
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The fluorescence quenching phenomenon in newly synthesized blue fluorescence protein molecule caused by anchoring group substitution: a DFT and TD-DFT study†
Chi Ma,
Yufang Liu *,
Chaozheng Li and
Yonggang Yang
*,
Chaozheng Li and
Yonggang Yang
College of Physics and Material Science, Henan Normal University, Xinxiang 453007, China. E-mail: yf-liu@htu.cn; Fax: +86 373 3329297; Tel: +86 373 3329297
First published on 28th February 2017
Abstract
The newly synthesized blue fluorescence protein (BFP) molecule combined with its derivatives were fully investigated using DFT and TD-DFT methods. The frontier molecular orbitals and NBO charges indicate that the intramolecular benzene ring in the BFP molecule can inhibit the charge redistribution after photo-excitation effectively. The substitution of malonic and malononitrile groups can create the fluorescence quenching phenomenon, which may be caused by their participation in the excited state charge redistribution and act as important electron-donating groups. Also, these substitutions can enhance the configuration stability in the S1 state and hinder the formation of a metastable structure. The malononitrile group substitution can significantly decrease the energy barrier in the S1 state and promote proton transfer.
1. Introduction
Excited state proton transfer (ESPT) chromophores play critical roles in many physical and biological systems, such as fluorescence sensors,1 laser dyes,2 UV filters,3 molecular switches4 and LEDs,5 as they can create some special photo-physical and photo-chemical phenomenon and undergo drastic structural reorganization and charge redistribution after photo-excitation. Since the excited-state proton transfer phenomenon was first observed in the experiment with methyl salicylate by Wille and co-workers,6 a vast number of scientists have directed their attention to clarify the specific excited state proton transfer mechanism.7–11 Several decades ago, people began to realize the fundamental importance of weak interaction in the excited state proton transfer process, especially the hydrogen bond12 with its directional character. Han et al.13 proposed that the intra- and intermolecular hydrogen bond strengthening after photo-excitation can facilitate the proton transfer effectively. As one of the most widely investigated excited state intermolecular proton transfer (ESIPT) biological chromophores, fluorescent proteins14,15 have drawn great attention due to their unique properties and numerous applications in molecular biology. For instance, the green fluorescence protein (GFP)16 chromophores have been used as powerful tools in bio-imaging technology due to their high fluorescence quantum yields. However, the overall performance, such as the brightness and stability, still need to be improved. Compared with the relative comprehensively investigated GFP chromophore, the previous studies about blue fluorescence proteins (BFP)17 chromophores were mainly focused on the spectroscopic properties, relative little is known about the specific reaction dynamic process and mechanism. Recently, Fang and co-workers18 have synthesized a new blue fluorescent protein chromophore analogue 2-BFP ((4Z)-4-[(1H-imidazol-2-yl)methylene]-1-methyl-2-phenyl-1H-imidazol-5-(4H)-one), which plays dual fluorescent emission and larger Stokes shift. In contrast with other fluorescent protein analogues, the nitrogen atoms were acted as both donor and acceptor moieties (N–H⋯N) in the 2-BFP molecule, it also contains greatly enhanced quantum yields and much slower proton-transfer rates. The previous investigation on ESPT structures,19,20 such as hydroxyphenylbenzoxazole (HBO)21 and hydroxyphenylimidazopyridine (HPIP),22 are typical O–H⋯N systems with the intramolecular hydrogen bond forming a six-membered ring. Therefore, the investigation on the newly synthesized 2-BFP chromophore with N–H⋯N hydrogen bond can propose further understanding about the underlying photophysical and photochemical mechanism of natural fluorescence proteins. In the present work, the newly synthesized blue fluorescent protein was set as the basic configuration under analyzing. The truncated BFP chromophore (T-BFP) was set as a comparison to investigate the influence of intramolecular benzene ring. And some classical anchoring groups were introduced to substitute the methyl group in the BFP chromophore to investigate the specific influence on spectral properties and excited state dynamic process. The potential energy curves were scanned with a series of fixed bond lengths and dihedral angles. The electronic spectra, frontier molecular orbitals and NBO charges were also been presented.2. Computational details
