
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAβ1–42 C-terminus fragment derived peptides prevent the self-assembly of the parent peptide†
Sunil Bansala,
Indresh Kumar Mauryab,
Kitika Shenmara,
Nitin Yadavc,
Chaitanya Kumar Thotac,
Vinod Kumard,
Kulbhushan Tikood,
Virander Singh Chauhanc and
Rahul Jain *a
*a
aDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, Sector 67, S. A. S. Nagar, Punjab 160 062, India. E-mail: rahuljain@niper.ac.in; Fax: +91 172 2214692; Tel: +91 172 2292024
bDepartment of Microbial Biotechnology, Panjab University, Sector 14, Chandigarh, 160 014, India
cInternational Center for Genetic Engineering and Biotechnology, Aruna Asif Ali Marg, New Delhi, 110 067, India
dDepartment of Pharmacology and Toxicology, National Institute of Pharmaceutical Education and Research, Sector 67, S. A. S Nagar, 160 062, Punjab, India
First published on 16th January 2017
Abstract
In an attempt to design Aβ aggregation inhibitors to combat Alzheimer's disease, herein we report a full peptide scan performed on a pentapeptide fragment (Aβ38–42) derived from the C-terminus of Aβ1–42 peptide. More than thirty new peptides were synthesized and tested for their inhibition activity towards Aβ self-assembly. In the cell viability assay, when co-incubated with Aβ, three peptides were found to completely prevent the toxicity induced by Aβ aggregation. Most active pentapeptides were also studied by ThT fluorescence assay and the results were well correlated to the MTT study. The inhibition potential of a pentapeptide (15) was further confirmed by CD spectroscopy and transmission electron microscopy.
Alzheimer's disease (AD), first reported by Alois Alzheimer in 1906,1 is manifested by the abnormal misfolding of proteins, amyloid-β (Aβ) and tau.2 An alarming number of patients, 44 million currently and set to triple by 2050, would create challenges for society on medical and economical fronts.3 Although a number of factors have been suggested to play a role in the etiology of AD,4 deposition of neuritic plaques due to the accumulation of Aβ have been the most widely accepted phenomenon.5
Acetylcholinesterase inhibitors such as tacrine, donepezil, rivastigmine and galantamine, and N-methyl-D-aspartate (NMDA)-receptor antagonist memantine were among the first drugs approved by FDA, however none could prevent or reverse the disease progression.6 Since then a number of other strategies such as secretase inhibitors,7–10 immunotherapy,11,12 metal chelators,13,14 tau aggregation regulators15 have been explored. However, out from more than two hundred molecules tested in the clinical trials from 2002–12, only one (memantine) is approved as a drug in 2003. Also, the therapeutics that are currently being tested in clinical trials, don't offer much hope. Due to low efficacy or side effects, a large number of promising candidates couldn't proceed beyond phase II, and very few progressed to the phase III clinical trials.
Aβ is known to play positive modulatory role in memory and neurotransmission,16 hence, the inhibition of Aβ aggregation, is currently being investigated.17,18 Reported to inhibit Aβ aggregation, a number of small molecules have reached in clinical trials.19,20 A number of studies have shown that modified fragments of Aβ inhibit the aggregation of parent peptide specifically.21–26 PPI-1019, a modified pentapeptide derivative of fragment Aβ17–21, has been tested up to phase II clinical trials.27 Similarly, N-methylated pentapeptide, SEN-606 has reached in pre-clinical developmental phases.
Since hydrophobic interactions are known to play a significant role in protein misfolding,28 and considering the importance of the peptide fragments in their specific binding with the main peptide chain, a number of Aβ aggregation inhibitors derived from the hydrophobic C-terminus of Aβ were reported. Fragments Aβ31–42 and Aβ39–42 were found to inhibit Aβ aggregation significantly.29 In a study, poly-N-methylated hexapeptides based on Aβ32–37 were reported as efficient inhibitors of Aβ self-assembly.30 We reported a hexapeptide based on the Aβ32–37 fragment that mitigate Aβ toxicity completely at sub-micromolar concentrations.31 Despite the current focus on the C-terminus of Aβ, the region still remains relatively less explored. In order to scrutinize the C-terminus region, we performed a full peptide scan on a C-terminus pentapeptide fragment, Aβ38–42. About 32 new peptides were synthesized by Fmoc solid phase peptide synthesis protocol. All amino acids of the pentapeptide fragment were substituted by physico-chemically analogous amino acid residues. Moreover, amino acids such as proline (Pro) and α-aminoisobutyric acid (Aib) were chosen because of their β-sheet breaking properties. Scheme 1 shows the general route for the synthesis of peptides on Wang resin.
