
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Replica exchange molecular dynamics study of the amyloid beta (11–40) trimer penetrating a membrane†
Son Tung Ngo‡
*ab,
Huynh Minh Hung‡c,
Khoa Nhat Trand and
Minh Tho Nguyen*abc
aComputational Chemistry Research Group, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: ngosontung@tdt.edu.vn
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: minh.nguyen@kuleuven.be
dDepartment of Biological Sciences, University of Maryland Baltimore County, 21250 Baltimore, Maryland, USA
First published on 23rd January 2017
Abstract
Alzheimer's disease is characterized by the interaction of neurotoxic Aβ oligomers with cellular membranes, which disturbs ion homeostasis. To determine the putative structures of the transmembrane 3Aβ11–40 oligomer, temperature replica exchange molecular dynamics (REMD) simulations with an explicit solvent have been employed to monitor the structural changes when interaction of the oligomer with the membrane DPPC lipid bilayer is induced. Although the initial conformation of the 3Aβ11–40 transmembrane was fibril-like, the obtained results are in good agreement with previous experiments, in which the β-structure of the Aβ oligomer represents ∼40% of the structure in the average of all considered snapshots. The statistical coil structure, which is located near and interacts with the membrane headgroups, amounts to almost 60% of the structure. The transmembrane Aβ oligomer helix structure basically disappears during the REMD simulations. Instead of the Asp23–Lys28 salt bridge, the polar contact between Asp23 and Asn27 has been found to be a factor stabilizing the structure of the Aβ oligomer. Although numerous polar contacts between lipid headgroups and the peptide have been found, free energy perturbation calculations indicated that van der Waals interactions are the key factor determining the binding between the Aβ trimer and the membrane. It may be argued that the Aβ11–40 trimer can be easily inserted into the membrane because the binding free energy between the trimer and the membrane reaches −70 kcal mol−1. The collision cross section of the optimized structures of 1341 ± 23 Å2 agrees well with the experimental values for the solvated Aβ trimer.
Introduction
Alzheimer's disease (AD) is known to be one of the most common neurodegenerative disorders1–5 and is frequently observed among elderly people. In fact, about a third of seniors are affected by AD or other dementias; as yet, there is no effective treatment for AD.1–6 Different mechanisms of AD have been proposed, including the cholinergic, tau and amyloid hypotheses.7–9Numerous previous studies have indicated that aggregation of amyloid beta (Aβ) peptides in the extracellular region of brain tissue is the main cause of AD.4,10–12 Furthermore, Aβ oligomers have recently been found to be more neurotoxic than the fibril forms.4,13,14 However, the mechanism by which Aβ oligomers damage neurons remains uncertain. Scientists currently suggest that these oligomers insert into lipid bilayers and establish ion channel-like structures.15 Consequently, Ca2+ dication homeostasis is perturbed, ultimately leading to cytotoxicity.16–18
In general, although Aβ peptides tend to favor interactions with the phosphate headgroups of zwitterionic lipids through electrostatic interactions,19–21 the interaction of Aβ with positively charged lipids is equivalent to that with negatively charged lipids.21,22 In addition, the oligomers appear to insert into the membrane more easily than their corresponding monomers.19 Several experiments investigating the effects of a membrane on Aβ peptides were reported under various conditions. The structure of an Aβ peptide is transformed into a helix–kink–helix structure when its C-terminus incompletely penetrates the membrane.23–25 It has been established that acidic phospholipids motivate Aβ peptides to adopt β-form coil structures.24,26 The kinetics and thermodynamics of the transformation of the random coil structure into the β-structure on the surface of the anionic membrane were studied.27 These experimental results were analyzed and supported by numerous subsequent theoretical studies.28–33
Due to the fact that the Aβ trimer is one of the most toxic forms of low weight oligomers,34 current studies are focused on screening potential inhibitors to prevent the trimer from forming.35 However, the equilibrated Aβ trimers exist in mixed environments consisting of monomers, dimers, higher order oligomers and mature fibrils; thus, experimental studies about them are few.36,37 Thus, it is difficult to design effective Aβ trimer inhibitors to treat AD. Moreover, the neurotoxicity and aggregate conformations of Aβ peptides rely upon the sequences and lengths of the peptides.38 Although several previous investigations indicated that the hydrophilic region of the Aβ peptide N-terminal alters peptide deposition,39–41 the effects of the hydrophobic core on the deposition of Aβ peptides are greater than the effects of the hydrophilic core. Consequently, previous studies have mainly focused on evaluating the structures of truncated Aβ peptides.42–44
In this context, a theoretical study of the transmembrane Aβ trimer and its interactions is thus of great interest. We thus determined to investigate the structures of the Aβ40 oligomers when they fully penetrate lipid bilayers. For this purpose, we considered the dipalmitoyl phosphatidylcholine (DPPC) lipid bilayer and inserted the 3Aβ11–40 oligomer into it. Moreover, it is likely impossible to study the Aβ trimer starting from the random coil form due to the high CPU time demand,45 which is in any case beyond our actual computational resources. Therefore, we used a fibril-like structure as the initial conformation of the 3Aβ11–40 transmembrane. To study the resulting solvated transmembrane Aβ peptide system, we used replica exchange molecular dynamics (REMD) simulations with an explicit solvent. This method allows the structural changes in the transmembrane 3Aβ11–40 peptide to be recorded during the simulation time and allows the mutual interactions between the membrane and the peptide to be probed.
Thus, the most stable structure of the transmembrane 3Aβ11–40 peptide corresponds to the energy global minimum of this peptide; this is explored through free energy landscape analyses. In accord with previous experiments and computations, the secondary structure of the oligomer has been predicted using the Define Secondary Structure of Proteins (DSSP) tool; α-content was lacking during our simulations, while the β-content and statistical coil structures comprised about 40% and 60% of the structure, respectively.24,26–33,46 The coil domains are located within the membrane surface and strongly interact with the phosphorus atoms of the lipid headgroups.24,26 The Asp23–Asn27 salt bridge was found to play an important role in stabilizing the structure of the Aβ peptide, instead of the Asp23–Lys28 salt bridge, as previously reported.30 Moreover, the binding free energy of the trimer to the membrane was determined using free energy perturbation calculations. The size of the trimer was determined through collision cross section prediction. The obtained results may enhance the search for an AD therapeutic agent and establish references for transmembrane mutant Aβ trimer studies.
Materials and methods
Replica exchange molecular dynamics (REMD) simulations
The 3Aβ11–40 oligomer44 was presented through the united atom GROMOS 53a6 force field.47 In this work, this Aβ peptide fully penetrated the membrane DPPC lipid bilayer.48 The transmembrane system was placed in a periodic boundary conditions box and was then solvated using the simple point charge water model.49 Three Na+ ions were added to maintain neutral conditions in the system. The crystal structure of the initial conformation employed is shown in Fig. 1.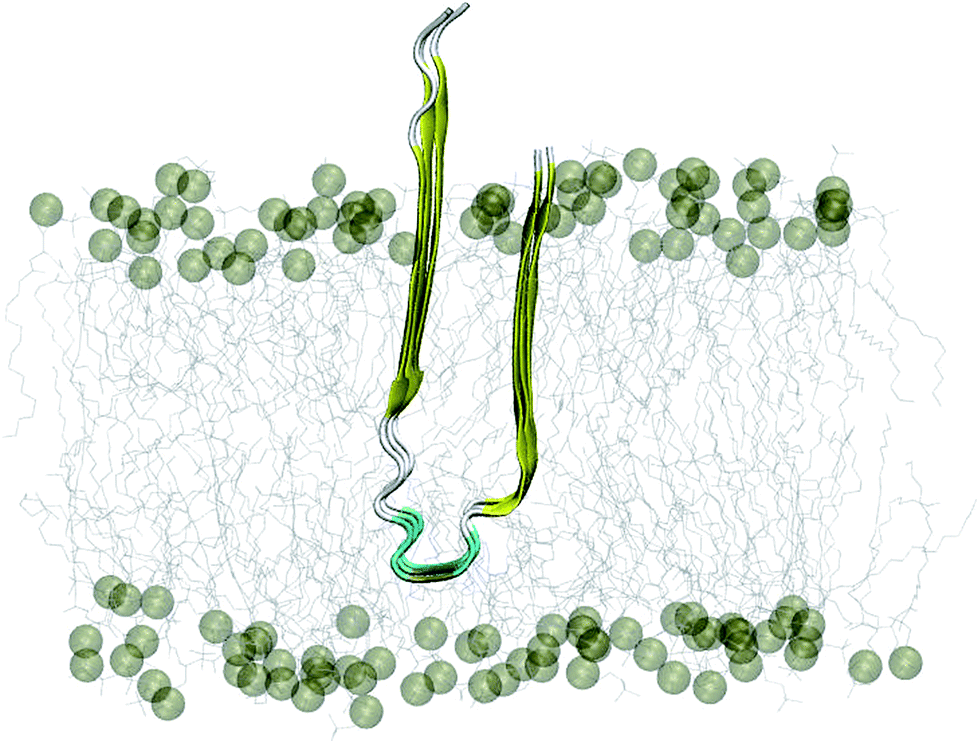 | ||
| Fig. 1 Starting conformation of the truncated transmembrane 3Aβ11–40 peptide inserted into the membrane DPPC lipid bilayer. Water and ion molecules are hidden in this figure. | ||
The solvated transmembrane oligomer system was used as the initial conformation of the computations using GROMACS version 5.0.7.50 The starting structure included the Aβ oligomer, 125 DPPC molecules, 3923 water molecules, and 3 Na+ ions. The steepest descent, conjugate gradient, and low-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) methods51 were employed to minimize the solvated transmembrane oligomer. The system reached an energy minimum when the maximum force recorded was smaller than 10−6 kJ (mol−1 nm). Consequently, the system was simulated over a time of 500 ps in the canonical (NVT) ensemble at 324 K with all atoms restrained using a weak harmonic force. The last snapshot of the NVT simulation was then used as the initial structure of the 500 ps isothermal–isobaric (NPT) simulation at 324 K.
