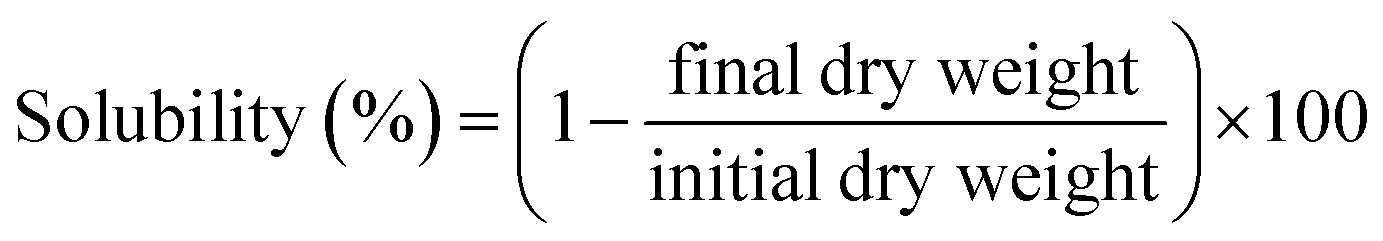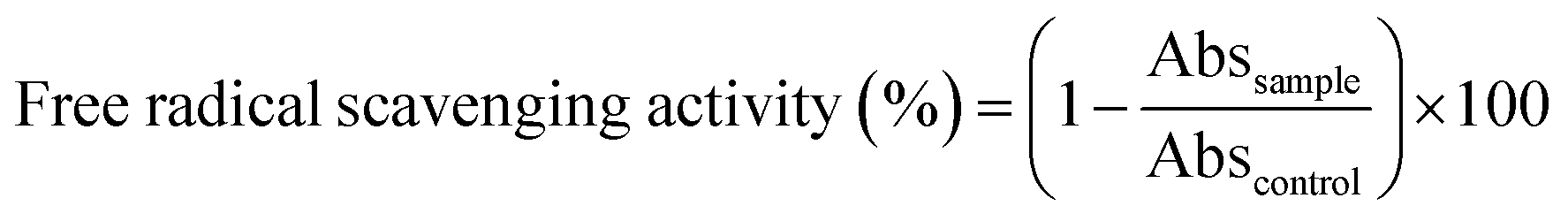DOI:
10.1039/C6RA26541C
(Paper)
RSC Adv., 2017,
7, 13707-13713
Preparation and performance of composite films based on 2-(2-aminoethoxy) ethyl chitosan and cellulose
Received
9th November 2016
, Accepted 14th February 2017
First published on 28th February 2017
Abstract
In this study, antioxidant active films were prepared from 2-(2-aminoethoxy) ethyl chitosan (AECs, a novel chitosan derivative) and cellulose under an environmentally friendly process using ionic liquids (ILs). AEC/cellulose composite films were prepared by efficiently blending AECs into cellulose N-methylmorpholine N-oxide (NMMO)/H2O solution, followed by shaping in a mold. The microstructure, physical, mechanical and antioxidant properties of the composite films were measured. The resulting composite films exhibited good physical properties, favorable mechanical properties and a high free radical scavenging activity against 2,2-diphenyl-1-picrylhydrazyl (DPPH) in methanol. Furthermore, a loading of AECs at 5% into cellulose resulted in significant improvement in antioxidant activity (86.5% ± 2.7%), while a slight decrease in the tensile strength (2.9% ± 0.2%) was noted. This result indicated that the oxidation resistance can be greatly improved by incorporation of AECs without significantly affecting the mechanical properties. The bioactive composite film is expected to be a promising material for potential applications in food packaging.
