
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous deposition of Ni(OH)2 onto cellulose-derived carbon aerogels for low-cost energy storage electrodes†
Guoqing Zu *abc,
Jun Shen*b,
Zhihua Zhangb,
Bin Zhoub,
Xiaodong Wangb,
Guangming Wub and
Yewen Zhangc
*abc,
Jun Shen*b,
Zhihua Zhangb,
Bin Zhoub,
Xiaodong Wangb,
Guangming Wub and
Yewen Zhangc
aTongji University, Shanghai, 200092, PR China. E-mail: guoqingzu@yahoo.com
bShanghai Key Laboratory of Special Artificial Microstructure Materials and Technology, Pohl Institute of Solid State Physics, Tongji University, Shanghai, 200092, PR China. E-mail: shenjun67@tongji.edu.cn
cSchool of Electronics and Information Engineering, Tongji University, Shanghai, 200092, PR China
First published on 8th February 2017
Abstract
Nano-architectured carbon aerogel/Ni(OH)2 composites have been prepared via a wet chemical approach that combines the sol–gel preparation of a highly porous carbon aerogel using microcrystalline cellulose, a low-cost and renewable polymer, as the carbon source and subsequent homogeneous deposition of Ni(OH)2 nanoparticles onto the backbone of the carbon aerogel via a two-step chemical precipitation process. The deposited Ni(OH)2 has small particle size (3–10 nm) and uniform dispersion and is well exposed to the electrolyte. The resulting composite possesses an interconnected, three-dimensional, high-surface-area (327 m2 g−1) nanostructure, which provides efficient transport of electrolyte ions and electrons and enables a fuller utilization of Ni(OH)2, thus leading to excellent electrochemical performance. The composite electrode exhibits high specific capacitance of 1906 and 1206 F g−1 at current density of 1 and 20 A g−1, respectively, which are much higher than those of Ni(OH)2. Moreover 89% capacitance is retained after 4000 cycles, implying a good cycling stability.
1. Introduction
Electrochemical energy storage devices have been widely studied in order to deal with the energy crisis. They are usually divided into two types, supercapacitors and batteries, in terms of their charge/discharge behaviors. Supercapacitors have drawn great interest as high-efficiency energy storage devices because of such advantages as long cycling life, high power density and short charging/discharging time.1,2 However, the energy density of the supercapacitor is much lower than that of batteries, which limits its applications. The most widely studied electrode materials for energy storage devices are such transition metal oxides or hydroxides as RuO2,3 MnO2,4 Co3O4,5 NiO6 and Ni(OH)2.7 Among these transition metal oxides or hydroxides, Ni(OH)2 is a promising candidate for high-performance energy storage electrodes due to its relatively low cost, natural abundance and high theoretical specific capacity.7–9However, the reported Ni(OH)2 usually exhibits poor cycling stability and low electric conductivity and its specific capacity is still far below the theoretical value.10,11 This is probably because active Ni(OH)2 is not effectively exposed to the electrolyte and the efficiency of the electrolyte ions and electron transport within the electrode material are relatively low, which limit the utilization of Ni(OH)2 for capacity generation. There are mainly two methods for enhancing the electrochemical performance of Ni(OH)2-based materials: the preparation of highly porous, high-surface-area Ni(OH)2, and the growth of Ni(OH)2 in a porous matrix of high electric conductivity such as activated carbon,12–14 carbon nanotubes (CNTs),8,15–18 graphene,19–23 conductive polymers,24 nickel foams,2 and some other metals (Au, Ag, Cu, etc.).25–27 The cycling stability of conductive polymers is usually unsatisfactory and the metals with excellent conductivities, such as Au and Ag, are expensive. Nickel foams are relatively low cost and can be directly used as binder-free electrodes. Among these matrixes, carbon-based matrixes demonstrate their advantage of high surface area, which enables more efficient charge and mass exchange, leading to high electrochemical performance. The interconnected, three-dimensional, conductive structure of the matrix, combined with the porous structure of Ni(OH)2, facilitates electrolyte ions and electron transport and capacity generation from Ni(OH)2 and effectively enhances the specific capacities and cycling stabilities. Recently, M. Xie et al. reported structurally stable Ni(OH)2, grown in situ on nickel foam, for supercapacitors with high energy density of 57.9 W h kg−1.28 The electrodes obtained from Ni(OH)2/ZnO and Ni(OH)2/Cu composites grown on nickel foam exhibited a high energy density of 42 W h kg−1 and a high areal capacitance of 8.66 F cm−2.27,29 The Ni(OH)2 electrodes with carbon-based matrixes also showed excellent electrochemical capacitive performances. The reported Ni(OH)2/graphene hydrogel electrodes exhibited a high specific capacitance of 1632 F g−1.30
Carbon aerogel (CA) is another type of promising porous matrix for electrode materials because of its numerous advantages such as high porosity, high surface area, low density, excellent electrical conductivity, and open, three-dimensional, interconnected network.31,32 Recently, several CA-based composites for energy storage electrodes have been reported, including CA/NiCo2O4 composite,1 CA/Mn3O4 composite,33 CA/Fe3O4 composite,34 etc. The carbon sources of CAs are mainly organic aerogels that are usually prepared from such precursors as resorcinol/formaldehyde, cresol/formaldehyde, melamine/formaldehyde and resorcinol/pyrocatechol in an inert atmosphere.35–38 Those traditional methods for the preparation of CAs involve harmful and expensive precursors and complex synthesis process, which limits the practical applications of CAs.39,40 Recently, cellulose, a natural, renewable and abundant polymer, has attracted increasing attention as a source for the preparation of CAs.41–43 Highly porous and high-surface-area CAs with high electric conductivities can be obtained from cellulose. It is reported that the electrochemical performance of the cellulose-derived CAs is comparable to that of the traditional CAs.44,45
In this study, high-surface-area CA/Ni(OH)2 composites are prepared, for the first time, via a two-step chemical precipitation method using microcrystalline cellulose as the carbon source. First of all, cellulose aerogels are prepared by dissolving microcrystalline cellulose in an aqueous solution of NaOH, followed by gelation, regeneration and supercritical CO2 drying. Subsequently, CAs are obtained by carbonizing the cellulose aerogels in an inert atmosphere. Finally, Ni(OH)2 nanoparticles are homogeneously deposited onto the backbone of the high-surface-area CAs via a two-step chemical precipitation route. The precursor, namely nickel chloride, is first deposited onto the CA backbone, followed by the growth of Ni(OH)2 nanoparticles on the backbone by introduction of water and an alkaline catalyst. This two-step chemical precipitation route ensures the deposition of Ni(OH)2 nanoparticles on the backbone of the CA matrix and affords cellulose-derived CA/Ni(OH)2 composites with high-surface-area, interconnected, three-dimensional and homogeneous nanostructures. This composite nanostructure enables Ni(OH)2 to be well exposed to the electrolyte and provides efficient transport of electrolyte ions and electrons, leading to the excellent electrochemical performance of the CA/Ni(OH)2 composites. The resulting composite materials may have application potentials in low-cost and high-performance energy storage devices.
