
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium(I) diphenylphosphine complexes supported on porous organic polymers as efficient and recyclable catalysts for alkene hydrogenation†
Cristian H. Campos *a,
Julio B. Belmarb,
Solange E. Jeriab,
Bruno F. Urbanoc,
Cecilia C. Torresd and
Joel B. Aldereteb
*a,
Julio B. Belmarb,
Solange E. Jeriab,
Bruno F. Urbanoc,
Cecilia C. Torresd and
Joel B. Aldereteb
aDepartamento de Físico-Química, Facultad de Ciencias Químicas, Universidad de Concepción, Concepción, Chile. E-mail: ccampos@udec.cl
bDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad de Concepción, Concepción, Chile
cDepartamento de Polímeros, Facultad de Ciencias Químicas, Universidad de Concepción, Concepción, Chile
dDepartamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andres Bello, Sede Concepción, Autopista Concepción-Talcahuano 7100, Talcahuano, Chile
First published on 13th January 2017
Abstract
This paper describes the synthesis and characterization of porous polymeric materials as a support for rhodium(I) cationic coordination compounds and their use as heterogeneous catalysts for alkene hydrogenation. The synthetic strategy was the insertion of a vinyl-moiety in a bis(2-chloroethyl)amine precursor to provide highly porous resins with an enriched modifiable surface. The precursors synthesized were N,N-bis(2-chloroethyl)prop-2-en-1-amine (Alk-POL) and N,N-bis(2-chloroethyl)acrylamide (Acy-POL). The resins were obtained through suspension polymerization of methyl acrylate and divinylbenzene as a co-polymer and cross-linker, respectively. The resin surfaces were functionalized with diphenylphosphine groups followed by Rh(I) metal deposition using [Rh(COD)2]BF4 (COD = 1,5-cyclooctadiene) as the catalyst precursor. The Rh-catalysts were characterized by different physicochemical techniques and assessed for their catalytic performances in the heterogeneous hydrogenation of styrene and its derivatives. It was found that the catalytic activities and selectivity of the heterogenized rhodium complex (Rh-Alk-POL and Rh-Acy-POL) in the hydrogenation reactions were comparable to its homogeneous analogue. Analysis of the spent homogeneous resin Rh-Alk-POL catalyst after the first reaction cycle showed the presence of metallic Rh nanoparticles arising from the reduction of the Rh complex. Extensive recycling and Rh leaching studies were carried out for the Rh-Acy-POL catalyst. Both the activity and selectivity could be maintained for at least seven reaction runs and without metal leaching during the reaction cycles. We have also studied the liquid-phase hydrogenation reaction of various styrene m-substituted derivatives. The Rh-Acy-POL catalyst exhibits excellent catalytic activity for hydrogenation of the substrates and only vinyl-group hydrogenation was detected. Finally, the presence of electron-donating/-withdrawing substituents at the meta-position resulted in different rates of vinyl group hydrogenation. This effect was quantified in terms of the Hammett relationship, in which the catalyst displayed a linear correlation between the Hammett substituent constant (σmeta) and the hydrogenation rate.
1. Introduction
Hybrid materials have become an excellent alternative for use as a support and/or catalyst in hydrogenation processes for fine chemicals because their organic/inorganic nature confers the ability to be used in very specific processes.1–5 Some examples of hybrid materials are metal organic frameworks (MOFs),6 hybrid-polymer resins,7,8 clay–polymer composites,9 among others. Especially in the use of synthetic hybrid-polymeric materials, Merrifield resins arise as an attractive type of support for the covalent immobilization of coordination metal complexes catalysts for selective hydrogenation10,11 due to their versatile surface functionalization. The main disadvantage of Merrifield materials is its low surface area which restrict the accessibility at the reactive moieties introduced into them as a result of the crosslinking density of the polymer matrix.5In recent years, suspension polymerization is used to produce crosslinked-polymers with a controllable pore structure and a high surface area compared to Merrifield resins.12–14 These materials contain well-developed micropores and mesopores, therefore should decrease the diffusion limitations and significantly improve the accessibility to the reactive pending groups and produce highly dispersed catalysts.15–17 In this way, Canali et al.18 have reported a porous polystyrene/polymethacrylate-based resin with supported Jacobsen's chiral Mn–saliciden–ethylenediamine complexes for alkene enantioselective oxidation. Drake et al. have explored the use of vinylbenzyl chloride-based resins which have been aminated using N,N,N′-trimethylethylenediamine to produce coordinated Pt(II) catalysts for the hydrosilylation of 1-octene using methyldichlorosilane at room temperature.19
Heterogeneous metal catalysts are widely used in the hydrogenation of alkenes.20,21 Liquid-phase hydrogenation of unsaturated organic compounds with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond typically involve transition metals such as Pd, Pt, Rh, Ni, Ru, among others. Palladium supported on carbonaceous materials is widely used as a catalyst in this reaction.22 However, problems in chemoselectivity are occasionally encountered when the substrate contains functional groups sensitive under the reaction conditions of hydrogenation.23–25 To address this issue, rhodium is the most employed noble metal as homogeneous catalyst for alkene hydrogenation reactions because possess the ability to activate the hydrogen molecule to provide a specific transfer of a hydride atom to the alkene moiety during the catalytic cycle.26,27 Phosphines molecules are used as ligands in the hydrogenation of several types of olefins by Rh catalysts4,28,29 and the literature contains several reports about the hydrogenation ability of cationic [Rh(diene)(PR3)2]+ complexes under relatively mild reaction conditions.27,30 From this background, we are interested in investigating the performance of diphenylphosphine–rhodium complexes based immobilized on the porous polymeric-resins in the hydrogenation of the styrene and its derivates. The aim of the present work is the design and synthesis of new polymeric-resins having functional moieties able to covalently bond Rh(I) diphenylphosphine catalysts. For this purpose we used two co-monomers derived from N,N-bis(chloroethyl)amine: N,N-bis(2-chloroethyl)-2-propen-1-amine (Alk-N(CH2CH2Cl)2) and N,N-bis(2-chloroethyl)acrylamide (Acy-N(CH2CH2Cl)2) as shown in Scheme 1. In fact, the polymer synthesis procedure including chloroethyl-monomers allows obtaining porous polymer-supports with an improved accessibility to its functionalities. Subsequently diphenylphosphine ligands were prepared into the polymer-resins and formed cationic rhodium(I) complexes, which were employed in heterogeneous liquid phase styrene hydrogenation. Herein, the best catalytic system was studied in seven recycles studies. Finally, the catalytic hydrogenation of a series of m-substituted styrene substrates was investigated and the catalytic data were subjected to Hammett treatment.
C bond typically involve transition metals such as Pd, Pt, Rh, Ni, Ru, among others. Palladium supported on carbonaceous materials is widely used as a catalyst in this reaction.22 However, problems in chemoselectivity are occasionally encountered when the substrate contains functional groups sensitive under the reaction conditions of hydrogenation.23–25 To address this issue, rhodium is the most employed noble metal as homogeneous catalyst for alkene hydrogenation reactions because possess the ability to activate the hydrogen molecule to provide a specific transfer of a hydride atom to the alkene moiety during the catalytic cycle.26,27 Phosphines molecules are used as ligands in the hydrogenation of several types of olefins by Rh catalysts4,28,29 and the literature contains several reports about the hydrogenation ability of cationic [Rh(diene)(PR3)2]+ complexes under relatively mild reaction conditions.27,30 From this background, we are interested in investigating the performance of diphenylphosphine–rhodium complexes based immobilized on the porous polymeric-resins in the hydrogenation of the styrene and its derivates. The aim of the present work is the design and synthesis of new polymeric-resins having functional moieties able to covalently bond Rh(I) diphenylphosphine catalysts. For this purpose we used two co-monomers derived from N,N-bis(chloroethyl)amine: N,N-bis(2-chloroethyl)-2-propen-1-amine (Alk-N(CH2CH2Cl)2) and N,N-bis(2-chloroethyl)acrylamide (Acy-N(CH2CH2Cl)2) as shown in Scheme 1. In fact, the polymer synthesis procedure including chloroethyl-monomers allows obtaining porous polymer-supports with an improved accessibility to its functionalities. Subsequently diphenylphosphine ligands were prepared into the polymer-resins and formed cationic rhodium(I) complexes, which were employed in heterogeneous liquid phase styrene hydrogenation. Herein, the best catalytic system was studied in seven recycles studies. Finally, the catalytic hydrogenation of a series of m-substituted styrene substrates was investigated and the catalytic data were subjected to Hammett treatment.