1. Introduction
In recent years, there has been a growing interest in the development of food packaging based on natural macromolecules that are environmentally friendly, biodegradable and renewable because the packaging can not only reduce plastic waste but also enhance food quality by preserving moisture, water vapor permeability and antioxidant activity.1,2 Cellulose, the most abundant biopolymer consisting of D-glucopyranose units connected by β-1,4-glycosidic bonds, is capable of forming a structural matrix with a sufficient cohesiveness due to its superior ability to form films and mechanical behavior.3,4 The excellent biological properties of cellulose show great promise for its application in food packaging.5,6 New trends in cellulose have focused on the improvement of its functionality through incorporation of active compounds, such as antioxidant agents.7–9
Chitosan (the cationic (1-4)-2-amino-2-deoxy-β-D-glucan) is a non-toxic and biodegradable polymer obtained from the deacetylation of chitin and has excellent biocompatibility, film forming ability and antioxidant activity.10,11 In order to improve the solubility, physicochemical and biological properties of chitosan, much effort has been made to introduce hydrophilic groups by covalent attachment to reactive amino groups.12–14 Various types of chitosan derivatives with good antioxidant activity have been investigated.15–17 Therefore, it is possible to prepare chitosan derivative/cellulose composite films, in which chitosan acts as an additive to improve the properties of cellulose and endows it good antioxidant ability.18–20
Previously, the preparation of cellulose films was usually carried out through the viscose process, which was harmful to the environment.21–23 As a result, ionic liquids (ILs), novel green solvents and catalysts, were regarded as promising alternatives to green solvents to replace the organic solvents in cellulose film formation.24,25 It was reported that several promising solvent systems, such as LiCl/N, N-dimethylacetamide (DMAc), dimethylformamide, and N-methylmorpholine N-oxide (NMMO)/H2O, have been utilized to prepare chitosan/cellulose blends.26,27 In particular, NMMO solution was technically feasible and has been widely applied in industrial production.28,29
The aim of this study was to develop a fully green approach for the preparation of bioactive films from renewable resources through the use of cellulose as the film matrix and the incorporation of various amounts of AECs, a novel chitosan derivative as an antioxidant agent. Extensive characterization of AEC/cellulose composite films revealed unique properties of these films and how the additive AECs affected the microstructure, physical and mechanical properties and antioxidant activity.
2. Experimental
2.1 Chemicals and materials
Chitosan (molecular weight 1.08 × 106, the degree of deacetylation was 0.85) was supplied by Qingdao Medicine Institute (Shandong, China) and degraded by γ-irradiation to various molecular weight samples. The molecular weight for the original chitosan was 1.9 × 105 g mol−1. Cellulose pulp (DP = 750) was provided by Tianjin Rayon Factory (Tianjin, China), which was dried at 80 °C for 24 h before use. N-Methylmorpholine-N-oxide (NMMO) aqueous solution (50 wt%) was purchased from BASF, Germany. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) was purchased from Sigma-Aldrich (St. Louis, MO). Other reagents were of analytical grade and provided by Jiangtian Chemical Technology (Tianjin, China).
2.2 Film preparation
2.2.1 Synthesis of AECs. Fig. 1 shows the synthetic process of AECs. Chitosan was suspended in sodium hydroxide (NaOH, 42 wt%) solution and then reacted with 2-chloroethanol at 25 °C for 12 h. After the filtration, hydroxyethyl chitosan (HECs) was dried at 60 °C under vacuum. Then, HECs (5 g) was dissolved in NaOH (42 wt%) solution and reacted with 2-chloroethylamine hydrochloride at 80 °C for 12 h. After the reaction, the pH of the solution was adjusted to 7.0 with hydrochloric acid. The crude product was dialyzed, lyophilized, and dried. The NH2% of AECs was determined by potentiometric titration (PT).
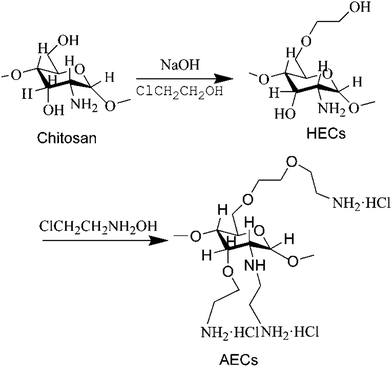 |
| | Fig. 1 Synthesis of AECs. | |
2.2.2 Preparation of AEC/cellulose co-solution. An AEC/cellulose co-solution was prepared as follows: 10 g cellulose was pulverized and dipped in 200 mL NMMO/H2O (50 wt%), heated to 110 °C and stirred in a vacuum until the content of H2O reduced to 13.3 wt% (wt% = H2O/(NMMO/H2O)).30 Then, the cellulose was filtered and completely dissolved in NMMO/H2O under stirring. AECs powder (5 g) was dissolved in 10 mL of sodium hydroxide solution (5 wt%). Then, the mixture was added into a cellulose NMMO/H2O solution under vigorous stirring to obtain an AEC/cellulose co-solution.
2.2.3 Preparation of AEC/cellulose composite films. Through co-solution of AECs and cellulose in the NMMO/H2O system, AEC/cellulose composite films were shaped in a mold at a stable temperature of 90 °C, and deionized water was used as a coagulation bath. The resultant films were washed three times to ensure complete coagulation and removal of NMMO. The thickness of the films was controlled to about 50 μm. The cellulose film was obtained using the same method.
2.3 Film microstructure
2.3.1 Fourier transform infrared spectroscopy (FTIR) analysis. FTIR spectra of AECs and AEC/cellulose composite films were obtained with a Nicolet 560 E.S.P. spectrometer. The samples were scanned from 400 to 4000 cm−1 with a resolution of 1 cm−1.