2. Experimental
2.1 Materials
Ethanol (EtOH), methanol (MeOH), NaOH, KOH, aqueous ammonia (NH3) (25–28 wt%), NiCl2·6H2O and microcrystalline cellulose were purchased from Sinopharm Chemical Reagent Corporation (China). Distilled water was purchased from the School of Environmental Science and Engineering of Tongji University. Polyvinylidene fluoride (PVDF) was purchased from Sigma-Aldrich, Co.2.2 Synthesis of cellulose-derived CA
The synthesis procedure for the cellulose-derived CA is similar to that of our previous reports.46 Distilled water was mixed with a certain amount of NaOH and microcrystalline cellulose at room temperature, separately. The temperatures of the mixtures of NaOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 w/w) and cellulose/H2O (1:7.5 w/w) were decreased to −10 °C and 5 °C, respectively, by putting them in a refrigerator. A transparent cellulose solution was obtained by mixing 24 g of NaOH/H2O and 10.2 g of cellulose/H2O with vigorous stirring for 30 min at room temperature. After the gel was formed, it was aged for 2 days, and then the wet gel was regenerated by immersing it in 120 mL of EtOH for 12 h. The regenerated wet gel was soaked 4 times in distilled water at 50 °C to remove the residual chemicals. Before drying, the wet gel was subjected to solvent exchange with ethanol 4 times to afford the alcogel. The cellulose aerogel was obtained after the alcogel was dried with supercritical CO2 at 33 °C and 10 MPa. The cellulose aerogel was put into a tube furnace, followed by nitrogen flushing at a rate of 600 mL min−1 for 1 h. The temperature was then raised to 800 °C at a rate of 5 °C min−1 and kept at this temperature for 2 h under nitrogen flow at a rate of 300 mL min−1. Finally, CA was obtained after the system was cooled to room temperature naturally.
:7 w/w) and cellulose/H2O (1:7.5 w/w) were decreased to −10 °C and 5 °C, respectively, by putting them in a refrigerator. A transparent cellulose solution was obtained by mixing 24 g of NaOH/H2O and 10.2 g of cellulose/H2O with vigorous stirring for 30 min at room temperature. After the gel was formed, it was aged for 2 days, and then the wet gel was regenerated by immersing it in 120 mL of EtOH for 12 h. The regenerated wet gel was soaked 4 times in distilled water at 50 °C to remove the residual chemicals. Before drying, the wet gel was subjected to solvent exchange with ethanol 4 times to afford the alcogel. The cellulose aerogel was obtained after the alcogel was dried with supercritical CO2 at 33 °C and 10 MPa. The cellulose aerogel was put into a tube furnace, followed by nitrogen flushing at a rate of 600 mL min−1 for 1 h. The temperature was then raised to 800 °C at a rate of 5 °C min−1 and kept at this temperature for 2 h under nitrogen flow at a rate of 300 mL min−1. Finally, CA was obtained after the system was cooled to room temperature naturally.
2.3 Synthesis of cellulose-derived CA/Ni(OH)2 composites
The CA/Ni(OH)2 composite was prepared via a two-step chemical precipitation method according to the following procedure. The monolithic CA was first immersed in the mixture of methanol (5.0 mL) and NiCl2·6H2O (2.4 or 6.5 g) at 50 °C for 12 h, followed by drying at 50 °C for 1 day, for deposition of NiCl2 on the CA backbone. Then, the dried CA composite was immersed in the mixture of methanol, aqueous ammonia and distilled water with the molar ratio of MeOH:NH3:H2O being 10.5:1:3.5 at room temperature for 1 day for the formation of Ni(OH)2 nanoparticles on the CA backbone. The CA/Ni(OH)2 composite was obtained after washing with water and methanol and drying at 100 °C for 1 day. The content of Ni(OH)2 in the composites deposited by 2.4 and 6.5 g NiCl2·6H2O was around 30 and 75 wt%, respectively, which was determined by the weight changes before and after the incorporation of Ni(OH)2. The CA/Ni(OH)2 composites with 30 and 75 wt% Ni(OH)2 were denoted as CA/Ni(OH)2/0.30 and CA/Ni(OH)2/0.75, respectively.
For comparison, pure Ni(OH)2 was also prepared according to the following two steps. First, NiCl2·6H2O (2 g) was dissolved in methanol (12 mL) at room temperature. Then, aqueous ammonia and distilled water (the molar ratio of MeOH:NH3:H2O was 10.5:1:3.5) were added to the precursor solution with stirring for 30 min at room temperature. Pure Ni(OH)2 was obtained after aging for 1 day, filtering, washing with water and methanol, and drying at 100 °C for 1 day.
2.4 Characterization
The morphologies of the samples were measured using a transmission electron microscope (TEM, JEOL-1230, JEOL, Japan) and a scanning electron microscope (SEM, XL30FEG, Philips, Netherlands). The compositions and crystallite structures were determined by powder X-ray diffraction (XRD) in a Rigata/max-C diffractometer using the Cu-Kα radiation (DX-2700, HaoYuan Instruments, China). The surface states of the samples were characterized by an X-ray photoelectron spectrometer (XPS, PHI-5000C ESCA, Perkin Elmer, USA) and a Raman spectrometer with 514.5 nm wavelength incident laser light (LabRAM HR800, HORIBA JOBIN YVON, France). The surface functional groups were investigated by Fourier transform infrared (FTIR) spectroscopy (TENSOR27, Bruker, Germany). The pore structure and SSA were measured by a N2 adsorption analyzer (TriStar 3000, Quantachrome Instruments, USA). The SSA was determined using the (Brunauer–Emmett–Teller) BET method. The pore volume was calculated from the amount of N2 adsorbed at a relative pressure of P/P0 = 0.99. The pore size distribution and mean pore diameter were obtained from the adsorption branch of the isotherm via the Barrett–Joyner–Halenda (BJH) method.2.5 Electrochemical measurement
The electrochemical measurements were carried out using an electrochemical working station (CHI660A, Shanghai, China) in a three-electrode system with a 6 M KOH solution as the electrolyte. The working electrode was prepared by loading the mixture of the active materials (carbon aerogel plus Ni(OH)2), poly(vinylidene fluoride) (PVDF) and carbon black with a mass ratio of 8:1:1 on a graphite substrate, followed by drying at 80 °C for 12 h. The working area of the electrode was 1 × 1 cm2 and the amount of the active materials was about 2 mg. A saturated calomel electrode (SCE) and Pt electrode served as the reference and counter electrodes, respectively. The cyclic voltammetry (CV) curves were measured within a potential range from −0.1 to 0.45 V and −0.3 to 0.6 V by varying the scan rate from 5 to 100 mV s−1 and 100 to 500 mV s−1, respectively. Galvanostatic charge/discharge measurements were conducted within a potential window of −0.1 to 0.4 V with a constant current density of 1–20 A g−1. The specific capacitance was calculated from the galvanostatic charge/discharge curve according to equation Q = (I × Δt)/(m × ΔV) where C is the specific capacitance (F g−1), I is the discharge current (A), Δt is the discharge time (s), ΔV is the discharge voltage, and m is the active material mass of the electrode (g).
