (Fig. 5B–D). No oxidation peak was seen when glucose was added to PBS at pH 7.2 (Fig. 5B). When the pH of the PBS was adjusted to 6, an oxidation peak was observed at 0.15 V (Fig. 5C). The acidic condition increased the proton concentration and the hydrogen evolution reaction rate (reaction (iii)), thereby increasing the overall reaction rate. Next, a double compartment separated by a Nafion membrane was used in the LSV experiment. The cathode compartment contained O2-saturated PBS (0.1 M, pH 3), while the anode compartment contained Ar-saturated PBS (0.1 M, pH 7.2). As shown in Fig. 5D, an obvious oxidation peak appeared at 0.4 V when glucose was added to the cathode compartment. This condition corresponds to the reaction of oxygen reduction on the Pt counter electrode (reaction (iv)), which proceeds more quickly than reaction (iii).40
| 4H+ + O2 → H2O | (iv) |
Therefore, the lack of a glucose response can be explained by the slow reaction rate on the Pt counter electrode. These results further demonstrate the glucose bioelectrocatalytic activity of CS/[GOx]5/[CNCs]5/GCE.
Fig. 6 shows the typical amperometric responses of CS/[GOx]5/[CNCs]5/GCE with the successive additions of 10 mM glucose at −400 mV. When glucose was added to PBS (0.1 M, pH 7.2, with stirring), the modified electrode responded rapidly to the substrate, and reduction currents increased stepwise at the CS/[GOx]5/[CNCs]5/GCE. The responses of glucose at CS/[GOx]5/[CNCs]5/GCE were linearly proportional to the glucose concentration from 0 to 1.9 mM (inset a of Fig. 6) with a detection limit of 0.16 mM. When the glucose concentration exceeded 1.9 mM, the response tended to plateau, indicating saturation of the GOx–glucose reaction. The apparent Michaelis–Menten constant (Kappm), an indicator of enzyme–substrate reaction kinetics, can be calculated by the electrochemical version of the Lineweaver–Burk equation,41
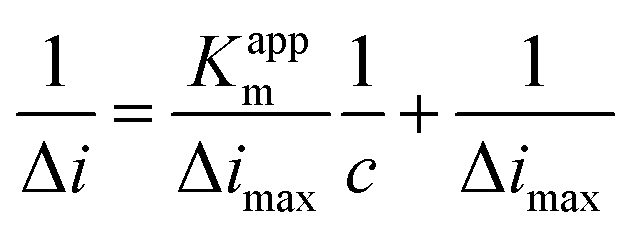 | (5) |
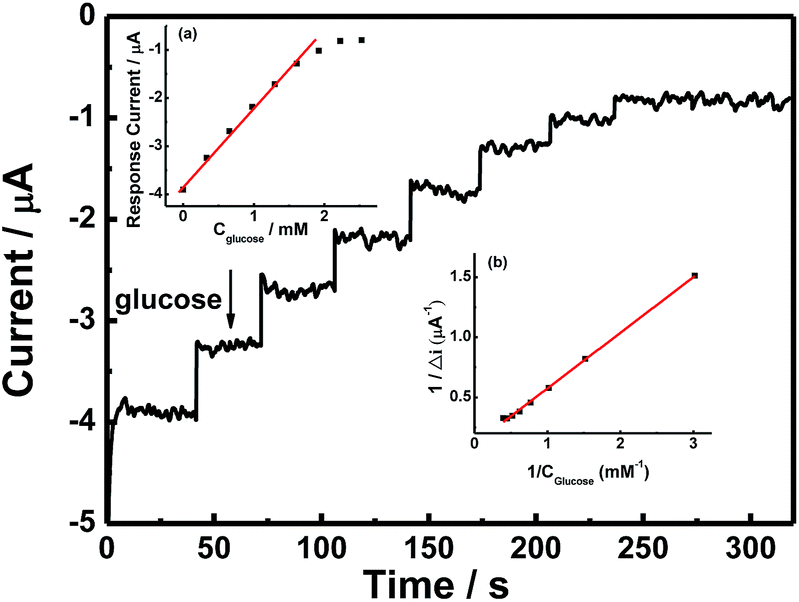 | ||
| Fig. 6 Amperometric responses of CS/[GOx]5/[CNCs]5/GCE at an operating potential of −400 mV with successive additions of 0.1 M glucose in PBS (0.1 M, pH 7.2). Inset (a): plot of catalytic current of the modified electrode vs. glucose concentration. Inset (b): Lineweaver–Burk plot for Kappm determination. | ||
3.4. BFC performance
