
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel cytotoxic steroidal saponins from the roots of Liriope muscari (Decne.) L.H. Bailey†
Yan Wua,
Xian-Min Wanga,
Su-Xia Bia,
Wen Zhangb,
Rui-Ming Liab,
Rui-Jing Wangb,
Bo-Yang Yu *a and
Jin Qi*a
*a and
Jin Qi*a
aJiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, Nanjing 211198, China. E-mail: boyangyu59@163.com; yaoyuelingxing@163.com; Fax: +86-025-86185158; Tel: +86-025-86185157
bTasly Research Institute, Tianjin Tasly Holding Group Co. Ltd., Tianjin 300410, China
First published on 28th February 2017
Abstract
Ten new steroidal saponins (1–10) and three known steroidal saponins (11–13) were isolated from a 70% EtOH extract of the roots of Liriope muscari (Decne.) L. H. Bailey. Their structures were determined by analyses of infrared, nuclear magnetic resonance and mass spectroscopic data. These compounds exhibited different levels of cytotoxic activity against MDA-MB-435, 95D, HepG2, HeLa, MCF-7 and A549 cell lines in an in vitro bioassay. The structure–activity relationship of these related compounds was also investigated.
Introduction
Liriope muscari (Decne.) L. H. Bailey, a commonly used herb of the Liliaceae family, has been used in China for a long time.1 The tuber of L. muscari exhibits clear therapeutic effects on cough, insomnia, acute and chronic inflammation and cardiovascular diseases.2–6 Previous phytochemical studies have indicated that saccharides and steroidal compounds are the predominant components of L. muscari tubers. The steroidal compounds are considered to be the main active ingredients responsible for the therapeutic effects of L. muscari because many of these steroidal saponins display a variety of biological properties, such as anti-inflammatory, hypoglycemic and cardiovascular activities.7–14 The steroidal constituents, which contain a spiro structure in their parent nucleus, are widely distributed in plants from Dioscoreaceae, Amaryllidaceae, Liliaceae and Agavaceae.15 Pharmacodynamic studies of the steroidal constituents have shown that they exhibit diverse biological properties, including anticancer, anti-inflammatory, antimicrobial, insecticidal and molluscicidal activities.16,17 Recently, the antitumorigenic properties of steroidal saponins have attracted attention for the development of antitumor therapies. Significant antitumor activities of some steroidal saponins from L. muscari have been demonstrated in vitro and in vivo.7,18–27 For example, DT-13, a mixture of 25 (R/S) steroidal saponins isolated from L. muscari, significantly suppresses adhesion and invasion of cancer cells, and has been considered a candidate drug for prevention of cancer metastasis.18–29Although some phytochemical investigations of L. muscari have been reported,7,30–35 the steroidal components of the herb are still not fully elucidated. To promote a better understanding of the steroidal components of L. muscari and enable further screening for potentially useful bioactive ingredients, a 70% EtOH extract of L. muscari was studied. As a result, 10 novel steroidal saponins (1–10) were identified, and three known steroidal saponins (11–13) were also obtained. The structures of these compounds were elucidated based on infrared (IR), nuclear magnetic resonance (NMR) and mass spectroscopic data. The cytotoxic activities of the compounds were evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The isolation, structural characterization and cytotoxic activities of the compounds are described in detail in this article. Furthermore, the structure–activity relationship of these related compounds is also discussed to better interpret the activities of these steroidal compounds.
Results and discussion
Compound 1 was obtained as a white amorphous powder and gave a positive Liebermann–Burchard reaction. Its molecular formula was determined as C44H70O18 according to HRESI-QTOF-MS data (m/z 909.4456 [M + Na]+, calcd 909.4454). The 1H and 13C spectra of 1 (Tables 1–4) displayed the following characteristic signals: two tertiary methyl groups (δH 1.34, s; δH 0.88, s), two secondary methyl groups (δH 1.14, d, J = 7.0 Hz; δH 1.08, d, J = 7.0 Hz), one quaternary carbon (δC 109.7) and an olefinic group (δC 139.4 and 124.7; δH 5.52, brs, 1H). Moreover, the 1H-NMR spectrum showed three anomeric proton signals [δH 5.04 (1H, d, J = 7.7 Hz), 5.52 (1H, d, J = 7.7 Hz) and 5.29 (1H, d, J = 7.7 Hz)], giving heteronuclear single quantum correlation (HSQC) correlations with three anomeric carbon signals at δC 100.1, 104.7 and 105.3, respectively. These observations together with the characteristic absorptions of a 25(S) spiroketal unit at 987, 920, 896, 848, and 920 > 896 cm−1 in the IR spectrum and chemical shifts at δC 26.4 (C-23), 26.2 (C-24), 27.6 (C-25), and 65.0 (C-26) in the 13C-NMR spectrum suggested that 1 was a (25S)-spirostanol derivative containing three sugar units.36| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on TCOSY, HSQC, and HMBC experiments. | ||||||||||
| 1 | 82.6 | 82.5 | 82.6 | 82.4 | 84.5 | 82.8 | 82.5 | 82.6 | 84.3 | 83.0 |
| 2 | 37.2 | 36.9 | 36.7 | 36.5 | 37.7 | 37.3 | 37.0 | 36.7 | 37.5 | 37.4 |