Subsequently, temperature REMD simulations were performed in which the input conformation was the last snapshot of the NPT simulations. The number of replicas was 48, ranging from 290 to 417 K (details are shown in the (ESI†) file). The temperatures of these replicas were determined using the temperature generator for the REMD simulations webserver.52 The acceptance ratio was chosen to be sufficiently larger than 20%. The exchanges between neighboring replicas were checked every 1 ps, which is sufficient to compare the coupling times of the heat bath. There were 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 replica exchange cycles during the computations. The data were collected every 10 ps.
000 replica exchange cycles during the computations. The data were collected every 10 ps.
The MD simulations were integrated using the accurate leap-frog stochastic dynamics integrator.53 A relaxation time of 0.1 ps was chosen; the system pressure was 1.0 atm and was controlled by the Parrinello–Rahman method.54 All bonds were constrained through the LINCS55 with an order of 4. In accord with previous computational studies on the folding/misfolding of Aβ peptides,39,56 the time step was selected as 2 fs. The non-bonded interaction pair list was renewed every 10 fs, with a cutoff of 1.0 nm. Electrostatic interactions were determined utilizing the fast smooth Particle-Mesh Ewald electrostatics method, with a cutoff of 1.0 nm.57 The Lennard–Jones interactions were computed with a cutoff equal to the cutoff of non-bonded interactions.
Free energy perturbation (FEP) method
The interactions between the protein and the membrane were determined through a free energy perturbation method.58 In this method, the obtained difference of free energy between two bound and unbound states is calculated through MD simulations; during these computations, the system changes from the bound Hamiltonian to the unbound Hamiltonian using λ intervals. The determination is performed when the system exists in the equilibrium state. The bound and unbound states correspond with the coupling parameters λ = 0 and 1, respectively. The Bennet's acceptance ratio (BAR) method59 was used to determine the change of free energy ΔGλi⇒λi+1 between neighbouring states, λi and λi+1. Consequently, the difference of free energy between the bound and unbound states is a summation over these values (eqn (1)):
 | (1) |
In the present work, we changed the coupling parameter λ from 0 to 1 to annihilate the trimer from the transmembrane and solvated systems by altering the non-bonded interactions, as shown in the thermodynamics diagram in Fig. 2. In particular, we used a total of 15 values of λ to reduce the non-bonded interactions from the full-interaction state to the non-interaction state over 15 independent MD simulations with lengths of 5 ns each. The independent MD simulations had the same initial crystal structures and starting velocities; however, they had different coupling parameters λ. In the latter, the Coulomb interaction was decreased through 6 values of the coupling parameter λ, including 0.00, 0.35, 0.55, 0.73, 0.88, and 1.00. Additionally, the van der Waals interactions were modified using 10 values of λ: 0.00, 0.10, 0.20, 0.25, 0.30, 0.40, 0.55, 0.70, 0.85, and 1.00. During these processes, the trimer was annihilated two times in different systems; thus, this method is called the double-annihilation binding free energy method (cf. Fig. 2).60–63 Overall, the binding free energy of the trimer to the membrane lipid bilayers was estimated through expression (2):
| ΔGbind = ΔG1 − ΔG2 | (2) |
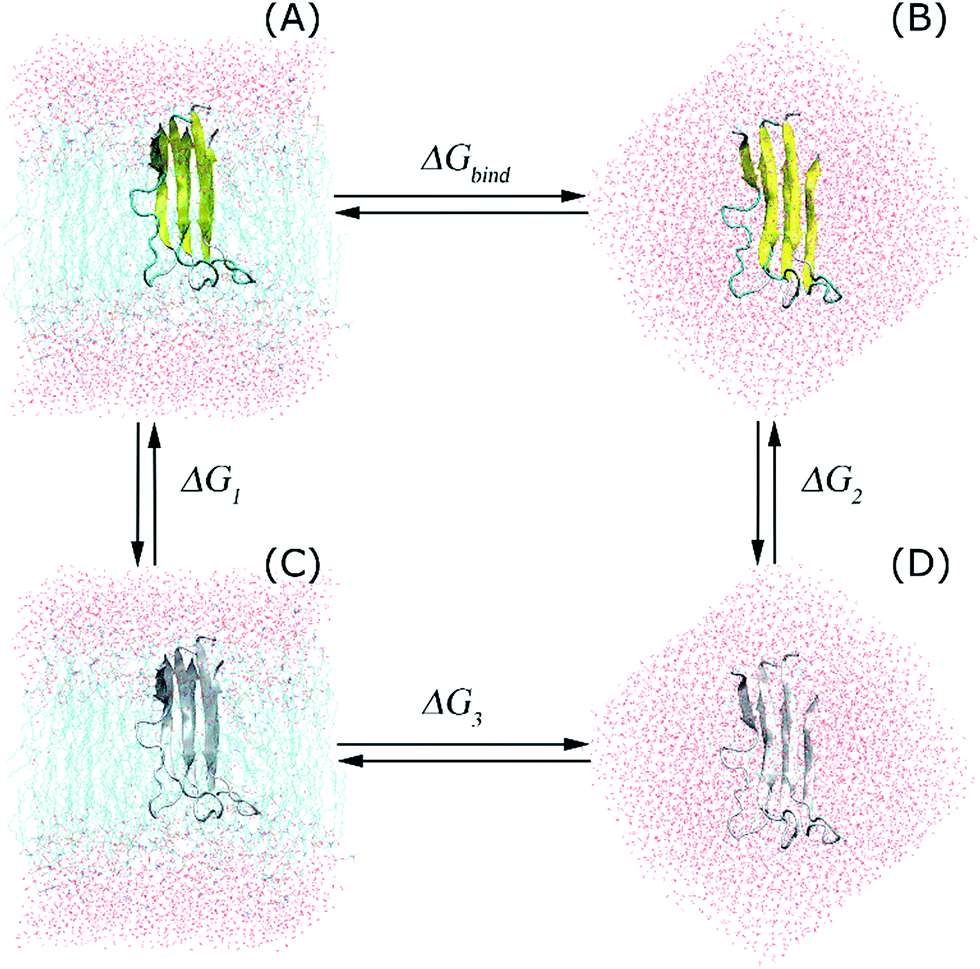 | ||
| Fig. 2 Thermodynamics diagram of the double-annihilation binding free energy method that was applied to determine the binding free energy of the trimer to the membrane DPPC lipid bilayer. In this diagram, (A) represents the full-interaction state of the Aβ peptide with the solvated membrane system. (B) presents the full-interaction state of the protein with the solution. (C) is a dummy protein penetrating the membrane, and (D) is the dummy protein in solution. The dummy protein represents the protein without any interaction with surrounding molecules. | ||
Secondary structures
The secondary structures of the transmembrane 3Aβ11–40 peptides were estimated using DSSP.64,65Free energy landscape (FEL)
The “gmx sham”66,67 is a tool in GROMACS that can be employed to determine the FEL of the trimer with two reaction coordinates, including the radius of gyration (Rg) and the root mean square deviation (RMSD). The clustering method68 was employed to find putative conformations of a protein which remained at a global minimum with a tolerance of 3.5 nm Cα RMSD.Collision cross section (CCS)
CCS is a high impact parameter that can be determined by the ion mobility projection approximation calculation tool (IMPACT).69Contact
The intermolecular contact between the heavy atoms of the residues and the phosphorus atoms of lipid headgroups was investigated through evaluation of the minimum distance of the corresponding atoms with a cutoff of 0.45 nm. Additionally, the distances between the sidechains of the neighbouring chains of the truncated peptide were considered. Thus, the sidechain contacts were counted when this distance was smaller than 0.45 nm.The order of the lipid bilayers
The lipid order parameters measure the oriented mobility of the carbon (C) and deuteron (D) bonds through the parameter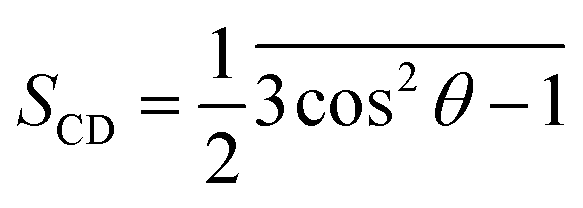 , where θ is the angle between the molecular axis given by the Ci−1 − Ci+1 vector and the bilayer normal, which is investigated over computational time. The results were averaged over the membrane during the simulation times.