In the present work, all the geometry configurations were fully optimized based on DFT23,24 method in ground state and TD-DFT25,26 method in excited state by Gaussian 09 (ref. 27) program package. The popular Becke's three-parameter hybrid exchange functional with Lee–Yang–Parr gradient-corrected correlation (B3LYP)28–31 as well as the standard 6-31++G(d,p) level were performed in both ground and excited states. For the previous investigations20,32–35 have proved the applicability on this kind of configurations. In order to reduce the influence of external environment, all the optimizations were performed in the adiabatic environment. Benzene was chosen as the solvent throughout based on the integral equation formalism variant of the polarizable continuum model (IEFPCM).36–38 The newly synthesized blue fluorescent protein chromophore and its anchoring group substituted derivatives were fully optimized in both S0 and S1 states without any constraint of bonds, angles and dihedral angles. The vertical excitation energies were calculated by TDDFT method with IEFPCM based on the ground state optimized structures. The theoretical calculation provided six low-lying absorbing transitions and all the local minima geometries were confirmed without any imaginary modes in the vibrational analysis. Meanwhile, the potential energy curves were scanned in both S0 and S1 states with a series of fixed N–H bonds and dihedral angles in benzene with adiabatic condition. Harmonic vibrational frequencies in S0 and S1 states were determined by diagonalization of the Hessian.39 The excited-state Hessian was obtained by numerical differentiation of the analytical gradients using central differences and default displacements of 0.02 bohr.403. Results and discussion
3.1 The influence of intramolecular benzene ring
In order to clarify the specific influence of intramolecular benzene ring of BFP chromophore, the truncated BFP (T-BFP) and BFP molecule were fully optimized using the B3LYP function with 6-31++G(d,p) basis set in vacuum (shown in Fig. 1). All the optimized structures mentioned above were confirmed with the minima energy and real frequencies. Some of the notable atoms involved in the analysis were marked and shown in Fig. 1. The bond lengths and angles in the hydrogen bond were presented in Table 1.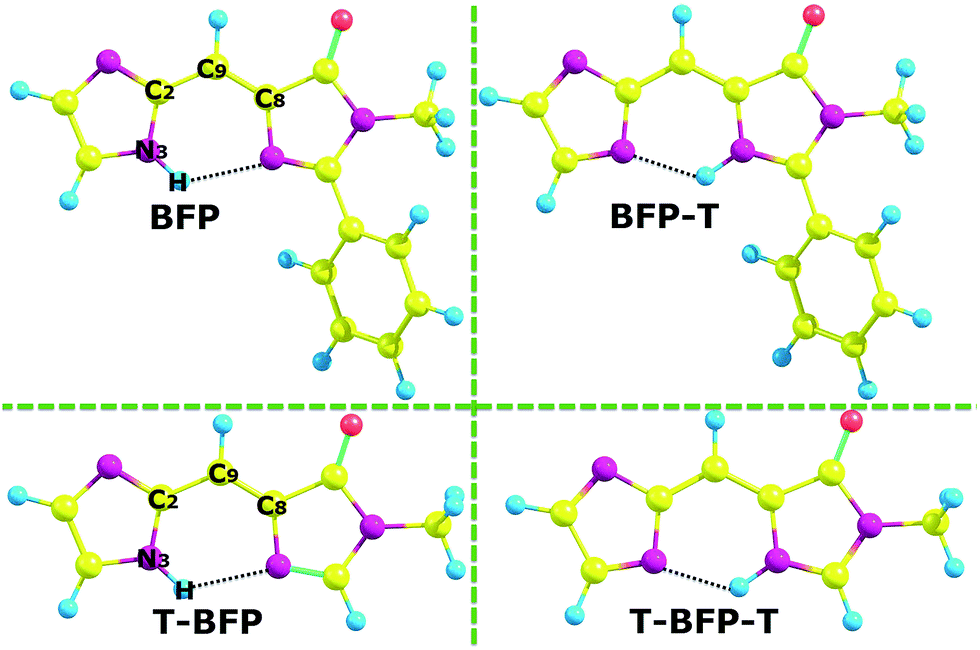 | ||
| Fig. 1 The optimized structures of BFP and T-BFP combined with their proton transferred configurations (-T). | ||
| N–H | H⋯N | δ(N–H⋯N) | ||||
|---|---|---|---|---|---|---|
| Bond length | Wiberg index | Bond length | Wiberg index | |||
| BFP | S0 | 1.017 | 0.7211 | 2.161 | 0.0253 | 123.11 |
| S1 | 1.020 | 0.7128 | 2.083 | 0.0333 | 125.10 | |
| BFP-T | S0 | 1.966 | 0.0601 | 1.031 | 0.6699 | 128.17 |
| S1 | 1.891 | 0.0619 | 1.033 | 0.6710 | 128.94 | |
| T-BFP | S0 | 1.017 | 0.7216 | 2.170 | 0.0243 | 122.90 |
| S1 | 1.019 | 0.7115 | 2.105 | 0.0315 | 124.54 | |
| T-BFP-T | S0 | 1.968 | 0.0590 | 1.030 | 0.6708 | 128.40 |
| S1 | 1.957 | 0.0719 | 1.026 | 0.6566 | 127.59 | |
As is shown in Table 1, the N–H bond in BFP chromophore slightly lengthens from 1.017 Å (S0) to 1.020 Å (S1). The intramolecular hydrogen bond H⋯N shortens from 2.161 Å (S0) to 2.083 Å (S1) combined with the δ(N–H⋯N) enlarge from 123.11° (S0) to 125.10° (S1) after photo-excitation. The similar configuration data and variation tendency can be found in the T-BFP molecule. Therefore, it can be concluded that the intramolecular hydrogen bonds were strengthened after photo-excitation in both BFP and T-BFP molecules. As for the proton transfer isomer, the hydrogen bond in BFP-T changes from 1.966 Å (S0) to 1.891 Å (S1) and that in T-BFP-T only changes from 1.968 Å (S0) to 1.957 Å (S1), which means that the reverse proton transfer is much easier happened in the BFP-T molecule (Table 2).
| Transition | λ (nm) | f | Composition | CI (%) | |
|---|---|---|---|---|---|
| BFP | S0 → S1 | 419 | 0.7287 | H → L | 99% |
| S0 → S2 | 343 | 0.0034 | H−1 → L | 6% | |
| T-BFP | S0 → S1 | 385 | 0.7117 | H → L | 99% |
| S0 → S2 | 342 | 0.0005 | H−1 → L | 5% |
The first excited states optimizations were performed by TD-DFT/B3LYP/6-31++G(d,p) and confirmed with real frequency. The absorption peak for BFP chromophore from ground state to first excite state was located at 419 nm with the large oscillator strength of 0.7287, while the S0 → S2 transition was calculated to be 343 nm with the oscillator strength of 0.0034. And the photo-excitation in T-BFP chromophore was also mainly from S0 to S1 state with large oscillator strength of 0.7117. Therefore, only the HOMO and LUMO orbitals were discussed in detail, which are shown in the ESI.† Both the BFP and T-BFP molecules clearly show π character for the highest occupied molecular orbital (HOMO) and π* character for the lowest unoccupied molecular orbital (LUMO), with a predominant ππ*-type transition from HOMO to LUMO. Except for the extension to the benzene ring, the electron distribution in BFP and T-BFP molecules have no clear difference.
In order to give a more accurate description on the charge redistribution after photo-excitation, the NBO charges was presented in Fig. 2. In T-BFP, the negative charge located on imidazole decreases from 0.082 (S0) to 0.112 (S1) together with that increasement on 4-oxygen-imidazole from −0.719 (S0) to −0.757 (S1). It means that the electron mainly transfer from imidazole to 4-oxygen-imidazole after photo-excitation. In BFP, the electron located on imidazole decrease from 0.073 (S0) to 0.088 (S1), however, that on 4-oxygen-imidazole only increases from −0.511 (S0) to −0.529 (S1). This means that the participation of benzene ring inhibit the electron density increasement on 4-oxygen-imidazole after photo-excitation. The charge redistribution on the atoms involved in the intramolecular hydrogen bond can be considered as a reliable evidence to study the excited state hydrogen bond. In T-BFP, the electron on H1 atom decreases from 0.477 (S0) to 0.480 (S1) together with that increasement on N2 atom from −0.562 (S0) to −0.583 (S1). In BFP, the electron on H1 atom maintain 0.476 in both S0 and S1 states, it only slightly increases from −0.554 (S0) to −0.566 (S1) on N2 atom.