| 3 | 68.1 | 67.9 | 68.1 | 67.9 | 68.0 | 68.4 | 68.1 | 68.1 | 67.8 | 68.3 |
| 4 | 43.7 | 43.5 | 43.5 | 43.4 | 43.4 | 43.7 | 43.4 | 43.5 | 43.4 | 43.6 |
| 5 | 139.4 | 139.2 | 139.4 | 139.2 | 139.2 | 140.0 | 139.6 | 139.4 | 140.0 | 139.8 |
| 6 | 124.7 | 124.7 | 124.6 | 124.4 | 124.8 | 124.3 | 124.1 | 124.6 | 124.6 | 124.4 |
| 7 | 31.9 | 31.7 | 31.8 | 31.7 | 31.8 | 32.1 | 31.8 | 31.8 | 31.5 | 32.0 |
| 8 | 32.9 | 32.8 | 32.9 | 32.7 | 33.0 | 33.0 | 32.7 | 32.9 | 33.0 | 33.2 |
| 9 | 50.3 | 50.2 | 50.5 | 50.1 | 50.2 | 50.3 | 50.0 | 50.2 | 50.0 | 50.4 |
| 10 | 42.7 | 42.6 | 42.8 | 42.6 | 42.6 | 43.0 | 42.7 | 42.8 | 42.4 | 42.9 |
| 11 | 23.9 | 23.8 | 23.7 | 23.4 | 24.1 | 23.6 | 23.4 | 23.7 | 23.9 | 23.7 |
| 12 | 40.3 | 40.2 | 40.2 | 40.0 | 40.3 | 40.5 | 40.2 | 40.1 | 40.0 | 40.3 |
| 13 | 40.2 | 40.1 | 40.0 | 39.9 | 40.2 | 40.3 | 40.0 | 40.1 | 40.0 | 40.4 |
| 14 | 56.8 | 56.7 | 56.6 | 56.4 | 56.8 | 57.0 | 56.8 | 56.6 | 56.7 | 57.0 |
| 15 | 32.3 | 32.2 | 32.2 | 32.0 | 32.3 | 32.4 | 32.2 | 32.2 | 32.0 | 32.4 |
| 16 | 81.2 | 81.0 | 81.1 | 80.8 | 81.0 | 81.3 | 80.9 | 81.3 | 81.1 | 81.5 |
| 17 | 62.8 | 62.8 | 62.7 | 62.7 | 62.9 | 63.0 | 62.8 | 62.8 | 62.7 | 63.1 |
| 18 | 16.7 | 16.6 | 16.6 | 16.4 | 16.8 | 16.8 | 16.5 | 16.5 | 16.6 | 16.7 |
| 19 | 15.0 | 14.9 | 14.9 | 14.7 | 15.0 | 15.1 | 14.9 | 14.9 | 14.8 | 15.1 |
| 20 | 42.5 | 41.9 | 42.3 | 41.7 | 41.9 | 42.6 | 41.8 | 41.7 | 41.6 | 41.9 |
| 21 | 14.8 | 14.8 | 14.7 | 14.7 | 14.9 | 14.9 | 14.8 | 14.8 | 14.6 | 14.9 |
| 22 | 109.7 | 109.2 | 109.7 | 109.0 | 109.2 | 109.8 | 109.0 | 109.4 | 109.3 | 109.5 |
| 23 | 26.4 | 31.6 | 26.3 | 31.5 | 31.7 | 26.4 | 31.6 | 33.1 | 32.8 | 33.0 |
| 24 | 26.2 | 29.1 | 26.1 | 28.9 | 29.2 | 26.3 | 29.0 | 28.8 | 28.6 | 29.0 |
| 25 | 27.6 | 30.5 | 27.4 | 30.3 | 30.5 | 27.6 | 30.4 | 144.3 | 144.2 | 144.6 |
| 26 | 65.0 | 66.7 | 64.9 | 66.5 | 66.7 | 65.1 | 66.6 | 64.9 | 64.6 | 65.0 |
| 27 | 16.3 | 17.2 | 16.2 | 17.0 | 17.2 | 16.3 | 17.1 | 108.6 | 108.4 | 108.6 |
On the basis of the HSQC and heteronuclear multiple-bond correlation (HMBC) correlations, the aglycone moiety of compound 1 was identified as (25S)-ruscogenin.37 The analyses of chemical shifts and coupling constants (J = 7.5–8.0 Hz) obtained from extensive 1D and 2D NMR experiments allowed the identification of two β-glucopyranosyl units and one β-xylopyranosyl unit in 1. The sugar residues were further confirmed by co-thin-layer chromatography (co-TLC) with standard sugars after hydrolysis of 1, and the D-configurations were confirmed by gas chromatography (GC) of their corresponding trimethylsilylthiazolidine derivatives. This procedure was also applied to the other new compounds (2–10). The sequence of the sugar chain and the glycosidic position of 1 were determined by HMBC, HSQC and total correlation spectroscopy (TOCSY) experiments. The HMBC correlations (Fig. 1) of the anomeric proton signal at δH 5.52 (d, J = 7.7 Hz, Glu H-1′′) to δC 81.6 (Glu C-2′), from δH 5.29 (d, J = 7.7 Hz, Xyl H-1′′′) to δC 86.0 (Glu C-3′) and from δH 5.04 (d, J = 8.0 Hz, Glu H-1′) to δC 82.6 (Agly C-1) proved that the sequence of the sugar chain was Glu-(1→2)-[Xyl-(1→3)]-Glu and that the glycosidic site was at C-1. Other key HMBC, HSQC and TOCSY correlations are shown in the ESI data.† The structure of compound 1 was therefore assigned as (25S)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylop yranosyl-(1→3)]-β-D-glucopyranoside.
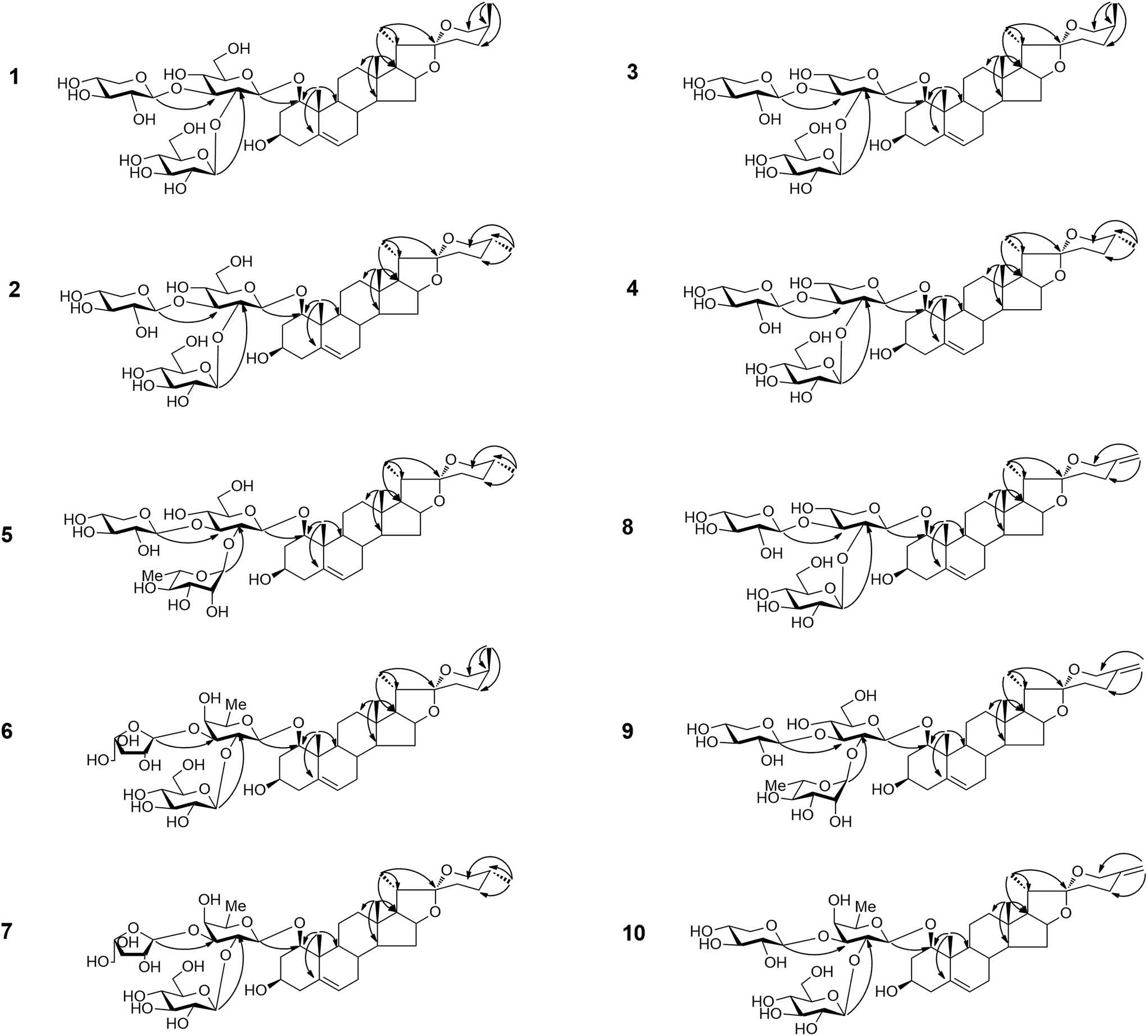 | ||
| Fig. 1 Key HMBC correlations ofcompounds 1–10. | ||
The HRESI-QTOF-MS spectrum of compound 2 (m/z 909.4454 [M + Na]+, calcd 909.4454) supported a molecular formula of C44H70O18, the same as compound 1. A detailed analysis of the NMR data of 2 (Tables 1–4) compared to those for 1 showed that 2 contained the same sugar chain at C-1 as compound 1. The major difference was that the chemical shifts at δC 31.58 (C-23), 29.13 (C-24), 30.47 (C-25) and 66.73 (C-26) in the 13C-NMR spectrum had lower field resonances than those of 1, indicating that the configuration at C-25 was R. This result was further confirmed by the characteristic absorptions of a 25(R) spiroketal unit at 982, 921, 902, 870 cm−1, and 902 > 921 cm−1 in the IR spectrum. The aglycone moiety of 2 was identified as (25R)-ruscogenin by comparison of spectroscopic data to those reported in the literature.37 Compound 2 was therefor assigned as (25R)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-glucopyranoside. Key HMBC, HSQC and TOCSY correlations are shown in Fig. 1 and the ESI data.†
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on TCOSY, HSQC, and HMBC experiments. | ||||||||||
| 1-Glc-1′ | 100.1 | 99.9 | 100.1 | 99.9 | ||||||
| 2′ | 81.6 | 81.5 | 76.1 | 75.9 | ||||||
| 3′ | 86.0 | 85.7 | 88.5 | 88.2 | ||||||
| 4′ | 70.1 | 70.0 | 70.1 | 69.8 | ||||||
| 5′ | 77.8 | 77.6 | 77.6 | 77.4 | ||||||
| 6′ | 63.2 | 63.0 | 63.1 | 62.9 | ||||||
| 1-Xyl-1′ | 100.2 | 100.0 | 100.2 | |||||||
| 2′ | 79.2 | 79.0 | 79.2 | |||||||
| 3′ | 81.6 | 81.4 | 81.5 | |||||||
| 4′ | 68.9 | 68.8 | 68.9 | |||||||
| 5′ | 66.4 | 66.2 | 66.3 | |||||||
| 1-Fuc-1′ | 99.9 | 99.6 | 100.5 | |||||||
| 2′ | 77.9 | 77.6 | 78.1 | |||||||
| 3′ | 82.6 | 82.2 | 83.2 | |||||||
| 4′ | 72.1 | 71.8 | 72.3 | |||||||