3. Results and discussion
3.1 Synthesis
Fig. 1 illustrates the fabrication process of the cellulose-derived CA/Ni(OH)2 composite. The cellulose microcrystalline powders (as shown in Fig. 2a) were first converted into a monolithic cellulose aerogel by a sol–gel process, followed by supercritical CO2 drying. Subsequently, the cellulose aerogel was carbonized in an inert atmosphere to afford monolithic cellulose-derived CA (Fig. 2b) as a nanoporous matrix. Finally, the cellulose-derived CA/Ni(OH)2 composite was obtained after a two-step chemical precipitation process. In the first step of the chemical precipitation process, the precursor was deposited into the porous CA matrix by immersing the CA in the precursor solution (NiCl2·6H2O/MeOH), followed by ambient drying. In the second step, the precursor was precipitated and Ni(OH)2 nanoparticles were deposited onto the CA backbone by immersing the CA in the alkaline solution (NH3/MeOH/H2O), followed by solvent exchange and ambient drying. This fabrication method enabled the active material, namely Ni(OH)2, to be deposited onto the backbone of the conductive nanoporous CA matrix obtained from a low-cost and renewable polymer, which is presumed to lower the cost and enhance the efficiency of capacity generation of Ni(OH)2.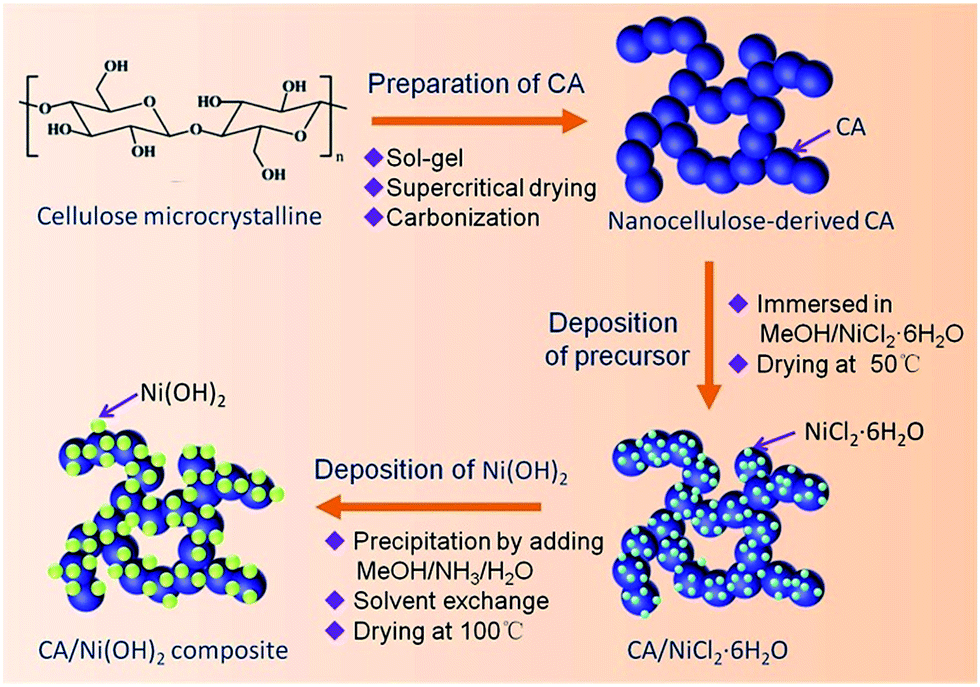 | ||
| Fig. 1 Schematic of the fabrication process of the cellulose-derived CA/Ni(OH)2 composite. | ||
 | ||
| Fig. 2 Photographs of (a) microcrystalline cellulose powder, (b) cellulose-derived CA, and (c) Ni(OH)2. (d) SEM images and (e and f) TEM images of cellulose-derived CA. (g) SEM images and (h and i) TEM images of Ni(OH)2. | ||
3.2 Morphology and appearance
As shown in Fig. S1,† the obtained cellulose aerogel is monolithic and exhibits randomly interconnected three-dimensional networks made up of fibrous nanoparticles with thickness of 40–60 nm. The SEM image (Fig. 2d) confirms that the cellulose-derived CA preserves the interconnected networks of the cellulose aerogel. Fig. 2e and f show the TEM images of the cellulose-derived CA. It can be seen that the CA exhibits randomly interconnected networks made up of fibrous particles with thicknesses in the range of 15–40 nm, which are smaller than that of the cellulose aerogel. The interconnected networks create abundant pores varying in size from several nanometers to around 100 nm, which confirms the nanoporous structure of the cellulose-derived CA. For comparison, pure Ni(OH)2 (Fig. 2c) was also prepared via a similar chemical precipitation method. As shown in the SEM image (Fig. 2g) and TEM image at low magnification (Fig. 2h), the obtained Ni(OH)2 exhibits randomly interconnected networks with numerous irregular aggregates larger than 100 nm. The TEM image at high magnification (Fig. 2i) shows that the aggregate is porous and made up of nanoparticles smaller than 20 nm. The corresponding selected area electron diffraction pattern (the inset of Fig. 2h) shows a set of concentric, diffuse rings indicating the polycrystalline structure of the obtained Ni(OH)2 sample.As shown in the SEM images (Fig. 3a, b, e and f), after the incorporation of Ni(OH)2, the cellulose-derived CA/Ni(OH)2 composite also exhibits the three-dimensional, interconnected, porous nanostructure. From the TEM images (Fig. 3c, d, g and h) we can further see that the morphology of the CA/Ni(OH)2 composite is quite different from those of the cellulose-derived CA and Ni(OH)2. The CA/Ni(OH)2 composite not only keeps the nanoporous structure of the CA but also shows embedded Ni(OH)2 nanoparticles on the interconnected CA backbone. The magnified TEM images (Fig. 3d and h) show that the small Ni(OH)2 nanoparticles in the ranges of 3–8 and 3–10 nm are homogeneously deposited onto the CA backbone for CA/Ni(OH)2/0.30 and CA/Ni(OH)2/0.75, respectively. Apparently, the particle size of Ni(OH)2 in the CA/Ni(OH)2 composite is much smaller than that of the pure Ni(OH)2 sample and there is no large Ni(OH)2 aggregate observed in the composite. This composite nanostructure would enable Ni(OH)2 to be well exposed to the electrolyte and make the charge travel distance within the Ni(OH)2 domain much shorter. The corresponding selected area electron diffraction pattern of CA/Ni(OH)2/0.75 (the inset of Fig. 3h) shows weaker concentric diffuse rings, compared to those of pure Ni(OH)2, which indicates the lower crystallinity of Ni(OH)2 in the CA/Ni(OH)2 composite, compared to that of the pure Ni(OH)2 sample. Fig. 3i and j show the appearance of CA/Ni(OH)2/0.30 and CA/Ni(OH)2/0.75, respectively. It can be seen that the CA/Ni(OH)2 composite maintains the initial shape of the CA and exhibits little shrinkage after the two-step chemical precipitation process.