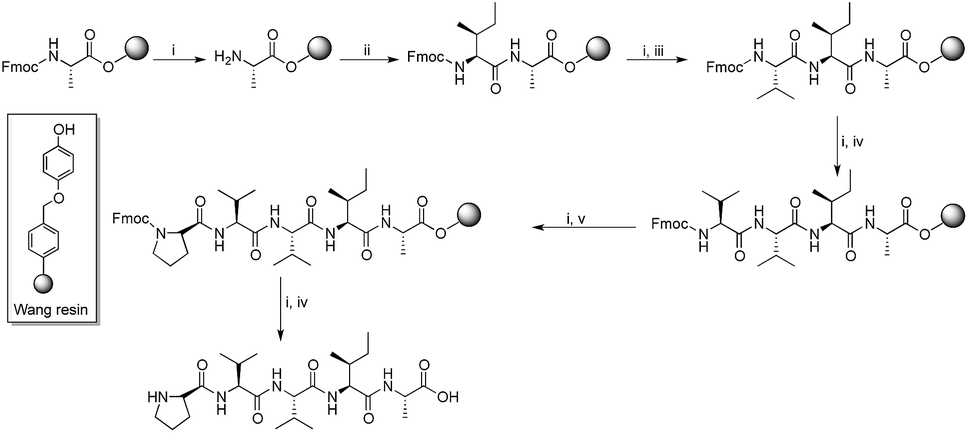 | ||
| Scheme 1 Synthesis of peptides exemplified by 6. Reaction conditions: (i) 20% piperidine in DMF, 15 min; (ii) Fmoc-Ile-OH, TBTU, DIEA, DMF, 2.5 h; (iii) Fmoc-Val-OH, TBTU, DIEA, DMF, 2.5 h; (iv) Fmoc-Val-OH, TBTU, DIEA, DMF, 2.5 h; (v) Fmoc-Pro-OH, TBTU, DIEA, DMF, 2.5 h; (vi) TFA, TIPS, H2O, 2.5 h. | ||
Since it is well known that protein misfolding is preceded by intramolecular as well as intermolecular interactions between the monomer units, the synthesized pentapeptide derivatives of fragment Aβ38–42 are supposed to exhibit high affinity to specifically interact with full-length Aβ peptide. MTT assay was performed to evaluate the newly synthesized pentapeptides for the inhibitory effects against Aβ1–42 aggregation and toxicity using PC-12 cells. Cell viability data for all the synthesized pentapeptides and the conversion in percentage inhibition of Aβ aggregation have been shown in Table 1. The lead peptide (1, Gly–Val–Val–Ile–Ala) previously reported to prevent Aβ1–42 toxicity by 70% when taken in ten-fold excess, was observed to exhibit significant activity in this study as well.30 In the cell viability assay, peptide 1 reduced the Aβ1–42 toxicity by 43% at 10 μM and 65% at 20 μM. Among the newly synthesized pentapeptide analogues, four peptides (6, Pro–Val–Val–Ile–Ala), (9, Gly–Gly–Val–Ile–Ala), (15, Gly–Phe–Val–Ile–Ala), and (21, Gly–Val–Aib–Ile–Ala) were found to prevent Aβ1–42-induced toxicity in the range of 90–100%. Cell samples treated with pre-monomerized Aβ1–42 (2 μM), showed a viability of about 70% relative to untreated cells (control, taken as 100%). However, in the presence of peptides 6 (10 μM), 9 (20 μM), and 15 (20 μM), incubated along with Aβ1–42, no loss of cell viability was observed. While, in the presence of the peptide 1, only 89.5% cells were observed to be viable. In the co-presence of peptide 21 (10 μM), about 97% cells were viable. Also, in the presence of peptides 13 (Gly–Pro–Val–Ile–Ala, 20 μM), 14 (Gly–Aib–Val–Ile–Ala, 20 μM), and 29 (Gly–Val–Val–Ile–Gly, 2 μM), about 88%, 93.4%, and 89.5% cells were observed to be viable, respectively. Table 1 shows the cell viability values of the control (untreated cells) and, PC-12 cells treated with Aβ, with and without the presence of pentapeptides. Cell viability values were correspondingly converted into the percentage inhibition values of Aβ toxicity.
| No. | Sequence | Tested concentration range of pentapeptides | |||||
|---|---|---|---|---|---|---|---|
| Cell viability | Inhibitiona (%) of Aβ1–42 (2 μM) | ||||||
| Peptides | 20 μM | 10 μM | 2 μM | 20 μM | 10 μM | 2 μM | |
| a Each experiment was performed in triplicates (n = 3). ODs (absorbance) of samples with untreated cells were set to 1. Taking the cell viability of untreated cells as 100, percentage cell viabilities were calculated for the cell samples treated with Aβ alone or plus the test peptides. Subsequently, the percentage inhibition of Aβ toxicity by each test peptide was calculated by using the formula: 100 × [OD570 (test peptide with Aβ1–42) − OD570 (Aβ1–42)]/[OD570 (control) − OD570 (Aβ1–42)]. Blank ODs were subtracted from each sample OD and the triplicate ODs were averaged. In a subset of triplicate wells, ODs did not deviate much from the mean and SD ranged between 1.68 and 4.40.b Aib denotes aminoisobutyric acid. | |||||||
| 1 | Gly–Val–Val–Ile–Ala | 89.5 | 82.9 | 73.6 | 65.0 | 43.0 | 12.0 |
| 2 | Val–Val–Val–Ile–Ala | 71.5 | 70.0 | 70.0 | 5.0 | 0 | 0 |
| 3 | Ala–Val–Val–Ile–Ala | 70.0 | 70.0 | 70.0 | 0 | 0 | 0 |
| 4 | Leu–Val–Val–Ile–Ala | 70.6 | 72.1 | 70.0 | 2.0 | 7.0 | 0 |
| 5 | Ile–Val–Val–Ile–Ala | 70.0 | 70.0 | 70.0 | 0 | 0 | 0 |
| 6 | Pro–Val–Val–Ile–Ala | 72.1 | 100 | 88.0 | 7.0 | 100 | 60.0 |
| 7 | Aibb–Val–Val–Ile–Ala | 70.0 | 70.0 | 70.3 | 0 | 0 | 1.0 |