2.3.2 Wide angle X-ray scattering (WAXS). WAXS experiments were performed for the films using a D/max-2500 X-ray diffractometer. The X-ray source was Ni-filtered CuKα radiation (25 kV, 10 mA). Samples were scanned at a range of 2θ from 5° to 40° at a scan rate of 4° min−1.
2.3.3 Scanning electron microscopy (SEM). SEM observations were made on the films. Films were coated with a small amount of gold and examined with a Philips SL-30 SEM.
2.4 Physical properties
2.4.1 Film thickness measurement. Thicknesses of the films were analyzed on a digital micrometer (RCBS Electronic Digital Micrometer). The thickness was tested at 12 randomly selected points of the same film, and then an average value was calculated.
2.4.2 Equilibrium moisture content. The water content of film samples was determined by measuring the weight loss of films. Films samples were conditioned under standard conditions (25 °C, 70% relative humidity) for 24 h before weighing (wet sample weight) and dried in triplicate at 105 °C for 24 h more in a vacuum oven until reaching a constant weight (dry sample weight). The equilibrium moisture content (%) was calculated from the following expression:| |
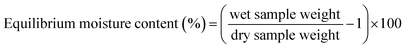 | (1) |
2.4.3 Film solubility in water. The water solubility of film samples was determined by immersing dried, weighed test samples (initial dry weight) into test beakers with distilled water for 24 h at room temperature. Then, each sample was dried at 105 °C for 24 h to determine the weight of the final dry matter. Every experiment was performed 5 times. The percentage of film solubility was calculated based on the following equation:| |
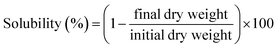 | (2) |
2.4.4 Water vapor permeability (WVP). The WVP of films was determined by a modified ASTM method as described in a previous study at a constant temperature of 20 °C.31 The film samples, cut into disks, were sealed to cups with an average diameter of 2 cm and a depth of 4.5 cm. After steady-state conditions were reached (about 2 h), the cups were weighed every 2 h over a period of 24 h. Tests were conducted in duplicate for each sample, and the slope, obtained from regression analysis of weight loss data as a function of time, was used to calculate WVP, according to ATSM.32
2.5 Mechanical properties
Mechanical properties of film samples were determined using M350-20KN Testometric. The tensile strength and elongation of the films were measured. The tests were carried out in accordance with the standard ISO 6239-1986 (E), and the stretching rate was 5 mm min−1. Surface tension of the films was measured by a dynamic contact angle measurement instrument and Tensionmeter, DataPhysics DCAT 21.
2.6 Antioxidant activity
The antioxidant activity of films was measured using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical method according to a procedure previously described.33,34 The films were cut and dipped in the methanol solution of DPPH (0.030 g mL−1) under magnetic stirring for 12 h at 25 °C. The decrease in absorbance was monitored by UV-Vis absorption spectrometry at 515 nm. The experiments were repeated 5 times for the evaluation of each antioxidant activity. DPPH radical scavenging activity was calculated as inhibition percentage as follows:| |
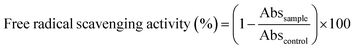 | (3) |
2.7 Statistical analysis
All values reported were from replicate determinations with basic mathematical calculations and analysis. A statistical analysis of data was obtained through a one-way analysis of variance. Homogeneous sample groups were performed by Fisher's protected least-significant difference (LSD) test and the significance level used was 0.05.
3. Results and discussion
3.1 Synthesis and solubility of AECs
The FTIR spectra of HECs and AECs are displayed in Fig. 2. In the FTIR spectrum of HECs, the main characteristic peaks at 1058 cm−1 were assigned to the stretching vibration of the C–O group, and 1313 cm−1 was assigned to the O–H bending vibration. The absorptions at 1650 cm−1 and 892 cm−1 showed evidence for the presence of N–H groups. The spectrum of AECs, on the other hand, exhibited an absorption of a –NH2 bending vibration at 1618 cm−1 and the peak for the –CH2–N– bending vibration at 1448 cm−1. In addition, the absence of the –OH absorption at 1058 cm−1 and the absorption of the secondary amino group at 1022 cm−1 indicated that the amination reaction occurred at both hydroxyl and amino groups.35 Due to the introduction of a number of –NH2 groups, the absorption of the –NH2 stretching vibration at 3428 cm−1 was enhanced.