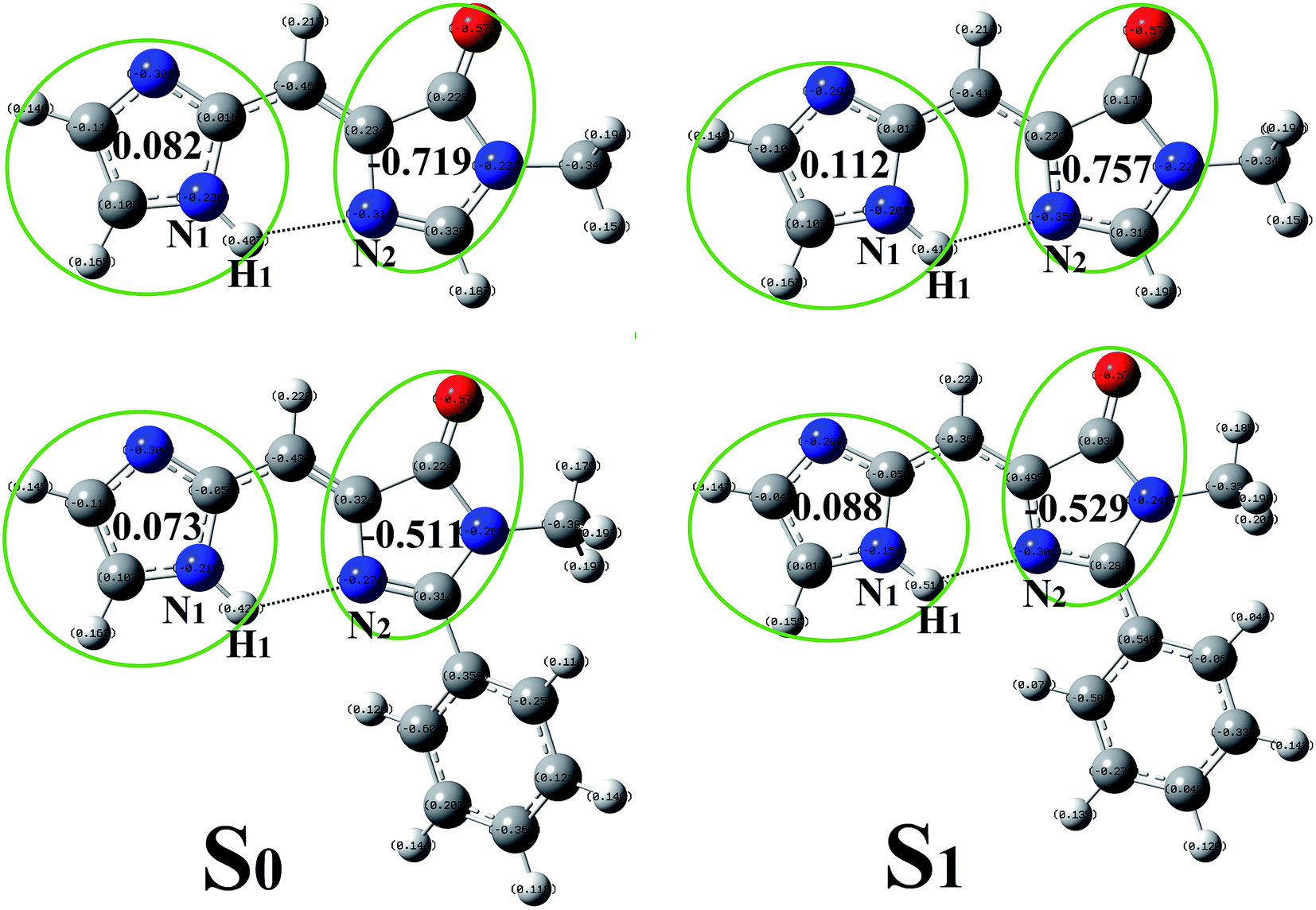 | ||
| Fig. 2 The NBO charges of T-BFP and BFP molecules. | ||
For describing the influence of intramolecular benzene ring on the electronic spectra, the corresponding absorption and emission spectra of T-BFP and BFP molecules were displayed in Fig. 3. The calculated absorption peaks of T-BFP and BFP are located at 385 nm and 420 nm, respectively. And the emission peak of BFP is located at 505 nm, which strongly redshift compared with that of T-BFP at 435 nm. The structural reorganization caused by excited state proton transfer makes the Stokes shift enlarges from 85 nm to 145 nm in BFP, however, it only changes from 50 nm to 79 nm in T-BFP. Therefore, it is reasonable to believe that the benzene ring has an appreciable impact on the spectral property.
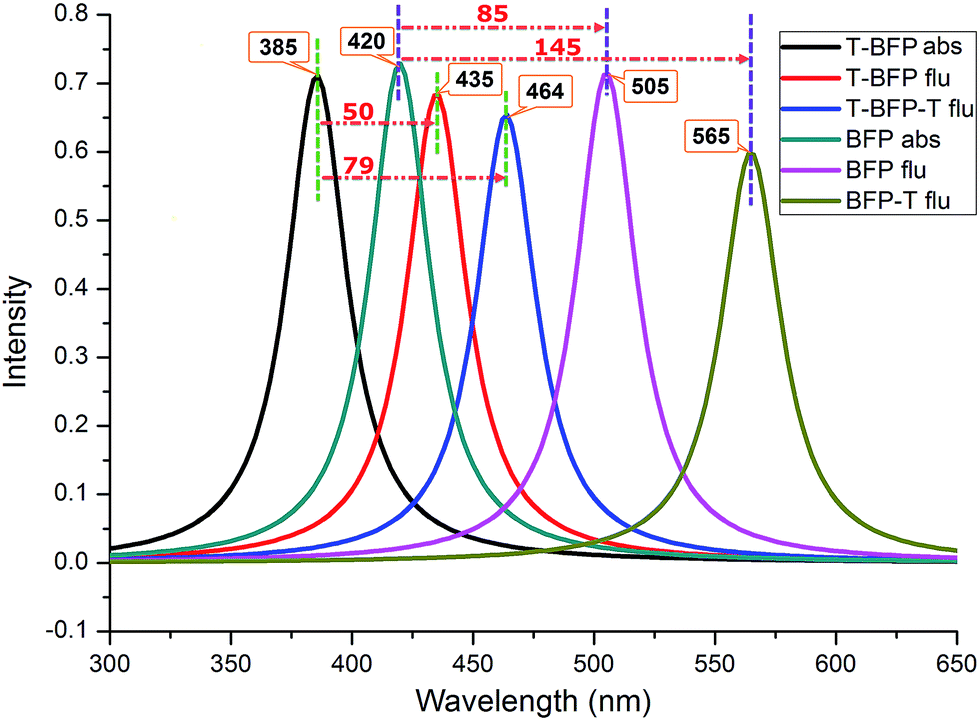 | ||
| Fig. 3 The absorption (abs) and fluorescence (flu) spectra of the molecules. | ||
In order to provide an unambiguous information about ESIPT process, the potential energies were scanned with a series of constrained N–H bond from 1.01 Å to 2.21 Å in step of 0.1 Å in both S0 and S1 states (shown in Fig. 4). In ground state, the potential energy barrier in T-BFP (15.40 kcal mol−1) is a little higher than that in BFP (14.62 kcal mol−1). After photo-excitation, the energy barriers are 10.28 and 9.08 kcal mol−1 in T-BFP and BFP, respectively. In brief, the energy barriers are relative lower in BFP molecule in both ground and first excited state, which means the intramolecular benzene ring may be conducive to proton transfer. It also can be noted that the overall energy distribution of BFP in S1 state is visible lower than that of T-BFP, indicating that the BFP molecule is more stable in first excited state.