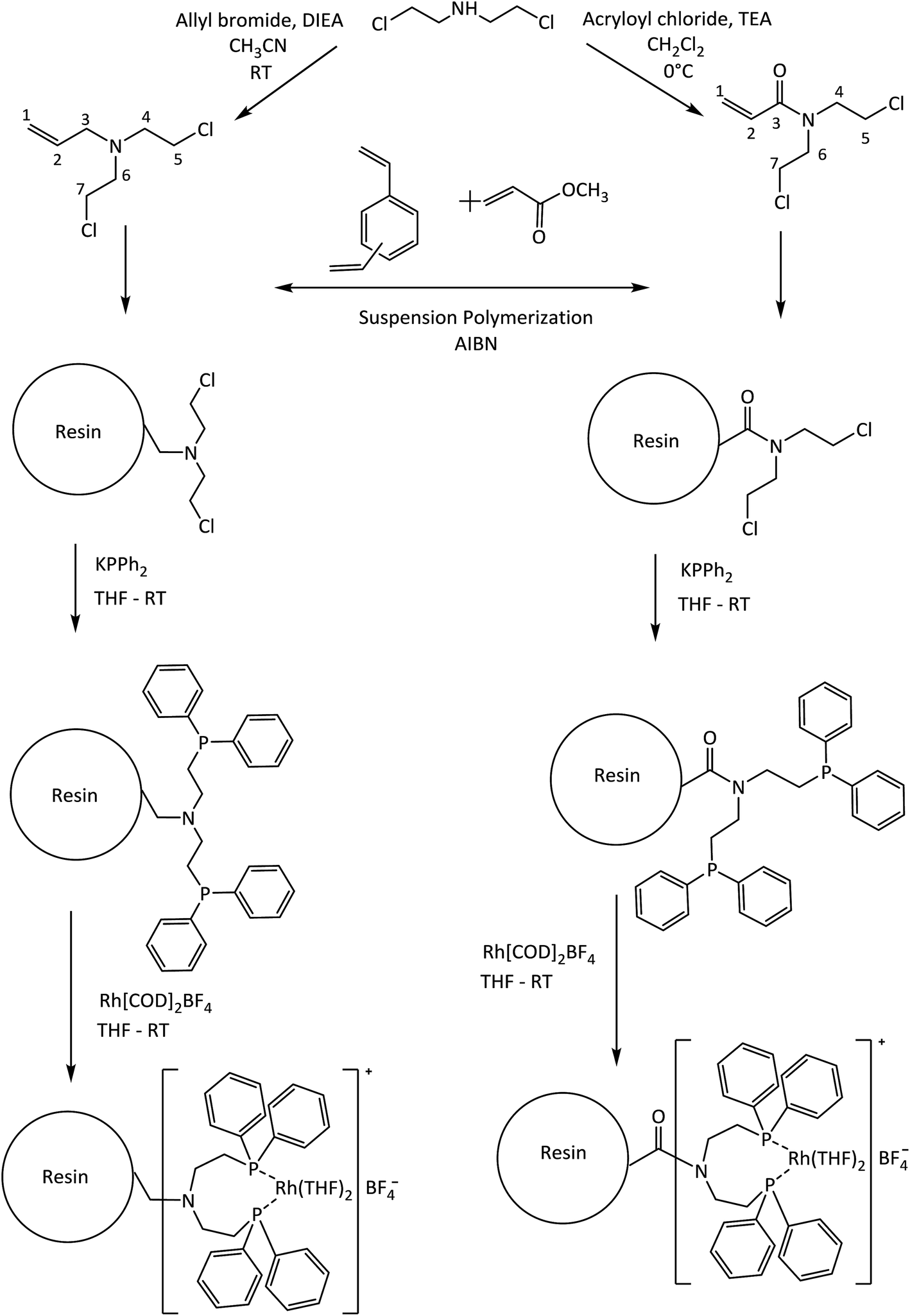 | ||
| Scheme 1 Immobilization rout for the Rh-based catalysts. | ||
2. Experimental section
2.1 Materials
All of the air-sensitive reactions were performed under an inert atmosphere (Ar or N2) in a Schlenk flask. Tetrahydrofuran (THF, Merck®) and toluene (Merck®) were dried in metallic sodium/benzophenone, dichloromethane (CH2Cl2, 99.8% anhydrous, Aldrich®) and acetonitrinile (CH3CN 99% Merck®) were dried as required by contact with calcium hydride (Sigma®) for several days and then distilled immediately before use. Triethylamine (TEA 95% Aldrich®), methyl acrylate (MA, 99% Aldrich®) and divinylbenzene (DVB, technical grade, 80% Aldrich®) were distilled at a reduced pressure prior to use: TEA with CaH2, both MA and DVB in presence of p-tert-butylcatechol as polymerization inhibitor. 2,2′-Azobis(isobutyronitrile) (AIBN, Aldrich®) was recrystallized from methanol. The compounds allyl bromide (98% Aldrich®), acryloyl chloride (99% Merck®), potassium diphenylphosphide (KPPh2 solution 0.5 M in THF, Aldrich®), bis(1,5-cyclooctadiene)rhodium(I) tetrafluoroborate (Aldrich®), diisopropylethylamine (DIEA, 99% Merck®), bis(2-chloroethyl)amine hydrochloride (98% Aldrich®), anhydrous sodium sulfate (Na2SO4, Merck®), hydrochloric acid (HCl, 37% Merck®), sodium chloride (NaCl, Merck®), sodium hydroxide (NaOH, Merck®), hydroxyethylcellulose (average Mw 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aldrich®), styrene (98%, Aldrich®), m-nitrostyrene (98%, Aldrich®), m-chlorostyrene (99%, Aldrich®), m-methylstyrene (99%, Aldrich®), m-aminostyrene (99%, Aldrich®) and the solvent n-hexanes (PA, Merck®) were used as received. The Scheme 1 shows the synthetic rout employed to prepare the immobilized catalysts.
000, Aldrich®), styrene (98%, Aldrich®), m-nitrostyrene (98%, Aldrich®), m-chlorostyrene (99%, Aldrich®), m-methylstyrene (99%, Aldrich®), m-aminostyrene (99%, Aldrich®) and the solvent n-hexanes (PA, Merck®) were used as received. The Scheme 1 shows the synthetic rout employed to prepare the immobilized catalysts.
2.2 Co-monomers synthesis
CH–), 5.76 (m, 1H, CH–CH2–); 13C NMR (CDCl3): δ 42.1 (s, C5 ± C7), 55.9 (s, C4 ± C6), 57.8 (s, C3), 118.0 (s, C1), 134.8 (s, C2).CH–), 6.34 (dd, 1H, CH2CH–); 6.62 (m, 1H, CH–CO–); 13C NMR (CDCl3): δ 42.7 (s, C5 ± C7), 52.9 (s, C4 ± C6), 126.2 (s, C1), 131.0 (s, C2), 162.8 (s, C3).2.3 Synthesis of porous polymer-resins
Porous polymer-resins were synthesized via suspension polymerization using a modified procedure reported in detail by Wei et al.14 The co-monomers (MA and x-N(CH2CH2Cl)2 where x: Alk or Acy) were first mixed well with the porogen (toluene) and DVB as crosslinker, in mass ratio 2:39:60, to form an organic phase in which the initiator, AIBN, was added (1 wt% monomers-based). The organic phase mixture was then added, at a 1:3 ratio (v/v), to the aqueous phase containing 0.2% hydroxyethyl cellulose and 20% NaCl, and the polymerization was allowed to proceed at 70 °C and 85–90 °C for 14 and 4 h, respectively. The resulting MA/x-N(CH2CH2Cl) co-polymer materials were washed with hot water and extracted thoroughly with acetone in a Soxhlet apparatus, and the acetone-swollen solid were finally dried in vacuum at 80 °C.