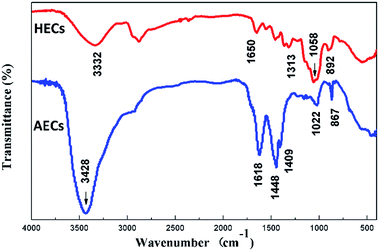 |
| | Fig. 2 FTIR spectrum of HECs and AECs. | |
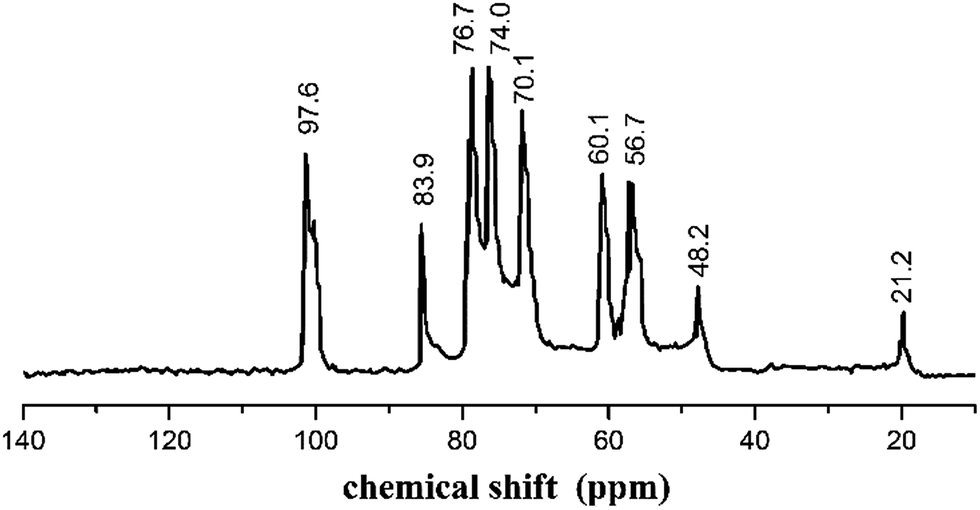
The structure of AECs was further explored by 13C-NMR (Fig. 3). The 13C-NMR spectrum was recorded on a Mercury-Vx300 spectrometer in D2O/CF3COOD 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v. The peaks observed at 97.6 ppm, 56.7 ppm, 70.1 ppm, 76.7 ppm, 74.0 ppm, 60.1 ppm corresponded to the pyranoid ring (C-1,2,3,4,5,6), respectively. The peak at 83.9 ppm was ascribed to –CH2NH2, another peak at 48.2 ppm was attributed to –CH2–O–, and the peak at 22.2 ppm was assigned to –CH3.36 FTIR and 13C NMR confirmed that the quaternary amino salt group was introduced onto chitosan, and AECs were successfully synthesized.
:5 v/v. The peaks observed at 97.6 ppm, 56.7 ppm, 70.1 ppm, 76.7 ppm, 74.0 ppm, 60.1 ppm corresponded to the pyranoid ring (C-1,2,3,4,5,6), respectively. The peak at 83.9 ppm was ascribed to –CH2NH2, another peak at 48.2 ppm was attributed to –CH2–O–, and the peak at 22.2 ppm was assigned to –CH3.36 FTIR and 13C NMR confirmed that the quaternary amino salt group was introduced onto chitosan, and AECs were successfully synthesized.
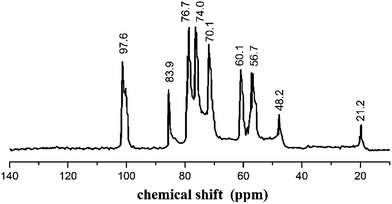 |
| | Fig. 3 13C-NMR spectrum of AECs. | |
The solubilities of AECs synthesized under various conditions in NMMO/H2O are given in Table 1. The results show that all samples obtained from the original chitosan could dissolve in aqueous NMMO/H2O except the one in experiment no. 1; in particular, the solubility of AECs in NMMO/H2O in experiment no. 5 reached 63.5 mg mL−1. Due to the hydration of the amino group in NMMO/H2O, AECs had good solubility, which benefited spinning and film formation. The incorporation of a polar group into the chitosan backbone offered excellent water solubility.