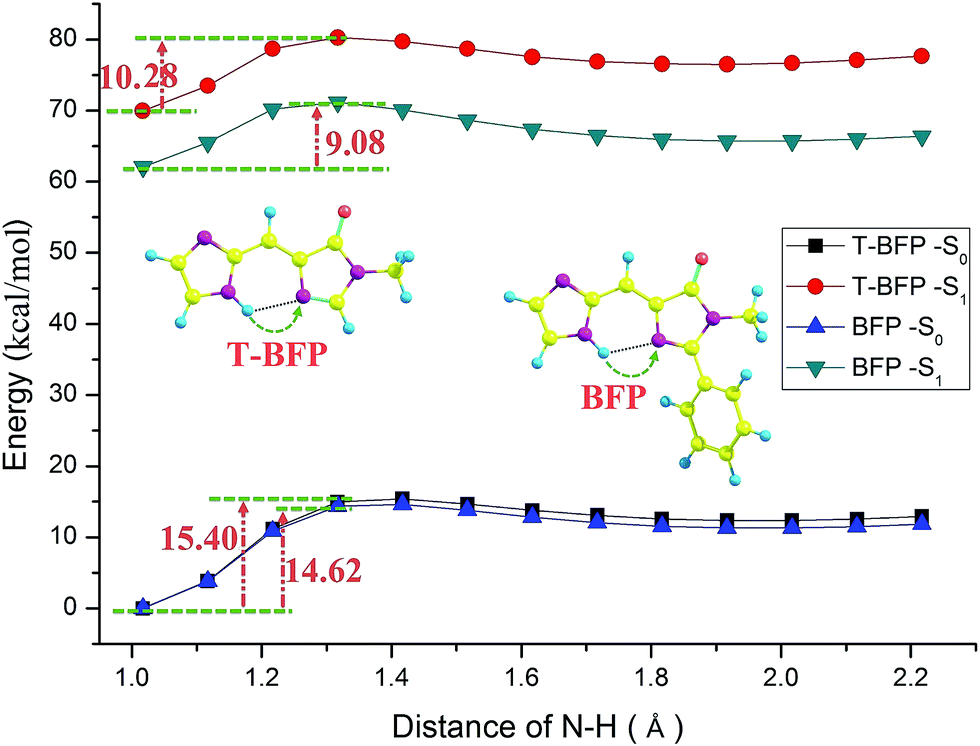 | ||
| Fig. 4 The constructed potential energy curves with constrained N–H bond lengths. | ||
As for the torsional stability, the potential energies were scanned with a series of constrained dihedral angles (N3–C2–C9–C8) from 0° to 180° in step of 10° in both S0 and S1 states (Fig. 5). In ground state, the energy barriers were calculated to be 14.26 and 14.55 kcal mol−1 in BFP and T-BFP, respectively. It is surprising to find that a metastable configuration was formed in both BFP and T-BFP molecules with the imidazole ring twist about 90° after photo-excitation. In BFP, it needs to conquer 12.94 kcal mol−1 to form the metastable configuration. And there is only 8.95 kcal mol−1 separate the original T-BFP molecule and its metastable structure. The intramolecular benzene ring can enhance the configuration stability in first excited state (Fig. 6).
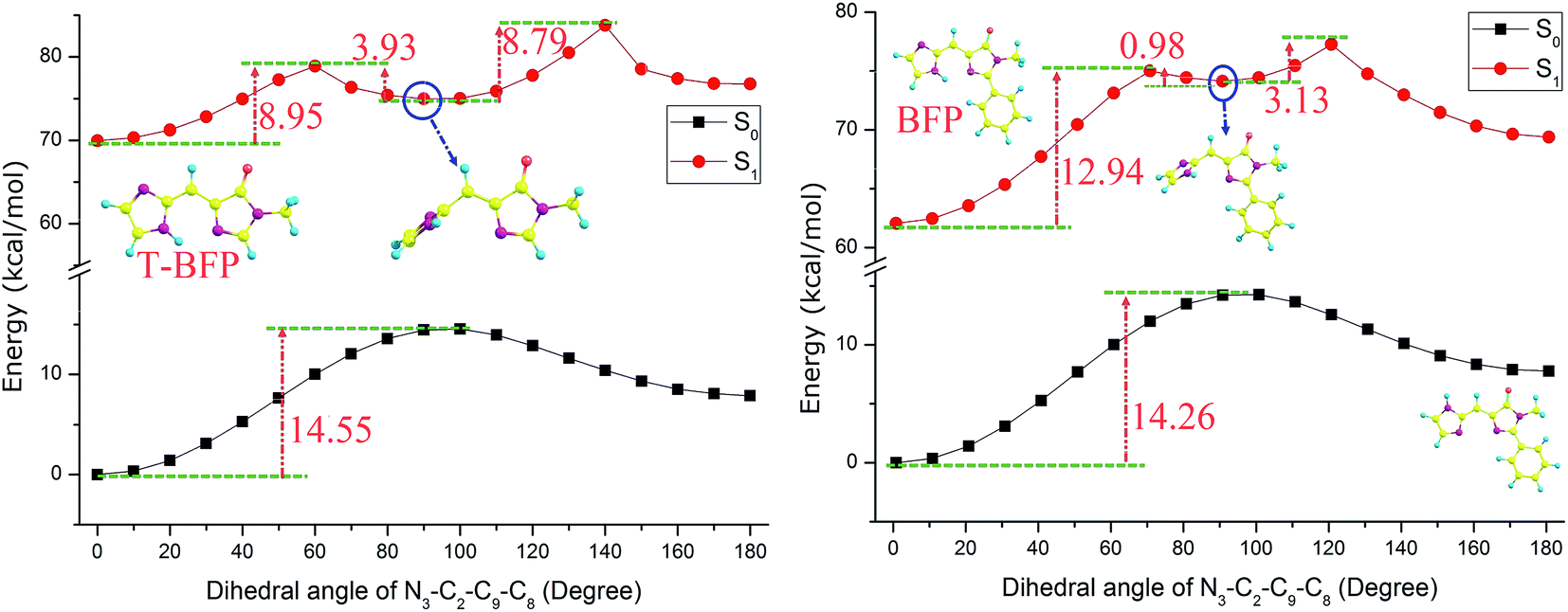 | ||
| Fig. 5 The constructed potential energy curves of T-BFP and BFP molecules with constrained dihedral angles. | ||
 | ||
| Fig. 6 The structures of anchoring groups: (1) carboxylic acid, (2) acrylic acid, (3) malonic acid, (4) cyanoacrylic acid and (5) malononitrile acid. | ||
3.2 The influence of anchoring group substitution
Some classical anchoring groups with weak electron pulling strength (-C, -AC, -MC) and strong electron pulling strength (-CC, -MN) were employed to substitute the methyl group in BFP chromophore. All the BFP derivatives were fully optimized in both S0 and S1 states with Gaussian 09 program. The bond lengths and angles involved in the hydrogen bond only have insignificant difference in both S0 and S1 states after substituted by several anchoring groups. The reasonable deducing can be presented that the anchoring groups only have a trifling influence on the hydrogen bond. As for the whole configuration, the anchoring group become vertical to the trunk in BFP-MC and BFP-MN in S1 state. The significant configuration variations may provide some unique properties in excited state.As is shown Fig. 7, the absorption peaks of these derivatives are spanning from 420 nm to 428 nm, which means the absorption property is not sensitive to anchoring groups. Due to the neglectful oscillator strength with 0.0004 (BFP-MC) and 0.0012 (BFP-MN), only three emission spectra (BFP-AC, -C, -CC) are presented in this figure. The fluorescence quenching phenomenon in the newly synthesized blue fluorescence protein induced by anchoring group substitution was firstly proposed in this work. In order to make clear the immanent cause of the fluorescence quenching phenomenon, the NBO charges were presented to be rigorously studied (Fig. 8).