2.4 Phosphination of porous polymer-resins and catalyst synthesis
Initially the a suitable amount of MA/x-N(CH2CH2Cl) co-polymer materials was charged into a cooled (0 °C) Schlenk flask (100 mL) containing dry and degasified THF (50 mL). Under dry N2, the required amount of KPPh2 was added and the mixture stirred for 2 h. The mixture was then left stirring for 48 h, at room temperature, under N2 gas. The quantity of KPPh2 used were such that a 1:2 molar ratio of porous polymer-resin-N(CH2CH2Cl) functionality:KPPh2. The reaction was quenched in degasified H2O (100 mL), filtered and the solids washed with both degasified H2O and THF, respectively. Each phosphinated resin (2.0 g) was placed in a Schlenk flask with dry and degasified THF (20 mL) and mixed with a suitable amount of [Rh(COD)2]BF4 dissolved in dry and degasified THF (5 mL). The quantity of the rhodium used was stoichiometrically equivalent to the diphenylphosphine content of the phosphinated resin used, and was calculated from the values detailed in Table 1. The mixture was stirred for 48 h, at room temperature, under N2 atmosphere. Finally the obtained materials were filtered and repeatedly washed with THF, acetone and water, respectively. The light-orange materials prepared were stored in a desiccator under an N2 atmosphere prior to the activity measurements. The catalysts obtained were labeled as Rh-Alk-POL and Rh-Acy-POL for both alkylated and acylated co-monomers precursors, respectively.
2.5 Characterization
The chemical composition of the catalysts was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, PerkinElmer 7300DV). The samples (10 mg) were chemically digested by dissolution in a mixture of 15 mL HCl and 5 mL HNO3 in an autoclave for 2 h at 150 °C. The 13C- and 31P-decoupled NMR spectra were obtained on a Bruker AMX-300 spectrometer (300 MHz for 1H, 75 MHz for 13C and 121 MHz for 31P) using trimethylsilane and H3PO4 as internal standards. The solid-state 13C and 31P CP-NMR spectra were recorded at 100 MHz and 162 MHz, respectively, uses a Bruker AV 400 WB spectrometer. The XRD patterns were recorded in a RigakuD/max-2500 diffractometer with the Cu Kα radiation at 40 kV and 100 mA. The N2 adsorption–desorption isotherms at −196 °C were performed in a Micromeritics ASAP 2010 apparatus. The specific surface areas were determined by the BET (Brunauer–Emmett–Teller) equation, using the adsorption data in the relative pressure range of 0.05 to 0.3, and the pore-size distributions were estimated using the BJH method. The catalysts morphology was examined by the transmission electron microscopy (TEM). The X-ray photoelectron spectra (XPS) were recorded using a VG Escalab 200 R spectrometer equipped with a hemispherical analyzer and using non-monochromatic Mg Kα X-ray radiation (hν = 1253.6 eV). The binding energies (BE) were calculated with respect to the C-component of the C 1s peak fixed at 284.8 eV. The data analysis was performed with the “XPS Peak” software. The peaks were decomposed into a sum of Gaussian/Lorentzian (G/L = 90/10) after the subtraction of a Shirley-type baseline. The surface P/C, Cl/C, Rh/C and Rh/P atomic ratios were estimated from the integrated intensities of the P 2p, Rh 3p, C 1s, and Cl 2p lines after the background subtraction and were corrected by the atomic sensitivity factors.312.6 Catalytic activity
The catalytic assays of the alkenes hydrogenation were performed in a stainless steel (100 mL) Parr-type semi-batch reactor at a substrate concentration of 0.01 mol L−1 using methanol (25 mL) as the solvent and stirring at 700 rpm under 5.0 bar of H2 pressure at 25 °C. All of the catalytic runs were conducted in the absence of external mass transfer limitations and were repeated three times for each experiment. The catalysts were in the form of fine powders (>30 μm), assuming a negligible effect of pore diffusion limitations. In the catalyst mass studies, the alkene/Rh molar ratio was varied, the concentration of PPD was maintained constant, and the mass of the catalyst was modified. No significant differences in the initial reaction rates were noted upon varying the catalyst mass over the range of 0.01 to 0.07 g because the reaction rate was proportional to the catalyst mass, indicating that the gas–liquid and liquid–solid external mass transfer limitations were absent.32,33 The pseudo-kinetic constants (k) were calculated using a pseudo-first-order kinetic model for a batch reactor under similar conditions, as reported in previous studies.34 The reactants and products were analyzed by gas chromatography and mass spectrometry using a GC-FID instrument (HP-4890) with a semi-capillary column HP-5 and N2 as the carrier gas. The recycling assays were performed by filtering the catalyst from the reaction medium. The filtered catalyst was washed three times consecutively with methanol (50 mL × 3) to clean the surface and then dried at 100 °C for 24 h. The activity of the catalyst was monitored by the following conversion levels and selectivity: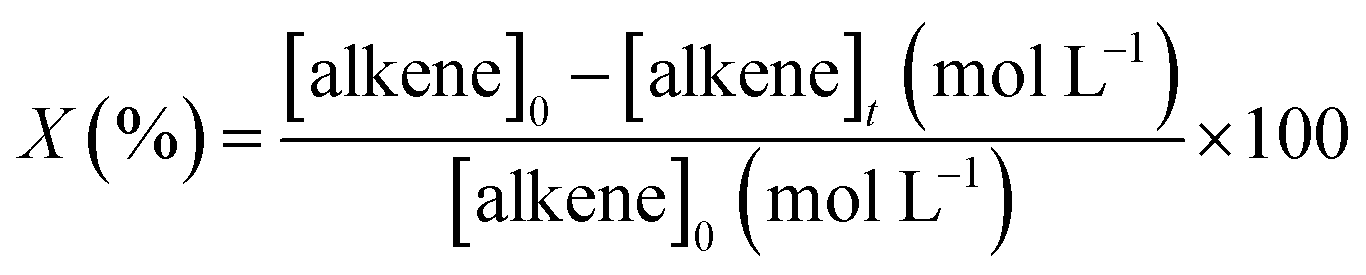