Table 1 Solubility of AECs in NMMO/H2O
| Experiment no. |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
| Time (h) |
6 |
12 |
24 |
6 |
12 |
24 |
6 |
12 |
24 |
| Temperature (°C) |
50 |
70 |
80 |
70 |
80 |
50 |
80 |
50 |
70 |
| NH2% |
8.4 |
9.51 |
9.85 |
10.31 |
10.7 |
9.36 |
9.85 |
10.5 |
10.2 |
| Solubility (mg mL−1) |
— |
12.3 |
40.8 |
14.6 |
63.5 |
35.7 |
53.1 |
25.3 |
60.1 |
3.2 Film microstructure
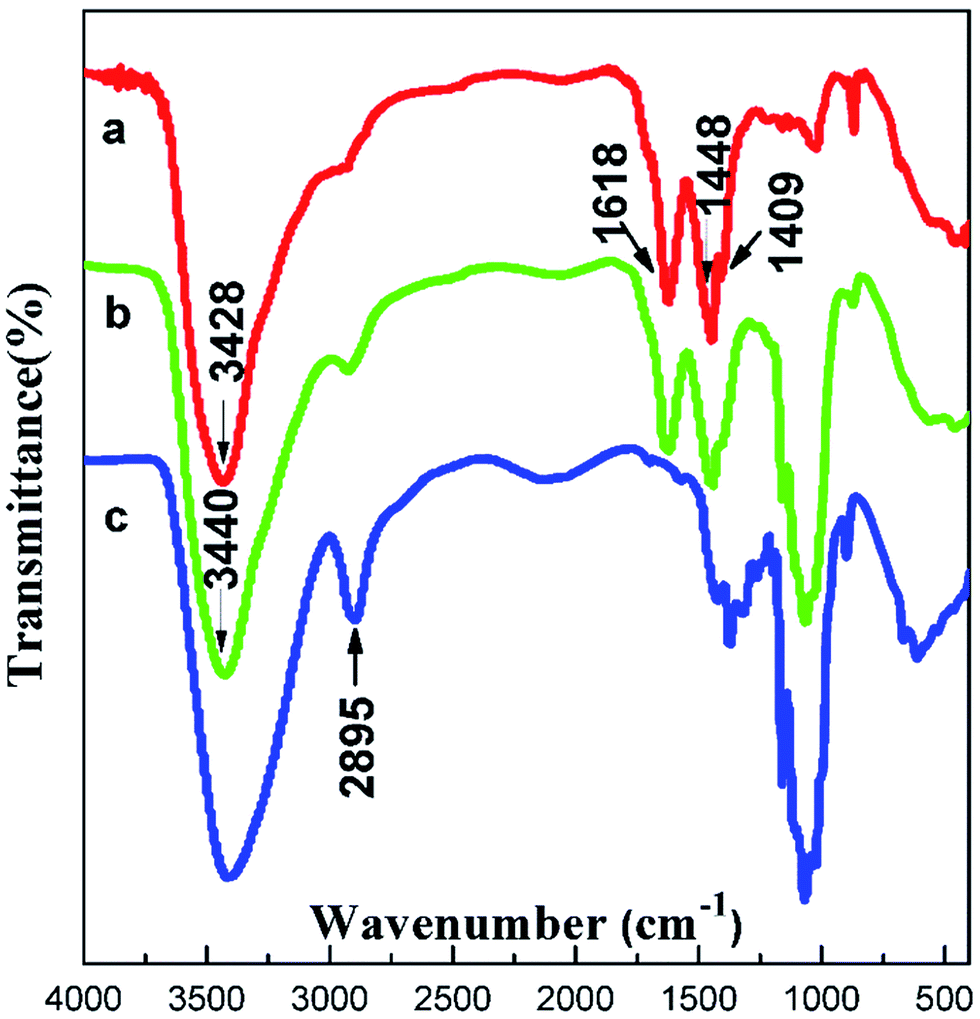
The structure of the composite films was studied via FTIR spectroscopy (Fig. 4). In the spectrum of cellulose, the absorption band of the hydroxyl group was wide and strong ranging from 3310 cm−1 to 3520 cm−1, and the peak at 2895 cm−1 was assigned to the stretching vibration of C–H. The spectrum of AEC/cellulose composite films was very similar to that of AECs. The strong absorption band at 1618 cm−1 was the deformation vibration of –NH2, and the peaks appearing at 1448 cm−1 and 1409 cm−1 corresponded to the deformation vibration and wagging vibration of –CH2 in the –CH2–N–, respectively. Compared with AECs, a slight motion at the high wavenumber of –NH2 was observed in the spectrum of AECs/cellulose composite films, indicating that an interaction between AECs and cellulose existed, proving the compatibility between AECs and cellulose.