, where θ is the angle between the molecular axis given by the Ci−1 − Ci+1 vector and the bilayer normal, which is investigated over computational time. The results were averaged over the membrane during the simulation times.
Results and discussion
Temperature REMD simulation of the transmembrane 3Aβ11–40 peptide
At the atomic computational scale, REMD simulation has become an essential computational method for treating biomolecular systems in general and Aβ aggregation problems in particular; it has been proved to be one of the most powerful enhanced sampling methods.28,70–72 This method has been validated for investigation of the structures of Aβ peptides in many previous studies.45,73–76 In contrast, this approach may become less productive compared to normal MD simulations if the maximum temperature chosen is too high,77 especially if the membrane becomes unstable at high temperature.78 This may lead to unstable Aβ11–40 trimer structures during simulations; however, here, the maximum temperature chosen of 417 K is not too high. However, in this case, although the β-structure of the Aβ trimer was never broken, it fluctuated within a large range, from ∼15% to ∼55% (Fig. 4C). This may be caused by the use of the GROMOS force field, which is known to favor formation of the β-structure.79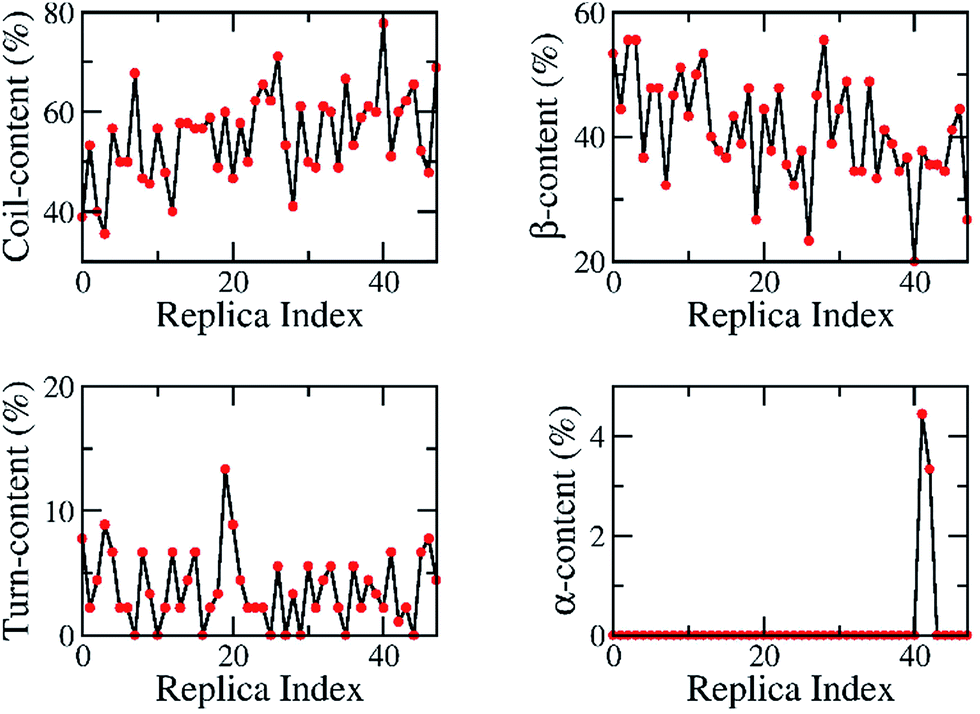 | ||
| Fig. 3 The secondary structures of 48 replicas of the truncated transmembrane Aβ11–40 trimer over 200 ns of REMD simulations, predicted using DSSP tools. The individual metrics are diffused over the entire wide range, suggesting that the computations did not focus on any special conformation. | ||
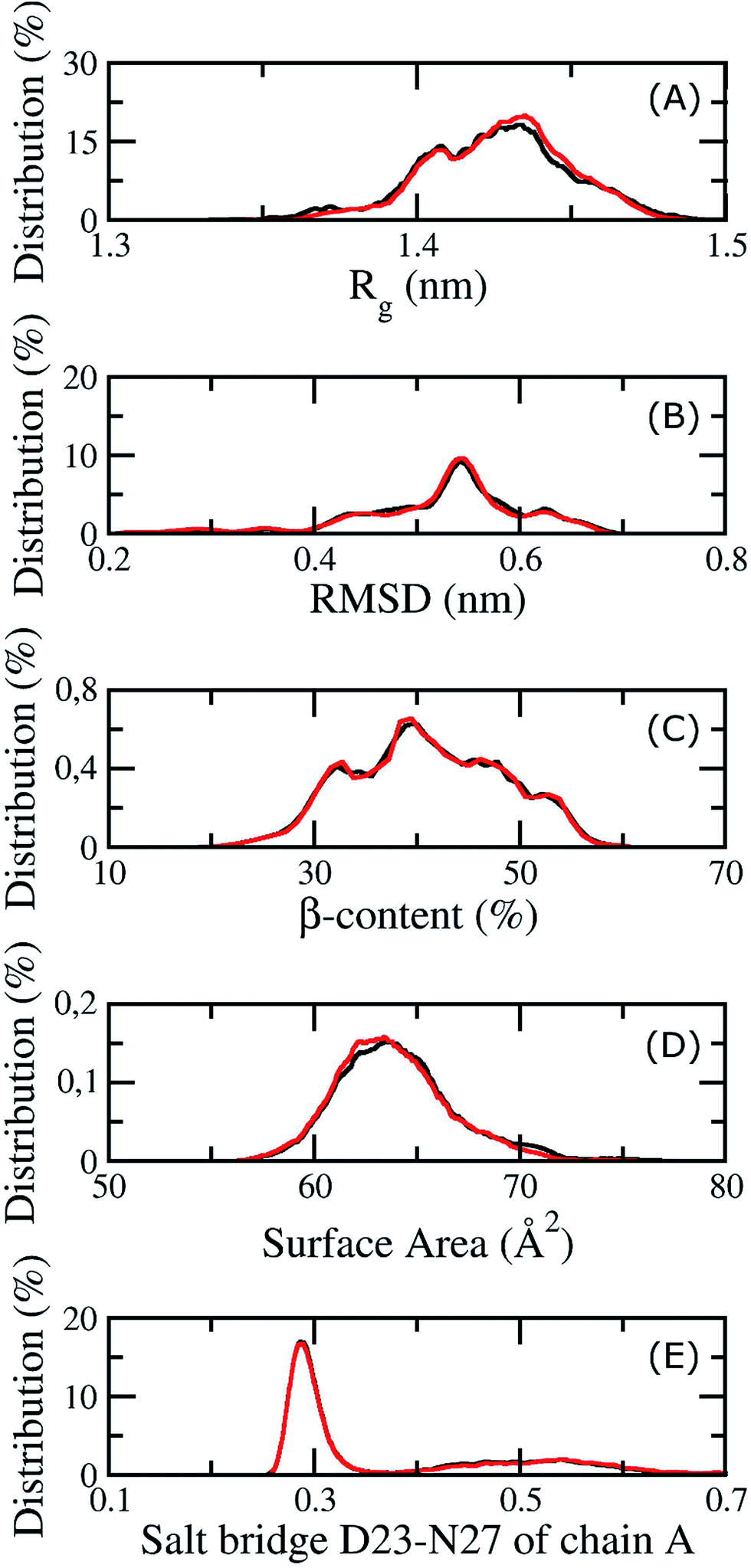 | ||
| Fig. 4 The convergence of the REMD simulations at 324 K. The black and red lines correspond to the values at different simulation intervals, 200 to 270 ns and 290 to 350 ns. (A) is the distribution of the radius of gyration of the transmembrane Aβ11–40 trimer, (B) is the population of the RMSD of the Aβ peptide, (C) is the distribution of the β-content of the trimer, (D) is the population of the surface accessible area of the Aβ peptide, and (E) is the distribution of the salt bridge D23–N27 of chain A. | ||
As the aggregation process of Aβ peptides is very slow, requiring up to several days, it is very difficult to obtain native structures of Aβ oligomers from random initial structures through extensive simulations. For example, in previous studies, the computations were proved to reach equilibrium even though the computed β-content was found to be ∼18% for the Aβ40 dimer;41 experiments indicated that the β-structure content of the dimer was ∼39%.80 These inconsistent data suggest that the initial conformations of Aβ peptides are very important and that there are several pathways and local minima in the Aβ aggregation process. We assumed that the conformations of the trimer oligomer were close to the those of the mature fibrils;45 thus, the initial conformation of the solvated 3Aβ11–40 peptide inserted into the membrane DPPC lipid bilayer was taken from the two-fold fibril form of the 12Aβ11–40 peptide,44 as shown in Fig. 1. Although the present computational study may be biased because it was performed with the initial conformation of a fibril-like structure, the metastable structures of the solvated Aβ11–40 trimer were obtained from the same initial structure.45
The solvated system was simulated using explicit solvent temperature REMD simulations involving 48 replicas in the range between 290 and 417 K. The temperature generator for the REMD simulations webserver52 was employed to choose temperatures for our simulation with the following parameters: exchange probability of 20%, tolerance of 10−4, fully flexible water molecules, constrained hydrogen bonds in proteins, and full hydrogen bonds in proteins. Details of all the replica temperatures are described in the ESI file.† 350 ns MD simulations were performed for each replica, amounting to a total of 16800 ns (16.8 μs) of completed MD simulations. The exchanges were attempted every 1 ps; the REMD simulation included 350000 replica exchange times. To avoid any initial bias, the first 200 ns of the REMD simulation was dismissed from the analyses. The parameters of compatibility of the computational simulations were subsequently evaluated. The results are averaged over individual snapshots.