where [alkene]0 corresponds to the initial concentration of the alkene, and [alkene]t is the concentration at different times. Target alkene corresponds to the hydrogenation of vinyl moiety of styrene or meta-substituted styrene substrates. The turnover frequency (TON) of the catalysts was calculated as follows:
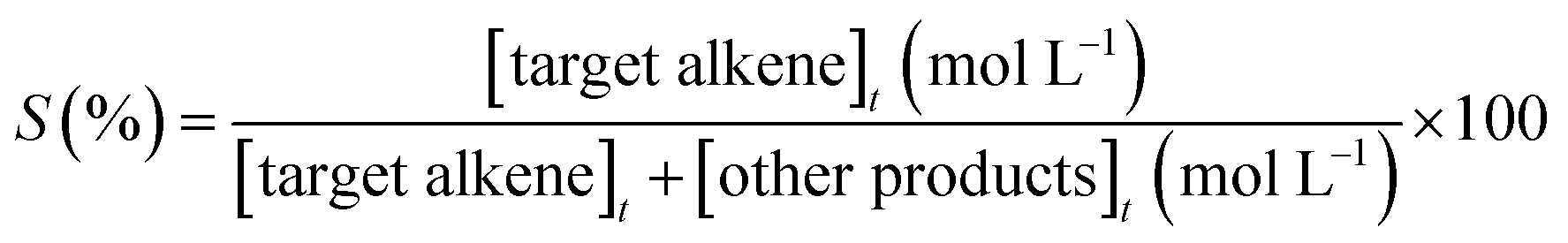
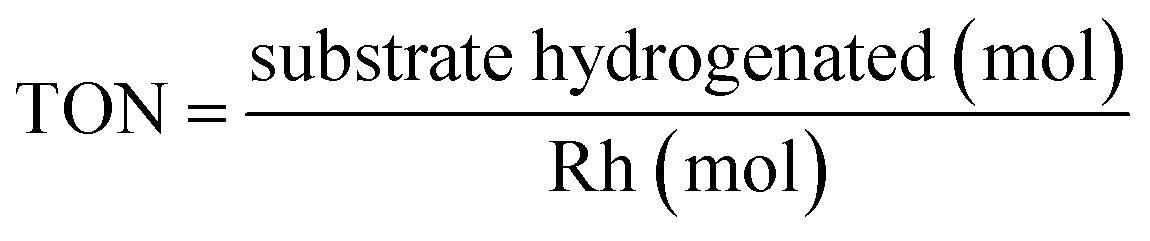
3. Results and discussion
3.1 Supports and catalysts characterization
Polymer supports were synthesized via the copolymerization of DVB with the as-prepared N,N-bis(2-chloroethyl)-2-propen-1-amine and N,N-bis(2-chloroethyl)acrylamide co-monomers, respectively, as illustrated in Scheme 1. These compound were inserted into the polymer matrices and then allow the synthesis of the catalyst systems as seen in Table 1. Both systems showed N2 adsorption–desorption isotherms with H1 hysteresis loop, which is a typical for mesoporous structure and cylindrical pores as shown in Fig. 1. In the same way, the catalysts isotherms remain a mesoporous structure of supports after the post-modification synthesis. However, the BET specific surface area of polymers and catalysts gradually decrease according to the parameters provided in Table 1. This phenomenon is mainly due to the introduction of bulky –P(Ph)2 ligands and Rh complex that partially block the pores and reduce the adsorption of N2.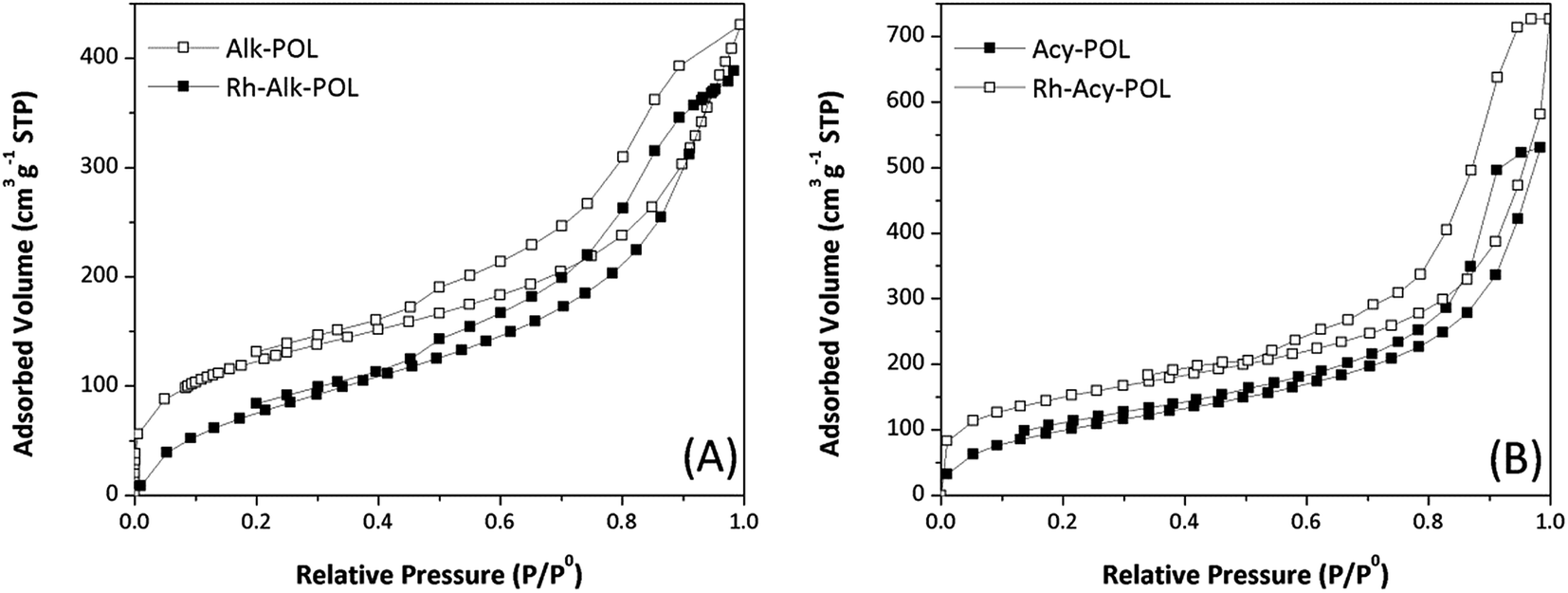 | ||
| Fig. 1 N2 adsorption/desorption isotherms of supports and Rh catalysts. (A) Alk-POL based materials and (B) Acy-POL based materials. | ||
The amount of rhodium determined by ICP-OES in the Rh-Alk-POL fresh catalysts is 0.67 wt%, corresponding to 65.1 μmol of complex per gram of sample, while the catalyst Rh-Acy-POL the amount is 0.95 wt% corresponding to 93 μmol of complex per gram of sample. According to these values the Rh-Alk-POL system showed both lower Rh loading and Rh/P mass ratio. This behavior could be attributed to the uncomplete chlorine substitutions during the support's modification exhibiting a decrease in the (Ph2)2P loading confirmed by XPS (vida infra). In the same way, the Rh/P mass ratio decreases compared with the nominal value, most likely due to uncoordinated P moieties remaining from the catalyst synthesis. Rh-Acy-POL catalyst showed the expected Rh and P contributions indicating that the support modifications were made according to the nominal catalyst loading.
The 13C and 31P solid-state NMR characterization are shown in Fig. 2. For all of the systems 13C NMR spectra (Fig. 2A) display the characteristic bands of the polymer backbone, –CH2 carbons from the ligands and –CH3 moiety attributed to MA methoxyl-group at chemical shifts between 20 and 60 ppm are detected. The signals at 120–160 ppm can be assigned to DVB crosslinker aromatic carbons. Both catalysts showed an increased contribution of the aromatics signals at 140 ppm attributed to the presence of diphenylphosphine ligands. Finally, the signal at 184 ppm ascribed to carbonyl contribution of MA is detected.
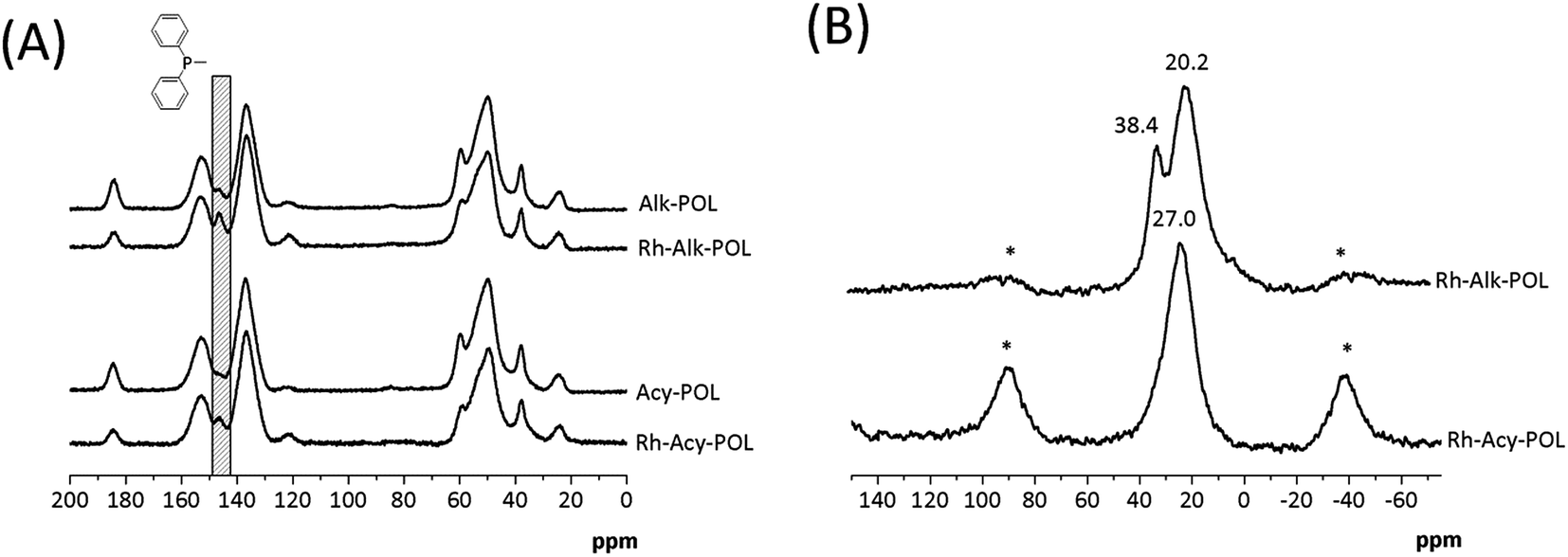 | ||
| Fig. 2 Solid state NMR spectra of polymer-based supports and Rh catalysts. (A) 13C CP/MAS and (B) 31P CP/MAS and rotational sidebands are denoted by asterisks (*). | ||
In 31P NMR characterization (Fig. 2B) for both catalysts, a broad signal at chemical shifts between 20 and 45 ppm is observed. In the case of system Rh-Alk-POL showed two contributions, the first one at 20.2 ppm attributed to the presence of species of P–Rh35 and the second one at 38.4 ppm attributed to PO species.36,37 This result is in agreement with the lower metal loading detected by ICP-AES. It must be noted that the NMR analysis of sample Rh-Alk-POL was conducted in air atmosphere, and hence the PO signal appears due to the oxidation of unreacted Ph2P groups with air to produce phosphine oxide. Rh-Acy-POL catalyst showed a signal at 27.0 ppm attributed to the coordinate P–Rh as was reported by Román-Martínez et al.38 whereas the signal of PO species do not appear. These results confirm the COD ligand displacement during the catalyst synthesis providing the complex (PP)–Rh(COD)BF4 complexes on the resins surface.
The XPS method has been used for qualitative characterizations of the obtained catalysts. The binding energies for Rh 3d5/2, Cl 2p, P 2p and C 1s as well as atomic ratios of Rh/P and Cl/C on the surface of the samples are given in Table 2. The Rh 3d5/2 binding energies (BE) confirm the presence of Rh(I) oxidation state.38,39 During the catalysts synthesis, Rh(COD)2BF4 precursor was not oxidized providing the ligand substitution pathway. In the same way, Rh-Alk-POL system showed BE for Cl 2p at 198.2 eV attributed to the organic chlorine contributions. In the synthesis design of Alk-POL and Acy-POL supports, a stoichiometric quantity of the co-monomers (Alk-NClCl or Acy-NClCl) to produce the desire immobilized catalysts were used. However, the Rh-Alk-POL catalyst showed a remained a fraction of CH2–Cl on the surface as a consequence to an uncomplete support's modification. The Rh-Acy-POL catalyst showed BE for Cl 2p at traces level as shown in Table 2. Both results are in agreement with ICP-AES characterization and confirm the Rh catalyst immobilization on the polymer's surface.
Table 2 includes also display the surface Rh wt% and F factor determined by XPS employing the same methodology reported by Román-Martínez et al.40,41 The values of F indicate a high dispersion degree in the location of the complex in both supports (where F = 1 correspond to the homogeneous distribution of the complex in the porous materials41). These results are in agreement with the decrease on the SBET as a consequence of Rh complex anchoraged in the resins.
For both Rh supported catalysts, P 2p BE at 132.2–132.4 eV were detected. Typically, these values are assigned to (Ph2)2P coordinated with Rh(I) species.38 The main advantage in the alkyl-phosphine immobilization is the P atom oxidation by O2.36,42,43 Unfortunately, it was not possible to distinguish between coordinated-phosphine (P–Rh) and phosphine oxide (PO) due to closer BE of the most intense peak P 2p at 132.0–132.6 eV (ref. 38 and 41) for P–Rh and 132.8–133.2 eV for PO.43 However, Rh-Alk-POL catalysts showed an Rh/P atomic ratio lower than in the nominal content meaning that unreacted/oxidized diphenylphopshine species are present, in agreement with 31P solid-state NMR characterization.
3.2 Catalytic activity
Fig. 3 shows the catalytic data obtained for diphenylphosphine-based Rh(I) catalyst at 30 minutes of reaction (see the synthesis details in ESI†) for hydrogenation of styrene (STY). Firstly, for comparison purposes a reference [Rh(COD)2]BF4 catalyst without the diphenylphosphine ligand was used. As can be observed in Fig. 3 the conversion obtained after 30 minutes of reaction (35%) shows the low activity for [Rh(COD)2]BF4 catalyst. Secondly, homogeneous catalyst Rh-Alk-L and Rh-Acy-L references were prepared in situ from the diphenylphosphines-based ligands and the [Rh(COD)2]BF4 precursor. These catalysts showed high activity under the same reaction conditions. However, a different selectivity on the products distribution was detected. The STY hydrogenation could be following the reaction pathways which are shown in Scheme 2. The [Rh(COD)2]BF4 and Rh-diphosphine catalysts are highly chemoselective towards alkene hydrogenation.44–47 The over-hydrogenated product could be attributed to the presence of metallic Rh nanoparticles indicating that the electronic state of rhodium in the complex was modified during the hydrogenation process.42,48,49 TEM analysis was employed to confirm that metallic Rh NPs are formed, for both homogeneous and heterogeneous diphenylphosphine-based Rh(I) catalyst, as shown in Fig. 4.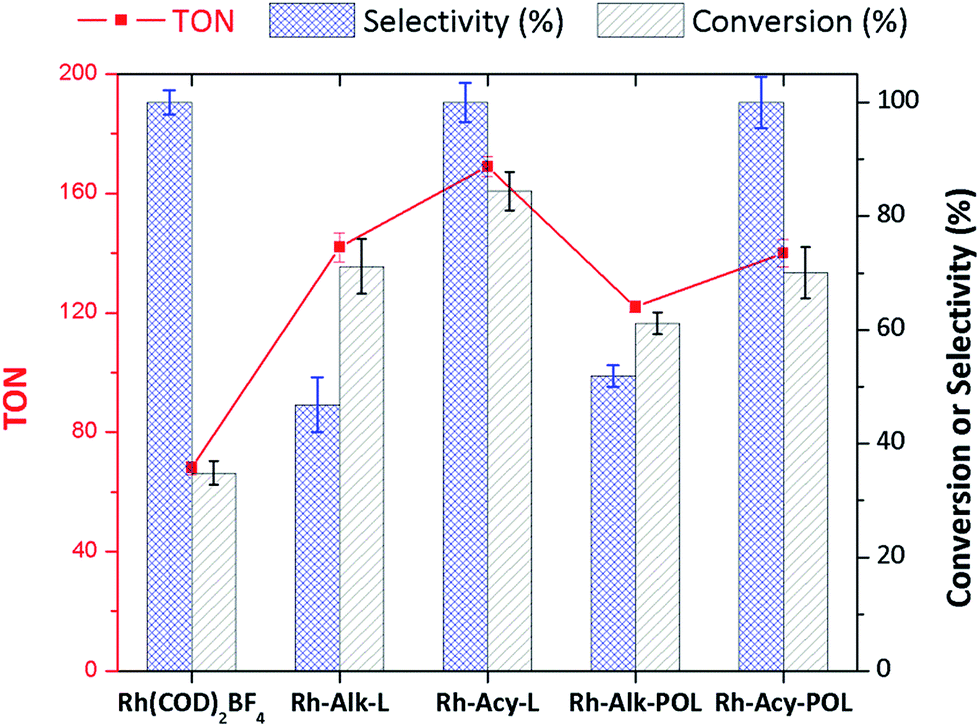 | ||