| 5′ | 70.8 | 70.5 | 70.7 | |||||||
| 6′ | 17.1 | 16.8 | 17.1 | |||||||
| 2′′-Glc-1′′ | 104.7 | 104.6 | 105.1 | 104.9 | 104.9 | 104.5 | 105.1 | 104.9 | ||
| 2′′ | 76.6 | 76.4 | 76.2 | 76.0 | 76.3 | 76.0 | 76.2 | 76.5 | ||
| 3′′ | 77.9 | 77.7 | 77.9 | 77.7 | 77.9 | 77.6 | 77.9 | 78.1 | ||
| 4′′ | 71.9 | 71.7 | 71.7 | 71.5 | 72.1 | 71.7 | 71.7 | 72.1 | ||
| 5′′ | 78.6 | 78.4 | 78.2 | 78.1 | 78.2 | 77.8 | 78.2 | 78.3 | ||
| 6′′ | 63.0 | 62.8 | 62.9 | 62.8 | 63.4 | 63.0 | 62.9 | 63.4 | ||
| 2′′-Rha-1′′ | 101.5 | 101.3 | ||||||||
| 2′′ | 72.3 | 72.1 | ||||||||
| 3′′ | 72.4 | 72.1 | ||||||||
| 4′′ | 74.1 | 73.9 | ||||||||
| 5′′ | 69.5 | 69.2 | ||||||||
| 6′′ | 19.2 | 19.0 | ||||||||
| 3′′′-Ara (f)-1′′′ | 111.2 | 110.8 | ||||||||
| 2′′′ | 83.0 | 82.7 | ||||||||
| 3′′′ | 78.0 | 77.7 | ||||||||
| 4′′′ | 86.6 | 86.2 | ||||||||
| 5′′′ | 62.9 | 62.5 | ||||||||
| 3′′′-Xyl-1′′′ | 105.3 | 105.1 | 105.9 | 105.7 | 105.1 | 105.9 | 104.9 | 106.5 | ||
| 2′′′ | 75.0 | 74.9 | 74.8 | 74.6 | 74.6 | 74.8 | 74.4 | 75.1 | ||
| 3′′′ | 78.8 | 78.6 | 78.1 | 78.0 | 78.2 | 78.2 | 78.0 | 78.6 | ||
| 4′′′ | 70.8 | 70.7 | 70.9 | 70.7 | 70.5 | 70.9 | 70.3 | 71.1 | ||
| 5′′′ | 67.4 | 67.2 | 67.1 | 66.9 | 67.1 | 67.0 | 66.9 | 67.3 | ||
The molecular formula of compound 3 was determined as C43H68O17 from the ion peak [M + Na]+ at m/z 879.4344 (calcd 879.4349) in the HRESI-QTOF-MS. The IR absorption bands at 3390 and 1072 cm−1 indicated the presence of hydroxyl and C–O groups, respectively. Comparison of the NMR data of 3 obtained from 1D and 2D NMR spectra (Tables 1–4) to those of 1 showed that they contained the same aglycone moiety ((25S)-ruscogenin), but differed slightly in the sugar moiety. The major difference was that the β-glucopyranoside unit was replaced by a β-xylopyranosyl unit in 3. The HMBC correlations (Fig. 1) between the anomeric proton signal at δH 5.43 (d, J = 7.7, Glu H-1′′) to δC 79.2 (Xyl C-2′), from δH 5.27 (d, J = 7.7 Hz, Xyl H-1′′′) to δC 81.6 (Xyl C-3′) and from δH 4.95 (d, J = 7.0 Hz, Xyl H-1′) to δC 82.6 (Agly C-1) proved that the sequence of the sugar chain was Glu-(1→2)-[Xyl-(1→3)]-Xyl and that the glycosidic site was at C-1. Thus, compound 3 was (25S)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-xylopyrano side.
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on TCOSY, HSQC, and HMBC experiments. | ||||||||||
| 1 | 3.96m | 3.98m | 3.88dd (11.5, 4.1) | 3.89dd (11.5, 4.2) | 3.90m | 3.90dd (10.9, 4.2) | 3.90dd (11.0, 4.0) | 3.89dd (11.8, 4.3) | 3.90m | 3.89dd (11.6, 4.3) |
| 2 | 2.32q-like (11.8) | 2.37q-like (12.0) | 2.35q-like (11.5) | 2.33q-like (11.5) | 2.44q-like (12.0) | 2.23q-like (11.4) | 2.25q-like (12.0) | 2.35q-like (11.8) | 2.46q-like (12.0) | 2.33q-like (11.6) |
| 2.76m | 2.80m | 2.79m | 2.74m | 2.70m | 2.68m | 2.68br d (12.0) | 2.79m | 2.71m | 2.73m | |
| 3 | 3.74m | 3.77m | 3.85m | 3.84m | 3.80m | 3.89m | 3.89m | 3.86m | 3.80m | 3.87m |
| 4 | 2.56dd (12.6, 5.2) | 2.59dd (11.8, 5.0) | 2.59dd (12.5, 5.2) | 2.58dd (11.5, 4.2) | 2.57dd (11.6, 4.5) | 2.60dd (12.5, 5.5) | 2.60dd (12.5, 5.5) | 2.59m | 2.58dd (12.8, 4.3) | 2.60dd (12.8, 5.3) |
| 2.68br d (12.6) | 2.71br d (11.8) | 2.70br d (12.5) | 2.69br d (11.5) | 2.71br d (11.6) | 2.69m | 2.69m | 2.70br d (12.1) | 2.71m | 2.70br d (12.8) | |
| 6 | 5.52br s | 5.54br s | 5.56d (5.6) | 5.57d (5.6) | 5.57d (5.7) | 5.57d (5.5) | 5.56d (5.5) | 5.57d (5.6) | 5.58d (5.7) | 5.57d (5.7) |
| 7 | 1.68m | 1.67m | 1.54m | 1.58m | 1.52m | 1.48m | 1.50m | 1.55m | 1.51m | 1.56m |
| 1.85m | 1.80m | 1.88m | 1.85m | 1.89m | 1.87m | 1.90m | 1.88m | 1.89m | 1.93m | |
| 8 | 1.54m | 1.54m | 1.57m | 1.55m | 1.61m | 1.57m | 1.61m | 1.57m | 1.77m | 1.78m |
| 9 | 1.45m | 1.46m | 1.41m | 1.42m | 1.67m | 1.39m | 1.41m | 1.41m | 1.67m | 1.45m |
| 11 | 1.56m | 1.56m | 1.56m | 1.54m | 1.67m | 1.60m | 1.62m | 1.56m | 1.66m | 1.56m |
| 2.80br d (10.3) | 2.80br d (10.3) | 2.87m | 2.84m | 2.90m | 2.84m | 2.85m | 2.87m | 2.90m | 2.87m | |
| 12 | 1.37m | 1.37m | 1.24m | 1.24m | 1.53m | 1.28m | 1.28m | 1.23m | 1.50m | 1.33m |
| 1.69m | 1.67m | 1.58m | 1.59m | 1.72m | 1.67m | 1.68m | 1.57m | 1.68m | 1.67m | |
| 14 | 1.07m | 1.08m | 1.08m | 1.10m | 1.22m | 1.11m | 1.13m | 1.09m | 1.20m | 1.17m |
| 15 | 1.42m | 1.44m | 1.55m | 1.45m | 1.50m | 1.44m | 1.50m | 1.56m | 1.50m | 1.47m |
| 1.97m | 1.98m | 2.00ddd (12.4, 7.3, 5.4) | 2.03ddd (12.3, 7.4, 5.5) | 1.99m | 2.01m | 2.04m | 2.00ddd (12.5, 7.5, 5.6) | 1.99m | 2.01m | |
| 16 | 4.45m | 4.48q-like (8.2) | 4.50m | 4.53m | 4.46m | 4.50m | 4.54m | 4.51m | 4.44m | 4.54q-like (7.3) |
| 17 | 1.76m | 1.80m | 1.73m | 1.75m | 1.84m | 1.77m | 1.82m | 1.74m | 1.83m | 1.83m |
| 18 | 0.88s | 0.88s | 0.85s | 0.87s | 0.93s | 0.87s | 0.89s | 0.85s | 0.92s | 0.89s |
| 19 | 1.34s | 1.33s | 1.38s | 1.38s | 1.44s | 1.37s | 1.37s | 1.38s | 1.44s | 1.38s |
| 20 | 1.90m | 1.97m | 1.87m | 1.93m | 1.99m | 1.89m | 1.94m | 1.93t-like (6.7) | 1.97m | 1.96t-like (6.8) |
| 21 | 1.14d (6.9) | 1.15d (6.9) | 1.11d (6.9) | 1.12d (7.0) | 1.13d (6.9) | 1.12d (6.9) | 1.10d (6.9) | 1.07d (6.9) | 1.08d (6.9) | 1.07d (7.0) |
| 23 | 1.44m | 1.44m | 1.41m | 1.52m | 1.69m | 1.43m | 1.43m | 1.56m | 1.58m | 1.60m |