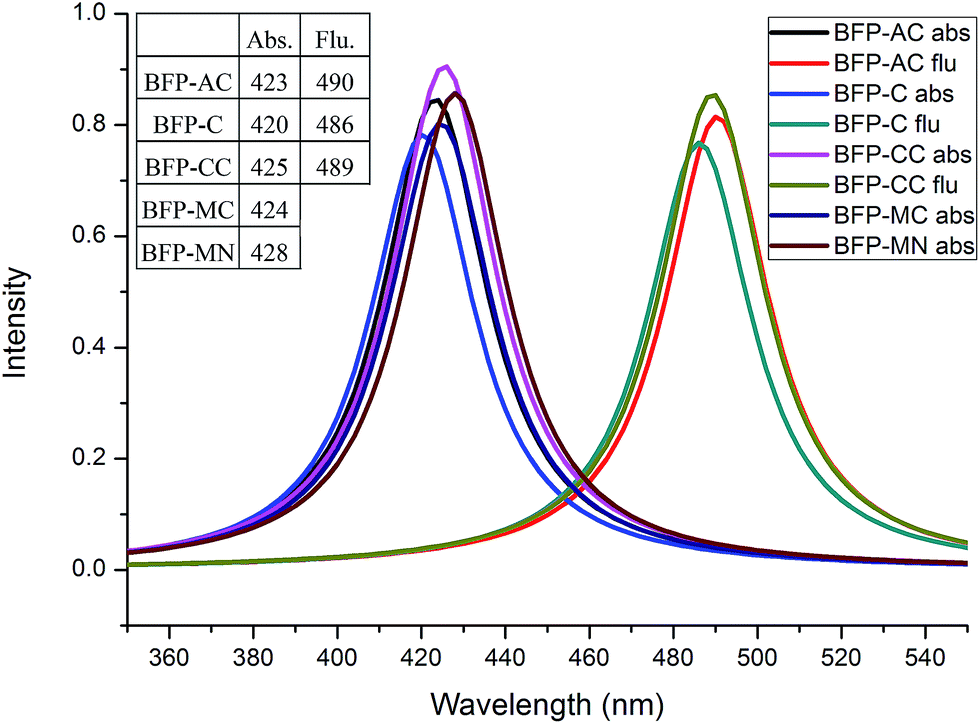 | ||
| Fig. 7 The absorption and emission spectra of BFP derivatives. | ||
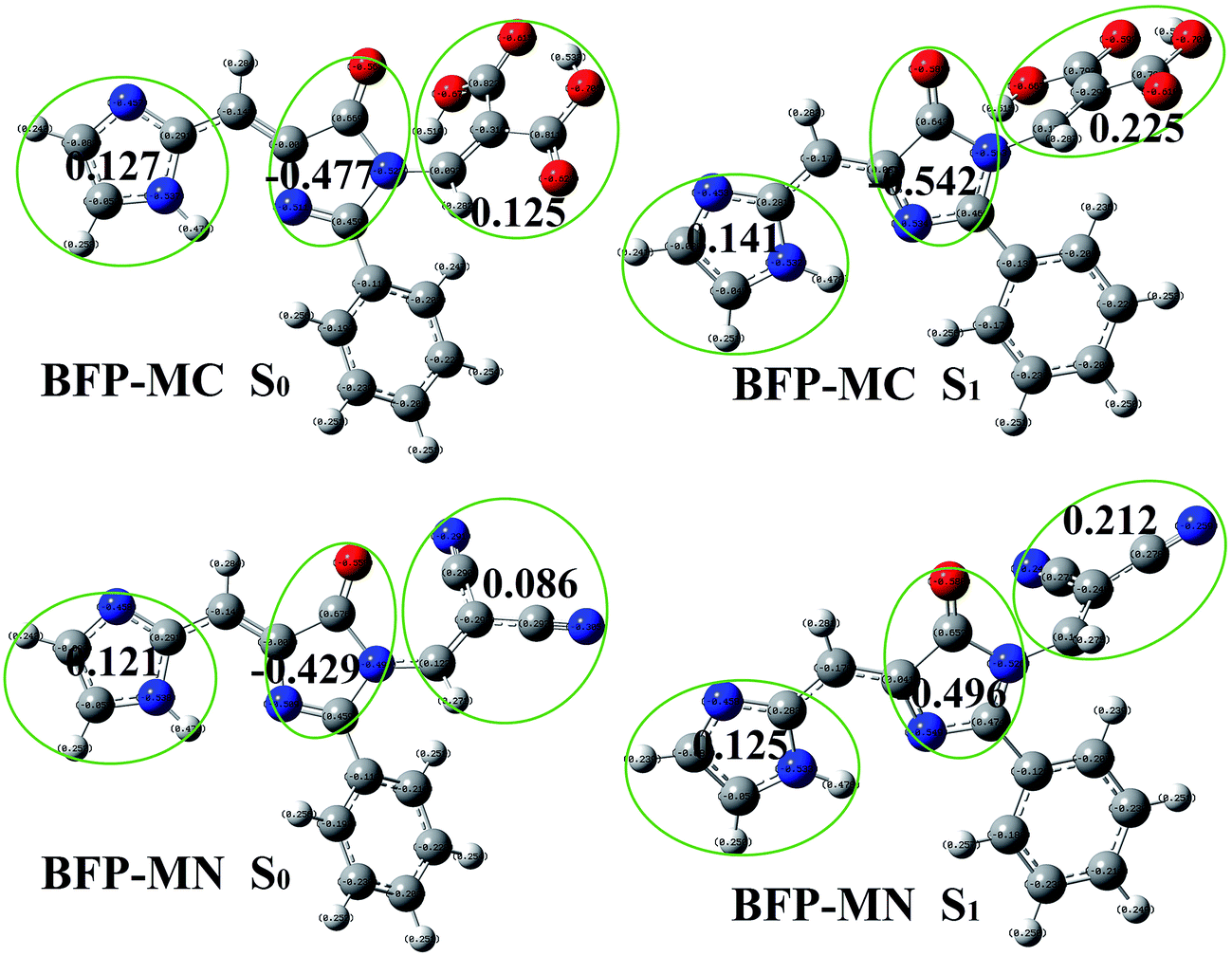 | ||
| Fig. 8 The NBO charges of BFP-MC and BFP-MN in both S0 and S1 states. | ||
As is shown in Fig. S2,† the electron density on N1, H1 and N2 atoms have no significantly change after photoexcitation, which is according with the insignificant change of hydrogen bond discussed above. The substituted anchoring group was set as part c to discuss the excited state charge redistribution. In BFP-C molecule, the electron density on imidazole decreases from 0.136 (S0) to 0.156 (S1) and that on 4-oxygen-imidazole increases from −0.542 (S0) to −0.558 (S1). However, the electron density on carboxylic acid only has a tiny decrease about 0.007 after photo-excitation. The similar changes also be found in BFP-CC and BFP-AC molecules. Therefore, it can be reasonably concluded that the electron was mainly transfer from imidazole to 4-oxygen-imidazole (Fig. 9).