| Fig. 3 Hydrogenation of STY with Rh-based catalysts. Reaction conditions: 25 °C, 5.0 bar H2, at STY/Rh = 200 mole ratio in methanol (25 mL) at 30 min of reaction. The homogeneous catalysts were prepared in situ adding Alk- or Acy-L into the reactor at diphosphine/Rh = 0.5 mole ratio. For the supported catalysts 100 mg of hybrid catalyst were used. The selectivity corresponds to EBZ production. | ||
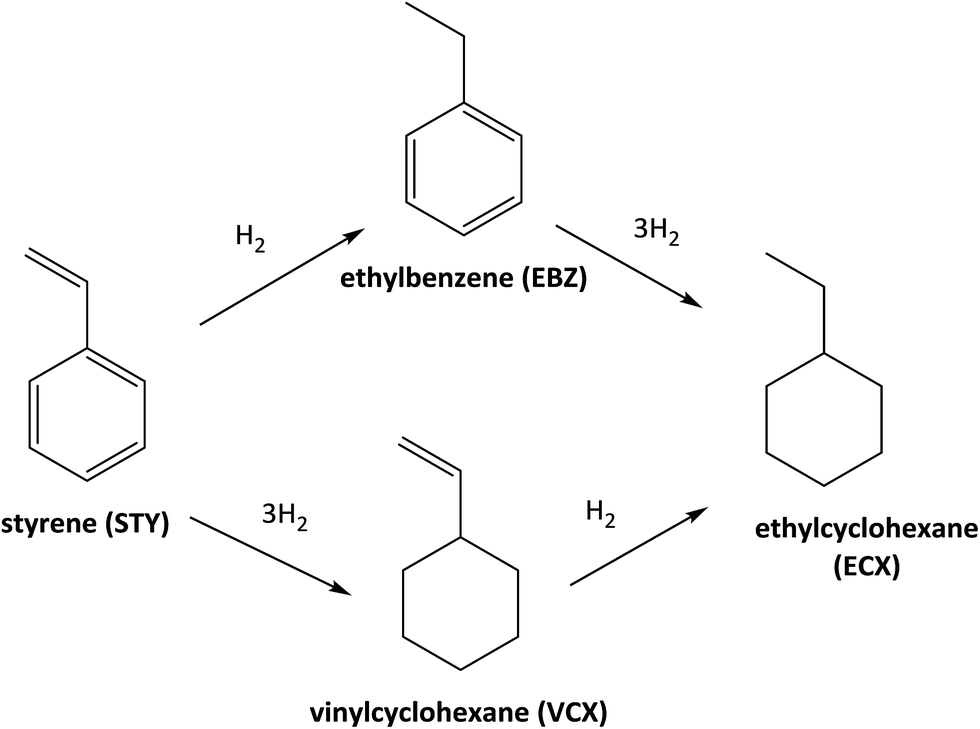 | ||
| Scheme 2 Reaction pathways in the catalytic hydrogenation of STY. | ||
 | ||
| Fig. 4 TEM characterization post-reaction. (A) Rh-Alk-L and (B) Rh-Alk-POL. | ||
Only Rh-Alk-L catalysts showed the presence of metallic Rh NPs mainly ascribed to Alk-L ligand which decrease the stability of the Rh-complex catalysts under reaction conditions at 30 minutes compared with Acy-L. This behavior could be explained by the nature of the N atom on the ligand, where the amide nature of Acy-L ligand provide greater stability to the Rh-center decreasing its reducibility under H2 atmosphere. Finally, Rh-Alk-L catalyst showed ECX and EBZ as hydrogenation products while Rh-Acy-L achieved the maximum selectivity to EBZ (100%).
A similar trend for the immobilized Rh-complex catalyst was detected. Both systems showed a decrease in the activity compared with the homogeneous catalysts which is a consequence of catalyst immobilization as was widely reported.7,18,30,36,38,40,41,44,45,47 Despite this, both systems showed a similar product distribution as their homogenous equivalents. Fig. 4B shows the TEM characterization where the Rh-Alk-POL showed the presence of metallic Rh NPs as a consequence of the Rh-complex reduction at the reactions conditions.
3.3 Recycling test
The catalyst Rh-Acy-POL showed the best results to be employed as recyclable catalyst. The stability of this catalyst was evaluated under reaction conditions (180 min) performing a run without the presence of the substrate. In this case, no leaching was observed as shown in Table 3. In Fig. 5, the STY conversion levels and recycling test for the Rh-Acy-POL catalyst is shown. The catalyst is active for the STY hydrogenation reaction reaching a conversion of 98% in 120 minutes. The conversion data fitted with pseudo-first order's kinetics with respect to the substrate.| Sample | Rh 3d5/2 (eV) | P 2p (eV) | Rh content (wt%) |
|---|---|---|---|
| Fresh | 309.1 | 132.4 | 0.95 |
| Used without STY | 309.0 | 132.3 | 0.96 |
| Used after last run | 308.9 | 132.2 | 0.92 |
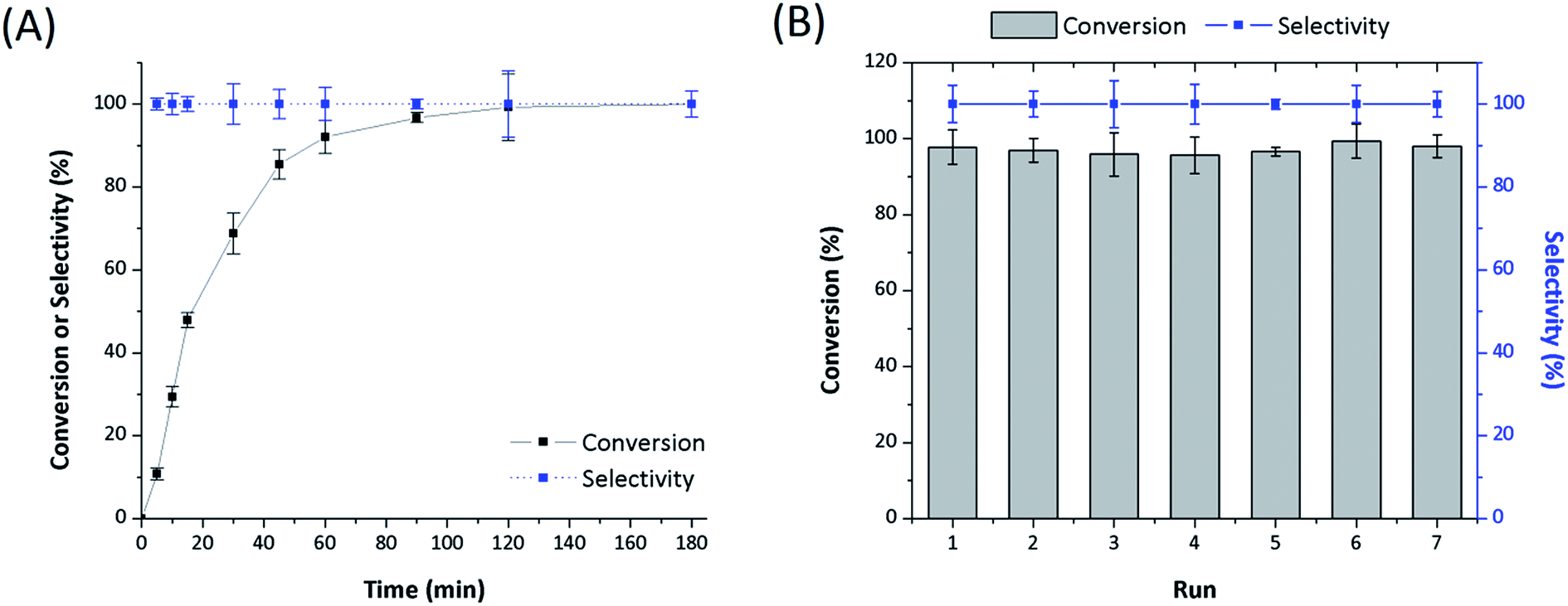 | ||
| Fig. 5 Reaction profiles for hydrogenation of STY and recycles studies using Rh-Acy-POL as catalyst. (A) Conversion level vs. time and (B) recycles runs at 120 minutes. Reaction conditions: 25 °C, 5.0 bar H2, at STY/Rh = 200 mole ratio in methanol (25 mL). | ||
The recycling studies, after the first run, the Rh-Acy-POL was recovered from the reaction mixture by filtration, washed with MeOH to remove traces of the previous mixture and engaged in a new catalytic run. A complete conversion of the substrate was obtained with the same selectivity to EBZ even at 7 consecutives cycles. The post-reaction characterization showed Rh 3d5/2 and P 2p BE similar than the fresh catalyst in agreement with the preservation of Rh metal loading even at seven recycles. These results probed that Rh-Acy-POL catalyst is stable and recyclable for its use as heterogeneous system for the hydrogenation of alkenes.