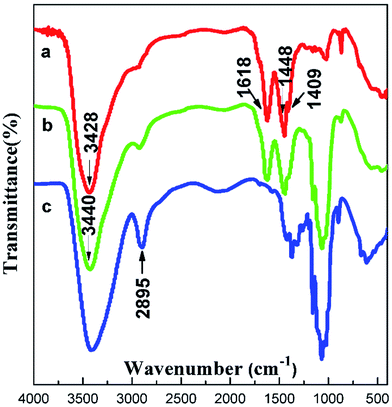 |
| | Fig. 4 FTIR spectra of (a) AECs, (b) AECs/cellulose composite films and (c) cellulose. | |
X-ray scattering patterns of cellulose pulp, cellulose film and AEC/cellulose composite films were also studied as shown in Fig. 5. Cellulose pulp showed a diffraction pattern for cellulose I, while cellulose film generally showed a diffraction pattern for cellulose II at 2θ = 12° for (101), 20° for (101), and 21.7° for (002).37 Compared with the cellulose film, the height of the peak at 12° for (101) in the diffraction patterns of AEC/cellulose composite films evidently declined with an increased content of AECs. This indicates that the interaction between AECs and cellulose broke the intermolecular hydrogen bonds between cellulose chains, which led to the weakness of the peak at 12° for (101).38
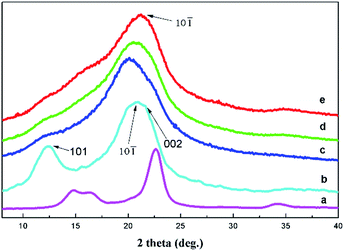 |
| | Fig. 5 X-ray diffraction patterns of the cellulose pulp (a), cellulose film (b) and the composite films with different contents of AECs: (c) 1%, (d) 3% and (e) 5%. | |
SEM microphotographs of the surfaces of cellulose and composite films are shown in Fig. 6, respectively. Because the cellulose and composite films were separated out from the solution during the longitudinal deformation and soft coagulation, there were holes in the surface of all the films (Fig. 6). Unlike the cellulose film, there were fewer and smaller holes for composite films. The cellulose film displayed a zigzag section, while the section of composite film was smooth and homogeneous. We attributed this morphological difference to the additive AECs, which may have changed the crystal structure of the cellulose film.
 |
| | Fig. 6 SEM images of (a) cellulose film's section 2000×, (b) cellulose film's section 5000×, (c) composite film's section 2000× and (d) composite film's section 5000×. | |
3.3 Physical properties
Homogeneous, thin and flexible films were obtained from the AECs/cellulose blending. Thicknesses of films varied between 50.46 ± 0.79 and 52.39 ± 3.25 μm, as shown in Table 2. As the amount of AECs in the films increased, the thicknesses of the films increased as well. This change in thickness may be caused by the compact difference between cellulose chains, groups of AECs and their interactions. When AECs was added to cellulose matrix, an interconnected bonding network could be formed by the hydrogen bonds.
Table 2 Thickness, equilibrium moisture content, film solubility in water and water vapor permeability (WVP) of films
| Content of AECs |
Thickness (μm) |
Equilibrium moisture content (%) |
Film solubility in water (%) |
WVP × 1011 (g m−1 s−1 Pa−1) |
| 0 |
52.39 ± 3.25 |
3.81 ± 0.64 |
2.22 ± 0.32 |
6.25 ± 0.33 |
| 1% |
51.61 ± 1.87 |
4.75 ± 0.53 |
4.35 ± 0.24 |
7.32 ± 0.20 |
| 3% |
51.04 ± 2.01 |
6.98 ± 0.92 |
5.91 ± 0.31 |
7.51 ± 0.35 |
| 5% |
50.46 ± 0.79 |
8.23 ± 0.89 |
7.07 ± 0.25 |
7.67 ± 0.74 |
The composite films with various amounts of AECs showed higher moisture content than cellulose films without AECs (Table 2), suggesting that AECs were more hydrophilic than cellulose. Cellulose and composite films had low solubility values, and the incorporation of AECs into the cellulose matrix significantly increased the solubility in water (Table 2). Hydrogen bonds between the cellulose chains and the quaternary amino salt group of AECs were the driving forces for the water solubility.39,40 Accordingly, AEC/cellulose composite films are not desirable for preserving liquid or liquid-containing food.
Table 2 also shows the water vapor permeability (WVP) values of the cellulose and composite films. The WVP values of the composite films, after adding AECs, increased to 7.32–7.67 × 10−11 g m−1 s−1 Pa−1 due to the higher water affinity of AEC/cellulose composite films. Since WVP is the contribution of diffusivity and solubility of the films through the matrix,41 composite films showed higher water vapor permeability than the cellulose film. Due to the greater number of hydrophilic groups of AECs and the presence of a greater amount of water molecules in the composite films, it is likely that the structural changes, induced by the addition of AECs, resulted in notable changes for the mass transfer rate of water molecules and greatly affected permeability values.
3.4 Characterization of surface tension
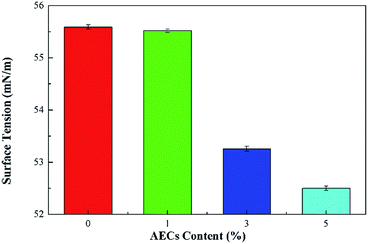
The effects of the content of AECs on the surface tension of cellulose and composite films are given in Fig. 7. The surface tension of the films decreased with an increasing content of AECs, which indicates that the moisture absorption of composite films was better than that of the cellulose film due to the significant moisture performance of AECs. This result agreed well with DSC analysis, which showed that the enthalpy of water loss increased with increased content of AECs.