| 8 | Phe–Val–Val–Ile–Ala | 70.0 | 70.0 | 70.0 | 0 | 0 | 0 |
| 9 | Gly–Gly–Val–Ile–Ala | 100 | 85.0 | 84.7 | 100 | 50.0 | 49.0 |
| 10 | Gly–Ala–Val–Ile–Ala | 70.0 | 70.0 | 70.0 | 0 | 0 | 0 |
| 11 | Gly–Leu–Val–Ile–Ala | 70.0 | 70.0 | 70.3 | 0 | 0 | 0 |
| 12 | Gly–Ile–Val–Ile–Ala | 70.0 | 70.0 | 72.1 | 0 | 0 | 7.0 |
| 13 | Gly–Pro–Val–Ile–Ala | 88.0 | 82.0 | 70.3 | 60.0 | 40.0 | 1.0 |
| 14 | Gly–Aib–Val–Ile–Ala | 93.4 | 75.1 | 73.6 | 78.0 | 17.0 | 12.0 |
| 15 | Gly–Phe–Val–Ile–Ala | 100 | 84.7 | 73.3 | 100 | 49.0 | 11.0 |
| 16 | Gly–Val–Gly–Ile–Ala | 70.0 | 70.0 | 70.3 | 0 | 0 | 0 |
| 17 | Gly–Val–Ala–Ile–Ala | 70.0 | 70.0 | 70.0 | 0 | 0 | 0 |
| 18 | Gly–Val–Leu–Ile–Ala | 78.1 | 77.2 | 74.8 | 27.0 | 24.0 | 16.0 |
| 19 | Gly–Val–Ile–Ile–Ala | 78.4 | 84.4 | 84.4 | 28.0 | 48.0 | 48.0 |
| 20 | Gly–Val–Pro–Ile–Ala | 72.7 | 71.2 | 71.8 | 9.0 | 4.0 | 6.0 |
| 21 | Gly–Val–Aib–Ile–Ala | 88.3 | 97.0 | 85.6 | 61.0 | 90.0 | 52.0 |
| 22 | Gly–Val–Phe–Ile–Ala | 71.8 | 70.0 | 70.0 | 6.0 | 0 | 0 |
| 23 | Gly–Val–Val–Val–Ala | 79.6 | 82.3 | 82.0 | 32.0 | 41.0 | 40.0 |
| 24 | Gly–Val–Val–Ala–Ala | 81.4 | 80.8 | 78.7 | 38.0 | 36.0 | 29.0 |
| 25 | Gly–Val–Val–Leu–Ala | 71.5 | 70.0 | 70.0 | 5.0 | 0 | 0 |
| 26 | Gly–Val–Val–Pro–Ala | 70.0 | 70.0 | 71.2 | 0 | 0 | 4.0 |
| 27 | Gly–Val–Val–Aib–Ala | 79.0 | 74.5 | 73.9 | 30.0 | 15.0 | 13.0 |
| 28 | Gly–Val–Val–Phe–Ala | 76.9 | 75.7 | 74.5 | 23.0 | 19.0 | 15.0 |
| 29 | Gly–Val–Val–Ile–Gly | 74.5 | 82.3 | 89.5 | 15.0 | 41.0 | 65.0 |
| 30 | Gly–Val–Val–Ile–Val | 74.8 | 73.3 | 72.7 | 16.0 | 11.0 | 9.0 |
| 31 | Gly–Val–Val–Ile–Leu | 70.4 | 72.0 | 70.0 | 4.0 | 1.0 | 0 |
| 32 | Gly–Val–Val–Ile–Ile | 70.6 | 70.0 | 72.1 | 2.0 | 0 | 7.0 |
| 33 | Gly–Val–Val–Ile–Phe | 84.7 | 82.6 | 82.3 | 49.0 | 42.0 | 41.0 |
| Control | 100 | ||||||
| Aβ1–42 | 70.0 | ||||||
Seven other pentapeptides 18 (Gly–Val–Leu–Ile–Ala), 19 (Gly–Val–Ile–Ile–Ala), 23 (Gly–Val–Val–Val–Ala), 24 (Gly–Val–Val–Ala–Ala), 27 (Gly–Val–Val–Aib–Ala), 28 (Gly–Val–Val–Phe–Ala), and 33 (Gly–Val–Val–Ile–Phe) showed moderate inhibition (21–50%) of Aβ1–42 toxicity while rest of the peptides did not show any inhibition activity. Pentapeptides 6, 9, and 15 showing complete protection for PC-12 cells against Aβ1–42 aggregation related toxicity were also tested against shorter and relatively lesser aggregating peptide Aβ1–40. However, none of the peptides showed inhibition potential towards Aβ1–40 toxicity in PC-12 cells (data not shown). The cell viabilities did not improve when the peptides 6, 9 or 15 were incubated with Aβ1–40 indicating their inefficiency towards attenuation of Aβ1–40 assembly. Fig. 1 represents effects of the most active pentapeptides on the restoration of cell viabilities against the toxicity of Aβ1–42 peptide.
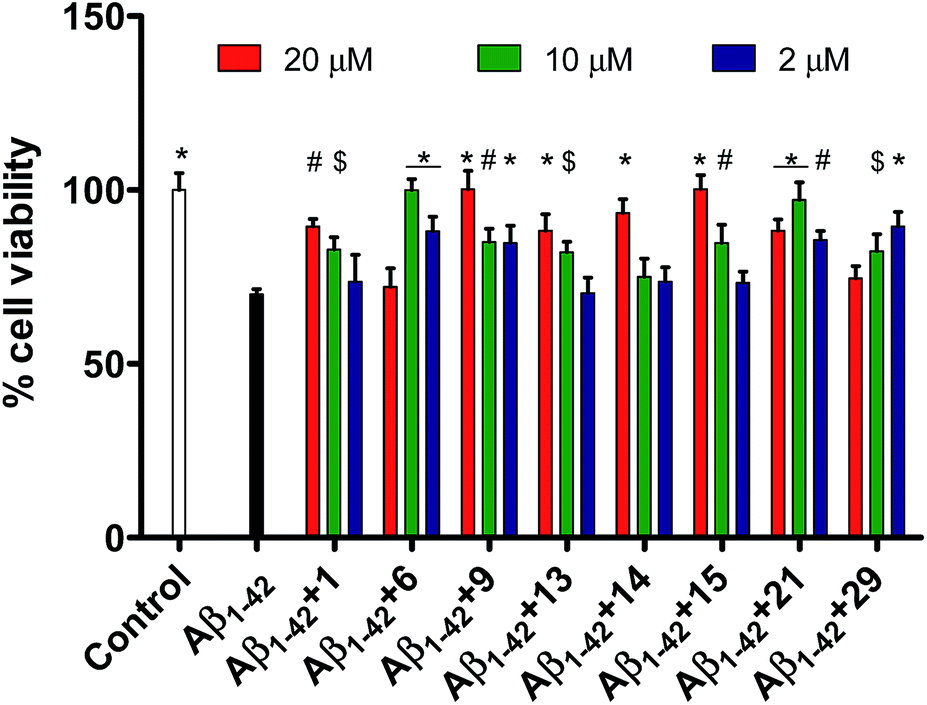 | ||
| Fig. 1 Effects of pentapeptides 1, 6, 9, 13, 14, 15, 21, and 29 on the Aβ1–42-induced cytotoxicity in PC-12 cells. Cells were treated with Aβ1–42 (2 μM) alone or Aβ1–42 plus 1, 6, 9, 13, 14, 15, 21, and 29 (2–20 μM) for 6 h, after which their ability to reduce the MTT was measured. Control represents the untreated cell samples while Aβ1–42 bar represents the cell viability upon treatment with Aβ1–42 peptide. Subsequent bars represent the cell samples where the pentapeptides 1, 6, 9, 13, 14, 15, 21, or 29 were co-incubated along with Aβ1–42 peptide. Viabilities are expressed as percentage of untreated cells (control). Error bars represent mean ± standard deviation (SD, n = 3). Data were analyzed by one way ANOVA test followed by Dunnett's multiple comparison test ($p < 0.05, #p < 0.01, *p < 0.001, vs. Aβ1–42) using software (Graph pad Prism, ISI, San Diego, CA). | ||