By using the currently proposed method, glucose can be directly and bioelectrocatalytically oxidized near the redox active potential of GOx on CS/[GOx]5/[CNCs]5/GCE via a DET reaction. This approach could be desirable for increasing the open circuit voltage (OCP) of the cell and for constructing a bioanode for use in DET-type BFCs. Voltages and currents of the BFC were measured by two multimeters with external resistance varying 100 Ω to 100 kΩ. The BFC was operated at room temperature and fed with a fuel solution of 10 mM glucose stock solution (0.1 M PBS, pH 6) with saturated air. Current and power densities of the BFC were calculated from the voltage and current. Polarization curves (Fig. 7) revealed that the bioanode afforded an OCP of 0.59 V, maximum power density of 55 μW cm−2, and maximum current density of 434 μA cm−2.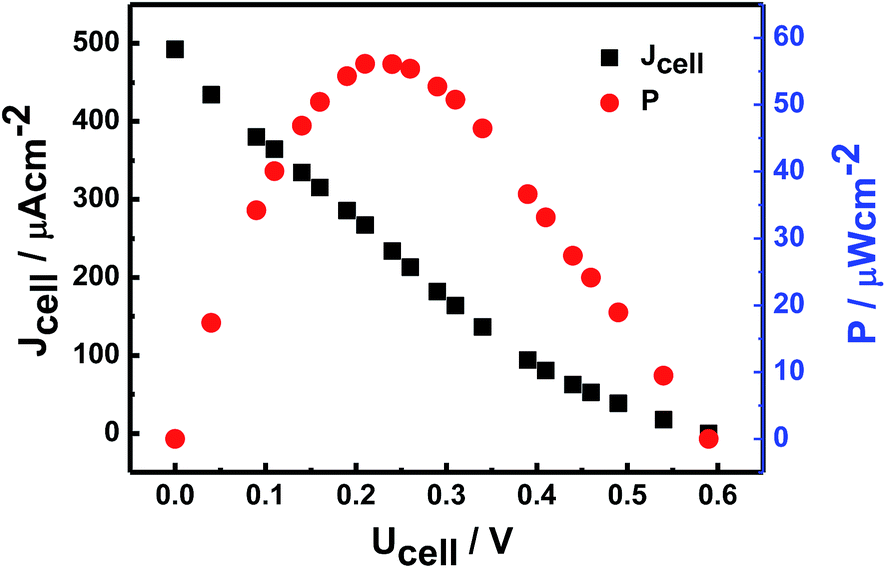 | ||
| Fig. 7 Typical power density vs. cell voltage and current density vs. cell voltage curves of CS/[GOx]5/[CNCs]5/GCE in air-saturated PBS (pH 7.2) containing 10 mM glucose against Pt electrode as cathode. | ||
4. Conclusions
In this study, a new class of graphite nanofibers, CNCs, was used to support the bioelectrocatalysis of GOx. Properties of GOx in CS/GOx/CNCs/GCE were characterized, that GOx retained a nearly native secondary structure in the CNC supports and displayed excellent DET properties. To investigate the effect of the [GOx]/[CNCs] volume ratio on electrochemical performance, the optimal CS/[GOx]5/[CNCs]5/GCE was determined via CV measurements. To investigate the direct electrocatalytic activity, LSV was used and the results indicate the immobilized GOx displayed good electrocatalytic activity in the oxidation of glucose, which had a linear current response from 0.07 to 1.9 mM with a detection limit of 0.054 mM and an apparent Michaelis–Menten constant of 0.149 mM. The polarization curve of the BFC showed that the bioanode afforded an OCP of 0.59 V, maximum power density of 55 μW cm−2, and maximum current density of 434 μA cm−2. These findings demonstrate that protein can be immobilized directly on the CNC surface to achieve DET, and provide a new perspectives toward understanding kinetics and thermodynamics of biological redox processes.Acknowledgements
Financial support for this work was provided by National Natural Science Foundation of China through grant 81301345.References
- Y. F. Gao, T. Yang, X. L. Yang, Y. S. Zhang, B. L. Xiao, J. Hong, N. Sheibani, H. Ghourchian, T. Hong and A. A. Moosavi-Movahedi, Biosens. Bioelectron., 2014, 60, 30–34 CrossRef CAS PubMed
.