The exchange rates between two neighbouring replicas were investigated; these are good values because they range from 26% to 38% (Fig. S1 of ESI†). Furthermore, the overlap of the sampling potential energies ensures the capability of exchange between the neighbouring replicas.81 The energetic overlap between different replicas is shown in Fig. S2 (ESI†), which indicates that the solvated transmembrane systems have good exchange probabilities according to the Metropolis Criterion. Consequently, the diffusion of the temperature space of the replica temperature index was monitored and is shown in Fig. S3 (ESI),† indicating the wide sampling of trajectories over the whole temperature space. Each replica moved through the entire temperature range, and no obstacles were present. Moreover, the secondary structures of the oligomer were estimated at 200 ns over all of the replicas, which are shown in Fig. 3. In particular, these metrics ranged from 36% to 78% of random coil, 20–56% of β-content, 0–13% turn structure, and 0–4% of α-content. These analyses demonstrate that our simulations were not biased by any distinct conformations.
Because membrane DPPC lipid bilayers have a phase transition at about 315 K, the thermodynamic properties of the 3Aβ11–40 peptide and the membrane were investigated at 324 K, including the structural changes of the peptide and the mutual interactions between the membrane and Aβ. Our computations were equilibrated at 324 K after 200 ns of REMD simulations because all values considered remained unchanged over two interval simulation times, 200 to 270 ns and 290 to 350 ns, including the gyrated radius, RMSD, β-content, surface area, and salt bridge of the peptide. The corresponding metrics in different time intervals are shown in Fig. 4. In the latter figure, the red lines correspond to the measured metrics at a simulated time interval of 200 to 270 ns, while the black lines highlight these values at the interval of 290 to 350 ns. In particular, the averages of Rg and RMSD are 0.47 ± 0.07 and 1.42 ± 0.02 nm, respectively. The β-content of the truncated trimer is 40 ± 7%, while the surface area of the peptide is estimated to be 64.73 ± 3.07 Å2. The distance between the charged groups of D23 and N27 of chain A is 0.40 ± 0.17 nm.
The root mean square fluctuation (RMSF) of the transmembrane 3Aβ11–40 peptide at 324 K was thus evaluated over the last 150 ns of the REMD simulations. The results presented in Fig. S4 (ESI†) are approximately arranged in two regions. The first region, including the sequences 11–13, 21–30, and 38–40, exhibits a high deviation over computational time, almost higher than 0.4 Å. The second region, consisting of the sequences 14–20 and 31–37, is a low deviation domain during the REMD simulations; all the deviations are smaller than 0.4 Å. These regions correspond to the domains where the secondary structure of the peptide regularly forms random coils and β-structures, respectively. The high deviation domain is caused by interactions within the surface regions of the membrane DPPC lipid bilayer, which will be described in the following subsection about the intermolecular interactions between the Aβ oligomer and the membrane DPPC lipid bilayer. The high fluctuation regions emerge as the main contributors to the structural changes of the transmembrane oligomer.
Second structure of transmembrane 3Aβ11–40 peptide
The average of the secondary structures of the protein which emerged at 324 K during the last 150 ns of the REMD simulation was predicted using the DSSP method.64,65 The averages of the random coil, beta, turn and helix structures are shown in Fig. 5. During our simulations, on average, the α-structure was seldom observed, only comprising ∼0.2% over the simulations. The obtained data confirmed that the α-structure is an intermediate step of the Aβ aggregation process.37,73,74,82,83 The turn structure was found to be ∼3%. The random coil form and β-content were dominant, with amounts of ∼57% and ∼40%, respectively. This finding is in good agreement with transmembrane Aβ oligomer24,46 experiments and the trimer of the Aβ40 peptide in solution.80 It should be noted that in experiments, the β-contents of transmembrane Aβ oligomers and the solvated Aβ trimer are almost the same, approximately 39–40%.24,46,80 | ||
| Fig. 5 The secondary structures of the transmembrane 3Aβ11–40 peptide, averaged from the last 150 ns of the REMD simulations at 324 K. The secondary structures were predicted using DSSP tools. | ||
In particular, chain C has fewer β-structures compared to both chains A and B. This is internally consistent with the salt bridge analysis given below because chain C was found to make a salt bridge between D23 and K28. This structural change leads to the appearance of the α-structure in the middle region of chain C (Fig. 5), although the α-structure appears less frequently.
The secondary structure patterns can roughly be divided into five domains: namely, sequences 11–13, 20–30, and 38–40 are mostly random coil structures, and sequences 14–19 and 31–37 are rigid β-structures in which most of the secondary structures have β-content. Overall, in analysing snapshots, the simulated structures of the transmembrane 3Aβ11–40 peptide appear to consist of two β-structure domains that are separate from the random coil regions. The β-structure regions are essentially penetrated by the DPPC lipid bilayer. Consequently, the random coil domains are regularly located and interact with the lipid headgroups. These results are in agreement with previous experiments which found that the Aβ peptide can adopt β conformations when penetrating into the membrane24,27,46 and can adopt coil conformations when located at the surface of the membrane.24,26 Both β-sheet domains are characterized by strong intermolecular interactions with each other through both fibril and hydrogen bond contacts. Moreover, the random coil domains have fewer contacts between the two neighbouring chains.
Mutual interaction contacts of the transmembrane 3Aβ11–40 peptide
The fibril and hydrogen bond contact maps between two adjacent chains of the 3Aβ11–40 peptide were obtained from an analysis of the computational time from 200 to 350 ns of the REMD simulations with explicit solvent molecules at 324 K. The details of the fibril and hydrogen bond contact maps of the 3Aβ11–40 oligomer are delineated in Fig. 6. The regions where its secondary structures are rigid β-sheets have frequent contact with each other. Particularly, the rigorous β-structure region of the C-terminal of chain B forms numerous intensive fibril and hydrogen bond contacts with the corresponding C-terminal domains of chains A and C (Fig. 6). Although a comparable N-terminal domain of chain B continued to produce a firm intermolecular interaction with the β-sheet region of the N-terminal of chain A, the fibril and hydrogen bond contacts between the corresponding domains of chains B and C decreased. This is consistent with the increasingly random coil structure of the N-terminal of chain C (Fig. 5). Overall, the obtained data indicate that the central hydrophobic cores of both the N- and C-terminals are essential to maintaining the stability of the Aβ oligomers.84 | ||
| Fig. 6 (A) + (B) and (C) + (D) show the fibril and hydrogen bond contact maps between the neighbouring chains of the transmembrane truncated 3Aβ11–40 peptide at 324 K over equilibrium snapshots of the REMD simulations, respectively. | ||