| 1.89m | 1.87m | 1.89m | 1.90m | 1.92m | 1.90m | 1.90m | 1.76m | 1.77m | 1.78m | |
| 24 | 1.36m | 1.58m | 1.36m | 1.55m | 1.58m | 1.35m | 1.57m | 2.26m | 2.24m | 2.27m |
| 2.13m | 1.69m | 2.14m | 1.66m | 1.68m | 2.14m | 1.66m | 2.72m | 2.72m | 2.72m | |
| 25 | 1.58m | 1.59m | 1.58m | 1.57m | 1.58m | 1.57m | 1.57m | — | — | — |
| 26 | 3.37br d (10.8) | 3.51t-like (10.5) | 3.35d (10.9) | 3.51d (10.9) | 3.49t (10.4) | 3.37br d (10.9) | 3.50d (10.3) | 4.03d (12.4) | 4.45d (12.0) | 4.03d (12.2) |
| 4.05m | 3.60dd (10.5, 3.5) | 4.05m | 3.58m | 3.59m | 4.07dd (10.9, 2.7) | 3.58m | 4.46br d (12.4) | 4.04d (12.0) | 4.47br d (12.2) | |
| 27 | 1.08d (7.1) | 0.70d (5.1) | 1.07d (7.1) | 0.70d (5.2) | 0.70d (5.0) | 1.09d (7.1) | 0.69d (4.9) | 4.81br s | 4.81br s | 4.82br s |
| 4.78br s | 4.79br s | 4.79br s | ||||||||
The HRESI-QTOF-MS spectrum of compound 4 (m/z 879.4344 [M + Na]+ calcd 879.4349) supported a molecular formula of C43H68O17, the same as compound 3. The NMR data of 4 (Tables 1–4) obtained from 1D and 2D NMR spectra were similar to those of 3. The chemical shifts at δC 31.50 (C-23), 28.94 (C-24), 30.27 (C-25) and 66.53 (C-26) in the 13C-NMR spectrum had lower field resonances than those of 3. Together with the characteristic absorptions of a 25(R) spiroketal unit at 982, 921, 902, 870 cm−1, and 902 > 921 cm−1 in the IR spectrum, these results indicated that the configuration at C-25 was R. The aglycone moiety of compound 4 was identified as (25R)-ruscogenin by comparison of the NMR data of 4 to those of compound 1. The sequence of the sugar chain and the glycosidic position of 4, determined d by HMBC and TOCSY experiments, showed that 4 contained the same sugar chain at C-1 as compound 3. The structure of compound 4 was therefore (25R)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-xy lopyranoside. Key HMBC, HSQC and TOCSY correlations are shown in Fig. 1 and the ESI data.†
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on TCOSY, HSQC, and HMBC experiments. | ||||||||||
| 1-Glc-1′ | 5.04d (8.0) | 5.05d (7.7) | 4.84d (7.7) | 4.83d (7.8) | ||||||
| 2′ | 4.45dd (4.2, 8.0) | 4.45m | 4.21d (7.7) | 4.21d (7.8) | ||||||
| 3′ | 4.23m | 4.23m | 4.09d (8.7) | 4.08d (9.0) | ||||||
| 4′ | 3.97m | 3.96m | 3.88m | 3.88m | ||||||
| 5′ | 3.83m | 3.85m | 3.81m | 3.81m | ||||||
| 6′ | 4.74dd (11.7, 2.7) | 4.77dd (11.7, 2.7) | 4.51m | 4.51m | ||||||
| 4.52dd (11.7, 3.5) | 4.54dd (11.7, 4.4) | 4.24m | 4.22m | |||||||
| 1-Xyl-1′ | 4.95d (7.0) | 4.95d (7.0) | 4.95d (7.0) | |||||||
| 2′ | 4.80dd (7.0, 9.0) | 4.79dd (7.0, 9.0) | 4.81m | |||||||
| 3 | 4.30m | 4.29m | 4.29m | |||||||
| 4 | 4.48m | 4.46m | 4.48m | |||||||
| 5′ | 3.71m | 3.72m | 3.72m | |||||||
| 4.28m | 4.26m | 4.28m | ||||||||
| 1-Fuc-1′ | 4.78d (7.9) | 4.79d (7.5) | 4.85d (7.6) | |||||||
| 2′ | 4.61dd (9.6, 7.5) | 4.64dd (9.6, 7.6) | 4.24m | |||||||
| 3′ | 4.13dd (9.6, 3.4) | 4.14dd (9.6,3.4) | 4.20m | |||||||
| 4′ | 4.23m | 4.24m | 4.24m | |||||||
| 5′ | 3.59m | 3.61m | 3.74m | |||||||
| 6′ | 1.48d (6.3) | 1.48d (6.3) | 1.54d (6.2) | |||||||
| 2′′-Glc-1′′ | 5.52d (7.7) | 5.52d (7.7) | 5.43d (7.8) | 5.42d (7.8) | 5.33d (7.8) | 5.36d (7.7) | 5.44d (7.8) | 5.45d (7.9) | ||
| 2′′ | 4.17m | 4.18m | 4.15m | 4.13m | 4.09m | 4.06m | 4.14m | 4.11m | ||
| 3′′ | 4.33m | 4.36m | 4.28m | 4.27dd (7.5, 3.1) | 4.26m | 4.23m | 4.28m | 4.24m | ||
| 4′′ | 4.31m | 4.35m | 4.27m | 4.26m | 4.23m | 4.23m | 4.27m | 4.19m | ||
| 5′′ | 4.10m | 4.10m | 3.95m | 3.95m | 3.86m | 3.84m | 3.95m | 3.93m | ||
| 6′′ | 4.50m | 4.23m | 4.46m | 4.45m | 4.38dd (11.4, 4.6) | 4.40br d (11.3) | 4.46m | 4.42dd (11.5, 4.6) | ||
| 4.23m | 4.50m | 4.61dd (11.6, 2.8) | 4.61br d (11.7) | 4.50m | 4.51m | 4.63br d (11.6) | 4.60dd (11.5, 2.9) | |||
| 2′′-Rha-1′′ | 6.47br s | 6.47br s | ||||||||
| 2′′ | 4.82m | 4.82m | ||||||||
| 3′′ | 4.61dd (8.7, 4.6) | 4.62br d (9.5) | ||||||||
| 4′′ | 4.35td (9.5, 3.8) | 4.34m | ||||||||
| 5′′ | 4.88dd (9.5, 6.2) | 4.86m | ||||||||
| 6′′ | 1.80d (6.2) | 1.80d (6.2) | ||||||||
| 3′′′-Ara (f)-1′′′ | 6.10d (2.3) | 6.10d (2.4) | ||||||||
| 2′′′ | 4.95m | 4.97m | ||||||||
| 3′′′ | 4.83m | 4.83m | ||||||||
| 4′′′ | 4.73m | 4.77m | ||||||||
| 5′′′ | 4.18m | 4.21m | ||||||||
| 4.28dd (11.8, 3.5) | 4.32br d (11.8) | |||||||||
| 3′′′-Xyl-1′′′ | 5.29d (7.7) | 5.28d (7.7) | 5.27d (8.0) | 5.26d (8.0) | 4.96d (8.0) | 5.28d (8.4) | 4.96d (7.6) | 5.26d (8.2) | ||
| 2′′′ | 4.02m | 4.05m | 4.00br d (8.0) | 4.00dd (8.0, 3.1) | 4.00dd (8.0, 3.5) | 3.98dd (8.4, 3.2) | 4.00m | 3.97br d (8.2) | ||
| 3′′′ | 4.04m | 4.04m | 4.15m | 4.15m | 4.10m | 4.15m | 4.10m | 4.12m | ||
| 4′′′ | 4.13m | 4.17m | 4.16m | 4.16m | 4.14m | 4.16m | 4.11m | 4.15m | ||
| 5′′′ | 4.28dd (12.4, 3.9) | 4.31dd (11.5, 5.1) | 4.31m | 4.31dd (11.1, 4.6) | 4.27dd (11.5, 5.2) | 4.31m | 4.27dd (11.4, 5.1) | 4.31dd (11.3, 5.2) | ||
| 3.73m | 3.75m | 3.71m | 3.74m | 3.70dd (11.5, 9.7) | 3.71m | 3.70t-like (11.4) | 3.72m | |||