 |
| | Fig. 7 Mechanical properties of the films with various contents of AECs. | |
3.5 Mechanical properties
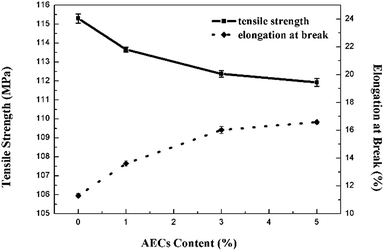
The tensile strength and elongation of cellulose and composite films in the dry state are shown in Fig. 8. The tensile strength of the films showed a continuous minor decrease with an increased content of AECs. Although the additive AECs may interfere with the orientation and crystallization of cellulose, the effect of small amounts of AECs was so weak that the performance of the films was not strongly influenced. Moreover, the films with AECs exhibited higher elongation, indicating that the addition of AECs could improve the flexibility of the composite films, which agreed well with the SEM microphotographs.
 |
| | Fig. 8 Surface tension of the films with various amounts of AECs. | |
3.6 Antioxidant activity
The antioxidant activity of films was evaluated by means of the dissolution of the film in a controlled amount of distilled water. Incorporating antioxidants into food packaging materials to control the oxidation of fatty components and pigments may contribute to the preservation of the quality of food products.42 The incorporation of AECs into cellulose matrix introduced a radical scavenging functionality in this biodegradable polymer. As shown in Fig. 9, cellulose film without AECs produced a small antioxidant response, which could be attributed to hydroxyl moieties along the cellulose chains. However, free radical scavenging activity by incorporating AECs at a concentration as low as 1% in cellulose was clearly seen. The scavenging activity was concentration dependent with a minimum of 55.4% ± 1.3% for 1% AECs to 86.5% ± 2.7% for 5% AECs. The increased free radical scavenging activity of AEC/cellulose composite films was in direct relation to the higher NH2% of incorporated AECs, which was in accordance with previous reports demonstrating that higher NH2% resulted in better scavenging activity.43,44
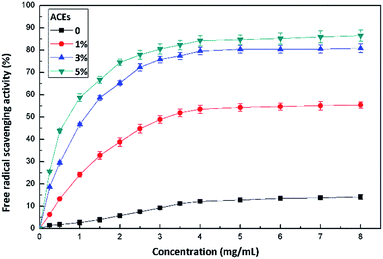 |
| | Fig. 9 DPPH radical scavenging activity of the composite films with various contents of AECs. | |
4. Conclusions
This study reported the preparation of bioactive composite films from AECs (a novel chitosan derivative) and cellulose using an environmentally friendly casting method. Addition of various amounts of AECs affected the moisture, water vapor permeability, mechanical strength and antioxidant activity of the prepared composite films. Moreover, these composite films with good antioxidant activity have the potential to tailor physical and mechanical properties. Results from this study indicated that incorporation of AECs into the films can lead to possible applications in the food industry, considering the bioactivity and antioxidant activity of both the natural polymers.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (no. 51473120).
Notes and references
- X. Hua, K. Wang, R. Yang, J. Kang and J. Zhang, Food Hydrocolloids, 2015, 44, 122–128 CrossRef CAS.
- Z. A. Nur Hanani, Y. H. Roos and J. P. Kerry, Int. J. Biol. Macromol., 2014, 71, 94–102 CrossRef CAS PubMed.
- A. Linder, R. Bergman, A. Bodin and P. Gatenholm, Langmuir, 2003, 19, 5072–5077 CrossRef CAS.
- M. Atef, M. Rezaei and R. Behrooz, Int. J. Biol. Macromol., 2014, 70, 537–544 CrossRef CAS PubMed.
- G. Seyhun, Y. Ahmet and A. Sacidealsoy, J. Food Eng., 2009, 90, 453–462 CrossRef.
- A. Safoura, K. Javad, N. Ali, H. Nasser, B. Tayebeh and A. Lionel, J. Appl. Polym. Sci., 2016, 133, 398–401 Search PubMed.
- C. Pastor, L. Sanchez-Gonzalez, A. Chiralt, M. Chafer and C. Gonzalez-Martinez, Food Hydrocolloids, 2013, 30, 272–280 CrossRef CAS.
- G. Davidov-Pardo and D. J. Mcclements, Trends Food Sci. Technol., 2014, 38, 88–103 CrossRef CAS.
- G. M. Raghavendra, T. Jayaramudu, K. Varaprasad, S. Ramesh and K. M. Raju, RSC Adv., 2013, 4, 3494–3501 RSC.
- D. Fong, M. B. Ariganello, J. Girard-Lauziere and C. D. Hoemann, Acta Biomater., 2014, 12, 183–194 CrossRef PubMed.
- A. M. El-Shafei, M. M. G. Fouda, D. Knittel and E. Schollmeyer, J. Appl. Polym. Sci., 2008, 110, 1289–1296 CrossRef CAS.