In ThT assay, significant reduction in the ThT fluorescence was observed when the pentapeptides 6, 9, 14, 15 or 21, were co-incubated with the monomeric Aβ1–42 peptide. Fig. 2A shows the bar graph representation of the relative fluorescence exhibited by ThT when incubated with Aβ1–42 alone, and plus the inhibitor pentapeptides. In the ThT assay, reporting % inhibition of Aβ1–42 along with RFU values provide better correlation of results. To convert the RFU values into % inhibition values, we also determined and reported RFU values of ThT alone. Relative to blank wells, the enhanced fluorescence shown by Aβ1–42 sample alone was taken as 100% and RFU values were calculated for the dye alone and samples containing inhibitor peptides co-incubated with Aβ1–42. ThT incubated alone (control) exhibited a RFU of nearly 27%. It was observed that the RFU values in the presence of the inhibitor peptides were similar to that of the control. For example, in the presence of the peptides, 6, 9, and 15, RFU values of 28.5%, 27%, and 27% were observed at 10, 20, and 20 μM, respectively. Similarly, samples containing 14 and 21 showed RFU of 34.3% and 49.7% at 20 and 10 μM, respectively (Table T1, ESI†). Reduction in ThT fluorescence in the co-presence of pentapeptides 6, 9, 14, 15 or 21 with Aβ1–42 clearly indicates the inhibition of self-assembly of Aβ1–42 and provides support to the results obtained in the MTT viability assay. Similar to the MTT assay, none of the tested pentapeptides showed inhibitory activity against the shorter form of amyloid peptide (Aβ1–40) in the ThT assay (data not shown). The RFU values remained close to that of Aβ1–40 alone, indicating the presence of aggregates and the inefficiency of the peptides to inhibit the aggregation of Aβ1–40 peptide. However, the inhibitor peptide 6 was found to reduce the aggregation of 11-residue Aβ peptide fragment (Aβ25–35) by 98%, while the other four pentapeptides (9, 14, 15 and 21) didn't show any significant activity (Fig. 2B, Table T2, ESI†). The control exhibited RFU as 57%, and in the co-presence of peptide 6 along with Aβ25–35, RFU value of 57.7% was observed. Using the ThT fluorescence measurement assay, we also studied the pentapeptide 15 in a time-dependent manner against the Aβ1–42 aggregation-mediated fluorescence. The peptide 15 was incubated with the Aβ1–42 peptide and fluorescence was measured at regular intervals for a total duration of 168 h. As shown in Fig. 3, in the absence of 15, when Aβ1–42 was aged alone with ThT dye, there was a dramatic enhancement in the fluorescence indicating the aggregation of Aβ1–42 peptide. The fluorescence reached a plateau at about 120 h. The enhancement in fluorescence observed in the Aβ1–42 sample relative to control (ThT alone) at saturation point was considered as 100%, and the relative fluorescence (RFU) values were calculated for the inhibitor peptide 15 co-incubated with the aggregating Aβ1–42 peptide. The fluorescence shown by the blank wells were subtracted from the test samples. In the presence of peptide 15, fluorescence measurements showed RFU of 62% at 12 h (Fig. 3).
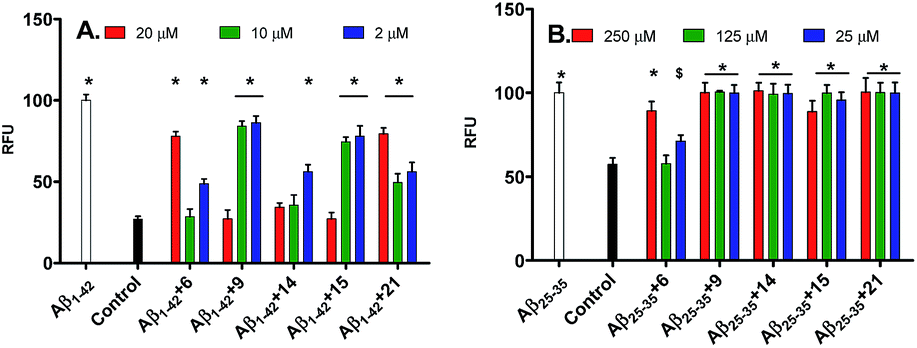 | ||
| Fig. 2 Effects of the pentapeptides 6, 9, 14, 15 and 21 on (A) Aβ1–42 and (B) Aβ25–35 aggregation-mediated ThT fluorescence. Control represents the dye alone while Aβ1–42 and Aβ25–35 represents the Aβ peptides incubated along with the dye. Subsequent bars represent the inhibitor peptides (indicated by numbers), co-incubated with the corresponding Aβ peptides and ThT dye. Error bars represent mean ± SD (n = 3). Data were analyzed by one way ANOVA test followed by Dunnett's multiple comparison test ($p < 0.05, #p < 0.01, *p < 0.001, vs. control) using software (Graph pad Prism, ISI, San Diego, CA). | ||
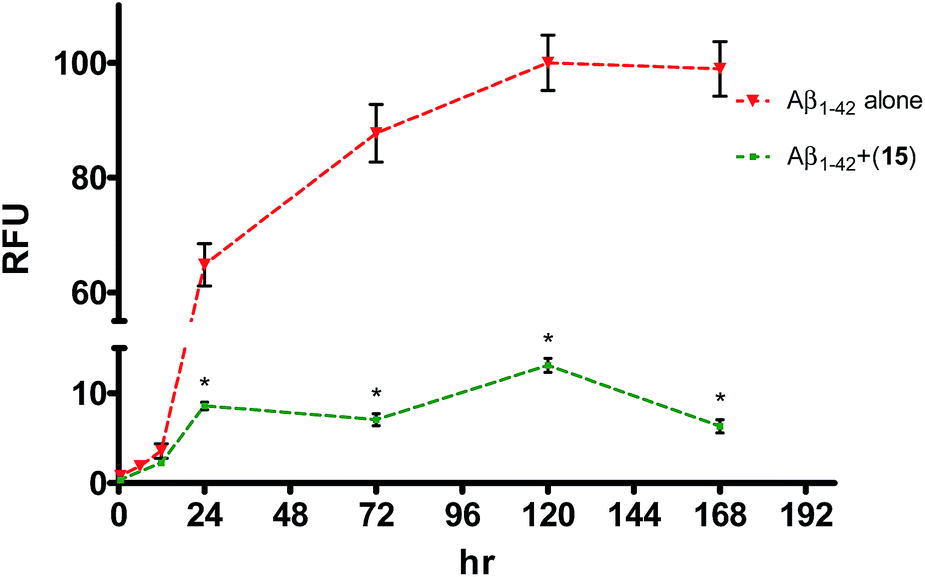 | ||
| Fig. 3 Effects of pentapeptide 15 on Aβ1–42 peptide aggregation, studied in a time-dependent manner for 7 days. Red line shows the Aβ1–42 peptide aged alone. Green line represents Aβ1–42 peptide co-incubated with peptide 15. Error bars represent mean ± SD (n = 3). Data were analyzed by t-test (*p < 0.001, vs. Aβ1–42) using software Graph pad Prism, ISI, San Diego, CA. | ||
At 24 h, peptide 15 co-incubated samples showed only a relative fluorescence of only 15%, while RFU values of 8%, 13% and 6%, were observed at the end of 72, 120, and 168 h, respectively. Summarily, in the presence of peptide 15, very low RFU values (6–15%) were observed especially once the fluorescence reached its saturation point (24–168 h). Since, the fluorescence values correspond to the aggregation state of the Aβ peptide, retardation in the fluorescence indicates the ability of peptide 15 to interfere with the Aβ1–42 peptide self-assembly.
CD spectroscopy was performed to study the inhibitory effects of pentapeptide 15 on the conformational transition of Aβ1–42 peptide. Monomeric Aβ1–42 incubated alone, showed a conformational change from random coil towards an increase in the β-sheet content indicated by the shift from a sharp minima at 198 nm to broad negative peak at 217–218 nm (Fig. 4). At the start, Aβ1–42 occupied a conformation as a mixture of 61.8% of random coil, 28.7% β-turn and only 9.5% of β-sheet, while at the end of 12 h of incubation, Aβ1–42 exhibited about 47.1% of β-sheet component, with the random coil being only 42.3%. However, after 12 h (t12 h) of co-incubation with peptide 15, Aβ1–42 exhibited only 16.2% of β-sheet conformation and 59% random coil form (Fig. 4). Thus the prevention of conformational transition towards β-sheet content is a direct indication of the inhibitory activity of the pentapeptide 15.
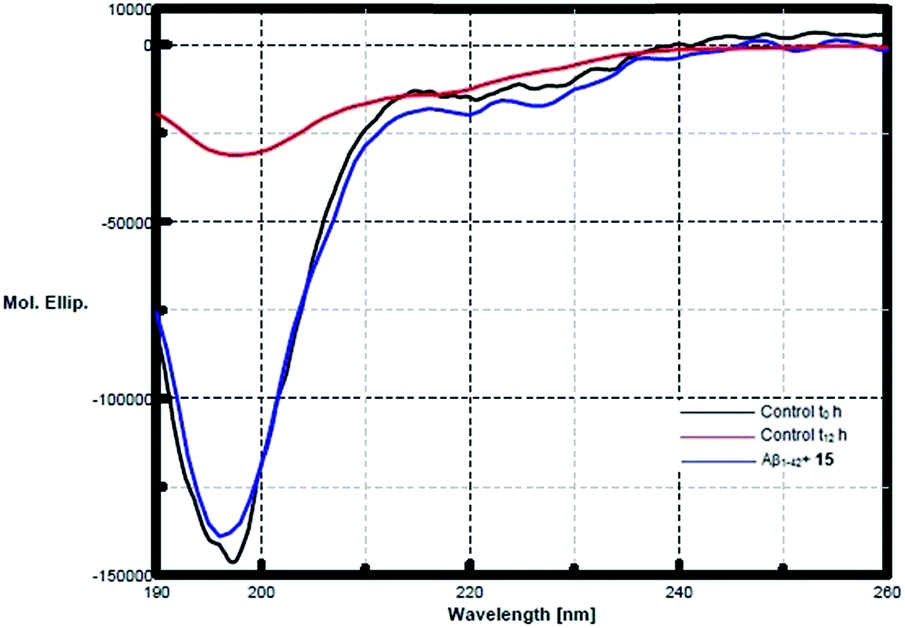 | ||
| Fig. 4 CD spectrum shows the inhibitory effects of pentapeptide 15 on the conformation of Aβ1–42. Conformational transitions of Aβ1–42 at t0 h and t12 h are represented by black and red spectral lines, respectively. The clear deepening of red curve after 12 h of incubation represents the increase in β-sheet structure relative to at zero hours. However, blue curve shows no such minima at 217 nm, when Aβ1–42 peptide was co-incubated with peptide 15 at 12 h, indicates the absence of any β-sheet aggregates. | ||
Since the secondary structural analysis presented by CD spectroscopy is a good indicator of the peptide conformational state, a study was performed to investigate the conformational behaviour of peptides 6 and 29 exhibiting inverse relationship of inhibition activity with increasing concentration (10-fold to Aβ1–42) in the MTT assay. Upon deconvolution of the CD spectra, it was observed that the peptide 6 showed an enhancement in the β-sheet content from an initial 13.1% to 46.6% at the end of incubation period. As shown in Fig. S3 (ESI†), the minima in the curve at 217 nm deepens at the 12 h (t12 h) of incubation, relative to the t0 h curve. At the same, time the random coil content decreased from 59.9% to 43.1%. Similarly, pentapeptide 29 also showed an increase in the β-sheet content during incubation (Fig. S4, ESI†). Starting from an initial value of 14%, 44.4% of peptide was observed to be in β-sheet conformation at the end of incubation period while a decrease in the random coil form was observed from 58.3% at t0 h to 45.5% at t12 h. The self-aggregation exhibited by these peptides also explains their higher activity at low concentration and vice versa. We envision that when taken at 10-fold excess, the peptides themselves aggregate, and thus may not be available to interact with the aggregating Aβ peptides in the solution. Hence, enhancement in the β-sheet content of the peptides 6 and 29 as shown by CD spectroscopy provides support to the pattern of results observed in the MTT and ThT.
Finally, visual investigation of the inhibitory effects of peptide 15 on the morphology and abundance of Aβ1–42 fibrils was performed by high resolution transmission electron microscopy (HR-TEM). Shapes and appearance of the fibrils were also examined using scanning transmission electron microscopy (STEM). Peptide 17 observed as inactive in the MTT cell viability assay was selected as a negative control. In the control sample when monomeric Aβ1–42 was incubated alone, we observed long, rod like cylindrical fibrils with no ends in successive images (Fig. 5A, HRTEM and Fig. 6A, STEM). However, no such fibrils were observed when the peptide 15 was co-incubated with the Aβ1–42 peptide (Fig. 5B, HRTEM and Fig. 6B, STEM).
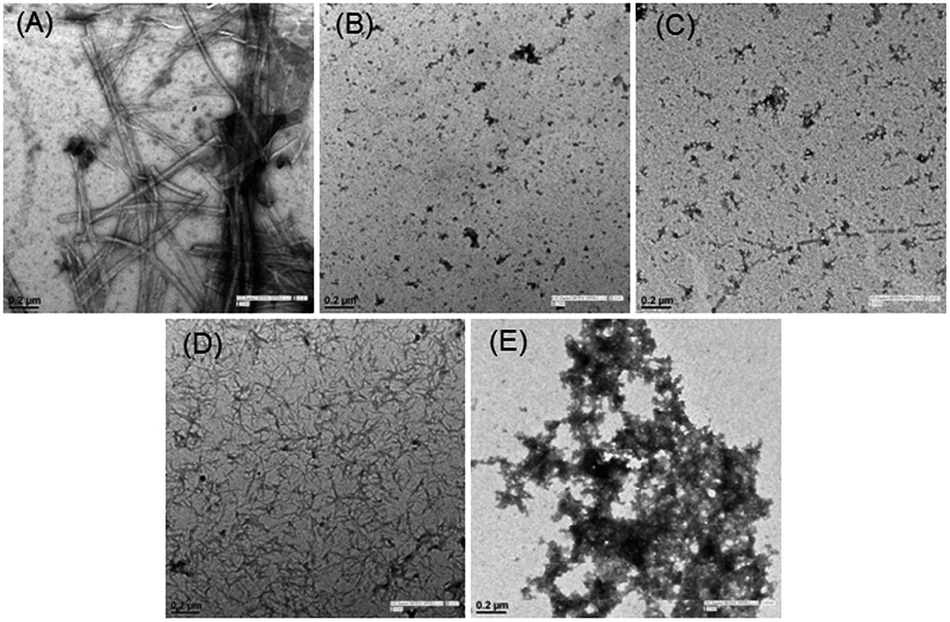 | ||
| Fig. 5 TEM analysis showing the effects of inhibitor peptide 15 and the inactive peptide 17 on aggregation of Aβ1–42 peptide. Aβ1–42 was incubated, (A) alone (B) with inhibitor peptide, 15 (D) with the inactive peptide, 17. Images (C) and (E) correspond to peptides 15 and 17, incubated alone, respectively. The scale bar shows 0.2 μm. | ||
 | ||
| Fig. 6 STEM analysis showing the effects of inhibitor peptide 15 and the inactive peptide 17, on aggregation of Aβ1–42 peptide. Aβ1–42 was incubated, (A) alone (B) with inhibitor peptide, 15 (D) with the inactive peptide, 17. Images (C) and (E) correspond to peptides 15 and 17, incubated alone, respectively. The scale bar shows 500 nm. | ||
Upon incubating peptide 15 alone under similar conditions, no aggregates of fibrillar morphology were observed. Instead very small globular structures were seen (Fig. 5C, HRTEM and Fig. 6C, STEM). An extensive network of very fine thread-like fibrils was observed when the peptide 17 was co-incubated with Aβ1–42 peptide (Fig. 5D, HRTEM and Fig. 6D, STEM). Though the thickness of the fibrils was much lesser than that observed in control (Aβ alone), the complex of fibrils was much more extensive. This probably might have resulted by the endwise association of Aβ monomers in the presence of peptide 17. The peptide 17 was also seen to form large clusters of amorphous aggregates when incubated alone under similar conditions (Fig. 5E, HRTEM and Fig. 6E, STEM). Constituted by highly hydrophobic amino acid residues, the plausible self-aggregation might be the reason for the inactivity of pentapeptide 17. Therefore, the absence of Aβ1–42 fibrils as observed in the electron microscopy study provides further support for the inhibitory activities of pentapeptide 15.
Being a part of the hydrophobic C-terminus of Aβ1–42, the most active pentapeptides 6, 9 and 15 were tested for the self-aggregation related toxicity by MTT cell assay. As described earlier for MTT viability procedure, sampling and assay was adopted for the activity study. The peptides did not show any toxic effect on the cells under study at the highest tested concentration of 20 μM (Fig. S1, ESI†). All sample wells containing peptides 6, 9 and 15 showed MTT reduction quite similar to that of untreated cells (control).