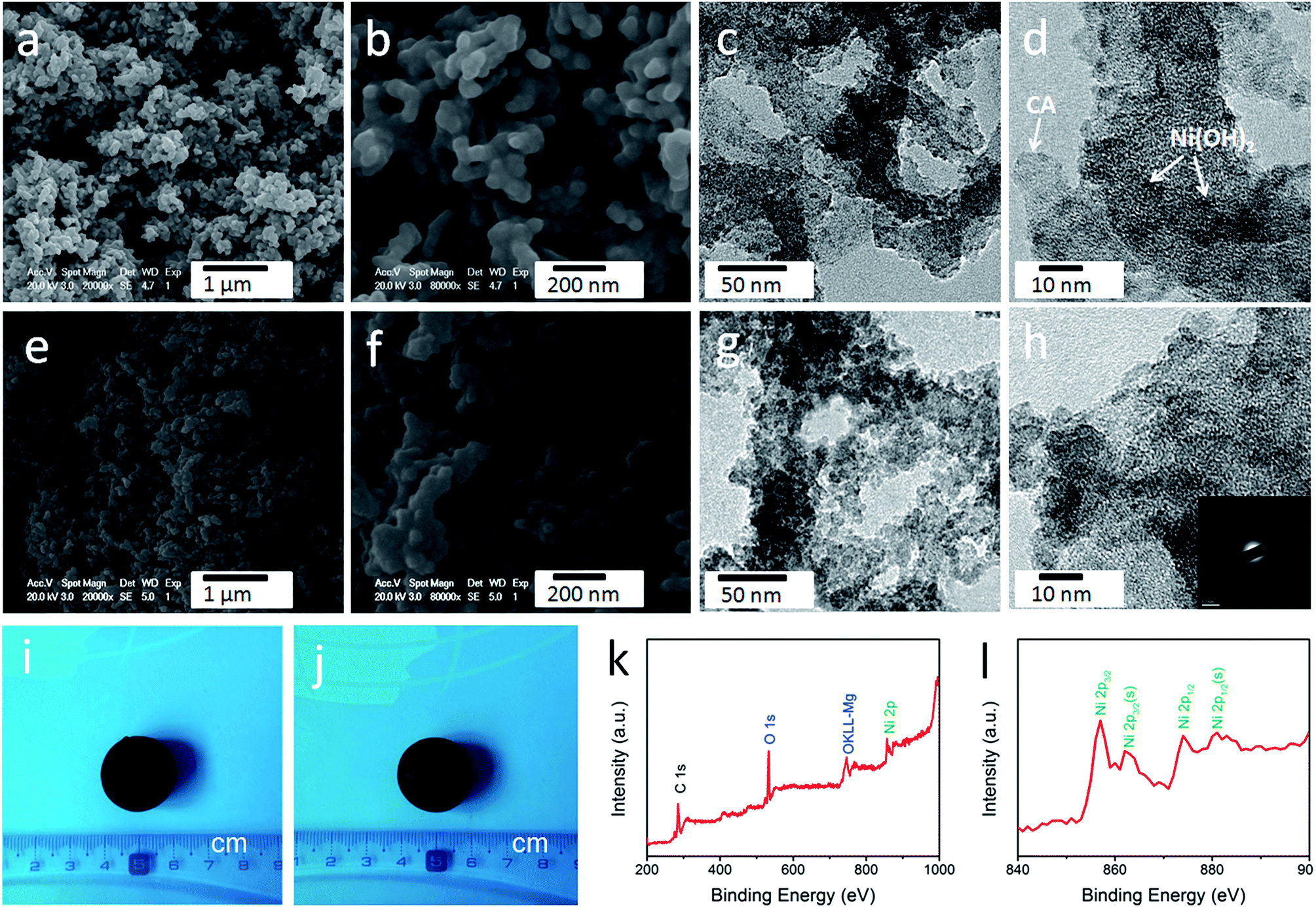 | ||
| Fig. 3 (a and b) SEM images and (c and d) TEM images of CA/Ni(OH)2/0.30. (e and f) SEM images and (g and h) TEM images of CA/Ni(OH)2/0.75. Photographs of (i) CA/Ni(OH)2/0.30 and (j) CA/Ni(OH)2/0.75. (k) XPS spectra and (l) Ni 2p XPS spectra of CA/Ni(OH)2/0.75. The inset is the selected area electron diffraction pattern of CA/Ni(OH)2/0.75. | ||
3.3 Composition
The composition of CA/Ni(OH)2/0.75 was examined by XPS measurement as shown in Fig. 3k and l. The XPS spectra confirm the presence of C, O, and Ni elements in the CA/Ni(OH)2 composite. It should be noted that the Ni 2p XPS spectra show two major peaks at around 856 and 874 eV, corresponding to Ni 2p3/2 and Ni 2p1/2, respectively, with a spin-energy separation of approximately 18 eV, which is characteristic of a Ni(OH)2 phase.47 The satellite peaks at around 862 and 880 eV, corresponding to Ni 2p3/2 and Ni 2p1/2, respectively, are also observed.The compositions of the cellulose-derived CA and CA/Ni(OH)2 composite were further investigated by Raman spectra and Fourier transform infrared (FTIR) spectral measurements. Fig. 4a shows the Raman spectra of the CA and CA/Ni(OH)2 composite. All the curves exhibit two broad peaks at around 1361 and 1583 cm−1, corresponding to the D and G bands of the CA. The G band represents the in-plane bond-stretching motion of the pairs of C sp2 atoms, whereas the D band is assigned to the breathing modes of rings or κ-point phonons of A1g symmetry.19 Probably due to the incorporation of Ni(OH)2, these two peaks of the CA/Ni(OH)2 composite are apparently weaker than those of the CA. It should be noted that the spectra of the CA/Ni(OH)2 composite exhibits two additional broad peaks at around 361 and 530 cm−1, which can be assigned to the Eu(T) and A2u(T) lattice vibrations of Ni(OH)2, respectively.20,48 Due to the higher loading of Ni(OH)2, the peaks at 361 and 530 cm−1 of CA/Ni(OH)2/0.75 are more apparent than those of CA/Ni(OH)2/0.30. The FTIR spectra of the CA and CA/Ni(OH)2 composite are shown in Fig. 4b. The broad band at 3420 cm−1 is attributed to the O–H stretching vibration, whereas the band at 1400 cm−1 is assigned to the C–H bond.49 The band at 1623 cm−1 can be assigned to H–O–H bending vibration of weakly bound H2O adsorbed on the nanoparticle surface of aerogels.50 The band at 1100 cm−1 can be ascribed to the C–O–C stretching vibration.34 These bands indicate that there are –OH, C–H, H2O, and C–O–C in the CA and the CA/Ni(OH)2 composite. In addition, unlike the spectrum of the CA, the spectrum of the CA/Ni(OH)2 composite shows two bands at around 665 and 457 cm−1 that are ascribed to the δOH and νNi–OH vibrations respectively, which become more intense with higher loading of Ni(OH)2.19
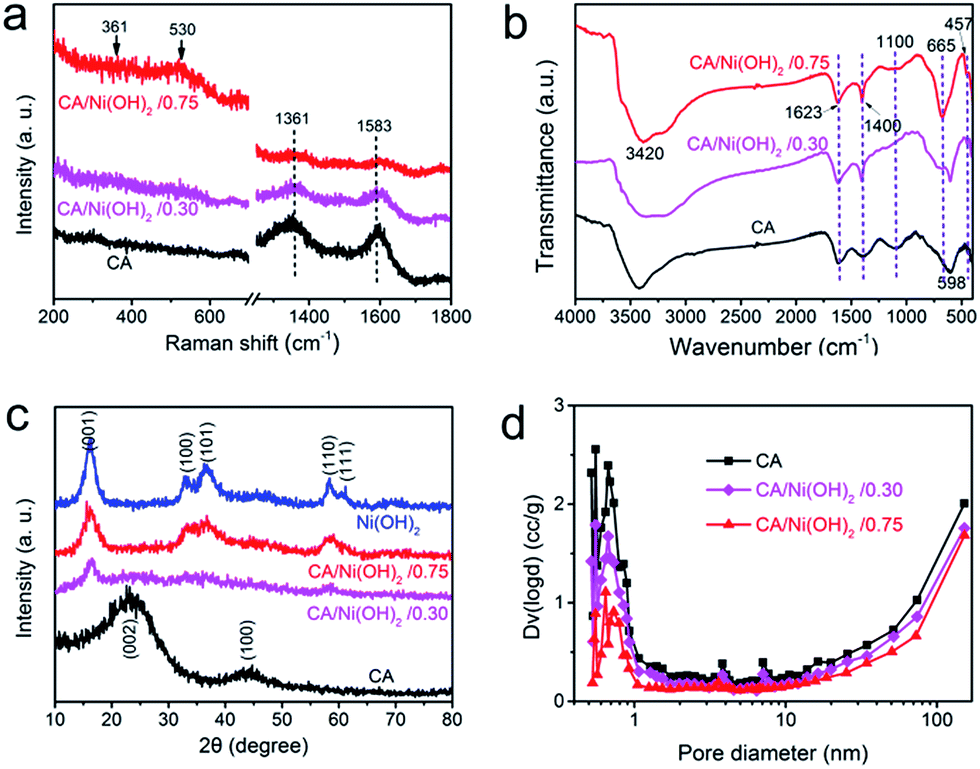 | ||
| Fig. 4 (a) Raman spectra and (b) FTIR spectra of the cellulose-derived CA and CA/Ni(OH)2 composite. (c) XRD patterns of cellulose-derived CA, Ni(OH)2, and CA/Ni(OH)2 composite. (d) Pore size distributions of cellulose-derived CA and CA/Ni(OH)2 composite. | ||
Fig. 4c shows the XRD patterns of the cellulose-derived CA, Ni(OH)2 and CA/Ni(OH)2 composite. The two broad diffraction peaks at around 24° and 43° in the XRD patterns correspond to the CA.1,41 The XRD pattern of Ni(OH)2 shows broad peaks at 16.2°, 32.9°, 36.6°, 58.4°, and 61.1°, which are assigned to the (001), (100), (101), (110), and (111) lattice planes of β-Ni(OH)2, respectively (JCPDS no: 14-0117).16,17 These five broad peaks can also be identified in the diffraction pattern of CA/Ni(OH)2/0.75. Moreover, the peaks at 16.2° and 58.4° can be identified in the diffraction pattern of CA/Ni(OH)2/0.30. These broad peaks indicate the successful incorporation of Ni(OH)2 and the polycrystalline structure of the CA/Ni(OH)2 composite. In addition, the weaker peaks of the CA/Ni(OH)2 composite compared to those of Ni(OH)2 reveal the lower crystallinity of Ni(OH)2 in the CA/Ni(OH)2 composite, compared to that of the pure Ni(OH)2 sample. These results are consistent with the abovementioned selected area electron diffraction pattern (the insets of Fig. 2h and 3h) analysis. The lower crystallinity is probably because the nanoporous CA matrix restrains the growth of large aggregates and enables the homogenous deposition of smaller Ni(OH)2 nanocrystals than those of the Ni(OH)2 sample prepared without the matrix, which was confirmed by the TEM results mentioned above.