The molecular formula of compound 5 was established unequivocally as C44H70O17 from the HRESI-QTOF-MS spectrum (m/z 871.4702 [M + H]+, calcd 871.4686). The IR spectrum indicated the presence of a hydroxyl and a 25(R) spiroketal unit. The aglycone moiety of 5 was established as 25(R)-ruscogenin by comparison of the NMR data of 5 (Tables 1–4) to those of 2, and the glycosidic site was identified as C-1. A detailed analysis of 1D and 2D NMR data of 5 compared to those reported for parisverticoside A,38 and parisyunnanoside G,39 showed that they shared the same sugar moiety. The sequence of the sugar chain was established as Rha-(1→2)-[Xyl-(1→3)]-Glu by HMBC, HSQC and TOCSY correlations. Thus, compound 5 was (25R)-ruscogenin-1-O-α-L-rhamnopyranosy l-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-glucopyranoside.
Compound 6 has a molecular formula of C44H70O17, as indicated by the HRESI-QTOF-MS spectrum (m/z 893.4461 [M + Na]+, calcd 893.4505). The aglycone moiety of 6 was established as (25S)-ruscogenin by comparison of the NMR data of 6 (Tables 1–4) to those of 3. The 1H-NMR spectrum showed three anomeric proton signals [δH 5.36 (1H, d, J = 7.7 Hz), 6.10 (1H, d, J = 2.4 Hz) and 4.79 (1H, d, J = 7.5 Hz)], giving HSQC correlations with three anomeric carbon signals at δC 104.5, 110.8 and 99.6, respectively. The HMBC correlations (Fig. 1) of the anomeric proton signal in 6 at δH 5.36 (d, J = 7.7, Glu H-1′′) to δC 77.6 (Fuc C-2′), from δH 6.10 (d, J = 2.4 Hz, Ara (f)H-1′′′) to δC 82.2 (Fuc C-3′) and from δH 4.79 (d, J = 7.5 Hz, Fuc H-1′) to δC 82.5 (Agly C-1) proved that the sequence of the sugar chain was Glu-(1→2)-[Ara (f)-(1→3)]-Fuc and that the glycosidic site was at C-1. HSQC and TOCSY correlations are shown in the ESI data.† The structure of 6 was therefore established as (25S)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[α-L-arabinofuranosyl-(1→3)]-β-D-fucopyranoside.
Compound 7 has the same molecular formula as compound 6 (C44H70O17) from the HRESI-QTOF-MS spectrum (m/z 893.4461 [M + Na]+, calcd 893.4505), and similar NMR data (Tables 1–4). The only difference in the chemical shifts at δC C-23, C-24, C-25 and C-26 of 7 was the lower field resonances compared to those of 6 in the 13C-NMR spectrum, indicating that 6 and 7 were a pair of C-25 epimers. The aglycone moiety of compound 7 was identified as (25R)-ruscogenin by comparison of the NMR data to those of 2. HMBC, HSQC and TOCSY experiments indicated that 7 had the same sequence of the sugar chain and glycosidic position as compound 6. Key HMBC and HSQC correlations are shown in Fig. 1 and the ESI data.† The structure of 7 was therefore established as (25R)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[α-L-arabinofuranosyl-(1→3)]-β-D-fucopyranoside.
Compound 8 has the molecular formula C43H66O17, as deduced from the HRESI-QTOF-MS data (m/z 877.4188 [M + Na]+, calcd 877.4192). The 1H- and 13C-NMR data (Tables 1–4) of 8 were similar to those of 3, with the exception of some differences in the carbon signals due to the presence of an exo-olefinic group (δH 4.81, 4.78; δC 144.3, 108.6) and the disappearance of signals for a secondary methyl (CH3-27) in 8. The location of an exo-double bond (Δ25(27)) was confirmed by the long-range correlations of H-27 (δH 4.81, 4.78) to C-24 (δC 28.8), C-25 (δC 144.3) and C-26 (δC 64.9) in the HMBC spectrum indicating that the aglycone moiety was neoruscogenin.37 The sugar moiety and glycosidic site of 8 were established to be the same as those of 3 by HMBC and TOCSY experiments. Key long-range correlations are shown in Fig. 1 and the ESI data.† Thus, 8 was characterized as neoruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylopyr-anosyl-(1→3)]-β-D-xylopyranoside.
The molecular formula of compound 9 was determined to be C43H68O17 from the pseudo-molecular ion peak [M + H]+ at m/z 869.4529 (calcd 869.4529) in the HRESI-QTOF-MS, differing from that of 5 by 2 Da, and corresponding to an additional double bond. The aglycone moiety of 9 was established as neoruscogenin by comparison of 1D and 2D NMR spectra (Tables 1–4) of compound 9 to those of 8. A detailed analysis of NMR data of 9 in comparison with those of 5 showed that 9 had the same sugar moiety and glycosidic site as 5. The sequence of the sugar chain and the glycosidic position of 9 were further confirmed by HMBC, HSQC and TOCSY experiments. Thus, compound 9 was established as neoruscogenin-1-O-α-L-rhamnopyranosy-l-(1→2)-[β-D-xylop yranosyl-(1→3)]-β-D-glucopyranoside.
The molecular formula of compound 10 was determined by HRESI-QTOF-MS to be C44H68O17 (m/z 891.4321 [M + Na]+, calcd 891.4349). The NMR data of 10 (Tables 1–4) based on HSQC, HMBC and TOCSY were similar to those of 11 {(25R)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-fucopyranoside, Fig. 2},30 except for the appearance of signals for an exo-olefinic group (δH 4.82, 4.79; δC 144.6, 108.6) and the disappearance of the signals for a secondary methyl (CH3-27) in 10. The location of an exo-double bond (Δ25(27)) was confirmed by the long-range correlations of H-27 (δH 4.82, 4.78) to C-24 (δC 29.0), C-25 (δC 144.6) and C-26 (δC 65.0) in the HMBC spectrum. The sequence of the sugar chain and the glycosidic position of 10 determined by HMBC and TOCSY experiments suggested that 10 had the same sugar moiety and glycosidic site as 11. Therefore, 10 was characterized as neoruscogenin-1-O-β-D-glucopyranosyl-(1 →2)-[β-D-xylopyranosyl-(1→3)]-β-D-fucopyranoside.
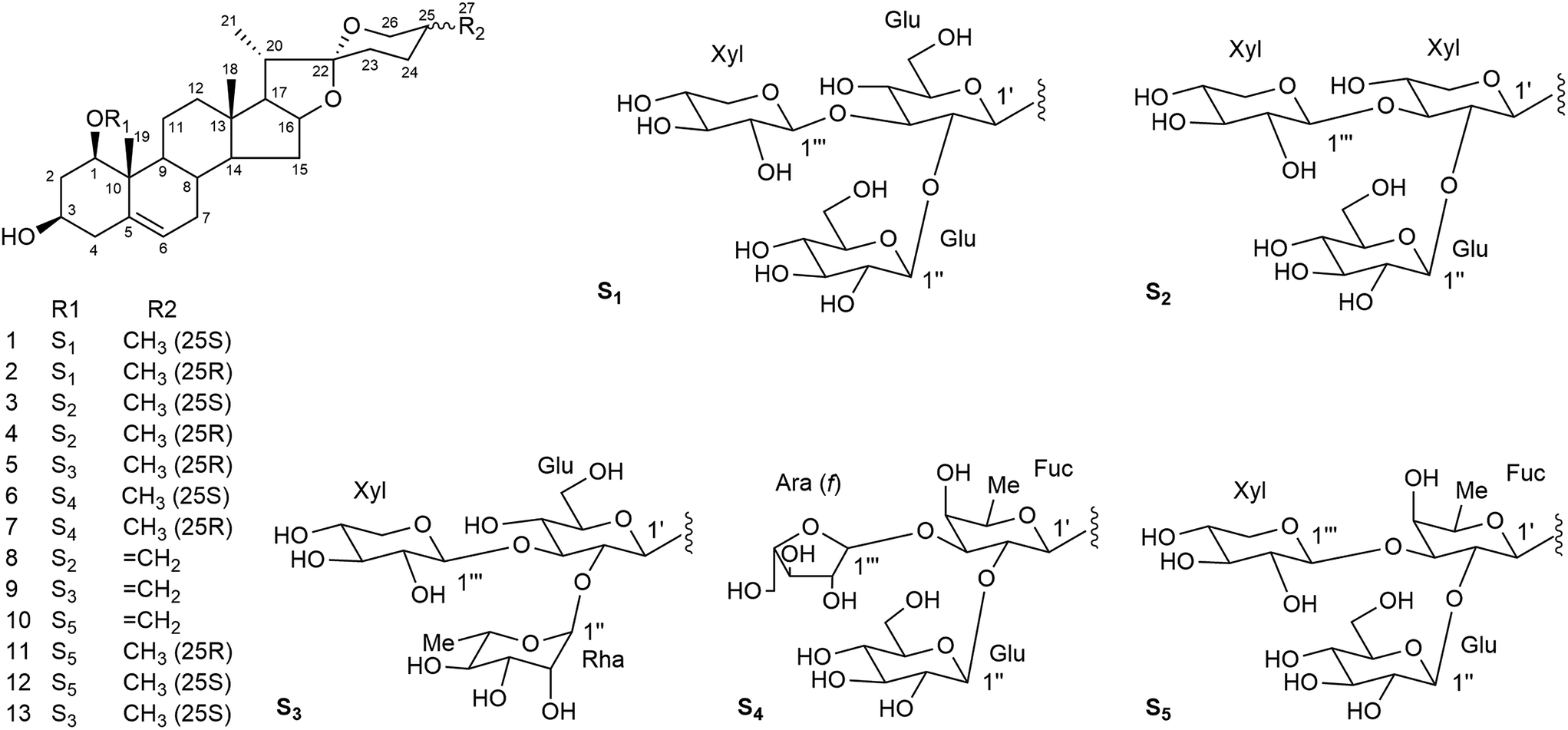 | ||
| Fig. 2 Chemical structures of compounds 1–13. | ||
The three known compounds were identified as (25R)-ruscogenin-1-O-β-D-glucopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-fucopyranoside (11),30 (25S)-ruscogenin-1-O-β-D-glucopyran osyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-fucopyranoside (12),30 and (25S)-ruscogenin-1-O-α-L-rhamnopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-glucopyranoside (13)40 by comparison of spectroscopic data with data reported in the literature.
According to the features of the chemical structures shown in Fig. 2, compounds 1–13 can be divided into three groups based on their aglycone moiety, including the (25S)-ruscogenin group (compounds 1, 3, 6, 12 and 13), (25R)-ruscogenin group (compounds 2, 4, 5, 7 and 11), and neoruscogenin group (compounds 8, 9 and 10). Compounds 3, 4 and 8 share the same A–E rings and Glu-(1→2)-[Xyl-(1→3)]-Xyl glycoside chain at C-1. The only difference between them is that the C-25 configurations of the aglycone moieties are 25S, 25R and 25, 27 double-bond for 3, 4 and 8, respectively. A similar situation prevails for compounds 13, 5 and 9 and for compounds 12, 11 and 10. Compounds 1 and 2, and compounds 6 and 7 are two pairs of C-25 S/R epimers.
All isolates were tested for their in vitro cytotoxic activity against MDA-MB-435, 95D, HepG2, HeLa, MCF-7 and A549 cell lines. As summarized in Table 5, compounds 11 and 12 exhibited the best cytotoxicity against the MDA-MB-435 cell line among all of the cytotoxicity data, with IC50 values of 4.71 and 5.91 μM, respectively. Compound 5 showed no significant cytotoxicity (IC50 > 50 μM) against HeLa and MCF-7 cell lines. Compounds 7, 8 and 11 were also inactive against different cell lines: 7 vs. MDA-MB-435, HepG2, MCF-7 and A549 cell lines; 8 vs. HepG2, MCF-7 and A549 cell lines; 11 vs. HepG2 and MCF-7 cell lines. For all tested cell lines, the configurations at C-25 (25R, 25S and 25, 27-double bond) in the aglycone can be deduced to be unrelated to the cytotoxicity of compounds containing β-D-glu-(1→2)-[β-D-xyl-(1→3)]-β-D-xyl or β-D-glu-(1→2)-[β-D-xyl-(1→3)]-β-D-glu sugar chains at C-1 because of the almost similar IC50 values of 1–4 and 8 vs. all cell lines. The C-25R compounds containing a β-D-glu-(1→2)-[β-D-ara-(1→3)]-β-D-fuc sugar chain were less active than their epimers (6 and 7 vs. all cell lines). Interestingly, our cytotoxicity data suggest that the C-25 configuration does not influence the cytotoxicity of compounds containing β-D-rha-(1→2)-[β-D-xyl-(1→3)]-β-D-glu or β-D-glu-(1→2)-[β-D-xyl-(1→3)]-β-D-fuc sugar chains against the 95D cell line (5, 9, 13, 10–12 vs. 95D). However, these C-25R compounds were less active than their epimers against HeLa, MCF-7 and A549 cell lines (5, 13; 11, 12 vs. HeLa, MCF-7 and A549). Additionally, for HeLa and HepG2 cell lines, C-25S compounds containing a β-D-glu-(1→2)-[β-D-xyl-(1→3)]-β-D-fuc sugar chain displayed similar cytotoxicity to those of 25, 27-double bond compounds (10 and 12 vs. HeLa and HepG2). In summary, the configuration at C-25 in the aglycone and the sugar chain may together determine the cytotoxicity of steroidal saponins, and further studies are required to define the underlying chemical and biological mechanisms.
| Compounds | IC50 (means ± SD, μM) | |||||
|---|---|---|---|---|---|---|
| MDA-MB-435 | 95D | HepG2 | HeLa | MCF-7 | A549 | |
| a No activity (IC50 > 50 μM).b Not measured due to insufficient amount of compounds. | ||||||
| 1 | 15.99 ± 1.03 | 20.13 ± 1.18 | 49.68 ± 1.57 | 39.98 ± 1.20 | 47.30 ± 1.56 | 36.35 ± 1.39 |
| 2 | 26.01 ± 0.85 | 30.00 ± 0.51 | 40.52 ± 0.96 | 33.42 ± 1.39 | 39.12 ± 1.02 | 36.01 ± 1.31 |
| 3 | 18.07 ± 1.34 | 25.67 ± 0.41 | 37.17 ± 1.71 | 21.58 ± 1.42 | 45.82 ± 1.44 | 43.53 ± 1.16 |
| 4 | 17.68 ± 2.50 | 17.83 ± 0.37 | 29.48 ± 1.64 | 22.23 ± 1.43 | 42.16 ± 1.26 | 43.20 ± 1.53 |
| 5 | 19.63 ± 0.76 | 10.82 ± 0.18 | 15.26 ± 1.29 | NAa | NAa | 35.56 ± 1.46 |
| 6 | 16.34 ± 0.60 | 14.34 ± 0.33 | 27.10 ± 0.84 | 14.76 ± 0.52 | 35.21 ± 2.02 | 24.69 ± 0.76 |
| 7 | NAa | 22.15 ± 1.41 | NAa | 42.56 ± 3.75 | NAa | NAa |
| 8 | 24.52 ± 0.91 | 36.12 ± 1.08 | NAa | 24.30 ± 1.55 | NAa | NAa |
| 9 | 17.54 ± 1.39 | 11.09 ± 0.15 | —b | —b | —b | —b |
| 10 | 9.74 ± 0.62 | 10.64 ± 0.21 | 15.48 ± 0.52 | 11.02 ± 0.42 | 10.02 ± 0.73 | 21.25 ± 1.42 |
| 11 | 4.71 ± 0.75 | 11.62 ± 2.00 | NAa | 26.36 ± 2.01 | NA | 23.56 ± 2.64 |
| 12 | 5.91 ± 0.27 | 11.20 ± 0.17 | 12.76 ± 0.74 | 8.00 ± 0.45 | 17.88 ± 0.97 | 8.226 ± 0.78 |
| 13 | 9.75 ± 0.34 | 19.58 ± 0.67 | 15.24 ± 1.53 | 14.03 ± 0.61 | 16.30 ± 0.73 | 13.99 ± 0.64 |
| 5-Fluorouracil | 116.8 ± 13.93 | 83.55 ± 10.66 | 91.9 ± 16.20 | 251.3 ± 19.93 | 568.3 ± 54.37 | 244.8 ± 21.23 |
Experimental section
General
Optical rotations were measured using a JASCO P-1020 digital polarimeter (JASCO Corporation, Easton, MD, USA). IR data (KBr disks, in cm−1) were recorded on a Perkin-Elmer FT-IR Spectra 400 (Perkin-Elmer, Norwalk, CT, USA). NMR spectra using pyridine-d5 as the solvent were recorded using a Bruker Avance 500 NMR spectrometer (Bruker Corporation, Faellanden, Switzerland) with tetramethylsilane as the internal standard. ESI-MS, HRESI-TOF-MS, and HRESI-QTOF-MS experiments were performed on an Agilent 1100 Series MSD Trap mass spectrometer (Agilent Technologies, Santa Clara, CA, USA), Agilent 6210 ESITOF spectrometer, and Agilent 6520 ESIQTOF spectrometer, respectively. Silica gel (100–200 mesh and 200–300 mesh; Qingdao Marine Chemical Factory, Qingdao, China), D101 macroporous resin (Tianjin Pesticide Co., Tianjin, China), and YMC RP-C18 (50 μm, YMC, Tokyo, Japan) were used for column chromatography (CC). Preparative high-performance liquid chromatography (HPLC) was carried out using an Agilent 1100 Series HPLC instrument equipped with a diode array detector. Fractions obtained from CC were analyzed by TLC using silica gel GF254 (Qingdao Marine Chemical Factory, Qingdao, China) plates. GC analysis was conducted on an Agilent 6890 gas chromatograph. L-Cysteine methyl ester hydrochloride, trimethylchlorosilane, hexamethyldisilazane, standard D-glucose, D-fucose, L-rhamnose, L-arabinose, and D-xylose were purchased from Sigma-Aldrich Trading Co. Ltd. (Shanghai, China).Plant material
Dried roots of L. muscari were purchased from Fujian Province, People's Republic of China, in October 2014, and authenticated by one of the authors (B. Y, Y.). A voucher specimen (No. 20141010) was deposited at the herbarium of Jiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, Nanjing, China.Extraction and isolation
Dried roots of L. muscari (10 kg) were crushed and extracted with 70% EtOH (2 h, 3 × 80 L), and then the extract was concentrated under vacuum to afford a residue. The residue was suspended in 20% EtOH and subjected to D101 macroporous resin CC, eluted successively with EtOH–H2O (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80, 85:15, 95:5, v/v) to afford a saponin-rich fraction (85% EtOH eluting fraction, 100 g).
:80, 85:15, 95:5, v/v) to afford a saponin-rich fraction (85% EtOH eluting fraction, 100 g).
The saponin-rich fraction was subjected to silica gel CC eluted with a gradient of CHCl3–MeOH–H2O (90:10:1 to 30:70:5, v/v) to afford three fractions (Fr. 1–3). Fr. 1 was chromatographed on a silica gel column, eluted with a gradient of CHCl3–MeOH–H2O (85:15:1.5 to 70:30:3, v/v), to give two sub-fractions (Fr. 1a, 1b). Fr. 2 was chromatographed on an ODS column with a gradient of MeOH–H2O (40:60 to 70:30, v/v) as the mobile phase to afford two sub-fractions (Fr. 2a, 2b). Fr. 1a was further purified using an Agela Venusil PAH Prep C-18 column, eluted with CH3CN–H2O (45:55, v/v) to yield compounds 1 (15 mg), 2 (20 mg), 3 (20 mg) and 4 (30 mg). Fr. 1b was further purified using an Agela Venusil PAH Prep C-18 column, eluted with CH3CN–H2O (50:50, v/v) to yield compounds 5 (50 mg), 6 (10 mg), 7 (30 mg) and 13 (40 mg). Fr. 2a was further purified using an Agela Venusil PAH Prep C-18 column, eluted with CH3CN–H2O (45:55, v/v) to yield compounds 8 (60 mg), 11 (300 mg) and 12 (200 mg). Compounds 9 (8 mg) and 10 (10 mg) were isolated from Fr. 2b using an Agela Venusil PAH Prep C-18 column, eluted with CH3CN–H2O (50:50, v/v).
Characterization of new compounds
Acid hydrolysis of compounds 1–10
Each compound (2 mg) was refluxed with 2 mL of 2 M HCl (dioxane–H2O, 1:1) at 100 °C for 4 h. The solution was diluted with H2O (2 mL) and extracted with EtOAc (1 mL × 3) after the removal of dioxane. The aqueous layer was evaporated under vacuum, and the residue was diluted with H2O (50 mL). This procedure was repeated until a neutral residue was obtained (10–15 times), and then it was analyzed by TLC over silica gel (Me2CO–n-BuOH–H2O, 6:3:1) together with authentic sugar samples. The remaining residue was dissolved in pyridine (300 μL), and L-cysteine methyl ester hydrochloride (4 mg) was added to it. The mixture was kept at 60 °C in an oil bath for 1.5 h. Then 300 μL of hexamethyldisilazane–trimethylchlorosilane (2:1) was added and the mixture was heated at 60 °C in an oil bath for a further 30 min. After centrifugation, the supernatant was analyzed by GC under the following conditions: capillary column, HP-5 (0.32 mm × 30 m × 0.5 μm); flame ionization detection; detector temperature, 280 °C; injection temperature, 250 °C; initial temperature, 200 °C and an initial time of 8 min, 6 °C min−1 to 260 °C and then held for 2 min; carrier, N2; split ratio, 1/50. The monosaccharides present in compounds 1, 2, 3, 4 and 8 were confirmed to be D-glucose and D-xylose by comparing the retention times (TR) of monosaccharide derivatives with the derivatives prepared similarly from standard sugars. In the same procedure, D-glucose, L-rhamnose and D-xylose were identified from compounds 6 and 7, D-glucose, L-arabinose and D-fucose for compound 5, and D-glucose, D-xylose and D-fucose from compound 10.
Cytotoxicity assay
Conclusions
In this work, ten new steroidal saponins (1–10) and three known steroidal saponins (11–13) were isolated from the root of L. muscari. Their structures were determined using IR, NMR and mass spectroscopic data. These compounds exhibited different cytotoxic activity against six cancer cell lines in vitro. Structure–activity analysis of these related compounds revealed that the C-25 configuration of the aglycone moiety and the sugar chain may together determine the cytotoxicity of steroidal saponins. These results provide a basis for evaluating the structure–activity relationships of other steroidal saponins, as well as for developing these compounds as potential anticancer drugs.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (No. 81473317 and 81673555), the Major National Science and Technology Project of China for Significant New Drugs Development (No. 2012ZX09102201-015), Qing Lan Project, the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 2011 Program for Excellent Scientific and Technological Innovation Team of Jiangsu Higher Education, and the Major Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMZZ201203).Notes and references
- B. Y. Yu, G. J. Xu and R. L. Jin, J. China Pharm. Univ., 1991, 3, 150–153 Search PubMed.
- X. C. Fang, B. Y. Yu, B. R. Xiang and D. K. An, J. Chromatogr. A, 1990, 514, 287–292 CrossRef CAS PubMed.
- B. Y. Yu and G. J. Xu, Chin. Tradit. Herb. Drugs, 1995, 26, 205–210 Search PubMed.
- B. Y. Yu, Chin. J. Nat. Med., 2007, 1, 10–14 Search PubMed.
- China Pharmacopoeia Committee, P. Chinese Pharmacopoeia, Chemical Industry Press, 2010 Search PubMed.
- ed. Pharmacopoeia Commission of PRC, Pharmacopoeia of the People's Republic of China, China Medical Science Press, 2010 Search PubMed.
- Y. W. Li, J. Qi, Y. Y. Zhang, Z. Huang, J. P. Kou, S. P. Zhou, Y. Zhang and B. Y. Yu, Chin. J. Nat. Med., 2015, 13, 461–466 CAS.
- Y. Tian, S. Ma, B. Lin, J. Kou and B. Yu, Indian J. Pharmacol., 2013, 45, 283–285 CrossRef CAS PubMed.
- Y. Q. Tian, J. P. Kou, L. Z. LI and B. Y. YU, Chin. J. Nat. Med., 2011, 9, 222–226 CAS.
- Q. Xu, R. Wang and B. Y. Yu, J. China Pharm. Univ., 1993, 24, 98–101 CAS.
- J. Liu, T. Chen, B. Yu and Q. Xu, J. Pharm. Pharmacol., 2002, 54, 959–965 CrossRef CAS PubMed.
- J. Song, J. Kou, Y. Huang and B. Yu, J. Pharmacol. Sci., 2010, 113, 409–413 CrossRef CAS.
- C. Qiu, L. Jozsef, B. Yu and J. Yu, Biochem. Biophys. Res. Commun., 2014, 443, 74–79 CrossRef CAS PubMed.
- J. Tao, H. Wang, H. Zhou and S. Li, Life Sci., 2005, 77, 3021–3030 CrossRef CAS PubMed.
- S. Man, W. Gao, Y. Zhang, L. Huang and C. Liu, Fitoterapia, 2010, 81, 703–714 CrossRef CAS PubMed.
- S. G. Sparg, M. E. Light and J. V. Staden, J. Ethnopharmacol., 2004, 94, 219–243 CrossRef CAS PubMed.
- A. V. Rao and D. M. Gurfinkel, Drug Metab. Drug Interact., 2000, 17(1–4), 211–235 CAS.
- J. L. Liu, T. Chen, B. Y. Yu and Q. XU, J. Pharm. Pharmacol., 2002, 54, 959–966 CrossRef CAS PubMed.
- J. Tao, H. Wang, J. Chen, H. Xu and S. Li, Am. J. Chin. Med., 2005, 33, 797–806 CrossRef CAS PubMed.
- L. Sun, S. Lin, R. Zhao, B. Yu, S. Yuan and L. Zhang, Biol. Pharm. Bull., 2010, 33, 1192–1198 CAS.
- Y. Zhang, J. Liu, J. Kou, J. Yu and B. Yu, Mol. Med. Rep., 2012, 6, 1121–1125 CAS.
- L. Sun, S. S. Lin, B. Y. Yu, S. T. Yuan and L. Y. Zhang, Chin. J. Nat. Med., 2010, 8, 466–470 CAS.
- Y. Y. Zhang, J. H. Liu, J. P. Kou, J. Yu and B. Y. Yu, Chin. J. Nat. Med., 2012, 10, 436–440 CAS.
- R. P. Zhao, S. S. Lin, S. T. Yuan, B. Y. Yu, X. S. Bai, L. Sun and L. Y. Zhang, Chin. J. Nat. Med., 2014, 12, 24–29 CAS.
- S. S. Lin, W. Fan, L. Sun, F. F. Li, R. P. Zhao, L. Y. Zhang, B. Y. Yu and S. T. Yuan, Chin. J. Nat. Med., 2014, 12, 833–840 CAS.
- H. Li, L. Sun, E. L. de Carvalho, X. Li, X. Lv, G. J. Khan, H. Semukunzi, S. Yuan and S. Lin, Eur. J. Pharmacol., 2016, 781, 164–172 CrossRef CAS PubMed.
- X. W. Yu, S. Lin, H. Z. Du, R. P. Zhao, S. Y. Feng, B. Y. Yu, L. Y. Zhang, R. M. Li, C. M. Qian, X. J. Luo, S. T. Yuan and L. Sun, Oncotarget, 2016, 7, 32990–33003 Search PubMed.
- S. Ma, J. Kou and B. Yu, Food Chem. Toxicol., 2011, 49, 2243–2251 CrossRef CAS PubMed.
- L. L. Chen, W. W. Yuan, Z. F. Hu, J. Qi, D. N. Zhu and B. Y. Yu, J. Pharm. Biomed. Anal., 2011, 56, 650–654 CrossRef CAS PubMed.
- B. Y. Yu, Y. Hirai, J. Shoji and G. J. Xu, Chem. Pharm. Bull., 1990, 38, 1931–1935 CrossRef CAS.
- B. Y. Yu, S. X. Qiu, K. Zaw, G. J. Xu, Y. Hirai, J. Shoji, H. H. Fong and A. D. Kinghorn, Phytochemistry, 1996, 43, 201–206 CrossRef CAS PubMed.
- D. Y. Lee, K. H. Son, J. C. Do and S. S. Kang, Arch. Pharmacal Res., 1989, 12, 295–299 CrossRef CAS.
- Z. H. Cheng, T. Wu, Y. L. Guo, B. Y. Yu and S. X. Luo, Chin. Chem. Lett., 2006, 17, 31–34 CAS.
- L. Jin, Y. Zhang, L. Yan, Y. Guo and L. Niu, Molecules, 2012, 17, 1797–1808 CrossRef PubMed.
- C. Jiang, Z. H. Liu, L. Li, B. B. Lin, F. Yang and M. J. Qin, J. Asian Nat. Prod. Res., 2012, 14, 491–495 CrossRef CAS PubMed.
- P. K. Agrawal, D. C. Jain and A. K. Pathak, Magn. Reson. Chem., 1995, 33, 923–953 CrossRef CAS.
- Y. Mimaki, Y. Takaashi, M. Kuroda, Y. Sashida and T. Nikaido, Phytochemistry, 1996, 42, 1609–1615 CrossRef CAS PubMed.
- C. L. Sun, W. Ni, H. Yan, Z. H. Liu, L. Yang, Y. A. Si, Y. Hua, C. X. Chen, L. He, J. H. Zhao and H. Y. Liu, Steroids, 2014, 92, 90–95 CrossRef CAS PubMed.
- L. P. Kang, Y. X. Liu, T. Eichhorn, E. Dapat, H. S. Yu, Y. Zhao, C. Q. Xiong, C. Liu, T. Efferth and B. P. Ma, J. Nat. Prod., 2012, 75, 1201–1205 CrossRef CAS PubMed.
- H. Y. Shen, W. J. Zuo, H. Wang, Y. X. Zhao, Z. K. Guo, Y. Luo, X. N. Li, H. F. Dai and W. L. Mei, Fitoterapia, 2014, 94, 94–101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26031d |
| This journal is © The Royal Society of Chemistry 2017 |