- C. Cai and J. Chen, Anal. Chem., 2004, 325(2), 85–292 Search PubMed
- Q. Liu, X. Lu, J. Li, X. Yao and J. Li, Biosens. Bioelectron., 2007, 22, 3203–3209 CrossRef CAS PubMed
- J. Filip, J. Šefčovičová, P. Gemeiner and J. Tkac, Electrochim. Acta, 2013, 87, 366–374 CrossRef CAS
- K. Hyun, S. W. Han, W.-G. Koh and Y. Kwon, Int. J. Hydrogen Energy, 2015, 40, 2199–2206 CrossRef CAS
- Z. Li, C. Xie, J. Wang, A. Meng and F. Zhang, Sens. Actuators, B, 2015, 208, 505–511 CrossRef CAS
- B. Liang, X. Guo, L. Fang, Y. Hu, G. Yang, Q. Zhu, J. Wei and X. Ye, Electrochem. Commun., 2015, 550, 1–5 CrossRef
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS PubMed
- M. Bankar and J. Fransaer, Electrochim. Acta, 2012, 81, 129–137 CrossRef
- C. Gutierrez-Sanchez, M. Pita, C. Vaz-Dominguez, S. Shleev and A. L. De Lacey, J. Am. Chem. Soc., 2012, 134, 17212–17220 CrossRef CAS PubMed
- K. Tian, S. Alex, G. Siegel and A. Tiwari, Mater. Sci. Eng., C, 2015, 46, 548–552 CrossRef CAS PubMed
- L. Fang, B. Liu, L. Liu, Y. Li, K. Huang and Q. Zhang, Sens. Actuators, B, 2016, 222, 1096–1102 CrossRef CAS
- S. El Ichi, A. Zebda, A. Laaroussi, N. Reverdy-Bruas, D. Chaussy, M. Naceur Belgacem, P. Cinquin and D. K. Martin, Chem. Commun., 2014, 50, 14535–14538 RSC
- W. Feng and P. Ji, Biotechnol. Adv., 2011, 29, 889–895 CrossRef CAS PubMed
- K. P. Prasad, Y. Chen and P. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 3387–3393 CAS
- L. Qian and L. Lu, RSC Adv., 2014, 4, 38273 RSC
- Y. Zhang, M. Chu, L. Yang, Y. Tan, W. Deng, M. Ma, X. Su and Q. Xie, ACS Appl. Mater. Interfaces, 2014, 6, 12808–12814 CAS
- C.-H. Kuo, W.-H. Huang, C.-K. Lee, Y.-C. Liu, C.-M. J. Chang, H. Yang and C.-J. Shieh, Int. J. Electrochem. Sci., 2013, 8, 9242–9255 CAS
- L. Sun, Y. Ma, P. Zhang, L. Chao, T. Huang, Q. Xie, C. Chen and S. Yao, Talanta, 2015, 138, 100–107 CrossRef CAS PubMed
- Z. Yu, H. Li, X. Zhang, N. Liu, W. Tan, X. Zhang and L. Zhang, Biosens. Bioelectron., 2016, 75, 161–165 CrossRef CAS PubMed
- L. B. Crepaldi, S. A. Neto, F. P. Cardoso, P. Ciancaglini and A. R. De Andrade, Electrochim. Acta, 2014, 136, 52–58 CrossRef CAS
- K. Zhang, X. Duan, X. Zhu, D. Hu, J. Xu, L. Lu, H. Sun and L. Dong, Synth. Met., 2014, 195, 36–43 CrossRef CAS
- S. D. Choi, J. H. Choi, Y. H. Kim, S. Y. Kim, P. K. Dwivedi, A. Sharma, S. Goel and G. M. Kim, Microelectron. Eng., 2015, 141, 193–197 CrossRef CAS
- T. Terse-Thakoor, K. Komori, P. Ramnani, I. Lee and A. Mulchandani, Langmuir, 2015, 31, 13054–13061 CrossRef CAS PubMed
- M. Ahmad, C. F. Pan, Z. X. Luo and J. Zhu, J. Phys. Chem. C, 2010, 114, 9308–9313 CAS
- M. Ammam and J. Fransaer, J. Power Sources, 2014, 257, 272–279 CrossRef CAS
- C. Hou, D. Yang, B. Liang and A. Liu, Anal. Chem., 2014, 86, 6057–6063 CrossRef CAS PubMed
- K. H. Hyun, S. W. Han, W.-G. Koh and Y. Kwon, J. Power Sources, 2015, 286, 197–203 CrossRef CAS
- C. E. La Rotta and E. R. Gonzalez, J. Electrochem. Soc., 2012, 160, G37–G45 CrossRef
- S. George and H. K. Lee, J. Phys. Chem. B, 2009, 113, 15445–15454 CrossRef CAS PubMed
- B. Singh, E. Dempsey and F. Laffir, Sens. Actuators, B, 2014, 205, 401–410 CrossRef CAS
- Inamuddin, K. Ahmad and M. Naushad, Int. J. Hydrogen Energy, 2014, 39, 7417–7421 CrossRef CAS
- R. Zhao, X. Liu, J. Zhang, J. Zhu and D. K. Wong, Electrochim. Acta, 2015, 163, 64–70 CrossRef CAS
- J. Liu, X. Zhang, H. Pang, B. Liu, Q. Zou and J. Chen, Biosens. Bioelectron., 2012, 31, 170–175 CrossRef CAS PubMed
- J. Chen, R. Zhu, J. Huang, M. Zhang, H. Liu, M. Sun, L. Wang and Y. Song, The Analyst, 2015, 140, 5578–5584 RSC
- M. Tasviri, H.-A. Rafiee-Pour, H. Ghourchian and M. R. Gholami, J. Mol. Catal. B: Enzym., 2011, 68, 206–210 CrossRef CAS
- A. S. Campbell, Y. J. Jeong, S. M. Geier, R. R. Koepsel, A. J. Russell and M. F. Islam, ACS Appl. Mater. Interfaces, 2015, 7, 4056–4065 CAS
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1974, 52, 395–402 CrossRef CAS
- A. Lasia, Handbook of fuel cells, 2010 Search PubMed
- B. C. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed
- Y. Huang, X. Qin, Z. Li, Y. Fu, C. Qin, F. Wu, Z. Su, M. Ma, Q. Xie, S. Yao and J. Hu, Biosens. Bioelectron., 2012, 31, 357–362 CrossRef CAS PubMed
- Y. Liu, Y. Du and C. M. Li, Electroanalysis, 2013, 25, 815–831 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |