
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOzonolysis of polycyclic aromatic hydrocarbons in participating solvents†
Anna Lundstedt ,
Matthew J. Webb and
Helena Grennberg*
,
Matthew J. Webb and
Helena Grennberg*
Uppsala University, Uppsala, Sweden. E-mail: Helena.Grennberg@kemi.uu.se
First published on 18th January 2017
Abstract
Seven polycyclic aromatic hydrocarbon (PAH) compounds that can be considered small models for graphene edges have been treated with ozone in solution. The presence of participating solvents such as water or methanol had a pronounced influence on conversion and identity of the functional groups formed, whereas the regioselectivity of the ozonation remained unaffected. Six previously unreported compounds have been isolated from the ozonolysis of pyrene 1, perylene 2 and benzo[e]pyrene 4. Comparison of the experimental data with calculated local ionization energy surfaces (IES) shows a good correlation, and indicates that this computational tool would be useful to predict the regioselectivity of ozone also for larger PAHs, including graphene and graphene nanoribbons.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) constitute a group of compounds of interest in many different fields such as organic dyes, ligands for asymmetric catalysis, pharmaceuticals, agrochemicals and have the potential to be used as both organic conductors or semiconductors,1–3 but also as models for graphene and graphene nanoribbons (GNRs).4 In previous work in our group, graphene on graphite and graphene on SiC was treated with ozone under dry and aqueous conditions, and an edge selectivity for the reaction is observed with introduction of ether/phenolic components and ester/carboxylic groups, leaving the bulk of the material unchanged.5–7 Calculations show that the differences in thermodynamic reactivity between the edges and the internal plane for graphene for cycloaddition reactions are large, with reaction at internal positions leading to significantly less stable states than reaction at edge positions.8,9PAHs are constructed of sp2 hybridized carbons in fused aromatic motifs, and can therefore be referred to as the smallest and most well-defined members of the nanographene family. Several graphene edge motifs are readily available (Chart 1), which opens for studies of structure-related reactivity and regioselectivity using soluble models.
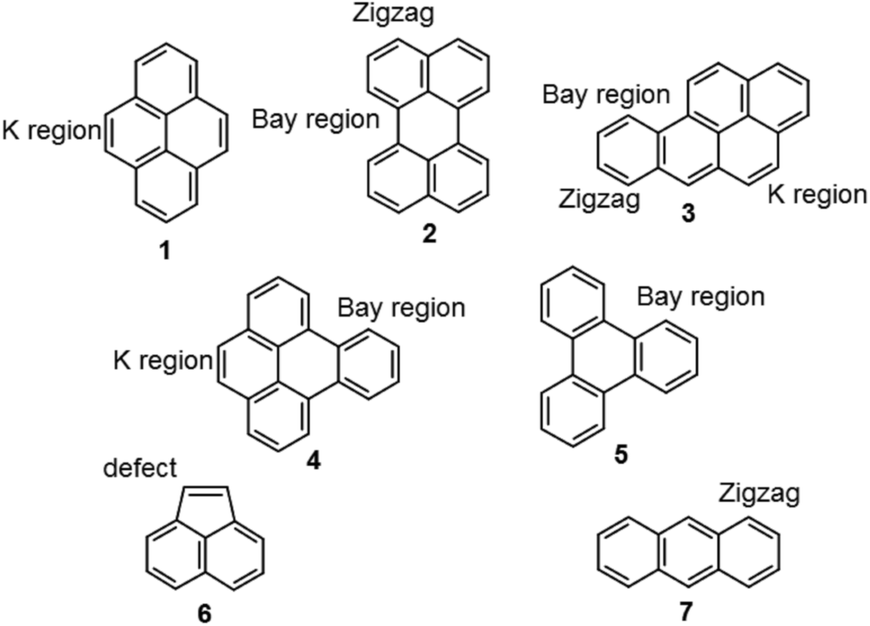 | ||
| Chart 1 Pyrene (1), perylene (2), benzo[a]pyrene (B[a]P, 3), benzo[e]pyrene (B[e]P, 4), triphenylene (5), acenaphthylene (6) and anthracene (7). | ||
In organic synthesis, ozone is used for oxidative cleavage (ozonolysis) of alkenes.10 Ozonation of PAHs, has been studied in the context of oxidative degradation in the environment,11 but also in a more synthetic perspective.12,13 Reactions between ozone and PAHs display regioselectivity as well as position-dependent variations in which functional groups that are formed.14–17 Extrapolating these observations to hydrogen-terminated graphene and graphene nanoribbons (GNRs) indicates a great potential for selective edge functionalizations leading to chemical differentiation of graphene edges. Selective functionalization of graphene is of interest both from a fundamental perspective as well as for application development.18,19
In order to evaluate and rationalize the influence of the edge structure1 seven different PAHs were chosen; pyrene (1), perylene (2), benzo[a]pyrene (B[a]P, 3), benzo[e]pyrene (B[e]P, 4), triphenylene (5), acenaphthylene (6) and anthracene (7) (Chart 1). Between them, PAHs 1–7 represent the most common edge motifs for graphene,20 namely; k region (armchair), bay region (armchair), zigzag region and five ring defect, in different combinations. Pyrene 1 has k regions, perylene 2 has zigzag and bay regions, B[a]P 3 has zigzag, k region and a bay region, B[e]P 4 has bay regions and a k region, triphenylene 5 has only bay regions, acenaphthylene 6 has a five ring with a localized double bond and anthracene 7 has zigzag edges.
Here, the regioselectivity of the reaction between PAHs and ozone in setups that can be adapted also to suspensions of graphene, the types of functionalities obtained and their dependence on edge structure and participatory solvents are discussed and correlated to calculated local ionization energy surfaces (IES) of the experimentally investigated PAHs.
Materials and methods
Ozone was generated by a corona discharge generator and transported in a dioxygen gas stream to the relevant reaction vessel (2 or 20 mL) via Teflon tubing and a stainless steel syringe. All the reactions were performed at 20 °C and the specified reaction time denotes the time that ozone was delivered to the vessel.Ozonides can undergo thermal decomposition and to minimize the risk, only small scale reactions (≤0.1 mmol) were performed and safety guidelines for working with peroxides were followed.21 Dichloromethane was used as solvent since it is ozone stable, dissolves PAHs 1–7 well and is a polar aprotic solvent. In the reactions with water as participating solvent, acetonitrile (which is ozone stable) was added to the mixture to support a homogeneous solution. Dichloromethane and methanol are miscible.
The overall conversion of starting material and the conversion to products were determined by analyzing the crude reaction mixtures by quantitative 1H NMR spectroscopy with 1,2-dichloroethane as internal standard (qNMR). The results reveal the formation of the initial products in the oxidative degeneration pathway for PAHs by ozone, as well as structure related differences in reactivity. The major products for each condition and PAH were isolated using preparative HPLC and structurally determined using NMR spectroscopy and HRMS or, if previously reported, NMR spectroscopy and MS. The structurally determined products are shown in Chart 2.
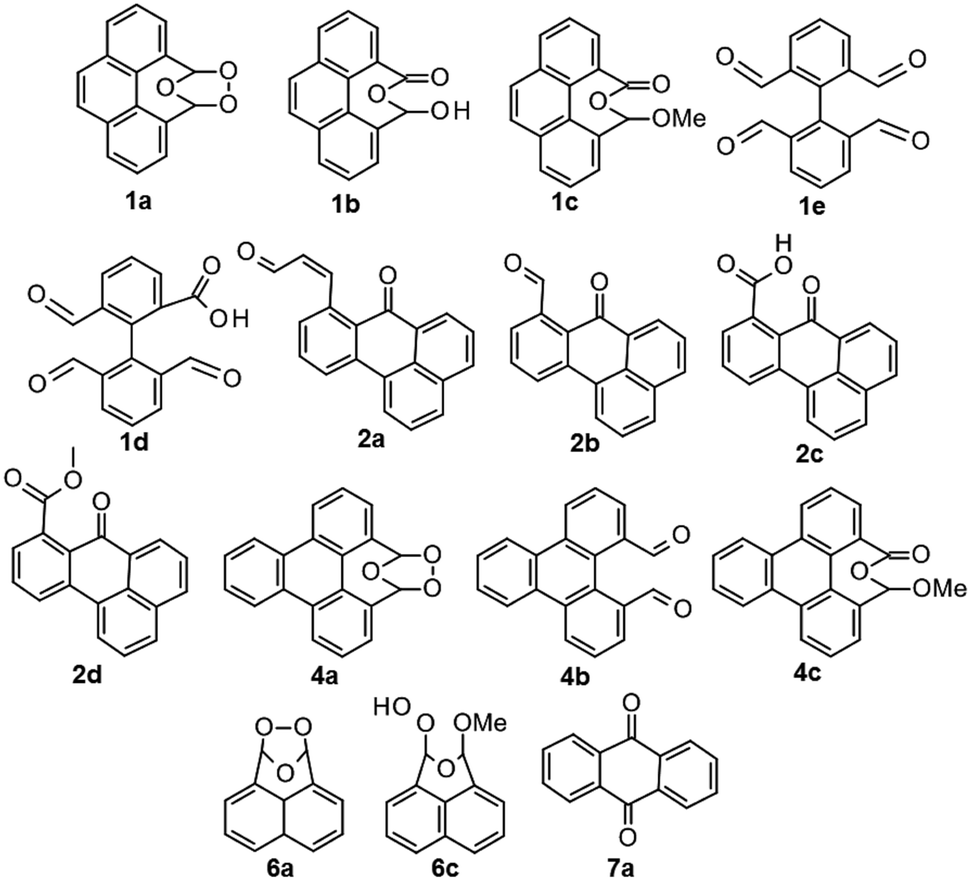 | ||
| Chart 2 The products from ozonolysis of PAHs 1, 2, 4, 6 and 7 using either water or methanol as participating solvents or under dry conditions in DCM. The previously unreported compounds are 1d, 2a–d, 4a. | ||
Results and discussion
Standard ozonolysis of PAHs
The baseline experimental procedure, without any participating solvents, involved ozonation in dry dichloromethane at 20 °C with no additional reagents during the work up procedure. This gave the mono-ozonides 1a and 4a as major products for pyrene 1 and B[e]P 4, both having k regions. The ozonolysis of perylene 2 lead to the generation of a black precipitate when the solvent was removed. This black solid was not soluble in any of the attempted solvents and are suspected to be a polymer (polyozonide) similar to the polymer that are reported to form upon treating acenaphthylene 6 with ozone.22 No perylene ozonide was detected in the crude reaction mixture (of what was still soluble). The soluble products was isolated using prep HPLC and identified as unsaturated aldehyde 2a, carboxylic acid 2c and aldehyde 2b from cleaving the inside of one of the bay regions.Triphenylene 5 was the most ozone-stable compound, but with longer reaction times than for the other PAHs, it too was eventually degraded but not to products in amounts detectable by 1H NMR spectroscopy. For acenaphthylene 6, a white precipitate formed during the reaction under dry conditions and full conversion was very fast but no products were detected by 1H NMR spectroscopy. According to literature,22 this white insoluble powder is a polyozonide. With B[a]P 3 a mixture of many products was quickly obtained. Since, the 1H NMR spectrum was very crowded, the crude mixtures were not analyzed by qNMR spectroscopy nor were any major products isolated (see ESI, Fig. S4†). The difficulty of obtaining products in isolatable amount by ozonolysis of B[a]P 3 have previously been noted by Fieser23 though, isolated compounds, mainly different quinones, have been reported in poor yields.17,24 Being highly carcinogenic and not providing easily interpreted information, B[a]P 3 was removed from further studies. Anthracene 7 was reactive, and almost 90% reacted directly and the only detectable product by 1H NMR spectroscopy was anthraquinone 7a in low amounts (15–16%).
Table 1 describes the conditions and results of the dry ozonolysis of selected PAHs for which products were isolated and characterized.
| Scale, mmol | V, mL | Ozone time, s | Substrate (conversion of %) | Product (conversion to %) |
|---|---|---|---|---|
| a The 0.01 mmol scale experiments were performed by dissolving 0.01 mmol PAH in 2 mL DCM-d2, followed by ozone treatment and leaving the sample for 30 min at 20 °C, then half the sample was transferred to a NMR tube and 1,2-DCE was added and half was treated with as much ozone again before it too was transferred to a NMR tube and 1,2-DCE added.b No new peaks in the 1H NMR spectrum. | ||||
| 0.01 | 2 | 5 | 1 (40) | 1a (41) |
| 0.025 | 4 | 25 | 1 (80) | 1a (77) |
| 0.01/2a | 2/2 | +5 | 1 (91) | 1a (73) |
| 0.025 | 4 | 25 | 2 (95) | 2a (12), 2b (2), 2c (12) + polyozonide |
| 3 | Too many products | |||
| 0.01 | 2 | 5 | 4 (40) | 4a (32) |
| 0.025 | 4 | 25 | 4 (88) | 4a (71) |
| 0.01/2a | 2/2 | +5 | 4 (98) | 4a (73) |
| 0.01 | 2 | 15 | 5 (29) | No detectableb product |
| 0.025 | 4 | 50 | 5 (54) | No detectableb product |
| 0.01/2a | 2/2 | +15 | 5 (63) | No detectableb product |
| 6 | Polyozonide | |||
| 0.01 | 2 | 5 | 7 (88) | 7a (15) |
| 0.01 | 2 | +5 | 7 (90) | 7a (16) |
From these results, it can be proposed that when treating defect-free graphene and GNRs with ozone under dry conditions, there will be ozonides on the armchair edges and corners and carbonyls that might tautomerize to phenols on the zigzag edges. In previous study where ozone was reacted with graphite5 no ketones were observed, only phenols.
Ozonolysis of PAHs with water as participating solvent
Ozone can decompose in water resulting in a number of reactive oxidative species (e.g. radicals).25 Water can also participate in the reaction by trapping the carbonyl oxide intermediate formed during ozonolysis yielding other ozonolysis products such as aldehydes, ketones, carboxylic acids, oxy-hydroperoxides than the ozonide.10,12,26,27With water as the participating solvent the ozonolysis of the PAHs progressed in a similar fashion as without, the ozonide was still the major product with pyrene 1 and B[e]P 4. From pyrene 1 also the aldehyde 1e, the carboxylic acid 1d and the hydroxy ester 1b are obtained. From B[e]P 4 also the aldehyde 4b is obtained. From perylene 2 the same products are obtained as for dry conditions, the unsaturated aldehyde 2a, carboxylic acid 2c and aldehyde 2b, and in the same low conversions. The black precipitate was also obtained and again no ozonide was detected in the crude mixture. For B[a]P 3 and triphenylene 5 again no major products were isolated. For perylene 2 and acenaphthylene 6 same results as observed from the experiments at dry conditions were obtained, and precipitates are formed during the reaction. Anthracene 7 reacted the same way as for under dry conditions, only with a bit lower conversion to 7a.
Table 2 describes the conditions and results of the ozonolysis of selected PAHs for which products were isolated and characterized when water was used as participating solvent.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN:water (0.025 mmol scale) or DCM-d2:MeCN-d3:D2O (0.01 mmol scale) at 20 °C. All conversions are from quantitative 1H NMR spectroscopy with 1,2-DCE as internal standard and the error is 1%
:MeCN:water (0.025 mmol scale) or DCM-d2:MeCN-d3:D2O (0.01 mmol scale) at 20 °C. All conversions are from quantitative 1H NMR spectroscopy with 1,2-DCE as internal standard and the error is 1%
| Scale, mmol | V, mL | Ozone time, s | Substrate (conversion of %) | Product (conversion to %) |
|---|---|---|---|---|
| a The 0.01 mmol scale experiments were performed by dissolving 0.01 mmol PAH in 2 mL DCM-d2, followed by ozone treatment and leaving the sample for 30 min at 20 °C, then half the sample was transferred to a NMR tube and 1,2-DCE was added and half was treated with as much ozone again before it too was transferred to a NMR tube and 1,2-DCE added.b No new peaks in the 1H NMR spectrum. | ||||
| 0.01 | 2 | 5 | 1 (35) | 1a (22), 1b (11), 1d (<1), 1e (1) |
| 0.025 | 4 | 25 | 1 (85) | 1a (37), 1b (15), 1d (3), 1e (5) |
| 0.01/2a | 2/2 | +5 | 1 (94) | 1a (60), 1b (14), 1d (8), 1e (12) |
| 0.025 | 4 | 25 | 2 (90) | 2a (5), 2b (10), 2c (16) |
| 0.01 | 2 | 5 | 4 (36) | 4a (21), 4b (0), 4d (8) |
| 0.025 | 4 | 25 | 4 (83) | 4a (44), 4b (0), 4d (20) |
| 0.01/2a | 2/2 | +5 | 4 (92) | 4a (36), 4b (5), 4d (0) |
| 0.01 | 2 | 15 | 5 (24) | No detectableb product |
| 0.025 | 4 | 50 | 5 (46) | No detectableb product |
| 0.01/2a | 2/2 | +15 | 5 (60) | No detectableb product |
| 6 | Polyozonide | |||
| 0.01 | 2 | 5 | 7 (87) | 7a (10) |
| 0.01 | 2 | +5 | 7 (90) | 7a (11) |
Mono-ozonide 1a has been reported in yields between 23–52%, and the reduced mono-ozonide 1a (the dialdehyde) in yields of 25–28% from reductive ozonolysis of pyrene 1.28–30 Also tetraaldehyde 1e has been reported as products from the reductive ozonolysis of pyrene 1, in yields between 46–55% (ref. 23 and 31–33) using common reductive work-up procedures and very similar ozonolysis procedures as for the mono-ozonide 1a and dialdehyde synthesis. Unlike the mono-ozonide 1a, the diozonide from ozonolysis of pyrene 1, which upon reduction would yield the tetraaldehyde 1e, is not to be found in the literature. Surprisingly, in our experiments neither the dialdehyde nor the diozonide from ozonolysis of pyrene 1 was observed in the crude reaction mixture under any conditions. An explanation for this discrepancy was found by Vollman et al.,24 who reported that ozonolysis of pyrene during longer times gives the tetraaldehyde 1e upon reduction and shorter times the aldehyde–carboxylic acid (as 1b) upon red work up of the mono-ozonide 1a. Here, no longer times were attempted and all experiments were stopped before 100% conversion of 1, explaining the lack of dialdehyde and di-ozonide.
From these results, it can be proposed that when treating defect free graphene and GNRs with ozone and water, there will be ozonides, aldehydes, hydroxy esters and carboxylic acids on the armchair edges and corners and carbonyls that might tautomerize to phenols on the zigzag edges. Ozone can also degrade in water, forming radical oxidative species,25 these radicals can also be expected to react. In this study with ozone and PAHs, no products from degenerated ozone were observed in the crude NMRs (when compared to NMR spectra predicted in MestReNova 8.1.4).
Ozonolysis of PAHs with methanol as participating solvent
When methanol is used as the participating solvent during ozonolysis of the PAHs significant variations on the standard procedure were observed. Pyrene 1 and B[e]P 4 reacted at the same position as without a participating solvent, however, the ozonides 1a and 4a were obtained in smaller amounts and were no longer the major product. Instead the methoxy ester 1c and 4c from the acid degraded peroxide (degrading in the HPLC by TFA buffer) were identified as the major products. For perylene 2, again the aldehyde 2b, saturated aldehyde 2a, carboxylic acid 2c were obtained, and also the acid-degenerated methanol trapped carbonyl oxide 2d is obtained in small amounts. The black precipitate from perylene 2 still formed. For triphenylene 5, still no major products were isolated. For acenaphthylene 6, the methoxy peroxide 6c was detected and now also the ozonide 6a was obtained and only traces of white polymer precipitate were observed. The mono-ozonide 6a was isolated as a white solid using prep HPLC in 11% yield (qNMR conversion was 17% to 6a). The peroxide 6c was detected in the crude 1H NMR spectrum, attempting to isolate it by preparative HPLC was not successful. Since this compound is known and its 1H NMR shifts,27 qNMR of the crude spectrum could still be done and a conversion of 40% to the methanol trapped peroxide 6c was obtained. Sugimoto et al.,27 reported yields of 13% of 6a and 66% of 6c at 0 °C, and 7% 6a and 69% 6c at −70 °C when performing the ozonolysis of 6 in DCM:MeOH. Ozonolysis of 1 in DCM:MeOH, was also reported in that study, and at 0 °C with a 66% conversion of 1, they isolated ozonide 1a in 9% and 1c in 35%. Anthracene 7 reacted the same way as for under dry conditions.
Table 3 describes the conditions and results of the ozonolysis of selected PAHs for which products were isolated and characterized when methanol was used a participating solvent.
:MEOH (1:1, 0.025 mmol scale) or DCM-d2:MeOD (1:1, 0.01 mmol scale) at 20 °C. All conversions are from quantitative 1H NMR spectroscopy with 1,2-DCE as internal standard and the error is 1%
| Scale, mmol | V, mL | Ozone time, s | Substrate (conversion of %) | Product (conversion to %) |
|---|---|---|---|---|
| a The 0.01 mmol scale experiments were performed by dissolving 0.01 mmol PAH in 2 mL DCM-d2, followed by ozone treatment and leaving the sample for 30 min at 20 °C, then half the sample was transferred to a NMR tube and 1,2-DCE was added and half was treated with as much ozone again before it too was transferred to a NMR tube and 1,2-DCE added.b No new peaks in the 1H NMR spectrum. | ||||
| 0.01 | 2 | 5 | 1 (38) | 1a (9), 1c (22), 1e (0) |
| 0.025 | 4 | 25 | 1 (78) | 1a (18), 1c (29), 1e (0) |
| 0.01/2a | 2/2 | +5 | 1 (88) | 1a (19), 1c (30), 1e (10) |
| 0.025 | 4 | 25 | 2 (92) | 2a (8), 2b (3), 2c (12), 2d (4) |
| 0.01 | 2 | 5 | 4 (38) | 4a (4), 4b (0), 4c (11), 4d (0) |
| 0.025 | 4 | 25 | 4 (78) | 4a (16), 4b (0), 4c (37), 4d (6) |
| 0.01/2a | 2/2 | +5 | 4 (92) | 4a (12), 4b (10), 4c (36), 4d (0) |
| 0.01 | 2 | 15 | 6 (100) | 6a (8), 6c (20) |
| 0.025 | 4 | 50 | 5 (24) | No detectableb product |
| 0.01/2a | 2/2 | +15 | 5 (48) | No detectableb product |
| 0.01 | 2 | 5 | 7 (85) | 7a (17) |
| 0.01 | 2 | +5 | 7 (86) | 7a (17) |
From these results, it can be proposed that when treating defect free graphene and GNRs with ozone and methanol, there will be ozonides, aldehydes, methoxy esters and carboxylic acids on the armchair edges and corners and carbonyls that might tautomerize to phenols on the zigzag edges.
Edge structure–reactivity correlation of ozonolysis of PAHs
In summary, ozone reacts k regions (part of armchair edges) and alkene ‘defect’ motifs, resulting in ozonides, aldehydes, esters and carboxcylic acids depending on reaction conditions (e.g. solvents) when ozone reacts with zigzag motifs, ketones form regardless of conditions.Zigzag edges has been reported to be more reactive towards oxidation than armchair edges.1 The acenes, a group of PAHs which are all linear and contain zigzag edges, have been reported to be very reactive towards ozone. Ozone can undergo electrophilic aromatic substitution (EAS) with zigzag edges and both B[a]P 3 and anthracene 7 have been reported to yield the quinones upon ozonolysis.15,17,24,34
Here, both B[a]P 3 and anthracene 7 was observed to be very reactive towards ozone and from PAH 7, anthraquinone 7a was observed. Pyrene 1 and B[e]P 4 (k region) have a similar reactivity towards ozone, triphenylene 5 (bay region) is less ozone reactive, and anthracene 7 (zigzag) is more reactive than 1 and 4. This corresponds nicely with the study by Pryor et al.16 were the relative ozone reactivity of PAHs, including 1, 2, 5 and 7 in dichloromethane was investigated. The least reactive was PAH 5, followed by 1, 2, then 7 being the most reactive PAH.
For hydrogen-terminated graphene, ozonation could possible lead to chemical differentiation of the edges which could be used for covalent stitching of graphene sheets and a study35 recently showed that the electron mobility of the connection depends on how the edges of two graphene sheets are connected (e.g. zigzag–zigzag or armchair–armchair etc.) as well as the entity connecting the sheets.
Calculated local ionization energy surfaces
Both regiochemical and functional group outcome from the ozone treatment will depend on if the ozone attaches to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (addition) or at a C atom (initial EAS) and ozone can react with sp2 carbon motifs by both,10,34 although the preferred route for alkenes is cycloaddition of the electrophilic ozone 1,3-dipole.36
C bond (addition) or at a C atom (initial EAS) and ozone can react with sp2 carbon motifs by both,10,34 although the preferred route for alkenes is cycloaddition of the electrophilic ozone 1,3-dipole.36
Predicting reactivity for PAHs and possible also for larger systems (e.g. graphene and GNRs) are of importance for development of safe and selective processes. Local ionization energy surfaces (IES) is one computational tool, by which the most nucleophile sites in PAHs can be predicted. Depending on the localization of the minimum, the likely reaction outcome will either be electrophilic aromatic substitution (if the minimum is localized over an atom) or electrophilic addition (if the minimum is localized between two atoms).37
Calculations of local IES for PAHs 1–7 was performed as described by Brown and Cockroft37 and the results compared to the experimental findings (previous sections). Generally, the calculated local IES correlates well to the experimental results. The major products from PAH 1, 4 and 6's were easily obtained under the right conditions; all have one double bond that is clearly more reactive than the others. The major product from perylene 2 was a polymer, possibly this formed from the initial product from ozone either adding to the bond with the lowest IE value or substituted to the atom with the lowest IE value. B[a]P 3, where many different products were observed, have four different bonds with similar IE values, and a zigzag edge. This correlates well with the observation of many different products forming already at short reaction times. Triphenylene 5 has six equal bonds with the same IE values and the observation of no major product, only slow degradation to many minor products is also in accordance with these calculations. Lastly, from anthracene 7 the quinone 7a was the only identified product, although the conversion of 7 was high.
PAHs 1–7 local IES are shown in Fig. 1, and the numbers are the most reactive positions for EAS and oxidative addition.
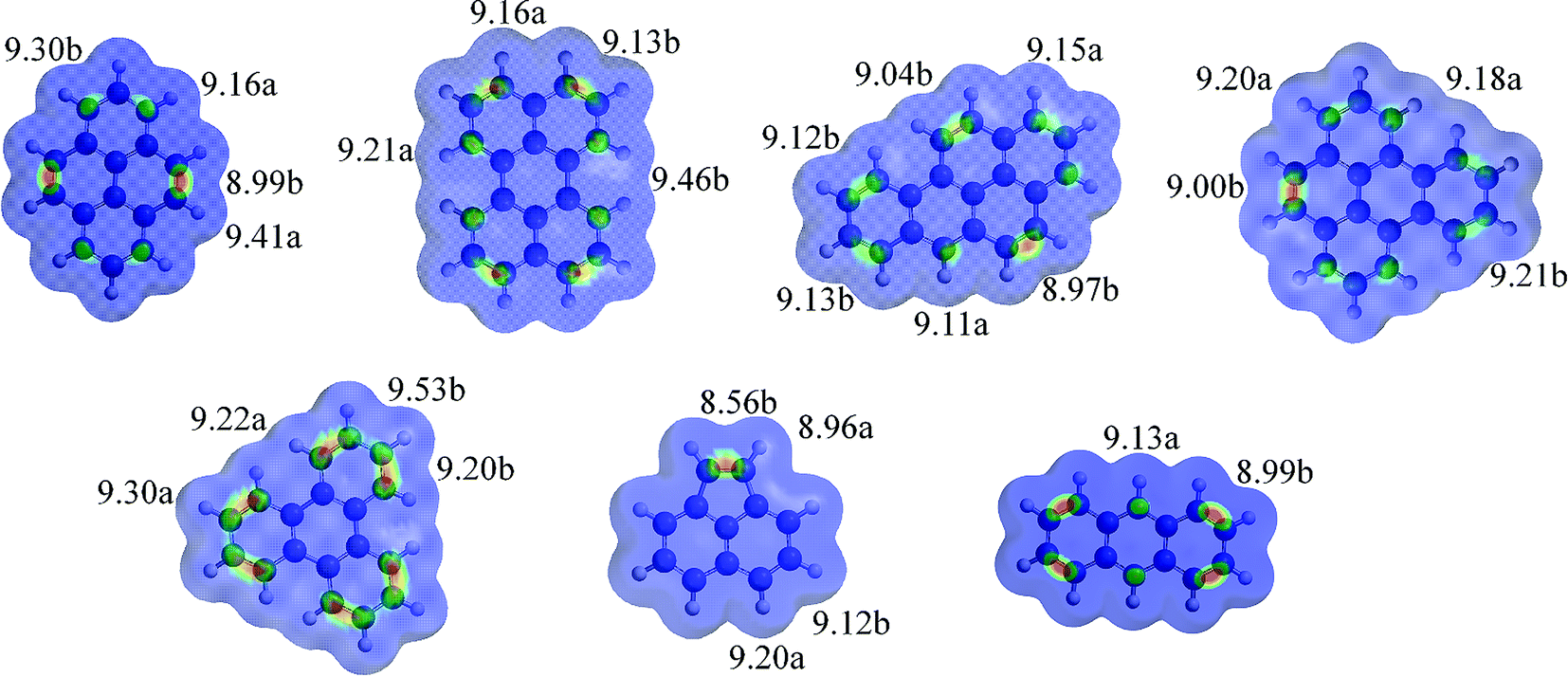 | ||
| Fig. 1 PAHs 1–6 calculate local ionization energy (IE, in eV) surfaces. The values for the two-three lowest IE values for bonds (b) and atoms (a) are shown. The brighter areas between two atoms are predicted to be more reactive toward oxidative addition. Property range: 9.0–9.4 eV. See ESI† for all values. | ||
Conclusions
Six previously unreported compounds have been isolated from the ozonation of pyrene 1, perylene 2, benzo[e]pyrene 4. Studying different PAHs reaction with ozone, calculated local IESs and the literature indicate that ozone reacts with zigzag regions forming ketones regardless of any co-reagents and with the k region of the armchair edge, resulting in ozonide-, aldehyde-, carboxylic acid- or ester-groups depending on co-reagents. Ozone has a preference for zigzag edges over armchair edges. In practice, since ozone reacts with both, the reaction time and relative abundance of the edge motif are key factors for achieving regioselectivity.The calculated local IESs correlates well against the experimental results indicating that this computational tool can be used for predicting regioselectivity of ozone addition to larger PAH systems e.g. hydrogen-terminated graphene and GNRs.
Experimental section
General
All reagents were commercially available and used without further purification. NMR spectra were recorded on a Varian UNITY (1H at 399.97 MHz, 13C at 100.58 MHz) or a Varian INOVA (1H at 499.93 MHz, 13C at 125.71 MHz) spectrometer. The chemical shifts are reported using the residual solvent signal as an indirect reference to TMS 1H: CHCl3: 7.26, acetone-d6: 2.05, DCM-d2: 5.32, MeCN-d3: 1.94 13C: CDCl3: 77.0. The vials used in all experiments were heavy-walled glass Emrys process vials (2–5 mL) sealed with aluminium crimp caps fitted with a septum. Ozone generator (Pilodist OGF 505) settings for all experiments were 5W, 0.25 bar, 0.5 L min−1. See ESI† for HPLC UV traces and NMR spectra.Calibration of the quantitative 1H NMR spectroscopy method
Mixed stock solutions in vials with screw cap. Stored all stock solutions in freezer until use. Stock solution A: weighted 20.0 mg pyrene into a 2 mL vial. Added 1000 μL DCM-d2 to the vial, sonicated until all pyrene dissolved. Stock solution B: added 3 × 1000 μL MeCN-d3 into a 4 mL vial (screw cap), added 10.0 μL 1,2-DCE. Stock solution C: added 4 mL MeCN-d3 into a 4 mL vial.Setting internal standard integral (4H) to 400 resulted in pyrene (4H) integral or pyrene (4H) + 1a (4H) integral values of: 1: 158 (140 + 18), 2: 157 (140 + 17), 3: 154 (138 + 16), 4: 154, 5: 156 (std dev 1%).
Acceptable inaccuracies for precise, accurate quantification using qNMR are 2%.38
Preparative HPLC
As solvents acetonitrile and water was used with 0.01% TFA.For all mixtures the same conditions were used.
After crude 1H NMR spectroscopy, the NMR solution was injected in the prep HPLC. Injected volume: 600 μL, collected volume: 5 mL, flow rate: 20 mL min−1, gradient: 20 to 90 (MeCN) over 20 min, 15 min isocratic (90% MeCN), UV detector: 254 nm, column: C18-PFP, 25 mm, 150 mm, 5 μ, 100 Å. Removed solvent from collected fractions in vacu, redissolved in 0.5 mL acetone-d6.
Calculation of local ionization energy surfaces
The structures was optimized using B3LYP/6-311G(d) with Gaussian 09 rev C.01 and Spartan 10. Local ionization energy surfaces were calculated at the same level using Spartan 10.General procedure “small scale” (0.01 mmol) for quantitative 1H NMR spectroscopy
Stock solutions of PAHs 1, 2, 4 and 5 (0.1 mmol ml−1) using DCM-d2 as solvent were prepared and of internal standard 1,2-DCE (20 μL in 4000 μL) using MeCN-d3 as solvent.PAH stock solution (100.0 μL, 0.01 mmol), internal standard stock solution (100 μL, 0.006 mmol 1,2-DCE) and deuturated solvent (tot. 1800 μL of D2O, MeOD, DCM-d2 and/or MeCN-d3) was added to the vial. The vial was sealed with a cap.
The cap was removed and the reaction mixture bubbled through with ozone (PAH 1: 5 s, 4: 5 s, 5: 15 s) at 20 °C. The mixture was left at 20 °C for 30 min and half the total volume was transferred to an NMR tube. The mixture in the vial was again bubbled through with ozone (1: 5 s, 4: 5 s, 5: 15 s) and left to stand at 20 °C for 30 min before being transferred to a NMR tube. Reference samples for the internal standard were done for all experiments, were O2 was used insted of O3. All NMR samples were analyzed by 1H NMR spectroscopy with a d1 of 30 s.
General procedure “medium scale” (0.025 mmol) for quantitative 1H NMR spectroscopy
To a vial, PAH (0.025 mmol) was added, the vial was sealed with a cap and solvents (tot 4 mL) was added via a syringe through the cap. The reaction mixture was bubbled through with ozone (PAH 1: 25 s, 2: 25 s, 4: 25 s 5: 50 s) at 20 °C. The mixture was left at 20 °C for 30 min and the solvent was removed in vacu and any isolated solid or liquid was re-dissolved in acetone-d6 (1 mL) and 1,2-DCE as internal standard and analyzed by 1H NMR spectroscopy with d1 = 30 s.General procedure “large scale” (0.1 mmol) for isolation of products
To a vial, PAH (0.1 mmol) was added, the vial was sealed with a cap and solvents (total volume: 20 mL) was added via a syringe through the cap. The reaction mixture was bubbled through with ozone (1: 90 s, 2: 90 s, 3: 70 s, 4: 90 s 5: 300 s). The mixture was left at 20 °C for 1–2 hours and left in the freezer (−18 °C) if left over night or longer. The solvent was removed in vacu and all isolated solid or liquid was re-dissolved in acetone-d6 (1.0 mL) and analyzed by 1H NMR spectroscopy.The solution was purified by preparative HPLC.
Additional procedures for the different conditions
:MeCN:H2O (8:11:1).:MeOH (1:1).Characterization data for isolated compounds
Monoozonide 1a. 1H NMR (500 MHz, acetone-d6) δ 8.15 (d, J = 7.2 Hz, 2H), 7.98 (s, 2H), 7.85 (d, J = 7.2 Hz, 2H), 7.73 (dd, J = 7.2 Hz, 2H), 6.89 (s, 1H) ppm.
13C NMR from HMBC (126 MHz, acetone) δ 106.8, 128.5, 128.8, 130.8, 132.8, 135.6, 135.9, 136.4 ppm.
Shifts and assignments in accordance with literature.39
m/z [APCI+]: 251 (M+), 235, 236, 237, 205, 206, 207.
Hydroxy ester 1b. 1H NMR (500 MHz, acetone-d6) δ 8.32 (d, J = 7.7 Hz, 2H), 8.14 (d, J = 7.7 Hz, 2H), 7.97 (s, 2H), 7.86 (dd, J = 7.7 Hz, 2H), 7.41 (s, 1H), 6.79 (s, 1H) ppm.
13C NMR from HMBC (126 MHz, acetone) δ 95.8, 124.1, 124.3, 127.2, 127.5, 127.7, 129.7, 130.6, 132.2, 133.2, 133.5, 133.6, 134.6, 134.8, 137.0, 168.4.
Shifts and assignments in accordance with literature (except the 7.41 PhCHOH shift in the 1H NMR spectrum which was reported at 3.35 and mixed up with the PhCHOH shift at 6.79).40
m/z [APCI+]: 251 (M+), 252, 253.
Methoxy ester 1c. 1H NMR (500 MHz, acetone-d6) δ 8.39–8.31 (m, 2H), 8.17 (dd, J = 7.9, 1.4 Hz, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.99 (ds, 2H), 7.87 (dd + dd, J = 22.3, 7.8 Hz, 2H), 6.40 (s, 1H), 3.79 (s, 3H) ppm.
Shifts and assignments in accordance with literature.41
m/z [APCI+]: 265 (M+), 266, 267.
Trialdehydecarboxylic acid 1d. 1H NMR (500 MHz, acetone-d6) δ 9.74 (s, 2H), 9.68 (s, 1H), 8.41 (d, J = 9.2 Hz, 1H), 8.26 (d, J = 7.7 Hz, 2H), 7.94–7.82 (m, 2H) ppm.
m/z [APCI+]: 283 (M+), 282, 267, 265.
HRMS [TOF MS ES+]: C16H10O5, calculated: 283.0606, found: 283.0597.
Tetraaldehyde 1e. 1H NMR (500 MHz, acetone-d6) δ 9.78 (s, 4H), 8.35 (d, J = 7.7 Hz, 4H), 7.98 (dd, J = 7.7 Hz, 2H) ppm.
Shifts and assignments in accordance with literature.31
m/z [APCI+]: 267(M+), 269.
Unsaturated aldehyde cis-2a. 1H NMR (500 MHz, acetone-d6) δ 9.92 (d, J = 7.8 Hz, 1H), 8.86 (d, J = 15.8 Hz, 1H), 8.81 (d, J = 7.3 Hz, 1H), 8.79 (d, J = 7.8 Hz, 1H), 8.69 (dd, J = 7.3, 1.3 Hz, 1H), 8.26 (d, J = 7.6, 1H), 7.93 (td, J = 7.6, 4.2 Hz, 2H), 7.85 (dd, 1H), 7.77 (d, J = 7.5 Hz, 1H), 6.54 (dd, J = 15.8, 7.8 Hz, 1H) ppm.
13C NMR from HMBC (126 MHz, acetone) δ 194.2, 185.5, 155.2, 139.2, 138.5, 138.3, 136.0, 132.9, 131.5, 130.8, 130.3, 129.5, 129.3, 129.2, 128.1, 128.0, 127.5, 127.0, 126.6, 124.9 ppm.
m/z [APCI+]: 285 (M)+, 286, 287.
HRMS [TOF MS ES+]: C20H12O2, calculated: 285.0916, found: 285.0910.
Aldehyde 2b. 1H NMR (500 MHz, acetone-d6) δ 10.74 (s, 1H), 8.85 (dd, J = 11.9, 8.2 Hz, 2H), 8.72 (dd, J = 7.3, 1.2 Hz, 1H), 8.51 (dd, J = 8.2, 1.2 Hz, 1H), 8.28 (d, J = 8.2 Hz, 1H), 7.97 (dt, J = 14.0, 7.6 Hz, 2H), 7.87 (dd, J = 7.9, 1H), 7.70 (dd, J = 7.7, 1.1 Hz, 1H) ppm.
m/z [APCI+]: 259 (M+), 260.
Carboxcylic acid 2c. 1H NMR (500 MHz, acetone-d6) δ 8.80 (d, J = 7.1 Hz, 1H), 8.72 (dd, J = 8.2, 1.0 Hz, 1H), 8.66 (dd, J = 7.3, 1.3 Hz, 1H), 8.46 (d, J = 9.1 Hz, 1H), 8.25 (d, J = 8.1 Hz, 1H), 7.91 (ddd, J = 8.2, 7.4, 2.1 Hz, 2H), 7.85 (dd, = 7.4, 1H), 7.58 (dd, J = 7.4, 1.0 Hz, 1H) ppm.
m/z [APCI+]: 275 (M)+, 276, 277.
HRMS [TOF MS ES−]: C18H10O3, calculated: 273.0552 (M − H−) found: 273.0553 (M − H−).
Methyl ester 2d. 1H NMR (500 MHz, acetone-d6) δ 8.83 (d, J = 7.4 Hz, 1H), 8.77 (d, J = 8.1 Hz, 1H), 8.69 (dd, J = 7.4, 1.4 Hz, 1H), 8.50 (dd, J = 8.1, 1.4 Hz, 1H), 8.29 (d, J = 8.1 Hz, 1H), 7.95 (dd, J = 8.1 Hz, 2H), 7.88 (dd, 1H), 7.58 (dd, J = 7.4, 1.1 Hz, 1H), 3.98 (s, 3H) ppm.
13C NMR from HMBC (126 MHz, acetone) δ 177.2, 165.5, 131.9, 131.1, 130.8, 130.7, 128.3, 126.2, 125.5, 124.7, 123.1, 123.0, 122.6, 122.6, 122.5, 121.4, 120.4, 119.9, 47.0, ppm.
m/z [APCI+]: 289 (M)+, 290, 291.
HRMS [TOF MS ES+]: C19H12O3, calculated: 289.0865 found: 289.0871.
Benzo[e]pyrene monoozonide 4a. 1H NMR (500 MHz, acetone-d6) δ 9.04 (dd, J = 8.2, 1.5 Hz, 2H), 8.82 (dd, J = 6.3, 3.3 Hz, 2H), 7.86 (dd, J = 8.2, 1.5 Hz, 2H), 7.79 (dd, J = 8.2, 7.2 Hz, 1H), 7.75 (dd, J = 6.3, 3.3 Hz, 1H), 6.87 (s, 1H) ppm.
13C NMR from HMBC (126 MHz, acetone) δ 130.6, 127.0, 122.9, 122.2, 120.7, 130.6, 118.8, 124.6, 122.1, 100.4.
HRMS [TOF MS ES+]: C20H12O3, calculated: 301.0865 found: 301.0866.
Benzo[e]pyrene dialdehyde 4b and benzo[e]pyrene acid-degenerated water trapped carbonyl oxide 4c only as 1H NMR spectra on crude products. Shifts used for qNMR spectroscopy; 4b: 8.79, 8.68, 8.03, 6.16, 4c: 8.74, 7.92 6.20, 5.60, 3.79.
Acenaphthylene monoozonide 6a. Isolated as a white solid (4.5 mg, 11%) by preparative HPLC by following the general procedure for larger scale, performing this in duplicates and pooling the crudes prior to purification.
1H NMR (500 MHz, acetone-d6) δ 8.00 (dd, J = 8.2, 1.2 Hz, 2H), 7.64 (dd, J = 6.9, 1.2 Hz, 2H), 7.59 (dd, J = 8.2, 6.9 Hz, 2H), 6.96 (s, 2H) ppm.