- H. Tamura, T. Furuike, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2011, 84, 820–824 CrossRef CAS.
- S. I. Hong, H. L. Jin, H. J. Bae, Y. K. Song, H. S. Lee and J. H. Choi, J. Appl. Polym. Sci., 2011, 119, 2742–2749 CrossRef CAS.
- L. Casettari, D. Vllasaliu, E. Castagnino, S. Stolnik, S. Howdle and L. Illum, Prog. Polym. Sci., 2012, 37, 659–685 CrossRef CAS.
- L. Zavaleta-Avejar, E. Bosquez-Molina, M. Gimeno, J. P. Perez-Orozco and K. Shirai, Food Hydrocolloids, 2014, 39, 113–119 CrossRef CAS.
- G. Q. Ying, W. Y. Xiong and H. Wang, Carbohydr. Polym., 2011, 83, 1787–1796 CrossRef CAS.
- W. Zhu and Z. Zhang, Int. J. Biol. Macromol., 2014, 70, 150–155 CrossRef CAS PubMed.
- M. Pereda, G. Amica and N. E. Marcovich, Carbohydr. Polym., 2012, 87, 1318–1325 CrossRef CAS.
- A. P. Martínez, Carbohydr. Polym., 2010, 82, 305–315 CrossRef.
- M. Srivastava, P. Rai, J. Singh and J. Singh, RSC Adv., 2014, 46, 30592–30597 RSC.
- H. P. Fink, J. Ganster and A. Lehmann, Cellulose, 2014, 21, 31–51 CrossRef.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- C. Yue, D. Fang, L. Liu and T. F. Yi, J. Mol. Liq., 2011, 163, 99–121 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2007, 10, 44–46 RSC.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- A. Takaragi, M. Minoda, T. Miyamoto, Q. L. Hai and N. Z. Li, Cellulose., 1999, 6, 93–102 CrossRef CAS.
- Z. Li, X. F. Liu, X. P. Zhuang, Y. Guan and K. Yao, J. Appl. Polym. Sci., 2002, 84, 2049–2059 CrossRef CAS.
- W. Dhaeze, C. Verplancke, V. Mironov and M. Holsters, Plasmid, 2002, 47, 88–93 CrossRef CAS PubMed.
- X. He and M. B. Hagg, Chem. Eng. J., 2012, 215, 440–448 Search PubMed.
- M. A. GarciA, A. Pinotti, M. N. Martino and N. E. Zaritzky, Carbohydr. Polym., 2004, 56, 339–345 CrossRef CAS.
- C. Andreuccetti, R. A. Carvalho, T. Galicia-Garcia, F. Martinez-Bustos and C. R. F. Grosso, J. Food Eng., 2011, 103, 129–136 CrossRef CAS.
- Y. C. Chen, S. H. Yu, G. J. Tsai, D. W. Tang, F. L. Mi and Y. P. Peng, J. Agric. Food Chem., 2010, 58, 6728–6734 CrossRef CAS PubMed.
- D. W. Tang, S. H. Yu, Y. C. Ho, B. Q. Huang, G. J. Tsai and H. Y. Hsieh, Food Hydrocolloids, 2013, 30, 33–41 CrossRef CAS.
- A. K. Giri, R. Patel and S. Mandal, Chem. Eng. J., 2012, 185, 71–81 CrossRef.
- X. Li, D. Yang, Y. Jiang and H. Fu, Green Chem., 2010, 12, 224–225 Search PubMed.
- R. E. Hunter and N. E. Dweltz, J. Appl. Polym. Sci., 1979, 23, 249–259 CrossRef CAS.
- S. Raymond, A. Kvick and H. Chanzy, Macromolecules, 2002, 28, 8422–8425 CrossRef.
- J. P. D. Mesquita, C. L. Donnici and F. V. Pereira, Biomacromolecules, 2010, 11, 473–480 CrossRef PubMed.
- M. Krumova, D. Lopez, R. Benavente, C. Mijangos and J. M. Perena, Polymer, 2000, 41, 9265–9272 CrossRef CAS.
- P. Hernandez-Munoz, R. Gavara and R. J. J. Hernandez, J. Membr. Sci., 1999, 154, 195–204 CrossRef CAS.
- N. Tammineni, B. Rasco, J. Powers, C. Nindo and G. J. Unlu, J. Agric. Food Chem., 2014, 2, 93–102 CAS.
- W. Xie, P. Xu and Q. Liu, Bioorg. Med. Chem. Lett., 2001, 11, 1699–1701 CrossRef CAS PubMed.
- M. S. Rao, R. Chander and A. Sharma, J. Food Sci., 2005, 70, 325–331 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a