3.4 meta-Substituted styrene hydrogenation
We also investigated the hydrogenation of meta-substituted styrene molecules (m-X-STY where X: NH2, CH3, Cl or NO2) over Rh-Acy-POL catalyst as shown in Table 4. All the kinetics showed pseudo-first order's kinetics with respect to the m-X-STY substrates consumption and only produced the hydrogenation of alkene moiety avoiding the hydrogenation of –NO2 or hydrodechlorination for m-NO2-STY and m-Cl-STY, respectively.| Substrate | Conversiona [±2%] | Selectivityb [±2%] | km-x-STYc [h−1 mmolRh−1] | σmetad |
|---|---|---|---|---|
| a 30 min of reaction.b Calculated for vinyl-group hydrogenation.c Pseudo-first-order constant.d Extract from Hansch et al.50 | ||||
| STY | 68 | 100 | 29 | — |
| m-NO2-STY | 83 | 100 | 46 | 0.71 |
| m-Cl-STY | 76 | 100 | 39 | 0.37 |
| m-CH3-STY | 65 | 100 | 26 | −0.07 |
| m-NH2-STY | 60 | 100 | 22 | −0.16 |
The kinetic data displayed dependence between reaction rate and the electronic nature of the substituent in meta position. Assuming the pseudo-first model, catalytic rate constant (km-X-STY and kSTY) were calculated as shown in Table 4. The m-X-STY substrates showed the tendency km-NH2-STY < km-CH3-STY < kSTY < km-Cl-STY < km-NO2-STY. For the electron-donating (EDG) groups the catalytic rate constant showed lower values in comparison with m-styrenes derivates with the electron withdrawing group (EWG). With these results, a correlation analysis between electronic nature of substrates and the experimental catalytic constants was performed. The kinetic data was correlated with Hammett σmeta constant, as shown in Fig. 6, since previous report has showed a linear correlation between σHammett of m-substituted nitroarenes and its the catalytic constants for hydrogenation reaction catalyzed by metallic Rh NPs.3 The empirical values of σmeta were defined by Hammett from the ionization constants of benzoic acids as: σmeta: log(Kx) − log(KH) where KH is the ionization constant for benzoic acid in water at 25 °C and Kx is the corresponding constant for a meta-substituted benzoic acid. The values used in our adjustment were compiled by Hansch et al.50 The rational of this analysis was based on the hydrogenation reaction mechanism proposed for Halpern.51 The [Rh(diene)Ln]+ catalysts operates in 4 stages: (1) oxidative addition of H2 promoting Rh(I) → Rh(III), (2) the substrate insertion into the coordination cloud, (3) migratory insertion of a hydride to the alkene substrate and (4) the reductive elimination of the hydrogenated substrate to retrieve Rh(I) in the catalyst. It is well known that the rate determining step (RDS) is the insertion of a hydride on the CC alkene moiety.26
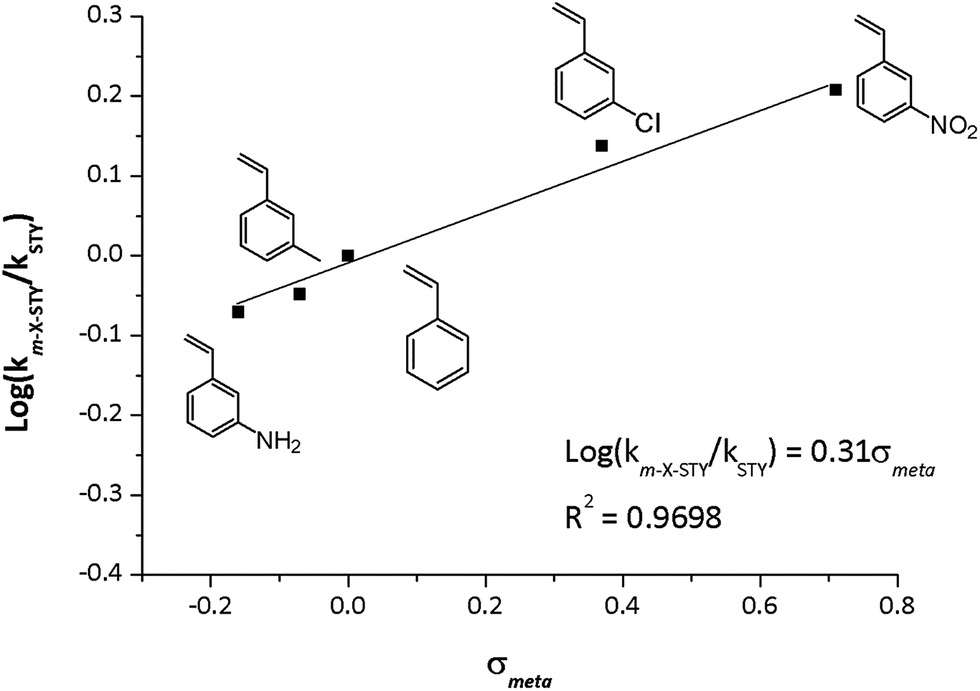 | ||
| Fig. 6 Experimental log(km-X-STY/kSTY) vs. σmeta constants of substrates. Reaction conditions: 25 °C, 5.0 bar H2, at m-X-STY/Rh = 200 mole ratio in methanol (25 mL). | ||
The hydride insertion could induce a carbanion character on CC bond at the transition state, hence the presence of EWG increases the affinity for the hydride, enhancing the catalytic activity and instead EDG decrease the hydrogenation rate of the vinyl group. A good linear correlation (R2 = 0.9698) between the experimental loglog(km-X-STY/kSTY) and the σmeta was found. The following logarithmic equation describes the relationship between km-X-STY/kSTY and the electronic nature of the substrates, showing to be a statistically accurate model with 99% confidence (p = 0.0072).
4. Conclusions
New heterogeneous rhodium-diphenylphosphine supported catalysts on porous resins have been prepared and characterized. The rhodium catalysts based in the precursor N,N-bis(2-chloroethyl)-2-propen-1-amine was a less stable than N,N-bis(2-chloroethyl)acrylamide and this difference was reasonably attributed to the instability of phosphinated ligand under the reaction conditions. As shown by TEM analysis, on the immobilized N,N-bis(2-(diphenylphosphino)ethyl)-2-propen-1-amine based catalyst, the rhodium(I) species are converted to rhodium(0) nanoparticles preventing the selective hydrogenation reaction. The amide based catalyst showed an excellent catalytic performance during the hydrogenation of styrene producing ethylbenzene as the only reaction product. The catalyst was highly stable at the reactions conditions and no-leaching was detected, even after 7 consecutive reaction cycles. Finally, the catalytic activity of this system for the hydrogenation of m-substituted styrene showed a chemoselective hydrogenation of alkene function due to the electronic nature of substituent group. Thus, substituted styrene with electron-withdrawing groups showed very high activities, while styrene derivatives with electron-donating groups presented lower activities. This catalyst proved to be active and highly chemoselective for the hydrogenation of alkenes and its immobilization via covalent immobilization on porous resin support may indeed be used as a strategy leading to stable catalysts.Acknowledgements
The authors thank CONICYT for the financial support (FONDECYT postdoctoral 3140130 and FONDECYT Initiation 11160468). We also gratefully acknowledge the help of Dr José Luis G. Fierro of the Instituto de Catálisis y Petroleoquímica (ICP-CSIC) – Sustainable Energy and Chemistry Group (Marie Curie 2 Cantoblanco Madrid – Spain) for the XPS analysis.References
- C. M. Eichenseer, B. Kastl, M. A. Pericàs, P. R. Hanson and O. Reiser, ACS Sustainable Chem. Eng., 2016, 4, 2698–2705 CrossRef CAS.
- E. Verde-Sesto, E. Merino, E. Rangel-Rangel, A. Corma, M. Iglesias and F. Sánchez, ACS Sustainable Chem. Eng., 2016, 4, 1078–1084 CrossRef CAS.