Shifts and assignments in accordance with literature.27
Acenaphthylene methoxyperoxide 6c. Not isolated. Used the NMR literature values for the integration of the qNMR spectrum of the crude product. 1H NMR: δ 3.69 (s, 3H), 6.09 (s, 1H), 6.49 (s, 1H), 7.2–7.9 (m, 6H), 9.45 (s, 1H) ppm.27
Anthraquinone 7a. Not isolated. Used the NMR values form a bought sample of 7a for the integration of the qNMR spectrum of the crude product. 1H NMR: δ 7.81 (m, 4H), 8.32 (m, 4H) ppm.
Conflict of interest
The authors declare no competing financial interests.Acknowledgements
The Swedish Research Council (Vetenskapsrådet), the Uppsala University KoF Priority Project on Graphene, and the Knut and Alice Wallenberg Foundation (KAW graphene) for financial support.References
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS.
- R. G. Harvey, Polycyclic aromatic hydrocarbons, Cambridge University Press, 1991 Search PubMed.
- O. Nachtigall, R. Lomoth, C. Dahlstrand, A. Lundstedt, A. Gogoll, M. J. Webb and H. Grennberg, Eur. J. Org. Chem., 2014, 2014, 966–972 CrossRef CAS.
- P. C. Mishra and A. Yadav, Chem. Phys., 2012, 402, 56–68 CrossRef CAS.
- M. J. Webb, P. Palmgren, P. Pal, O. Karis and H. Grennberg, Carbon, 2011, 49, 3242–3249 CrossRef CAS.
- M. J. Webb, C. Polley, K. Dirscherl, G. Burwell, P. Palmgren, Y. Niu, A. Lundstedt, A. A. Zakharov, O. J. Guy, T. Balasubramanian, R. Yakimova and H. Grennberg, Appl. Phys. Lett., 2014, 105, 081602 CrossRef.
- T. Yager, M. J. Webb, H. Grennberg, R. Yakimova, S. Lara-Avila and S. Kubatkin, Appl. Phys. Lett., 2015, 106, 063503 CrossRef.
- I. K. Petrushenko, Monatsh. Chem., 2014, 145, 891–896 CrossRef CAS.
- Y. Cao, S. Osuna, Y. Liang, R. C. Haddon and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 17643–17649 CrossRef CAS PubMed.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- L. N. Ukiwe, U. U. Egereonu, P. C. Njoku, C. I. A. Nwoko and J. I. Allinor, Int. J. Chem., 2013, 5, 43–55 Search PubMed.
- P. S. Bailey, Chem. Rev., 1958, 58, 925–1010 CrossRef CAS.
- P. S. Bailey, Ozonation in Organic Chemistry V2: Nonolefinic Compounds, Academic Press, Inc., 1982 Search PubMed.
- P. S. Bailey, J. Am. Chem. Soc., 1956, 78, 3811–3816 CrossRef CAS.
- P. Bailey and J. Ashton, J. Org. Chem., 1957, 22, 98 CrossRef CAS.
- W. A. Pryor, G. J. Gleicher and D. F. Church, J. Org. Chem., 1983, 48, 4198–4202 CrossRef CAS.
- E. J. Moriconi, B. Rakoczy and W. F. O'Connor, J. Am. Chem. Soc., 1961, 83, 4618–4623 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 6156–6214 CrossRef CAS PubMed.
- K. P. Loh, Q. L. Bao, P. K. Ang and J. X. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- M. Acik and Y. J. Chabal, Jpn. J. Appl. Phys., 2011, 50, 070101 CrossRef.
- P. H. Dussault, Organic peroxides (and ozone): safety-oriented overview, http://digitalcommons.unl.edu/chemistryperoxides/1).
- R. H. Callighan, M. F. Tarker and M. H. Wilt, J. Org. Chem., 1961, 26, 1379–1382 CrossRef CAS.
- L. F. Fieser and F. C. Novello, J. Am. Chem. Soc., 1940, 62, 1855–1859 CrossRef CAS.
- H. Vollmann, H. Becker, M. Corell and H. Streeck, Justus Liebigs Ann. Chem., 1937, 531, 1–159 CrossRef CAS.
- J. Staehelin and J. Hoigne, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef CAS PubMed.
- C. W. Lee and T. S. Huh, Bull. Korean Chem. Soc., 1999, 20, 969–972 CAS.
- T. Sugimoto, M. Nojima and S. Kusabayashi, J. Org. Chem., 1990, 55, 3816–3820 CrossRef CAS.
- T. Caronna, S. Gabbiadini, A. Mele and F. Recupero, Helv. Chim. Acta, 2002, 85, 1–8 CrossRef CAS.
- M. G. Sturrock and R. A. Duncan, J. Org. Chem., 1968, 33, 2149–2152 CrossRef CAS.
- B. L. Van Duuren, G. Witz and S. C. Agarwal, J. Org. Chem., 1974, 39, 1032–1035 CrossRef CAS PubMed.
- S. Yu, X. Zhang, Y. Yan, C. Cai, L. Dai and X. Zhang, Chem.–Eur. J., 2010, 16, 4938–4943 CrossRef CAS PubMed.
- I. Agranat, M. Rabinovitz and W.-C. Shaw, J. Org. Chem., 1979, 44, 1936–1941 CrossRef.
- J. Vachon, S. Rentsch, A. Martinez, C. Marsol and J. Lacour, Org. Biomol. Chem., 2007, 5, 501–506 CAS.
- P. S. Bailey, P. Kolsaker, B. Sinha, J. B. Ashton, F. Dobinson and J. E. Batterbee, J. Org. Chem., 1964, 29, 1400–1409 CrossRef CAS.
- I. A. Pshenichnyuk, P. B. Coto, S. Leitherer and M. Thoss, J. Phys. Chem. Lett., 2013, 4, 809–814 CrossRef CAS PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry – Part B, Reactions and Synthesis, Springer, 5th edn, 2007 Search PubMed.
- J. J. Brown and S. L. Cockroft, Chem. Sci., 2013, 4, 1772–1780 RSC.
- S. K. Bharti and R. Roy, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS.
- H. S. Shin, C. won Lee, J. Y. Lee and T. S. Huh, Eur. J. Org. Chem., 2000, 2000, 335–348 CrossRef.
- R. Gillis and Q. Porter, Aust. J. Chem., 1989, 42, 1007–1010 CrossRef CAS.
- K. Bowden and A. M. Last, J. Chem. Soc., Perkin Trans. 2, 1973, 1144–1150, 10.1039/P29730001144.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26248a |
| This journal is © The Royal Society of Chemistry 2017 |