3.4 Pore structure and specific surface area
The pore size distributions of the cellulose-derived CA and CA/Ni(OH)2 composite were measured by a N2 adsorption analyzer using the Brunauer–Emmett–Teller (BET) N2 adsorption/desorption technique as shown in Fig. 4d. It was observed that both the CA and CA/Ni(OH)2 composite exhibit micropores (<2 nm), mesopores (2–50 nm) and macropores (>50 nm), and show a pore size peak located in the micropore region. The pore volume, particularly the micropore volume, decreased after the incorporation of Ni(OH)2, which further decreased with higher loading of Ni(OH)2. Table 1 details the pore structural parameters of the CA and CA/Ni(OH)2 composite. The specific surface area and total pore volume decreased from 883 m2 g−1 and 1.74 cm3 g−1 for the CA to 327 m2 g−1 and 1.23 cm3 g−1, respectively for CA/Ni(OH)2/0.75. The corresponding micropore specific surface area and micropore volume were reduced from 506 m2 g−1 and 0.202 cm3 g−1 for the CA to 125 m2 g−1 and 0.054 cm3 g−1, respectively for CA/Ni(OH)2/0.75. There are probably two main reasons for the decrease in pore volume and specific surface area, particularly micropore volume and micropore specific surface area. On one hand, nanopores in the CA matrix are partially filled with Ni(OH)2 nanoparticles during the precipitation process. On the other hand, some nanopores of the CA may collapse during the ambient drying process. According to the report,51 the deposition of polymer follows the hierarchical structure of aerogels. Thus, Ni(OH)2 forms a conformal coating around primary particles and then fills secondary particles, followed by accumulating mostly on and around secondary particles. This deposition process results in the significant decrease of micropores in the hierarchically porous CAs. Mainly due to the decrease in micropores, after the incorporation of Ni(OH)2, the mean pore diameter increases from 9.48 nm for the CA to 13.1 nm for CA/Ni(OH)2/0.30 and 15.0 nm for CA/Ni(OH)2/0.75. The N2 adsorption/desorption isotherms of the cellulose-derived CA and CA/Ni(OH)2/0.75 are additionally provided in Fig. S2.† Both of the aerogels exhibit type IV isotherms and type H3 hysteresis loops, indicating the nanoporous 3D network structure. This high-surface-area, highly porous composite nanostructure would enable active Ni(OH)2 to be well exposed to the electrolyte and enhance the efficiency of the electrolyte ions and electron transport within the electrode material, which could significantly enhance the efficiency of capacity generation from Ni(OH)2.| Sample | SBETa (Smicb) (m2 g−1) | Dc (nm) | Vtd (Vmice) (cm3 g−1) |
|---|---|---|---|
| a Specific surface area.b Micropore specific surface area from t-method micropore analysis.c Mean pore diameter.d Total pore volume.e Micropore volume from t-method micropore analysis. | |||
| Cellulose-derived CA | 883 (506) | 9.48 | 1.74 (0.202) |
| CA/Ni(OH)2/0.30 | 529 (238) | 13.1 | 1.42 (0.097) |
| CA/Ni(OH)2/0.75 | 327 (125) | 15.0 | 1.23 (0.054) |
3.5 Electrochemical performances
The electrochemical performances of the CA/Ni(OH)2 composite as well as the cellulose-derived CA and Ni(OH)2 were investigated using cyclic voltammetric (CV) measurements within the potential window from −0.1 to 0.45 V and −0.3 to 0.6 V in 6 M KOH aqueous solution with a two-electrode system. The CV curves of the CA, Ni(OH)2, and CA/Ni(OH)2 composite at a scan rate of 25 mV s−1 are shown in Fig. 5a. It can be seen that the CV curve area of the CA is much smaller than those of Ni(OH)2 and CA/Ni(OH)2 composite, indicating the negligible capacity of the CA compared to those of Ni(OH)2 and CA/Ni(OH)2 composite. The maximum CV curve area of CA/Ni(OH)2/0.75 indicates the better electrochemical performance of CA/Ni(OH)2/0.75 than those of other samples. It should be noted that the CV curve of the CA/Ni(OH)2 composite shows a pair of oxidation and reduction peaks, which result from the reversible reaction of Ni(OH)2 + OH− ↔ NiOOH + H2O + e−, involving the intercalation and deintercalation of protons.2,8 From the CV curves at different scan rates (Fig. 5b) we can see that the oxidation peak of CA/Ni(OH)2/0.75 shifts to a more positive position and the reduction peak shifts to a more negative position with an increase in scan rate. This is probably due to the increase of the internal diffusion resistance within the active material with an increase of scan rate.52 In addition, the CV curves of the CA at different scan rates are given in Fig. S3.