- C. H. Campos, E. Rosenberg, J. L. G. Fierro, B. F. Urbano, B. L. Rivas, C. C. Torres and P. Reyes, Appl. Catal., A, 2015, 489, 280–291 CrossRef CAS.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed.
- C. Jimeno, S. Sayalero and M. A. Pericàs, in Heterogenized Homogeneous Catalysts for Fine Chemicals Production: Materials and Processes, ed. P. Barbaro and F. Liguori, Springer Netherlands, Dordrecht, 2010, pp. 123–170 Search PubMed.
- I. E. Ertas, M. Gulcan, A. Bulut, M. Yurderi and M. Zahmakiran, J. Mol. Catal. A: Chem., 2015, 410, 209–220 CrossRef CAS.
- A. Popa, R. Ene, D. Visinescu, E. S. Dragan, G. Ilia, S. Iliescu and V. Parvulescu, J. Mol. Catal. A: Chem., 2015, 408, 262–270 CrossRef CAS.
- M. T. Gokmen and F. E. Du Prez, Prog. Polym. Sci., 2012, 37, 365–405 CrossRef CAS.
- J. Xiong, C. Hang, J. Gao, Y. Guo and C. Gu, Chem. Eng. J., 2014, 254, 276–282 CrossRef CAS.
- E. Breysse, C. Pinel and M. Lemaire, Tetrahedron: Asymmetry, 1998, 9, 897–900 CrossRef CAS.
- F. J. L. Heutz and P. C. J. Kamer, Dalton Trans., 2016, 45, 2116–2123 RSC.
- Y. Jun, X. Rongnan and Y. Juntan, J. Appl. Polym. Sci., 1989, 38, 45–54 CrossRef.
- J. Wei, X. Y. Bai and J. Yan, Macromolecules, 2003, 36, 4960–4966 CrossRef CAS.
- J. Wei, X. Zhao and J. Yan, J. Appl. Polym. Sci., 2004, 92, 2681–2688 CrossRef CAS.
- F. Bildik, E. Yavuz, T. Sismanoglu and B. Filiz Senkal, Macromol. Symp., 2015, 352, 66–71 CrossRef CAS.
- I.-H. Park, J. M. Rhee and Y. S. Jung, Angew. Makromol. Chem., 1999, 267, 27–34 CrossRef CAS.
- A. Blasig, J. Tang, X. Hu, S. P. Tan, Y. Shen and M. Radosz, Ind. Eng. Chem. Res., 2007, 46, 5542–5547 CrossRef CAS.
- L. Canali, E. Cowan, H. Deleuze, C. L. Gibson and D. C. Sherrington, J. Chem. Soc., Perkin Trans. 1, 2000, 2055–2066 RSC.
- R. Drake, D. C. Sherrington and S. J. Thomson, React. Funct. Polym., 2004, 60, 65–75 CrossRef CAS.
- S. Bulut, Z. Fei, S. Siankevich, J. Zhang, N. Yan and P. J. Dyson, Catal. Today, 2015, 247, 96–103 CrossRef CAS.
- J. Su and J.-S. Chen, Microporous Mesoporous Mater., 2017, 237, 246–259 CrossRef CAS.
- G. Busca, Heterogeneous Catalytic Materials, Elsevier, Amsterdam, 2014, pp. 297–343 Search PubMed.
- F. Jiang, J. Cai, B. Liu, Y. Xu and X. Liu, RSC Adv., 2016, 6, 75541–75551 RSC.
- P. Pospiech, J. Chojnowski, U. Mizerska, T. Makowski, K. Strzelec and N. Sienkiewicz, Appl. Organomet. Chem., 2016, 30, 399–407 CrossRef CAS.
- C. Liu, J. Liu, S. Yang, C. Cao and W. Song, ChemCatChem, 2016, 8, 1279–1282 CrossRef CAS.
- L. A. Oro and D. Carmona, The Handbook of Homogeneous Hydrogenation, Wiley-VCH Verlag GmbH, 2006, pp. 2–30 Search PubMed.
- E. de Wolf, A. L. Spek, B. W. M. Kuipers, A. P. Philipse, J. D. Meeldijk, P. H. H. Bomans, P. M. Frederik, B.-J. Deelman and G. van Koten, Tetrahedron, 2002, 58, 3911–3922 CrossRef CAS.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- R. Haag, W. Bannwarth, F. Koç, F. Michalek and L. Rumi, Synthesis, 2005, 3362–3372 Search PubMed.
- P. B. Webb and D. J. Cole Hamilton, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons, Ltd, 2012, pp. 497–532 Search PubMed.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- V. Nieminen, A. Taskinen, M. Hotokka and D. Y. Murzin, J. Catal., 2007, 245, 228–236 CrossRef CAS.
- E. Toukoniitty, P. Mäki-Arvela, N. Kumar, T. Salmi and D. Y. Murzin, Catal. Today, 2003, 79–80, 189–193 CrossRef CAS.
- C. H. Campos, C. C. Torres, A. B. Dongil, D. Ruiz, J. L. G. Fierro and P. Reyes, Catal. Today, 2014, 235, 226–236 CrossRef CAS.
- S. Sahoo, H. Lundberg, M. Edén, N. Ahlsten, W. Wan, X. Zou and B. Martín-Matute, ChemCatChem, 2012, 4, 243–250 CrossRef CAS.
- K. J. Stanger, J. W. Wiench, M. Pruski and R. J. Angelici, J. Mol. Catal. A: Chem., 2003, 195, 63–82 CrossRef CAS.
- C. R. Hilliard, N. Bhuvanesh, J. A. Gladysz and J. Blumel, Dalton Trans., 2012, 41, 1742–1754 RSC.
- M. C. Román-Martínez, J. A. Díaz-Auñón, C. Salinas-Martínez de Lecea and H. Alper, J. Mol. Catal. A: Chem., 2004, 213, 177–182 CrossRef.
- C. M. Standfest-Hauser, T. Lummerstorfer, R. Schmid, H. Hoffmann, K. Kirchner, M. Puchberger, A. M. Trzeciak, E. Mieczyńska, W. Tylus and J. J. Ziółkowski, J. Mol. Catal. A: Chem., 2004, 210, 179–187 CrossRef CAS.
- L. J. Lemus-Yegres, M. C. Román-Martínez, I. Such-Basáñez and C. Salinas-Martínez de Lecea, Microporous Mesoporous Mater., 2008, 109, 305–316 CrossRef CAS.
- M. Pérez-Cadenas, L. J. Lemus-Yegres, M. C. Román-Martínez and C. Salinas-Martínez de Lecea, Appl. Catal., A, 2011, 402, 132–138 CrossRef.
- D. Dehe, L. Wang, M. K. Müller, G. Dörr, Z. Zhou, R. N. Klupp-Taylor, Y. Sun, S. Ernst, M. Hartmann, M. Bauer and W. R. Thiel, ChemCatChem, 2015, 7, 127–136 CrossRef CAS.
- R. Yerushalmi, J. C. Ho, Z. Fan and A. Javey, Angew. Chem., Int. Ed., 2008, 47, 4440–4442 CrossRef CAS PubMed.
- K. J. Stanger, Y. Tang, J. Anderegg and R. J. Angelici, J. Mol. Catal. A: Chem., 2003, 202, 147–161 CrossRef CAS.
- A. Crosman and W. F. Hoelderich, Catal. Today, 2007, 121, 130–139 CrossRef CAS.
- P. Kleman and A. Pizzano, Tetrahedron Lett., 2015, 56, 6944–6963 CrossRef CAS.
- R. Sayah, M. Le Floch, E. Framery and V. Dufaud, J. Mol. Catal. A: Chem., 2010, 315, 51–59 CrossRef CAS.
- H.-Y. Jiang and X.-X. Zheng, Appl. Catal., A, 2015, 499, 118–123 CrossRef CAS.
- J. L. Castelbou, P. Blondeau, C. Claver and C. Godard, RSC Adv., 2015, 5, 97036–97043 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. Halpern, Inorg. Chim. Acta, 1981, 50, 11–19 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26104c |
| This journal is © The Royal Society of Chemistry 2017 |