In addition, the D23–K28 salt bridge has been shown in several previous studies39,42,43,85 to play an important role in stabilizing the structures of the monomers and the fibril structures of Aβ peptides in solution; the distribution of the intramolecular D23–K28 salt bridge of the transmembrane 3Aβ11–40 peptide was further analysed. This distribution is shown in Fig. 7. At the starting point, no salt bridge appeared. After simulation over a long period of time using REMD, chain C was found to form a salt bridge between Asp23 and Lys28 with a very high probability. However, the Asp23–Lys28 salt bridges of chains A and B was rarely observed. This is due to the fact that both Lys28A and Lys28B form hydrogen bonds with lipid headgroups located within this region. These interactions are the main element altering the secondary structures of the peptides at the loop region. This result is consistent with available computational studies about the effects of lipid bilayers on the structure of the Aβ1–40 monomer.86,87 Instead, in our present simulation, the polar contact between Asp23 and Asn27 of both chains A and B was found to be a stabilizing contact (Fig. 7B). This contact replaces the salt bridge Asp23–Lys28 in order to secure the turn region, in accord with a previous computational study showing that the membrane alters this salt bridge of the Amyloid precursor protein.88
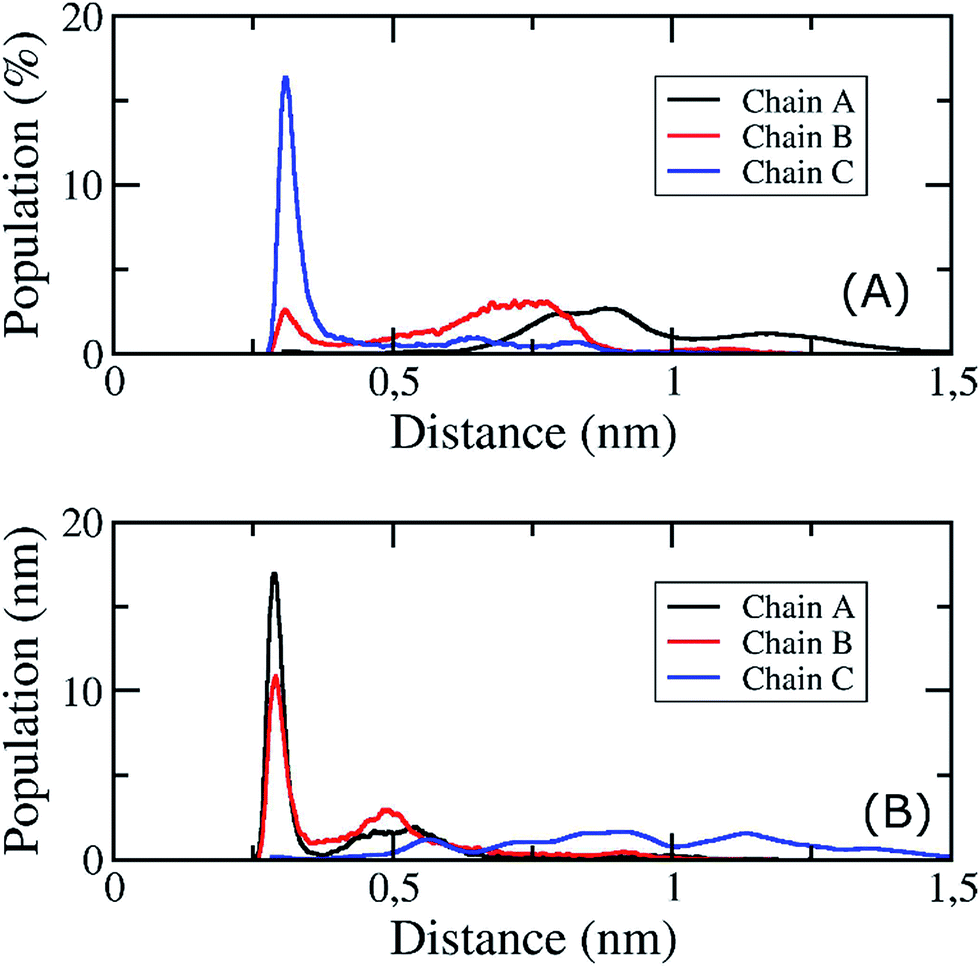 | ||
| Fig. 7 The distribution of the Asp23–Lys28 salt bridge (A) and the Asp23–Asn27 salt bridge (B) of the transmembrane 3Aβ11–40 oligomer at 324 K during the simulation time interval of 200 to 350 ns of the REMD simulations. | ||
The optimized structure of the transmembrane Aβ11–40 trimer
In an attempt to identify the putative structures of the 3Aβ11–40 peptide penetrating the membrane DPPC lipid bilayer, the two-dimensional free energy landscape was constructed using the “gmx sham” tool. The RMSD of the protein was chosen as the first reaction coordinate. The radius of gyration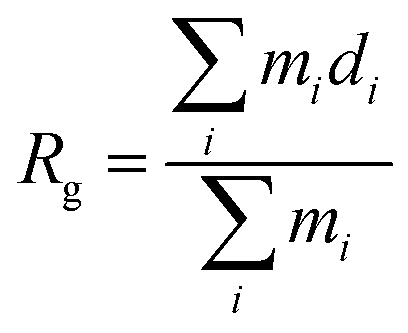 is defined as the average of the mass-weighted squared distances of all atoms to the center of mass; this was chosen as the second reaction coordinate. The clustering method was then used to search for the stable structures of the peptide which remain at the minima.
is defined as the average of the mass-weighted squared distances of all atoms to the center of mass; this was chosen as the second reaction coordinate. The clustering method was then used to search for the stable structures of the peptide which remain at the minima.
Fig. 8 shows the FEL of the truncated 3Aβ11–40 peptide at 324 K. In total, there are five representative structures, which are noted as M1 to M5. Four of these structures (i.e. M1 to M4) have the same magnitudes of Rg and RMSD, approximately 1.43 and 0.55 nm, respectively; they have the lowest free energy value of −13.9 kJ mol−1. M5 is located in a different free energy hole, with a free energy of −11.59 kJ mol−1. This conformation has Rg and RSMD values of ∼1.40 and ∼0.63 nm, respectively. The details of the secondary structures of these conformations are shown in Table 1. On average, the coil structure occupies 54% and the β-content occupies 44%. The turn structure represents approximately 2%, and the helix structure completely disappears. This finding may be due to the fact that the putative structures tend to be Aβ fibril formations because the helix structure is the intermediate step of Aβ aggregation.37,73,74,82,83 These observations are in accord with previous experimental findings that lipids alter the secondary structure of Aβ to approximately 40–60% β-content,24,46 whereas the random coil structure is found near the lipid headgroups.24,26
 | ||
| Fig. 8 Free energy landscape of the transmembrane 3Aβ11–40 peptide as a function of the first two principal components, Rg and RMSD. M1 to M5 are minima of the transmembrane truncated 3Aβ11–40 peptide inserted into the DPPC lipid bilayer; these were obtained from FEL analysis during the last 150 ns of the REMD simulations at 324 K. | ||
| Minima | Coil content (%) | Beta content (%) | Turn content (%) | Helix content (%) | CCS (Å2) |
|---|---|---|---|---|---|
| M1 | 58 | 40 | 2 | 0 | 1361 |
| M2 | 45 | 53 | 2 | 0 | 1311 |
| M3 | 62 | 38 | 0 | 0 | 1371 |
| M4 | 56 | 40 | 4 | 0 | 1317 |
| M5 | 51 | 49 | 0 | 0 | 1343 |
In particular, the optimized conformation M2 adopts the highest possibility of beta structure, with an amount of 53%. The other metrics, including the alternate random coil and turn structures, occupy 45% and 2%, respectively. It is interesting to note that M2 is the most common conformation of the trimer in the lowest free energy state, with a population of ∼29% of the total snapshots, located in the lowest free energy minimum; this was predicted through the clustering method with a cutoff of 0.35 nm. Therefore, we chose M2 as the initial conformation to estimate the annihilation free energy of the transmembrane Aβ11–40 trimer.
In this free energy hole, the populations of M1, M3, and M4 amount to 9%, 21% and 13%, respectively. Meanwhile, M5 is located in a different free energy hole, with a population of 38% of all snapshots at this state. The rest are other clusters whose populations are quite low. The putative structure M3 has the lowest probability of β-content, at 38%, and the highest probability of random coil structure, with an amount of 62%. There is no turn structure in the M3 minimum.
Additionally, both the M1 and M4 conformations form the same amounts of β-structures, at 40%. However, M4 has more turn structure at 4%, whereas M1 only forms 2%. Thus, the random coil structures of M1 and M4 attain 58% and 56%, respectively, due to the lack of helix structures. The optimized structure M5 contains 49% beta structure and 51% random coil. The other metrics, α- and turn-content, have zero percentages.
Although several previous studies are available on the screening of potential inhibitors for AD, including candidates to inhibit Aβ peptides,56,89–93 an effective drug for the treatment of AD remains elusive. This may be because the structures of solvated Aβ peptides are employed as targets in most screening of potential inhibitors for Aβ peptides.56,89–93 However, as discussed above, the major mechanism of Aβ neurotoxicity arises from the membrane penetration of Aβ oligomers. Strong inhibitors of Aβ peptides in solution appear not to effectively inhibit these peptides when they penetrate the membrane. Therefore, the putative structures of the transmembrane trimer Aβ11–40 peptide that are determined using REMD simulations can be used as targets for computer-aided drug design that may lead to the discovery of an effective inhibitor of Aβ oligomers.
It is known that the CCS is an important parameter for characterizing proteins. This metric can be determined not only in experiments, such as ion mobility mass spectrometry, but also by computation. The IMPACT method has successfully estimated the CCS of proteins with high correlation to experiments.69,94 Thus, the CCS of the optimized structure of the transmembrane Aβ trimer was investigated using IMPACT. The obtained results are shown in Table 1; the mean of the metrics is 1341 ± 23 Å2. As far as we know, the experimental CCS of the transmembrane Aβ trimer is still unavailable. However, the CCS of the Aβ trimer in solution was determined in previous experiments, with values of 1265 ± 16 and 1386 ± 145 Å2.95,96 Overall, our calculated CCS result is in good agreement with these experiments.