Considering the activity of the lead peptide (1, 65% inhibition at 20 μM) as the reference, inhibition potential of the newly synthesized pentapeptides was correlated with the replacement amino acid residues and structure–activity relationship was established (Fig. 7). The replacement of the first amino acid residue (Gly38), by proline resulted in a very efficient Aβ1–42 polymerization inhibitor. The resulting peptide (Pro–Val–Val–Ile–Ala, 6) prevented the Aβ1–42 aggregation completely. The substitutions at the Val39 residue yielded the best inhibitor peptides. The peptide wherein Val39 was replaced with proline (Gly–Pro–Val–Ile–Ala, 13) showed 60% inhibition of Aβ1–42 fibrillation; the replacement by Aib afforded a peptide (Gly–Aib–Val–Ile–Ala, 14) with significant (78%) inhibition potential. Two more highly promising peptides were discovered by modification at this position with Gly (Gly–Gly–Val–Ile–Ala, 9) and Phe (Gly–Phe–Val–Ile–Ala, 15) residues, exhibiting 100% inhibition of Aβ1–42 aggregation. The peptide synthesized by replacing Val40 with Aib (Gly–Val–Aib–Ile–Ala, 21) showed 90% inhibitory activity. Lastly, most of the replacements at the next two amino acids (Ile41 and Ala42) resulted in the peptides that were only moderately active, showing a maximum of 40% and 65% inhibition, respectively. Conclusively, the substitutions, specifically at the residues, Aβ39→Pro/Aib and Aβ40→Pro/Aib, have resulted in peptides with significant inhibition potential towards the Aβ1–42 peptide self-assembly.
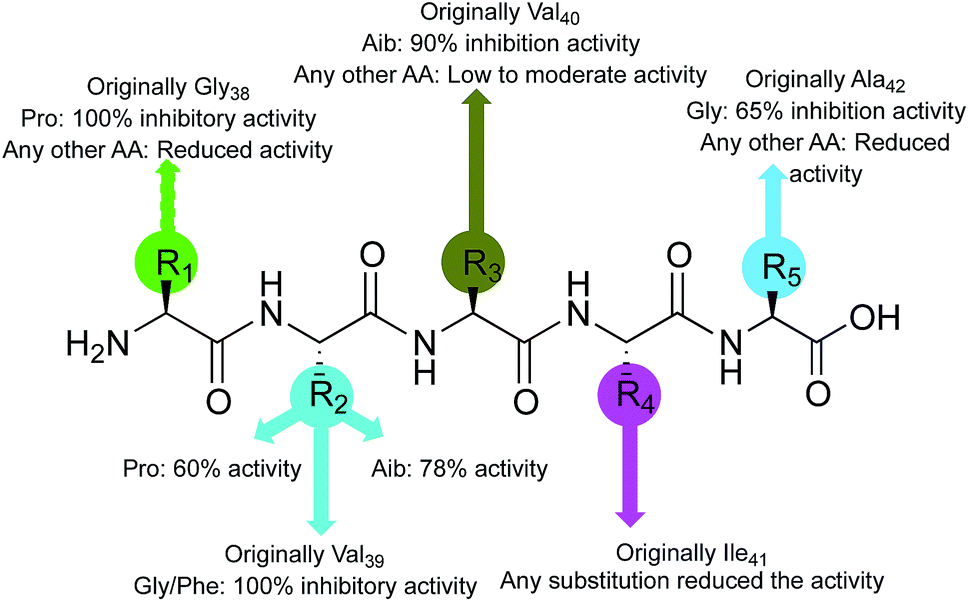 | ||
| Fig. 7 Structure activity relationship of pentapeptides. | ||
Conclusions
More than thirty new peptides were synthesized by performing a full peptide scan on fragment Aβ38–42 of Aβ1–42. The synthesized pentapeptides were bio-evaluated for their inhibitory activity against the aggregation of Aβ peptides. Three of the best peptides identified in the initial screening by MTT assay, showing significant to complete reduction in amyloid toxicity were taken forward for further studies. ThT fluorescence assay results were found to be in good agreement with the cell-based MTT assay. CD spectral analysis and electron microscopy further provided confirmative supports to the observations. As observed in several studies earlier, the selective activity of pentapeptides (6, 9, 15) against the longer form (Aβ1–42) than shorter amyloid peptide (Aβ1–40) suggests their probable interactions with the last two residues (Ile41 and Ala42) of hydrophobic C-terminus of Aβ1–42. Further, it is well known that hydrophobic interactions play an important role in peptide aggregation. Two best peptides [GGVIA (9) and GFVIA (15)] consist of only hydrophobic amino acid residues, therefore it is very probable that the interaction of pentapeptide fragments with hydrophobic C-terminus of Aβ may outrace that between Aβ monomers and intercalate between them, leading to the formation of non-fibrillar structures. Another active peptide (PVVIA, 6) with an anti-β-sheet residue (proline) probably interact with Aβ and prevent the formation of cross-β-sheet structures, which is a pre-requisite for Aβ aggregation. We envision that the peptides discovered herein by peptide scan approach, may, with further modifications, yield promising therapeutics to reach to advanced studies for treatment against Alzheimer's disease.Abbreviations
| CD | Circular dichroism |
| DIEA | N,N-Diisopropylethylamine |
| FDA | Food and drug administration |
| Fmoc | 9-Fluorenylmethoxycarbonyl |
| HPLC | High-performance liquid chromatography |
| HR-TEM | High resolution transmission electron microscopy |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2‚5-diphenyltetrazolium]bromide |
| OD | Optical density |
| PC-12 cells | Pheochromocytoma-12 cells |
| RFU | Relative fluorescence unit |
| SD | Standard deviation |
| STEM | Scanning transmission electron microscopy |
| TBTU | O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate |
| TIPS | Triisopropylsilylether |
| ThT | Thioflavin T |
Acknowledgements
Sunil Bansal thanks the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for the award of senior research fellowship.Notes and references
- A. Alzheimer, R. A. Stelzmann, H. N. Schnitzlein and F. R. Murtagh, Clin. Anat., 1995, 8, 429–431 CrossRef CAS PubMed.
- G. S. Bloom, JAMA Neurol., 2014, 71, 505–508 CrossRef PubMed.
- Dementia and risk reduction: an analysis of protective and modifiable factors, Alzheimer's disease international, World Alzheimer Report, 2014, http://www.alz.co.uk/research/WorldAlzheimerReport2014.pdf.
- T. Jiang, J. T. Yu and L. Tan, Curr. Alzheimer Res., 2013, 10, 852–867 CrossRef CAS PubMed.
- S. Sadigh-Eteghad, B. Sabermarouf, A. Majdi, M. Talebi, M. Farhoudi and J. Mahmoudi, Med. Princ. Pract., 2015, 24, 1–10 CrossRef PubMed.
- E. D. Roberson and L. Mucke, Science, 2006, 314, 781–784 CrossRef PubMed.
- B. Vellas, O. Sol, P. Snyder, P. Ousset, R. Haddad, M. Maurin, J.-C. Lemarié, L. Désiré and M. Pando, Curr. Alzheimer Res., 2011, 8, 203–212 CrossRef CAS PubMed.
- https://Merck.ClinicalTrials.gov [website on the Internet] Bethesda, MD, US national library of medicine, 2013, Accessed November 22, 2013, efficacy and safety trial of MK-8931 in participants with prodromal Alzheimer's disease (MK-8931-019) (APECS) updated November 19, 2013. Available from, http://www.clinicaltrials.gov/ct2/show/NCT01953601?term=MK-8931%26rank=3, NLM identifier: NCT01953601.
- R. S. Doody, R. Raman, M. Farlow, T. Iwatsubo, B. Vellas, S. Joffe, K. Kieburtz, F. He, X. Sun, R. G. Thomas and P. S. Aisen, N. Engl. J. Med., 2013, 369, 341–350 CrossRef CAS PubMed.
- G. K. Wilcockemail, S. E. Black, A. H. Balch, D. A. Amato, A. P. Beelen, L. S. Schneider, R. C. Green, E. A. Swabb and K. H. Zavitz, Alzheimer's Dementia, 2009, 5, 86 CrossRef.
- J. M. Orgogozo, S. Gilman, J. F. Dartigues, B. Laurent, M. Puel, L. C. Kirby, P. Jouanny, B. Dubois, L. Eisner, S. Flitman, B. F. Michel, M. Boada, A. Frank and C. Hock, Neurology, 2003, 61, 46–54 CrossRef CAS PubMed.
- G. Munch and S. R. Robinson, J. Neural Transm., 2002, 109, 1081–1087 CrossRef CAS PubMed.
- C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q. X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi and C. L. Masters, Arch. Neurol., 2003, 60, 1685–1691 CrossRef PubMed.
- L. Lannfelt, K. Blennow, H. Zetterberg, S. Batsman, D. Ames, J. Harrison, C. L. Masters, S. Targum, A. I. Bush, R. Murdoch, J. Wilson and C. W. Ritchie, Lancet Neurol., 2008, 9, 779–786 CrossRef.
- Safety and efficacy study evaluating TRx0237 in subjects with mild-to-moderate Alzheimer's disease,https://www.clinicaltrials.gov/ct2/show/NCT01689233.
- D. Puzzo, L. Privitera, E. Leznic, M. Fa, A. Staniszewski, A. Palmeri and O. Arancio, J. Neurosci., 2008, 28, 14537–14545 CrossRef CAS PubMed.
- Q. Wang, X. Yu, L. Li and J. Zheng, Curr. Pharm. Des., 2014, 20, 1223–1243 CrossRef PubMed.
- Q. Nie, X.-G. Du and M.-Y. Geng, Acta Pharmacol. Sin., 2011, 32, 545–551 CrossRef CAS PubMed.
- S. Salloway, R. Sperling, R. Keren, A. P. Porsteinsson, C. H. van Dyck, P. N. Tariot, S. Gilman, D. Arnold, S. Abushakra, C. Hernandez, G. Crans, E. Liang, G. Quinn, M. Bairu, A. Pastrak and J. D. Cedarbaum, Neurology, 2011, 77, 1253–1262 CrossRef CAS PubMed.
- A. Mahler, S. Mandel, M. Lorenz, U. Ruegg, E. E. Wanker, M. Boschmann and F. Paul, EPMA J., 2013, 4, 5 CrossRef PubMed.
- E. Gazit, FEBS J., 2005, 272, 5971–5978 CrossRef CAS PubMed.
- A. F. Susanne and W. Dieter, Curr. Pharm. Des., 2012, 18, 755–767 CrossRef.
- A. Esteras-Chopo, M. T. Pastor, L. Serrano and M. Lopez de la Paz, J. Mol. Biol., 2008, 377, 1372–1381 CrossRef CAS PubMed.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. A. Kumar, E. M. Castaño and B. Frangione, Nat. Med., 1998, 4, 822–826 CrossRef CAS PubMed.
- S. Bieler and C. Soto, Curr. Drug Targets, 2004, 5, 553–558 CrossRef CAS PubMed.
- C. Soto and L. Estrada, Subcell. Biochem., 2005, 38, 351–364 CAS.
- Safety Study of PPI-1019 in Patients With Mild–Moderate Alzheimer's Disease, 2004, https://clinicaltrials.gov/ct2/show/NCT00100282.
- C. Hilbich, B. Kisters-Woike, J. Reed, C. L. Masters and K. Beyreuther, J. Mol. Biol., 1992, 228, 460–473 CrossRef CAS PubMed.
- E. A. Fradinger, B. H. Monien, B. Urbanc, A. Lomakin, M. Tan, H. Li, S. M. Spring, M. M. Condron, L. Cruz, C. W. Xie, G. B. Benedek and G. Bitan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14175–14180 CrossRef CAS PubMed.
- P. Pratim Bose, U. Chatterjee, C. Nerelius, T. Govender, T. Norström, A. Gogoll, A. Sandegren, E. Göthelid, J. Johansson and P. I. Arvidsson, J. Med. Chem., 2009, 52, 8002–8009 CrossRef PubMed.
- S. Bansal, I. K. Maurya, N. Yadav, C. Thota, V. Kumar, K. Tikoo, V. S. Chauhan and R. Jain, ACS Chem. Neurosci., 2016, 7, 615–623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and purification methods, characterization data and, HRMS and HPLC chromatograms of few representative peptides. General method, assay and statistical calculations for the MTT cell viability and thioflavin T fluorescence assay, circular dichroism and transmission electron microscopy. Bar graph showing cytotoxicity profile of peptides 6, 9, 15 and, CD spectra of peptides 6 and 29 alone, and a zoomed section of CD spectrum of peptide 15 are provided in ESI. See DOI: 10.1039/c6ra26295c |
| This journal is © The Royal Society of Chemistry 2017 |