† The CV curves of CA/Ni(OH)2/0.75 in a wide potential window of −0.3 to 0.6 V at scan rates of 100, 200, 300, 400 and 500 mV s−1 were also carried out (Fig. 5c). With a further increase in the scan rate, the oxidation and reduction peaks further shift to the positive and negative directions, respectively.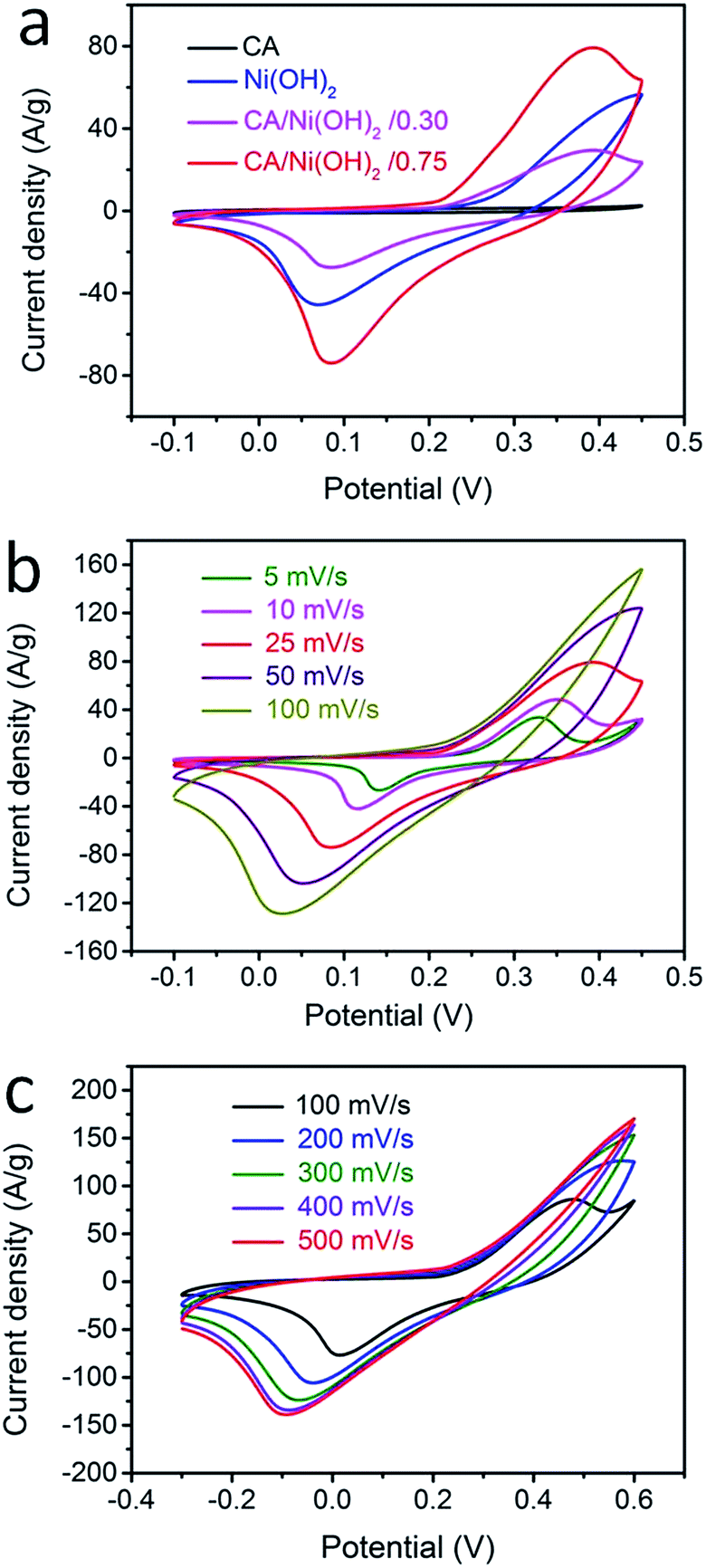 | ||
| Fig. 5 (a) CV curves of cellulose-derived CA, Ni(OH)2, and CA/Ni(OH)2 composite at a scan rate of 25 mV s−1. (b) CV curves of CA/Ni(OH)2/0.75 at different scan rates ranging from 5 to 100 mV s−1. (c) CV curves of CA/Ni(OH)2/0.75 in a potential window of −0.3 to 0.6 V at scan rates of 100, 200, 300, 400 and 500 mV s−1. | ||
The galvanostatic charge/discharge performances of the CA/Ni(OH)2 composite and Ni(OH)2 were also investigated at current densities ranging from 1 to 20 A g−1, as shown in Fig. 6a and b. The charge/discharge curves of both Ni(OH)2 and CA/Ni(OH)2/0.75 show a significant deviation from the typical triangular shape of electric double layer capacitances, indicating the faradaic redox reaction characteristics of the charge storage. This result is consistent with the abovementioned analysis of CV curves. The specific capacities of CA/Ni(OH)2/0.75 from the discharge curves based on the mass of the active material Ni(OH)2 are calculated to be 1906 and 1206 C g−1 at a current density of 1 and 20 A g−1, respectively, approximately 700 F g−1 higher than those of Ni(OH)2 (1164 and 540 C g−1, respectively). In addition, the specific capacities of CA/Ni(OH)2/0.75 based on the mass of the entire CA/Ni(OH)2 electrodes (1430 and 904 C g−1 at 1 and 20 A g−1, respectively) are also higher than those of Ni(OH)2. Both the samples show decreased specific capacity with increasing current density as shown in Fig. 6c. This is because ion transport within the porous structure lags behind the potential variation, and the internal resistances for charge carriers retard the electrochemical performance of the active materials at higher current densities.33 However, when the current density is increased from 1 to 20 A g−1, about 63% specific capacity of CA/Ni(OH)2/0.75 is retained, well above that of Ni(OH)2 (46%), indicating a better diffusion of electrolyte ions and electrons of the CA/Ni(OH)2 composite compared to that of Ni(OH)2 at high current densities. The higher specific capacity of CA/Ni(OH)2/0.75 compared to that of Ni(OH)2 is probably attributed to the enhanced efficiency of capacity generation due to the fuller utilization of the well-exposed Ni(OH)2 nanoparticles on the interconnected, three-dimensional, high-surface-area CA matrix. The specific capacity is further compared to that of the reported Ni(OH)2-based materials. The specific capacity of CA/Ni(OH)2/0.75 at a current density of 1 A g−1 is lower than that of Ni foam/Ni(OH)2 (2384 F g−1), but higher than that of carbon fiber/Ni(OH)2 (1416 F g−1).53,54 The specific capacity of CA/Ni(OH)2/0.75 at a current density of 2 A g−1 is higher than that of graphene/CNT/Ni(OH)2 (1212 F g−1).21
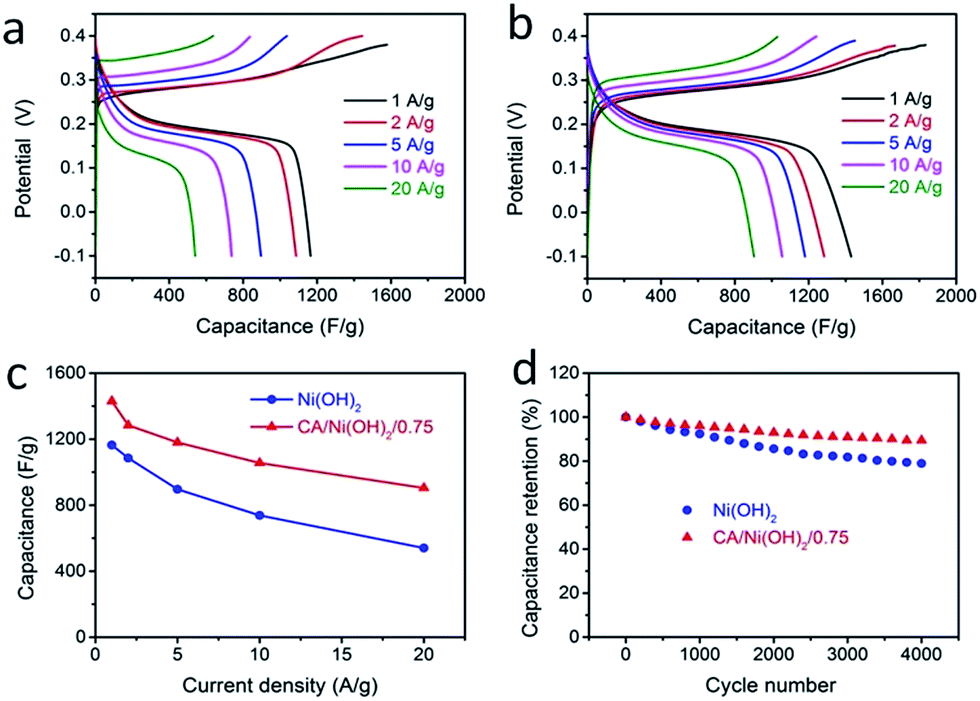 | ||
| Fig. 6 Galvanostatic charge/discharge curves of (a) Ni(OH)2 and (b) CA/Ni(OH)2/0.75 at different constant current densities, ranging from 1 to 20 A g−1. (c) Specific capacitances at various discharge current densities. Here, the capacitances of CA/Ni(OH)2/0.75 are based on the mass of the entire CA/Ni(OH)2 electrodes. (d) Cycling performance measured at a current density of 20 A g−1 in a potential range of −0.1 to 0.4 V. | ||
The cycling stabilities of Ni(OH)2 and CA/Ni(OH)2 composite were further investigated at a current density of 20 A g−1 as shown in Fig. 6d. After 4000 cycles, the specific capacitance of CA/Ni(OH)2/0.75 maintains 89% of the initial capacitance, higher than that of Ni(OH)2 (79%). This confirms that the cycling stability of Ni(OH)2 is effectively enhanced by introducing the CA as the conductive and nanoporous matrix. The capacitance loss is probably due to the loss of the electrical contact between the porous CA and the active materials, namely Ni(OH)2, resulting from the volume change in the active material during cycling.