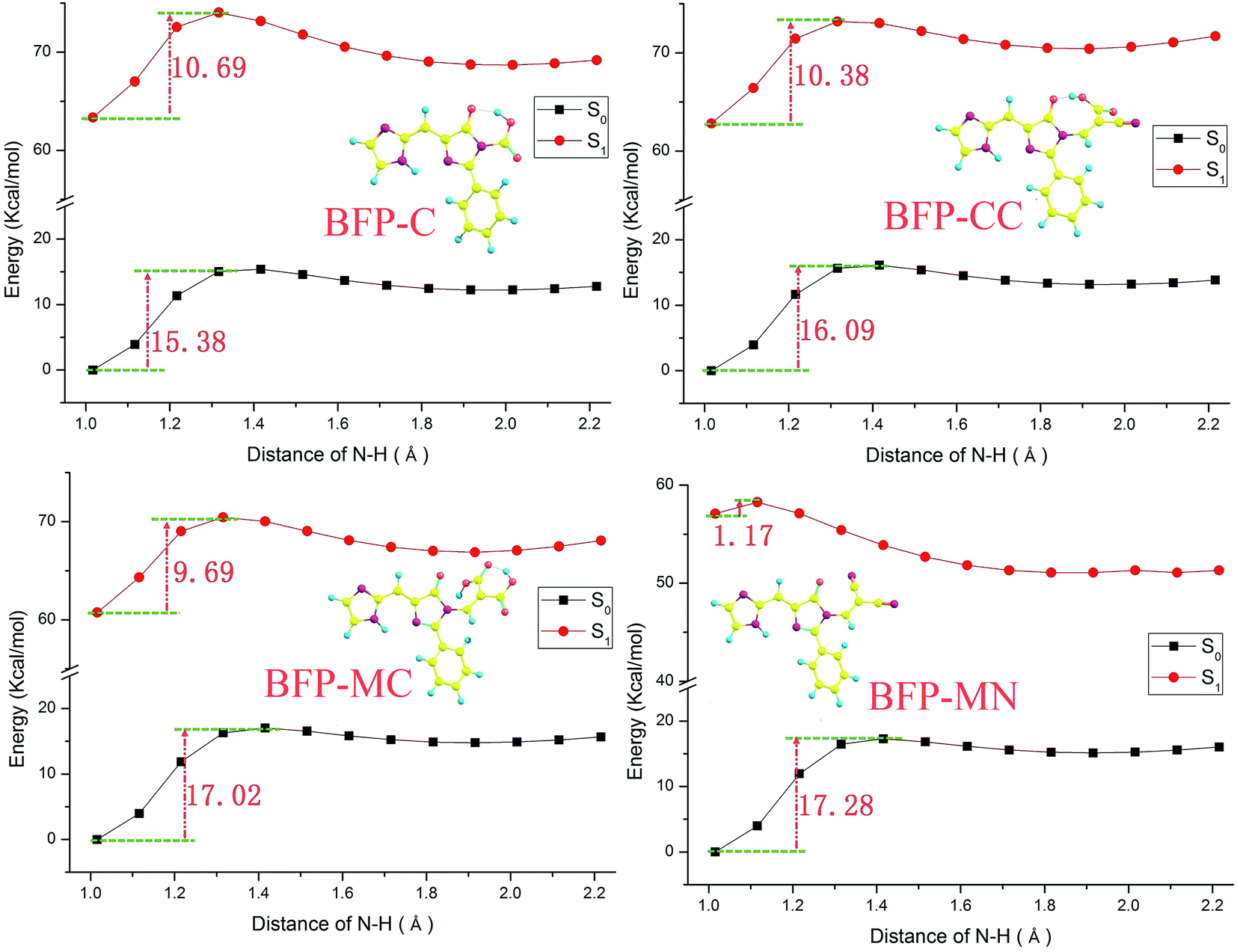 | ||
| Fig. 9 The potential energy curves with a series of fixed N–H bond lengths. | ||
In BFP-MC molecule, the electron density on imidazole decreases 0.014 from 0.127 (S0) to 0.141 (S1) and that on 4-oxygen-imidazole increases 0.065 from −0.477 (S0) to −0.542 (S1). The electron on the malonic acid clearly decreases 0.1 from 0.125 (S0) to 0.225 (S1), which means the anchoring group is considered to be an important electron-donating group. As for BFP-MN molecule, it also found the diminution on the malononitrile acid is 0.126 from 0.086 (S0) to 0.212 (S1). In brief, the unusual fluorescence properties of BFP-MC and BFP-MN can be explained by the participation of anchoring group in charge redistribution.
The potential energy curves were scanned with a series of fixed N–H bonds in both S0 and S1 states to clarify the influence of anchoring groups on intramolecular proton transfer. Some incidental information are listed in the ESI and shown as Fig. S3 and Table S1.† In ground state, the energy barriers of the substitution derivatives have distinct increasement compared with that of the original BFP molecule, which means the anchoring groups can hinder the proton transfer in ground state. The result also reveal that the proton transfer probability of BFP substitution derivatives in ground state follow the sequence BFP < BFP-C < BFP-AC < BFP-CC < BFP-MC < BFP-MN. After photo-excitation, the substitution derivatives, except for BFP-MN molecule, also show proton transfer restrain phenomenon with the compared increasement of potential energy barrier. However, it is clearly shown that the energy barrier of BFP-MN molecule distinctly decreased to 1.17 kcal mol−1 in first excited state. Hence, the substitution of malononitrile group can significantly promote the proton transfer after photo-excitation. Also, a similar sequence of proton transfer probability in first excited state can be presented, which is BFP-MN < BFP < BFP-MC < BFP-C < BFP-AC < BFP-CC.
As is shown in Fig. 10, the BFP substitution derivatives were scanned with a series of fixed dihedral angles in both S0 and S1 states. Compared with the original BFP molecule, the energy barriers of the derivatives in ground state are increased after being substituted by anchoring groups. Among these derivatives, the BFP-CC molecule has the most obvious enhancement, which increase about 1.85 kcal mol−1. It can be deduced that the anchoring group will hinder the configuration twist and maintain the molecule in the same plane. As discussed above, in the S1 state, the imidazole ring can twist about 90° to produce the metastable configuration. It can be noted that the BFP-C, BFP-AC and BFP-CC molecules are much easier transferring to the metastable state, for their potential energies are visibly decreased compared with the original BFP molecule in S1 state, which means their stability are weakened in excited state. However, it is surprised to note that the potential energies of BFP-MC and BFP-MN molecules sharply raised to 23.31 and 23.98 kcal mol−1, respectively. And the metastable configuration are not existed in these molecules. In brief, the participation of anchoring groups enhance the ground state stability. Particularly, the malonic and malononitrile group significantly enhance the configuration stability in first excited state and hinder the formation of metastable configuration in excited state.
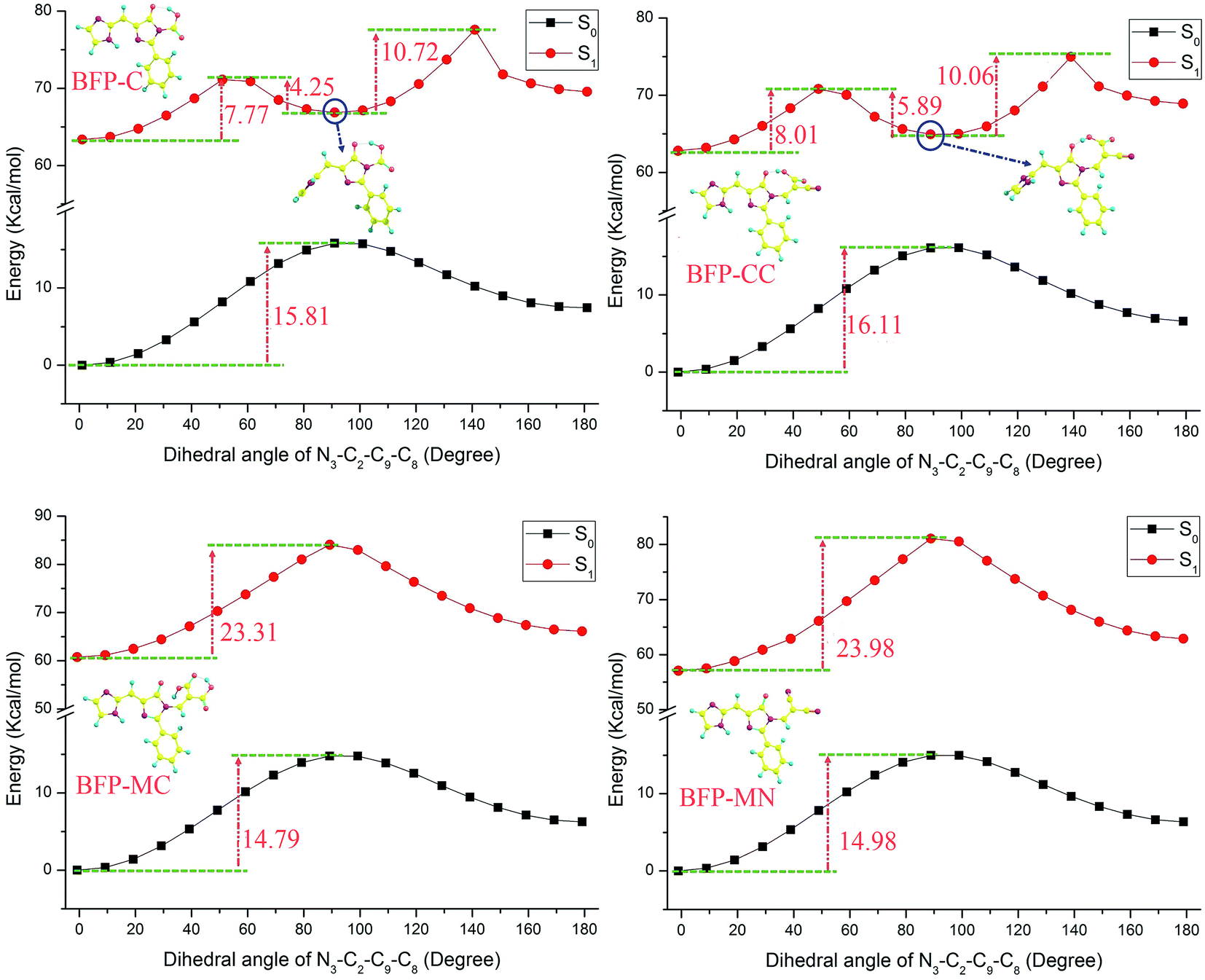 | ||
| Fig. 10 The potential energy curves with a series of fixed dihedral angles. | ||
4. Conclusion
In the present work, the BFP molecule was truncated to investigate the effect of intramolecular benzene ring. The bond and angle analysis indicates that the benzene ring only has a trifling influence on the intramolecular hydrogen bond. Based on the frontier molecular orbitals and NBO charges, it found that the intramolecular benzene ring can inhibits the charge redistribution after photo-excitation. We also found the absorption peak redshift from 385 nm (T-BFP) to 420 nm (BFP) and the Stokes shift enlarge from 79 nm (T-BFP) to 145 nm (BFP), which means the benzene ring can influence the electronic spectra of BFP molecule. The potential energy curves of BFP and T-BFP molecule were scanned with a series of fixed hydrogen bonds and dihedral angles in both ground and first excited states. The clear comparison indicates that the benzene ring can lower the energy barrier in first excited state and promote the intramolecular proton transfer. It also help enhancing the configuration stability and maintain the molecule in the same plane.Some classical anchoring groups, such as carboxylic acid, acrylic acid, malonic acid, cyanoacrylic acid and malononitrile acid, were introduced to substitute the methyl group in the BFP molecule. It is found that these substitution groups only have a trifling influence on the intramolecular hydrogen bond. However, the substitution of malonic and malononitrile groups can create the fluorescence quenching phenomenon. Comparisons of the NBO charges in ground and first excited states show that these two anchoring groups can participate the charge transfer and act as an important electron-donating group. The malononitrile group substitution can lower the energy barrier significantly in first excited state and promote the intramolecular proton transfer. The malonic and malononitrile groups can enhance the stability in first excited state and hinder the formation of metastable configuration.
Acknowledgements
This work is supported by National Natural Science Foundation of China (Grant no. 11274096) and supported by Program for Innovative Research Team (in Science and Technology) in University of Henan Province (Grant no. 13IRTSTHN016), Supported by The High Performance Computing Center of Henan Normal University.References
- Y. Wu, X. Peng, J. Fan, S. Gao, M. Tian, J. Zhao and S. Sun, Fluorescence sensing of anions based on inhibition of excited-state intramolecular proton transfer, J. Org. Chem., 2007, 72, 62–70 CrossRef CAS PubMed.
- J. H. Richardson, L. L. Steinmetz, S. B. Deutscher, W. A. Bookless and W. L. Schmelzinger, Measurement of Fluorescence Lifetimes of Coumarin Laser Dyes with a Mode-Locked Krypton Ion Laser, Z. Naturforsch., A: Phys. Sci., 2014, 33, 1592 Search PubMed.
- D. L. Giokas, A. Salvador and A. Chisvert, UV filters: From sunscreens to human body and the environment, TrAC, Trends Anal. Chem., 2007, 26, 360–374 CrossRef CAS.
- F. M. Raymo, Digital Processing and Communication with Molecular Switches, Adv. Mater., 2002, 14, 401–414 CrossRef CAS.
- F. Steuber, J. Staudigel, M. Stössel, J. Simmerer, A. Winnacker, H. Spreitzer, F. Weissörtel and J. Salbeck, White Light Emission from Organic LEDs Utilizing Spiro Compounds with High-Temperature Stability, Adv. Mater., 2000, 12, 130–133 CrossRef CAS.
- D.-C. E. Wille and P. D. W. Lüttke, 4,4,4′,4′-Tetramethyl-Δ2,2′-bipyrrolidine-3,3′-dione, a Compound having the Basic Chromophore System of Indigo, Angew. Chem., Int. Ed., 1971, 10, 803–804 CrossRef.
- D. Yang, F. Zhao, R. Zheng, Y. Wang and J. Lv, A detailed theoretical investigation on the excited-state intramolecular proton-transfer mechanism of 3-BTHPB chemosensor, Theor. Chem. Acc., 2015, 134(5), 62 CrossRef.
- H. Li, Y. Yang, D. Yang, Y. Liu and J. Sun, TDDFT study on the excited-state hydrogen bonding of N-(2-hydroxyethyl)-1,8-naphthalimide and N-(3-hydroxyethyl)-1,8-naphthalimide in methanol solution, J. Phys. Org. Chem., 2014, 27, 170–176 CrossRef CAS.
- J. Catalán, J. C. D. Valle, F. Fabero and N. A. Garcia, The influence of molecular conformation on the stability of ultraviolet stabilizers toward direct and dye-sensitized photoirradiation: the case of 2-(2′-hydroxy-5′-methyphenyl)benzotriazole (tin p), Photochem. Photobiol., 1995, 61(2), 118–123 CrossRef.
- Y. Kubo, S. Y. Maeda, S. Tokita and M. Kubo, Colorimetric chiral recognition by a molecular sensor, Nature, 1996, 382, 522–524 CrossRef CAS.
- T. He and Y. Songjie, Recent progresses on diarylethene based photochromic switches, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- H. Li, Y. Liu, Y. Yang, D. Yang and J. Sun, Excited-state intramolecular hydrogen bonding of compounds based on 2-(2-hydroxyphenyl)-1,3-benzoxazole in solution: A TDDFT study, Spectrochim. Acta, Part A, 2014, 133, 818–824 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, Site-specific solvation of the photoexcited protochlorophyllide a in methanol: formation of the hydrogen-bonded intermediate state induced by hydrogen-bond strengthening, Biophys. J., 2008, 94, 38–46 CrossRef CAS PubMed.
- Y. Mochizuki, H. Nakanishi, Y. Kodera, S. Ito, Y. Yamamura, T. Kato, K. Hibi, S. Akiyama, A. Nakao and M. Tatematsu, TNF-alpha promotes progression of peritoneal metastasis as demonstrated using a green fluorescence protein (GFP)-tagged human gastric cancer cell line, Clin. Exp. Metastasis, 2004, 21, 39–47 CrossRef CAS PubMed.
- W. Y. Huang, J. Aramburu, P. S. Douglas and S. Izumo, Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy, Nat. Med., 2000, 6, 482–483 CrossRef CAS PubMed.
- A. Tomura, K. Goto, H. Morinaga, M. Nomura, T. Okabe, T. Yanase, R. Takayanagi and H. Nawata, The subnuclear three-dimensional image analysis of androgen receptor fused to green fluorescence protein, J. Biol. Chem., 2001, 276, 28395–28401 CrossRef CAS PubMed.
- A. E. Jablonski, R. B. Vegh, J. C. Hsiang, B. Bommarius, Y. C. Chen, K. M. Solntsev, A. S. Bommarius, L. M. Tolbert and R. M. Dickson, Optically modulatable blue fluorescent proteins, J. Am. Chem. Soc., 2013, 135, 16410–16417 CrossRef CAS PubMed.
- X. Fang, Y. Wang, D. Wang, G. Zhao, W. Zhang, A. Ren, H. Wang, J. Xu, B. R. Gao and W. Yang, Synthesized Blue Fluorescent Protein Analogue with Tunable Colors from Excited-State Intramolecular Proton Transfer through an N–H⋯N Hydrogen Bond, J. Phys. Chem. Lett., 2014, 5, 92–98 CrossRef CAS PubMed.
- C. Ma, Y. Yang, C. Li and Y. Liu, TD-DFT Study of the Double Excited-State Intramolecular Proton Transfer Mechanism of 1,3-Bis(2-pyridylimino)-4,7-dihydroxyisoindole, J. Phys. Chem. A, 2015, 119, 12686–12692 CrossRef CAS PubMed.
- J. Zhao, H. Yao, J. Liu and M. R. Hoffmann, New excited-state proton transfer mechanisms for 1,8-dihydroxydibenzo[a,h]phenazine, J. Phys. Chem. A, 2015, 119, 681–688 CrossRef CAS PubMed.
- H. Roohi, F. Hejazi, N. Mohtamedifar and M. Jahantab, Excited state intramolecular proton transfer (ESIPT) in 2-(2′-hydroxyphenyl)benzoxazole and its naphthalene-fused analogs: A TD-DFT quantum chemical study, Spectrochim. Acta, Part A, 2014, 118, 228–238 CrossRef CAS PubMed.
- A. Brenlla, M. Veiga, M. C. Rodríguez, M. Mosquera and F. Rodríguez-Prieto, Fluorescence of methylated derivatives of hydroxyphenylimidazopyridine. Resolution of strongly overlapping spectra and a new ESIPT dye showing very efficient radiationless deactivation, Photochem. Photobiol. Sci., 2011, 10, 1622–1636 CAS.
- R. G. Parr, Density functional theory, Annu. Rev. Phys. Chem., 1983, 34, 631–656 CrossRef CAS.
- E. K. Gross and R. M. Dreizler, Density functional theory, Springer Science & Business Media, 2013 Search PubMed.
- E. Runge and E. K. Gross, Density-functional theory for time-dependent systems, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- M. Marques, Time-dependent density functional theory, Springer Science & Business Media, 2006 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hrat-chian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dan-nenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowskiand and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
- J. Paier, M. Marsman and G. Kresse, Why does the B3LYP hybrid functional fail for metals?, J. Chem. Phys., 2007, 127(2), 024103 CrossRef PubMed.
- J. E. Del Bene, W. B. Person and K. Szczepaniak, Properties of Hydrogen-Bonded Complexes Obtained from the B3LYP Functional with 6-31G(d,p) and 6-31+G(d,p) Basis Sets: Comparison with MP2/6-31+G(d,p) Results and Experimental Data, J. Phys. Chem., 1995, 99(27), 10705–10707 CrossRef CAS.
- J. Tirado-Rives and W. L. Jorgensen, Performance of B3LYP density functional methods for a large set of organic molecules, J. Chem. Theory Comput., 2008, 4(2), 297–306 CrossRef CAS PubMed.
- L. A. Curtiss, K. Raghavachari and P. C. Redfern, et al., Investigation of the use of B3LYP zero-point energies and geometries in the calculation of enthalpies of formation, Chem. Phys. Lett., 1997, 270(5–6), 419–426 CrossRef CAS.
- R. Rondao, J. Seixas de Melo, M. Melo and A. Parola, Excited-State Isomerization of Leuco Indigo, J. Phys. Chem. A, 2012, 116, 2826–2832 CrossRef CAS PubMed.
- S. Chai, G. J. Zhao, P. Song, S. Q. Yang, J. Y. Liu and K. L. Han, Reconsideration of the excited-state double proton transfer (ESDPT) in 2-aminopyridine/acid systems: role of the intermolecular hydrogen bonding in excited states, Phys. Chem. Chem. Phys., 2009, 11, 4385–4390 RSC.
- J. Zhao, J. Chen, Y. Cui, J. Wang, L. Xia, Y. Dai, P. Song and F. Ma, A questionable excited-state double-proton transfer mechanism for 3-hydroxyisoquinoline, Phys. Chem. Chem. Phys., 2015, 17, 1142–1150 RSC.
- Y. Shi and H. Wei, A theoretical study about three organic semiconductor based on oligothiophenes, Journal of Atomic and Molecular Sciences, 2015, 6, 161–171 CrossRef.
- B. Mennucci, E. Cancès and J. Tomasi, Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Theoretical Bases, Computational Implementation, and Numerical Applications, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
Theoretical Bases, Computational Implementation, and Numerical Applications, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS. - E. Cances, B. Mennucci and J. Tomasi, New Integral Equation Formalism For The Polarizable Continuum Model: Theoretical Background And Applications To Isotropic And Anisotropic Dielectrics, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- R. Cammi and J. Tomasi, Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: Iterative versus matrix-inversion procedures and the renormalization of the apparent charges, J. Comput. Chem., 1995, 16, 1449–1458 CrossRef CAS.
- O. Treutler and R. Ahlrichs, Efficient molecular numerical integration schemes, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS.
- F. Furche and R. Ahlrichs, Adiabatic time-dependent density functional methods for excited state properties, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26470k |
| This journal is © The Royal Society of Chemistry 2017 |