Intermolecular interaction between the DPPC lipid bilayer and the Aβ peptide
Intermolecular interactions between the peptide and the membrane were also computed through evaluation of the minimum distance between the heavy atoms of the Aβ residues and the phosphorus atoms of the headgroups. A contact is recorded whenever the distance becomes smaller than 0.45 nm. The probability of existence of these contacts is demonstrated in Fig. 9. Consistent with the investigation of the RMSF and secondary structures, the residues that have high deviations and solid coil structures frequently form non-bonded contacts with the membrane surface. Additionally, the β-structure domains of the N-terminals of the peptides are commonly found to interact with lipid headgroups. Moreover, both Lys16 and Lys28 have the most regular contact with lipid phosphate headgroups, which is in good agreement with previous studies on the interaction between a membrane and the Aβ1–40 monomer.86,87 Although the observed results imply that electrostatic interactions are the main factor in the binding process between the Aβ trimer and the membrane DPPC lipid bilayer, this qualitative finding is inconsistent with the quantitative analysis of binding free energy calculations, which is mentioned below, indicating that van der Waals interactions emerge as the key element of interaction between the trimer and the membrane. These inconsistent results thus suggest that a study on the interactions of Aβ peptides and membranes using evaluation of the distance between the different heavy atoms of the different molecules could enable artificial observation.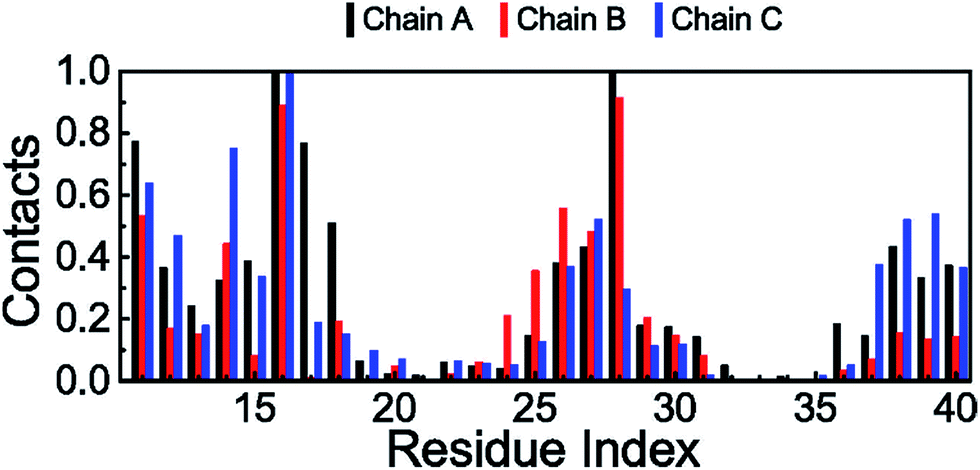 | ||
| Fig. 9 Probability of intermolecular contacts between the phosphorus atoms of bilayer DPPC lipids and the heavy atoms of the truncated 3Aβ11–40 peptide. The results were investigated at 324 K throughout the last 150 ns of the REMD simulations. | ||
Free energy difference of binding between the DPPC lipid bilayer and the Aβ trimer
In order to investigate the nature of binding between the trimer and the lipid bilayer, the double-annihilation binding free energy method61,63 was used to estimate for the first time to estimate the binding free energy of the Aβ trimer to the DPPC lipid bilayer, because it is one of the most accurate methods currently known for this purpose.97–99 In this scheme, the protein is annihilated from both the transmembrane and solvated systems. To determine the binding free energy, in the first step, the putative structure of the transmembrane Aβ trimer in M2 is chosen as the initial conformation of the first annihilation simulation; it is annihilated from the complex, and subsequently ΔG1 is determined. In the second step, the trimer is solvated and then equilibrated in 30 ns of MD simulations. The RMSD analysis indicates that the system reaches equilibrium after 0.2 ns (cf. Fig. S5†). The last snapshot of the MD is chosen as the initial conformation for the free energy calculation using the FEP method. In this process, the protein is annihilated from solution; then, ΔG2 is determined. The free energy of this process was determined using the BAR method59 in every ps, and the results are shown in Fig. 10. The free energies reached equilibrium after 2 ns; then, the binding free energy ΔGbind of the oligomer to the DPPC lipid bilayer was determined to be −70 ± 18 kcal mol−1, which is averaged from the differences of the two annihilation free energies. In particular, the contribution of electrostatic energy (ΔGcou) to the binding free energy amounts to −114 ± 18 kcal mol−1, and the difference in free energy of the van der Waals interactions (ΔGvdW) is −184 ± 3 kcal mol−1. These results indicate that although intermolecular polar contacts between the Aβ peptide and the membrane are very important, according to the analysis given above and also from previous studies,21,28 the natures of the binding differ from each other. The van der Waals interactions emerge as dominant over the Coulomb interactions in the binding process of the Aβ peptide to the membrane DPPC lipid bilayer.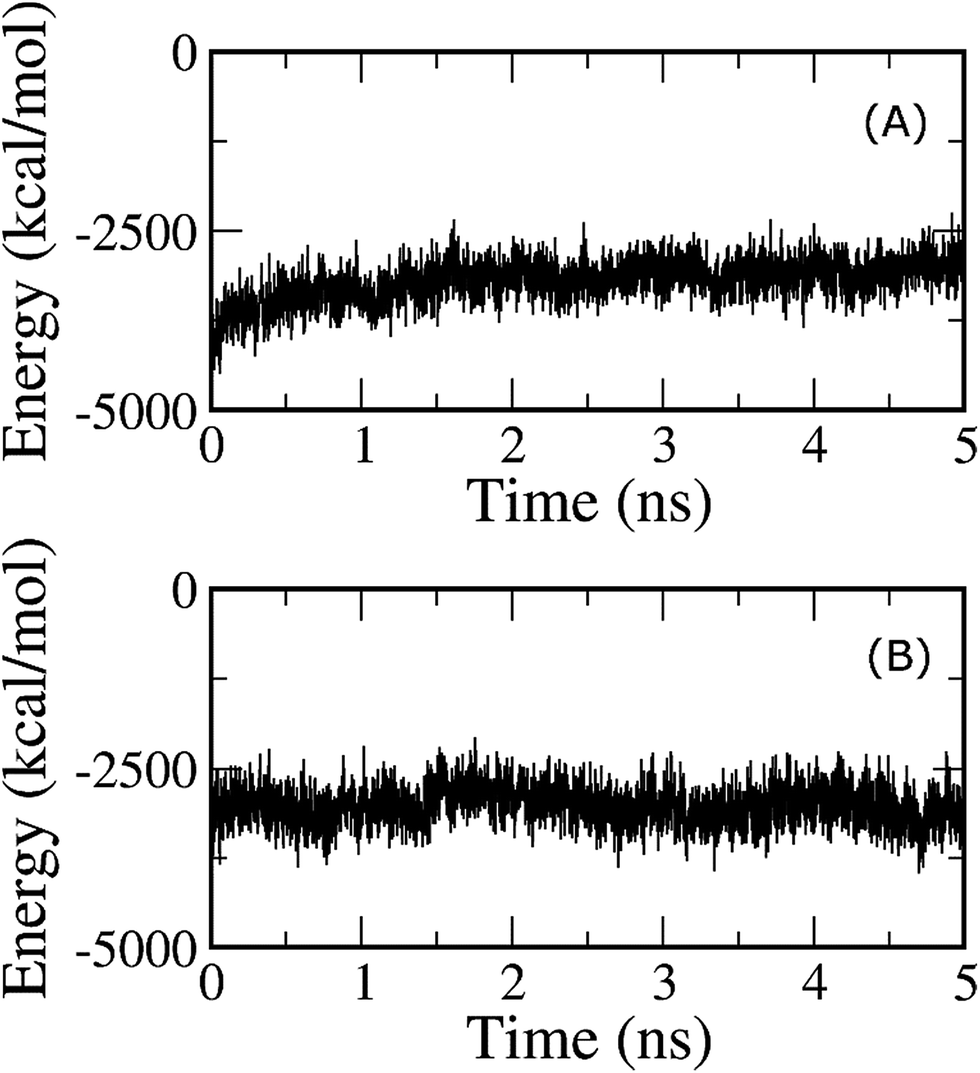 | ||
| Fig. 10 (A) Annihilation free energy of the Aβ trimer from the solvated membrane system, and (B) annihilation free energy of the Aβ trimer from the solution. The free energy was determined using the BAR method at every ps. | ||
Stabilization of the membrane over simulation time
In order to further detect the effects of the Aβ oligomer on the oriented mobility of the membrane DPPC lipid bilayer, the averages of the lipid order parameters were computed for both carbon atoms of the acyl chains sn-1 and sn-2, as shown in Fig. 11. The membrane DPPC lipid bilayer without the induced oligomer is simulated in solution. The lipid order parameters of the solvated DPPC lipid bilayer are indicated by the green and blue lines in Fig. 11. MD simulation of the pure lipid bilayer was carried out for a length of 200 ns. Our pure solvated DPPC lipid bilayer observations are in good agreement with both previous experimental and computational results.100–102 In addition, the lipid order parameters of the membrane with the oligomer induced are also investigated, as illustrated by the black and red lines in Fig. 11. Generally, the lipid order parameter curves of the two saturated DPPC chains are quite different from each other for both systems with and without the oligomer. The results obtained here are consistent with the view that the interaction between the Aβ peptides and the lipid headgroups leads to fluctuation of the lipid order parameters.26,103,104 However, the obtained results also indicate that the membrane DPPC lipid bilayer is stabilized upon protein insertion.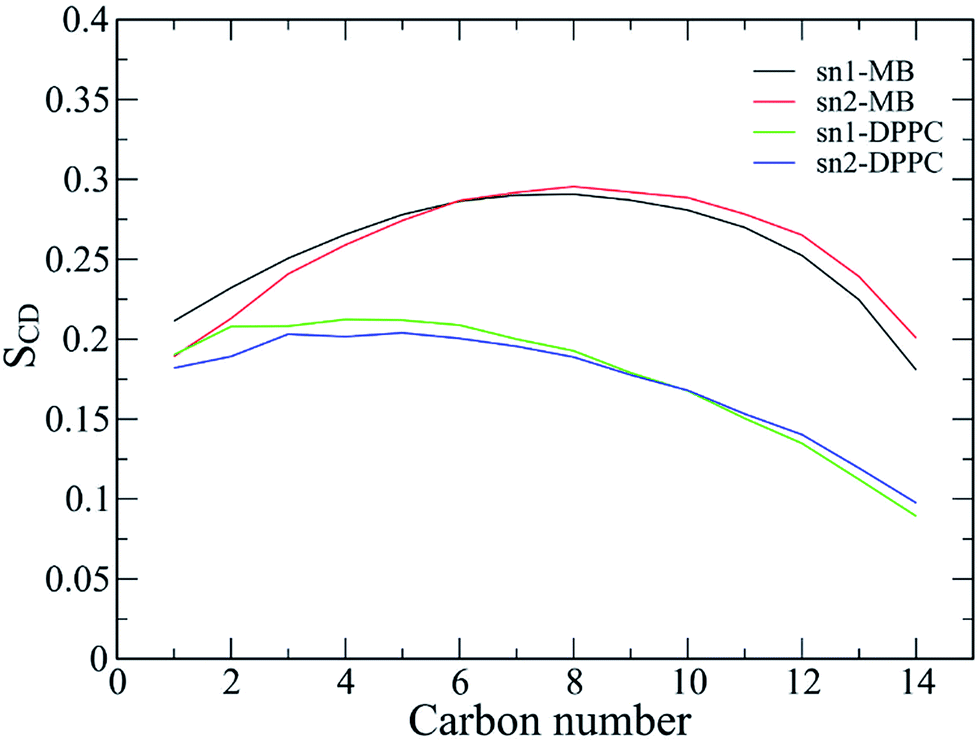 | ||
| Fig. 11 The lipid order parameters for both carbon atoms of acyl chains sn-1 and sn-2. The results are averaged over the last 150 ns of the temperature REMD simulations. | ||
Conclusions