The Nyquist plots of the cellulose-derived CA, Ni(OH)2, and CA/Ni(OH)2 composite obtained using AC electrochemical impedance spectroscopy in the frequency range from 0.5 Hz to 0.1 MHz are shown in Fig. S4.† In the low frequency region, the Nyquist plots of CA/Ni(OH)2/0.75 show a more vertical curve compared to that of Ni(OH)2, indicating better capacitive behavior.21 The equivalent series resistance of the energy storage devices can be obtained from the intercept at the real axis in the Nyquist plot at high frequency55 and it is composed of the ionic resistance of the electrolytes, electronic resistance of the electrode materials, and interface resistance. The obtained equivalent series resistance of CA/Ni(OH)2/0.75 is 0.69 Ω, which is higher than that of the CA (0.63 Ω) but lower than that of Ni(OH)2 (0.95 Ω). This demonstrates that the internal resistance in the energy storage devices of the CA/Ni(OH)2 composite is smaller than that of Ni(OH)2, indicating better electrochemical performance of the CA/Ni(OH)2 composite compared to that of Ni(OH)2. Considering the renewable carbon source, high specific capacity and good cycling stability, the cellulose-derived CA/Ni(OH)2 composite materials may have application potential in low-cost and high-performance energy storage devices.
4. Conclusion
A wet chemical approach has been applied to the synthesis of nano-architectured carbon aerogel/Ni(OH)2 composites, which combines the sol–gel preparation of highly porous carbon aerogel using microcrystalline cellulose as the carbon source and the subsequent homogeneous deposition of Ni(OH)2 nanoparticles onto the backbone of the obtained carbon aerogel via a two-step chemical precipitation process. The deposited Ni(OH)2 on the carbon aerogel shows much smaller size and more uniform dispersion, compared to the Ni(OH)2 prepared by a conventional method. The specific capacitance of the resulting composite was 1906 and 1206 F g−1 at a current density of 1 and 20 A g−1 respectively, much higher than that of Ni(OH)2. The composite also showed good cycling stability with 89% of the capacitance retention after 4000 cycles. The good electrochemical performance is attributed to the enhanced efficiency of capacitance generation from Ni(OH)2 due to the better utilization of the well-exposed Ni(OH)2 nanoparticles on the interconnected, three-dimensional, high-surface-area carbon aerogel matrix. The cellulose-derived carbon aerogel/Ni(OH)2 composite materials may have application potentials in low-cost and high-performance energy storage devices.Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (No. U1230113), National key Technology R&D Program of China (No. 2013BAJ01B01), National High Technology Research and Development Program of China (863) (No. 2013AA031801), Shanghai Aerospace Science and Technology Innovation Fund (No. SAST201469) and Project Funded by China Postdoctoral Science Foundation (No. 0800229072).Notes and references
- H.-C. Chien, W.-Y. Cheng, Y.-H. Wang and S.-Y. Lu, Adv. Funct. Mater., 2012, 22, 5038–5043 CrossRef CAS.
- J. Ji, L. L. Zhang, H. Ji, Y. Li, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef CAS PubMed.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- Z. S. Wu, W. C. Ren, D. W. Wang, F. Li, B. L. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed.
- T.-Y. Wei, C.-H. Chen, K.-H. Chang, S.-Y. Lu and C.-C. Hu, Chem. Mater., 2009, 21, 3228–3233 CrossRef CAS.
- J.-W. Lang, L.-B. Kong, W.-J. Wu, Y.-C. Luo and L. Kang, Chem. Commun., 2008, 44, 4213–4215 RSC.
- Y. Yang, L. Li, G. Ruan, H. Fei, C. Xiang, X. Fan and J. M. Tour, ACS Nano, 2014, 8, 9622–9628 CrossRef CAS PubMed.
- Z. Tang, C. Tang and H. Gong, Adv. Funct. Mater., 2012, 22, 1272–1278 CrossRef CAS.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- J. Li, F. Luo, X. Tian, Y. Lei, H. Yuan and D. Xiao, J. Power Sources, 2013, 243, 721–727 CrossRef CAS.
- J. H. Park, O. O. Park, K. H. Shin, C. S. Jin and J. H. Kim, Electrochem. Solid-State Lett., 2002, 5, H7–H10 CrossRef CAS.
- Q. Huang, X. Wang, J. Li, C. Dai, S. Gamboa and P. J. Sebastian, J. Power Sources, 2007, 164, 425–429 CrossRef CAS.
- G. Hasegawa, T. Deguchi, K. Kanamori, Y. Kobayashi, H. Kageyama, T. Abe and K. Nakanishi, Chem. Mater., 2015, 27, 4703–4712 CrossRef CAS.
- R. R. Salunkhe, J. Lin, V. Malgras, S. X. Dou, J. H. Kim and Y. Yamauchi, Nano Energy, 2015, 11, 211–218 CrossRef CAS.
- D. P. Dubal, G. S. Gund, C. D. Lokhande and R. Holze, ACS Appl. Mater. Interfaces, 2013, 5, 2446–2454 CAS.
- M. Shahid, J. Liu, I. Shakir, M. F. Warsi, M. Nadeem and Y. Kwon, Electrochim. Acta, 2012, 85, 243–247 CrossRef CAS.
- X. Ma, J. Liu, C. Liang, X. Gong and R. Che, J. Mater. Chem. A, 2014, 2, 12692–12696 CAS.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- J. Yan, W. Sun, T. Wei, Q. Zhang, Z. Fan and F. Wei, J. Mater. Chem., 2012, 22, 11494–11502 RSC.
- Y. Xu, X. Huang, Z. Lin, X. Zhong, Y. Huang and X. Duan, Nano Res., 2013, 6, 65–76 CrossRef CAS.
- D. Fang, Z. Chen, X. Liu, Z. Wu and C. Zheng, Electrochim. Acta, 2012, 81, 321–329 CrossRef CAS.
- J. Xie, X. Sun, N. Zhang, K. Xu, M. Zhou and Y. Xie, Nano Energy, 2013, 2, 65–74 CrossRef CAS.
- X. H. Xia, D. L. Chao, Z. X. Fan, C. Guan, X. H. Cao, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 1651–1658 CrossRef CAS PubMed.
- J. Zhu, Z. Xu and B. N. Lu, Nano Energy, 2014, 7, 114–123 CrossRef CAS.
- Y. H. Li, Z. Y. Wang and Y. F. Zhang, J. Alloys Compd., 2015, 644(644), 47–53 CrossRef CAS.
- D. Shi, L. Zhang, X. Yin, T. J. Huang and H. Gong, J. Mater. Chem. A, 2016, 4, 12144–12151 CAS.
- M. Xie, S. Duan, Y. Shen, K. Fang, Y. Wang, M. Lin and X. Guo, ACS Energy Lett., 2016, 1, 814–819 CrossRef CAS.
- P. Xu, C. Miao, K. Cheng, K. Ye, J. Yin, D. Cao, G. Wang and X. Zhang, Electrochim. Acta, 2016, 216, 499–509 CrossRef CAS.
- R. Wang, A. Jayakumar, C. Xu and J.-M. Lee, ACS Sustainable Chem. Eng., 2016, 4, 3736–3742 CrossRef CAS.
- J. Biener, M. Stadermann, M. Suss, M. A. Worsley, M. M. Biener, K. A. Rose and T. F. Baumann, Energy Environ. Sci., 2011, 4, 656–667 CAS.
- X. Wu and A. Xu, J. Mater. Chem. A, 2014, 2, 4852–4864 CAS.
- Y.-H. Lin, T.-Y. Wei, H.-C. Chien and S.-Y. Lu, Adv. Energy Mater., 2011, 1, 901–907 CrossRef CAS.
- X. Wu, T. Wen, H. Guo, S. Yang, X. Wang and A. Xu, ACS Nano, 2013, 7, 3589–3597 CrossRef CAS PubMed.
- Y. J. Lee, J. C. Jung, J. Yi, S. H. Baeck, J. R. Yoon and I. K. Song, Curr. Appl. Phys., 2010, 10, 682–686 CrossRef.
- R. W. Pekala, J. C. Farmer, C. T. Alviso, T. D. Tram, S. T. Mayer, J. M. Miller and B. Dunn, J. Non-Cryst. Solids, 1998, 225, 74–80 CrossRef CAS.
- Y. Zhu, H. Hu, W. C. Li and X. Zhang, J. Power Sources, 2006, 162, 738–742 CrossRef CAS.
- C. Moreno-Castilla, M. B. Dawidziuk, F. Carrasco-Marin and Z. Zapata-Bennabithe, Carbon, 2011, 49, 3808–3819 CrossRef CAS.
- Z. Y. Wu, C. Li, H. W. Liang, J. F. Chen and S. H. Yu, Angew. Chem., 2013, 125, 2997–3001 CrossRef.
- A. G. Sadekar, S. S. Mahadik, A. N. Bang, Z. J. Larimore, C. A. Wisner, M. F. Bertino, A. K. Kalkan, J. T. Mang, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2012, 24, 26–47 CrossRef CAS.
- L. Wang, C. Schutz, G. Salazar-Alvarez and M. M. Titirici, RSC Adv., 2014, 4, 17549–17554 RSC.
- E. Guilminot, F. Fischer, M. Chatenet, A. Rigacci, S. Berthon-Fabry, P. Achard and E. Chainet, J. Power Sources, 2007, 166, 104–111 CrossRef CAS.
- B. Grzyb, C. Hildenbrand, S. Berthon-Fabry, D. Begin, N. Job, A. Rigacci and P. Achard, Carbon, 2010, 48, 2297–2307 CrossRef CAS.
- K. Gao, Z. Shao, X. Wang, Y. Zhang, W. Wang and F. Wang, RSC Adv., 2013, 3, 15058–15064 RSC.
- Z. Y. Wu, H. W. Liang, C. Li, B. C. Hu, X. X. Xu, Q. Wang, J. Chen and S. Yu, Nano Res., 2014, 7, 1861–1872 CrossRef CAS.
- G. Zu, J. Shen, L. Zou, F. Wang, X. Wang, Y. Zhang and X. Yao, Carbon, 2016, 99, 203–211 CrossRef CAS.
- S. Bag and C. R. Raj, J. Mater. Chem. A, 2014, 2, 17848–17856 CAS.
- S. R. Shieh and T. S. Duffy, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 134301–134308 CrossRef.
- G. Zu, J. Shen, W. Wang, L. Zou, Y. Lian and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 5400–5409 CAS.
- G. S. Li, L. P. Li, J. Boerio-Goates and B. F. Woodfield, J. Am. Chem. Soc., 2005, 127, 8659–8666 CrossRef CAS PubMed.
- D. P. Mohite, Z. J. Larimore, H. Lu, J. T. Mang, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2012, 24, 3434–3448 CrossRef CAS.
- U. M. Patil, K. V. Gurav, V. J. Fulari, C. D. Lokhande and O. S. Joo, J. Power Sources, 2009, 188, 338–342 CrossRef CAS.
- X. Xiong, D. Ding, D. Chen, G. Waller, Y. Bu, Z. Wang and M. Liu, Nano Energy, 2015, 11, 154–161 CrossRef CAS.
- N. A. Alhebshi, R. B. Rakhi and H. N. Alshareef, J. Mater. Chem. A, 2013, 1, 14897–14903 CAS.
- J. W. Lang, X. B. Yan, X. Y. Yuan, J. Yang and Q. J. Xue, J. Power Sources, 2011, 196, 10472–10478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photograph and SEM images of cellulose aerogel, N2 adsorption/desorption isotherms for typical aerogels, CV curves of nanocellulose-derived CA, Nyquist curves for typical aerogels. See DOI: 10.1039/c6ra26566a |
| This journal is © The Royal Society of Chemistry 2017 |