In the present theoretical study, the high efficiency sampling method of REMD simulations with explicit solvent was applied to investigate the structural changes of the 3Aβ11–40 oligomer inserted in the membrane of a DPPC lipid bilayer.We firstly generated the stable structures of the Aβ11–40 trimer when it is inserted into the membrane DPPC lipid bilayer. On average, the percentages of the putative trimer secondary structure were estimated to be 44% β-structure, 54% random coil structure, 2% turn structure, and absolutely 0% α-structure. There was a small shift compared with the averaged metrics of all snapshots, which were 40% β-content, 57% random coil, 3% turn structure and 0% α-structure. Consequently, the β-content is slightly higher than the β-content of the solvated Aβ11–40 trimer.45 However, this difference may be caused by the tendency of the GROMOS force field to favor β formation.79 Moreover, we found that the transmembrane Aβ trimer structure is arranged in two domains, including the favored β-structure region that is fully inserted into the membrane, involving the sequences 14–19 and 31–37. The mostly random coil region was found to be located close to the surface of the membrane and consists of the sequences 11–13, 20–30 and 38–40. These two domains correspond to the stable and fluctuating regions of the oligomer, respectively. This finding is in agreement with both previous experimental24,26,27,46 and computational28–33 studies. We hope that new knowledge of these structures can enhance the search for an Aβ therapy.
The membrane of the DPPC lipid bilayer was in addition found to be stable during the REMD simulations, and the intermolecular interaction contacts between the oligomer and the DPPC lipid bilayer indicated that the random coil domain favors interaction with the membrane headgroups. Moreover, the van der Waals interaction energy is predominant over the electrostatic energy in the binding of the Aβ trimer to the membrane, based on double-annihilation binding free energy calculations. Consequently, the trimer experiences a strong attraction with the membrane, with a large binding free energy of −70 ± 18 kcal mol−1.
The size of the transmembrane Aβ11–40 trimer is approximately equal to the size of the Aβ trimer in solution, with a cross section (CCS) of 1341 ± 23 Å2; this compares well with the experimental values of the solvated trimer of 1265 ± 16 and 1386 ± 145 Å2.95,96
The membrane DPPC lipid bilayer induces a strong effect on chain C, which leads to a smaller number of non-bonded contacts with chain B compared to the contacts between chains A and B. Although the membrane of the DPPC lipid bilayer was stable during all the MD computations, the lipid order parameters fluctuated when the oligomer was induced.26,103,104
The polar contact Asp23–Asn27 was also found to be the stabilizing factor of the oligomer structure, rather than the Asp23–Lys28 salt bridge as reported in a previous study by Miyashita et al.30 on the transmembrane amyloid precursor protein.
Acknowledgements
STN thanks Ton Duc Thang University (TDTU-DEMASTED) for support. HMH is grateful to the Vietnam Ministry of Education and Training for a scholarship (911 program). MTN is indebted to the KU Leuven Research Council (GOA program).References
- A. S. Henderson and A. F. Jorm, Dementia, John Wiley & Sons Ltd, 2002 Search PubMed.
- D. J. Selkoe, Neuron, 1991, 6, 487–498 CrossRef CAS PubMed.
- J. L. Cummings, N. Engl. J. Med., 2004, 351, 56–67 CrossRef CAS PubMed.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N.-V. Buchete, S. Coté, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS PubMed.
- Alzheimer's_association, Journal, 2015.
- D. J. Selkoe, Science, 2002, 298, 789–791 CrossRef CAS PubMed.
- I. Khlistunova, J. Biernat, Y. Wang, M. Pickhardt, M. von Bergen, Z. Gazova, E. Mandelkow and E.-M. Mandelkow, J. Biol. Chem., 2006, 281, 1205–1214 CrossRef CAS PubMed.
- K. SantaCruz, J. Lewis, T. Spires, J. Paulson, L. Kotilinek, M. Ingelsson, A. Guimaraes, M. DeTure, M. Ramsden, E. McGowan, C. Forster, M. Yue, J. Orne, C. Janus, A. Mariash, M. Kuskowski, B. Hyman, M. Hutton and K. H. Ashe, Science, 2005, 309, 476–481 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- M. Citron, Nat. Rev. Neurosci., 2004, 5, 677–685 CrossRef CAS PubMed.
- A. Aguzzi and T. O'Connor, Nat. Rev. Drug Discovery, 2010, 9, 237–248 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed.
- A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10427–10432 CrossRef CAS PubMed.
- T. L. Williams and L. C. Serpell, FEBS J., 2011, 278, 3905–3917 CrossRef CAS PubMed.
- Y. Nakazawa, Y. Suzuki, M. P. Williamson, H. Saitô and T. Asakura, Chem. Phys. Lipids, 2009, 158, 54–60 CrossRef CAS PubMed.
- H. Jang, J. Zheng and R. Nussinov, Biophys. J., 2007, 93, 1938–1949 CrossRef CAS PubMed.
- C. Ege and K. Y. C. Lee, Biophys. J., 2004, 87, 1732–1740 CrossRef CAS PubMed.
- H. Zhao, E. K. J. Tuominen and P. K. J. Kinnunen, Biochemistry, 2004, 43, 10302–10307 CrossRef CAS PubMed.
- C. Poojari, A. Kukol and B. Strodel, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 327–339 CrossRef CAS PubMed.
- J. J. Kremer, D. J. Sklansky and R. M. Murphy, Biochemistry, 2001, 40, 8563–8571 CrossRef CAS PubMed.
- H. Shao, S.-c. Jao, K. Ma and M. G. Zagorski, J. Mol. Biol., 1999, 285, 755–773 CrossRef CAS PubMed.
- E. Terzi, G. Hölzemann and J. Seelig, J. Mol. Biol., 1995, 252, 633–642 CrossRef CAS PubMed.
- O. Crescenzi, S. Tomaselli, R. Guerrini, S. Salvadori, A. M. D'Ursi, P. A. Temussi and D. Picone, Eur. J. Biochem., 2002, 269, 5642–5648 CrossRef CAS PubMed.
- J. McLaurin and A. Chakrabartty, Eur. J. Biochem., 1997, 245, 355–363 CAS.
- M. Meier and J. Seelig, J. Mol. Biol., 2007, 369, 277–289 CrossRef CAS PubMed.
- B. Strodel, J. W. L. Lee, C. S. Whittleston and D. J. Wales, J. Am. Chem. Soc., 2010, 132, 13300–13312 CrossRef CAS PubMed.
- C. Lockhart and D. K. Klimov, J. Phys. Chem. B, 2014, 118, 2638–2648 CrossRef CAS PubMed.
- N. Miyashita, J. E. Straub and D. Thirumalai, J. Am. Chem. Soc., 2009, 131, 17843–17852 CrossRef CAS PubMed.
- Q. Wang, J. Zhao, X. Yu, C. Zhao, L. Li and J. Zheng, Langmuir, 2010, 26, 12722–12732 CrossRef CAS PubMed.
- Z. Chang, Y. Luo, Y. Zhang and G. Wei, J. Phys. Chem. B, 2011, 115, 1165–1174 CrossRef CAS PubMed.
- H. Jang, J. Zheng, R. Lal and R. Nussinov, Trends Biochem. Sci., 2008, 33, 91–100 CrossRef CAS PubMed.
- M. K. Jana, R. Cappai, C. L. L. Pham and G. D. Ciccotosto, J. Neurochem., 2016, 136, 594–608 CrossRef CAS PubMed.
- Y. Sun, W. Xi and G. Wei, J. Phys. Chem. B, 2015, 119, 2786–2794 CrossRef CAS PubMed.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS PubMed.
- M. D. Kirkitadze, M. M. Condron and D. B. Teplow, J. Mol. Biol., 2001, 312, 1103–1119 CrossRef CAS PubMed.
- L. K. Thompson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 383–385 CrossRef CAS PubMed.
- M. H. Viet, P. H. Nguyen, S. T. Ngo, M. S. Li and P. Derreumaux, ACS Chem. Neurosci., 2013, 4, 1446–1457 CrossRef CAS PubMed.
- B. Murray, M. Sorci, J. Rosenthal, J. Lippens, D. Isaacson, P. Das, D. Fabris, S. Li and G. Belfort, Proteins: Struct., Funct., Bioinf., 2016, 84, 488–500 CrossRef CAS PubMed.
- P. H. Nguyen, F. Sterpone, J. M. Campanera, J. Nasica-Labouze and P. Derreumaux, ACS Chem. Neurosci., 2016, 7, 823–832 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- A. T. Petkova, W. M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS PubMed.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef CAS PubMed.
- S. T. Ngo, H. M. Hung, D. T. Truong and M. T. Nguyen, Phys. Chem. Chem. Phys., 2017 10.1039/c6cp05511g.
- E. Terzi, G. Hoelzemann and J. Seelig, Biochemistry, 1994, 33, 7434–7441 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- J. F. Nagle, Biophys. J., 1993, 64, 1476–1481 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and a. J. Hermans, Intermolecular Forces, Reidel, Dordrecht, Jerusalem, Israel, 1981 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- D. F. Shanno, Math. Comput., 1970, 24, 647–656 CrossRef.
- A. Patriksson and D. van der Spoel, Phys. Chem. Chem. Phys., 2008, 10, 2073–2077 RSC.
- W. F. Van Gunsteren and H. J. C. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- M. H. Viet, S. T. Ngo, N. S. Lam and M. S. Li, J. Phys. Chem. B, 2011, 115, 7433–7446 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS.
- C. H. Bennett, J. Comput. Phys., 1976, 22, 245–268 CrossRef.
- W. L. Jorgensen, J. K. Buckner, S. Boudon and J. Tirado-Rives, J. Chem. Phys., 1988, 89, 3742–3746 CrossRef CAS.
- G. Jayachandran, M. R. Shirts, S. Park and V. S. Pande, J. Chem. Phys., 2006, 125, 084901 CrossRef PubMed.
- H. Fujitani, Y. Tanida, M. Ito, G. Jayachandran and C. D. Snow, J. Chem. Phys., 2005, 123, 084108 CrossRef PubMed.
- S. T. Ngo, B. K. Mai, D. M. Hiep and M. S. Li, Chem. Biol. Drug Des., 2015, 86, 546–558 CAS.
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef PubMed.
- R. P. Joosten, T. A. H. te Beek, E. Krieger, M. L. Hekkelman, R. W. W. Hooft, R. Schneider, C. Sander and G. Vriend, Nucleic Acids Res., 2011, 39, D411–D419 CrossRef CAS PubMed.
- Y. Mu, P. H. Nguyen and G. Stock, Proteins: Struct., Funct., Bioinf., 2005, 58, 45–52 CrossRef CAS PubMed.
- E. Papaleo, P. Mereghetti, P. Fantucci, R. Grandori and L. De Gioia, J. Mol. Graphics Modell., 2009, 27, 889–899 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. P. Benesch, Structure, 2015, 23, 791–799 CrossRef CAS PubMed.
- A. Morriss-Andrews and J. E. Shea, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, 2015, vol. 66, pp. 643–666 Search PubMed.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- C. H. Davis and M. L. Berkowitz, J. Phys. Chem. B, 2009, 113, 14480–14486 CrossRef CAS PubMed.
- S. Jang and S. Shin, J. Phys. Chem. B, 2006, 110, 1955–1958 CrossRef CAS PubMed.
- S. Jang and S. Shin, J. Phys. Chem. B, 2008, 112, 3479–3484 CrossRef CAS PubMed.
- A. Okamoto, A. Yano, K. Nomura, S. i. Higai and N. Kurita, Chem. Phys. Lett., 2013, 577, 131–137 CrossRef CAS.
- A. Yano, A. Okamoto, K. Nomura, S. i. Higai and N. Kurita, Chem. Phys. Lett., 2014, 595–596, 242–249 CrossRef CAS.
- H. Nymeyer, J. Chem. Theory Comput., 2008, 4, 626–636 CrossRef CAS PubMed.
- T. Mori, J. Jung and Y. Sugita, J. Chem. Theory Comput., 2013, 9, 5629–5640 CrossRef CAS PubMed.
- A. K. Somavarapu and K. P. Kepp, ChemPhysChem, 2015, 16, 3278–3289 CrossRef CAS PubMed.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS PubMed.
- M. Meli and G. Colombo, Int. J. Mol. Sci., 2013, 14, 12157–12169 CrossRef PubMed.
- Y. Fezoui and D. B. Teplow, J. Biol. Chem., 2002, 277, 36948–36954 CrossRef CAS PubMed.
- D. K. Klimov and D. Thirumalai, Structure, 2003, 11, 295–307 CrossRef CAS PubMed.
- W. Han and K. Schulten, J. Am. Chem. Soc., 2014, 136, 12450–12460 CrossRef CAS PubMed.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Doeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS PubMed.
- J. A. Lemkul and D. R. Bevan, Arch. Biochem. Biophys., 2008, 470, 54–63 CrossRef CAS PubMed.
- J. A. Lemkul and D. R. Bevan, Protein Sci., 2011, 20, 1530–1545 CrossRef CAS PubMed.
- N. Miyashita, J. E. Straub, D. Thirumalai and Y. Sugita, J. Am. Chem. Soc., 2009, 131, 3438–3439 CrossRef CAS PubMed.
- S. T. Ngo and M. S. Li, J. Phys. Chem. B, 2012, 116, 10165–10175 CrossRef CAS PubMed.
- P. D. Huy, Y. C. Yu, S. T. Ngo, T. V. Thao, C. P. Chen, M. S. Li and Y. C. Chen, Biochim. Biophys. Acta, 2013, 1830, 2960–2969 CrossRef CAS PubMed.
- S. T. Ngo and M. S. Li, Mol. Simul., 2013, 39, 279–291 CrossRef CAS.
- S.-H. Wang, F.-F. Liu, X.-Y. Dong and Y. Sun, J. Phys. Chem. B, 2010, 114, 11576–11583 CrossRef CAS PubMed.
- W.-J. Du, J.-J. Guo, M.-T. Gao, S.-Q. Hu, X.-Y. Dong, Y.-F. Han, F.-F. Liu, S. Jiang and Y. Sun, Sci. Rep., 2015, 5, 7992 CrossRef CAS PubMed.
- Y. Sun, S. Vahidi, M. A. Sowole and L. Konermann, J. Am. Soc. Mass Spectrom., 2016, 27, 31–40 CrossRef CAS PubMed.
- E. Sitkiewicz, J. Olędzki, J. Poznański and M. Dadlez, PLoS One, 2014, 9, e100200 Search PubMed.
- M. Kłoniecki, A. Jabłonowska, J. Poznański, J. Langridge, C. Hughes, I. Campuzano, K. Giles and M. Dadlez, J. Mol. Biol., 2011, 407, 110–124 CrossRef PubMed.
- J. Aqvist, C. Medina and J.-E. Samuelsson, Protein Eng., 1994, 7, 385–391 CrossRef CAS PubMed.
- J. Srinivasan, T. E. Cheatham, P. Cieplak, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 1998, 120, 9401–9409 CrossRef CAS.
- S. T. Ngo, H. M. Hung and M. T. Nguyen, J. Comput. Chem., 2016, 37, 2734–2742 CrossRef CAS PubMed.
- H. I. Petrache, S. W. Dodd and M. F. Brown, Biophys. J., 2000, 79, 3172–3192 CrossRef CAS PubMed.
- D. P. Tieleman, S. J. Marrink and H. J. C. Berendsen, BBA, Biochim. Biophys. Acta, Rev. Biomembr., 1997, 1331, 235–270 CrossRef CAS.
- H. M. Hung, V. P. Nguyen, S. T. Ngo and M. T. Nguyen, Biophys. Chem., 2016, 217, 1–7 CrossRef PubMed.
- C. E. Dempsey, BBA, Biochim. Biophys. Acta, Rev. Biomembr., 1990, 1031, 143–161 CrossRef CAS.
- S. H. White, W. C. Wimley and M. E. Selsted, Curr. Opin. Struct. Biol., 1995, 5, 521–527 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Include the list of additional figures consist of the mean exchange rates between neighbouring replicas, the distribution of potential energy of all replicas, the diffusion of temperature space of 1st and 48th replicas, the RMSF of the trimer, and the RMSD of the solvate Aβ11–40 trimer. See DOI: 10.1039/c6ra26461a |
| ‡ Contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |