
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of polypropylene/Mg–Al layered double hydroxides nanocomposites through wet pan-milling: formation of a second-staging structure in LDHs intercalates
Yilei Zheng and
Yinghong Chen *
*
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: johnchen@scu.edu.cn; Fax: +86-28-85402465; Tel: +86-28-85405136
First published on 5th January 2017
Abstract
In this paper, polypropylene/MgAl-layered double hydroxide nanocomposites were in situ prepared by using a novel solid state shear milling (S3M) technology without use of any compatibilizer and any prior organic treatment of raw LDHs. The obtained composite powders and nanocomposites were characterized using X-ray diffraction (XRD), Fourier transform infrared (FT-IR) spectroscopy, scanning electron microscopy (SEM) and transmission electron microscopy (TEM), respectively. The results show that under the very strong shear force field of pan-milling, the anion-exchange reaction of CO32−-LDHs with sodium dodecyl sulfate (SDS) can be realized under non-acidic conditions. In addition, the in situ modification of LDHs with SDS and the co-intercalation of PP macromolecular chains into LDH interlayers were found to be completed in one process in aqueous medium. XRD results demonstrate that there are two types of layered structures in the prepared PP/LDHs co-powders and the occurrence of the transformation from layered structure I to II with increasing milling cycles and SDS loading. Staging is proposed to illustrate the anion-exchange reaction of intercalation of the SDS anion into CO32−-LDH layers and the corresponding transformation, showing the second stage intermediate corresponding to layered structure I and the first stage product corresponding to structure II. Both XRD analysis and TEM observation show the formation of exfoliated LDH layers at low LDH loading and concomitant intercalated/exfoliated LDH layers at high LDH loading in the prepared PP/LDHs nanocomposites. The dynamic mechanical properties and crystallization behavior of the obtained nanocomposites were also investigated using dynamic mechanical analysis (DMA) and differential scanning calorimetry (DSC), respectively.
Introduction
In recent years, polymer/layered crystal nanocomposites have been recognized as one of the most promising polymer composites due to their remarkably improved mechanical performances, thermal stability, flammability, gas barrier properties, and other physico-chemical properties by addition of only a small amount of layered fillers.1–4 Such an improvement is hard to be achieved by blending fillers of micrometer particle size with the polymers. In the past several years, most emphases have been given to polymer layered silicate nanocomposites,5 such as polymer/montmorillonite system. Recently, a new promising class of inorganic layered materials, e.g., layered double hydroxides (LDHs), have been attracting much attention because of their wide applications in catalysts, catalyst precursors, anion exchangers, fire retardants, hydrogenation reaction, acid absorbents, bioactive nanocomposites, electroactive and photoactive materials, etc.6–10Layered double hydroxides (LDHs), also known as anionic or hydrotalcite-like clays, are a class of lamellar compounds having overall charge neutrality due to the existence of positively charged brucite-like hydroxide layers with typical thickness 0.48 nm and hydrated exchangeable anions located in the interlayer gallery.6 The positive charge of the brucite-like layers arises from the partially isomorphous substitution of trivalent cations for divalent metal ones. The general formula can be expressed as [M1−x2+Mx3+(OH)2]x+(An−)x/n·mH2O, where M2+ and M3+ are divalent (Mg2+, Zn2+, Ca2+, Co2+, Ni2+, Cu2+, Mn2+, etc.) and trivalent (Al3+, Cr3+, Fe3+, Co3+, Ga3+, Mn3+, etc.) cations occupying the octahedral central positions within the hydroxide layers, respectively; x is equal to the ratio M3+/(M2+ + M3+) and A is the corresponding anion with valence n (Cl−, CO32−, SO42−, NO3−, C12H25SO4−, etc.); the remaining free space of the interlayer is occupied by water of crystallization. Due to their highly tunable properties and unique anion exchange properties, LDHs are considered as a new emerging and the most favorable class of layered crystals for fabricating of multifunctional polymer/layered inorganic nanocomposites.1,11,12 However, the integrated hydrogen-bonding networks and the strong electrostatic interactions between hydroxide layers and intercalated anions/water molecules, which results from the high charge density of LDH layers associated with a high content of intercalated hydrated anions, make the intercalation or exfoliation very difficult in polymer matrix. This may explain the reason for the relatively less reported LDHs-based nanocomposites. Moreover, pure inorganic LDHs are generally incompatible with organic non-polar polymers. On the other hand, the interlayer spacing between the metal hydroxide layers is not big enough for large polymer chains intercalation. Therefore, the organic modification of pristine LDHs is a key factor in the preparation of polymer/LDHs nanocomposites. In the organic modification process, many organic anions are introduced into the interlayer galleries. In this way, on one hand, the galleries above mentioned would become more lipophilic and on the other hand, the interlayer distances were correspondingly increased, hence weakening the electrostatic forces between the hydroxide sheets to facilitate the intercalation of polymer. Based on this, several methods have been developed for the synthesis of organo-LDHs, including direct synthesis by co-precipitation, anion-exchange of a precursor, rehydration of a calcined LDHs precursor and thermal reaction.6 So far, a large number of contributions are focused on organically modified LDHs prepared via co-precipitation and anion-exchange of NO3−. In order to achieve a good dispersion and exfoliation of LDHs in polymer matrix, various intercalation methods have been utilized in the literatures. For example, melt intercalation, solution intercalation, in situ polymerization, exfoliation–adsorption process, and template synthesis13–16 are found to be used to prepare the nanocomposites of LDHs with a series of polymers.17–20 Also, delamination of LDHs into single nanosheets in suspension was recently attracting much attention due to their new nanostructure and potential applications,21 which sometimes tends to be an important step in preparing polymer nanocomposites.
The natural LDHs generally exist as hydrotalcite (HT) [Mg6Al2(OH)16CO3·H2O]. However, the affinity of carbonate anions in the LDH interlayer space to metal cations is so strong that they cannot easily be exchanged with organic anions, which is a major impediment for LDH layers exfoliation.22 So, the delamination of the CO32−-LDHs in suspension is believed to be almost impossible and the traditional anion-exchange method is not suitable for preparation of organically-modified CO32−-LDHs. To overcome this problem, deintercalation of carbonate has been effectively achieved using acid–salt mixed solution, successfully realizing conversion of CO32−-LDHs into other organic anions contained LDHs based on a mechanism of dissolution recrystallization.23,24 Another method is rehydration.19,25 The unique property of LDHs has been referred to as the “memory effect” and provides an effective route to incorporate organic anions in the galleries of the layered double hydroxides. However, the disadvantage of this method is that the original structure and crystal shape of LDHs may be damaged during heat treatment process, hence making it difficult to prepare well crystalline nanocomposite.
Until now, only few articles have reported the preparation and properties of polypropylene/layered double hydroxides nanocomposites.26–28 It is usually necessary to use polar compatibilizer to enhance the interactions between non-polar polypropylene (PP) and LDHs to obtain good LDH dispersion in matrix at nanometer scale. In our previous articles, polymer/layered inorganic fillers nanocomposites were prepared using solid state shear milling (S3M) without prior organic treatment of filler.29–31 In the present study, we further develop the novel S3M method, i.e. wet pan-milling, to in situ prepare PP/LDHs nanocomposites using commercial hydrotalcite without any compatibilizer and any prior organic treatment. During pan milling process, the presence of staging phenomenon of LDHs contained composite powder (co-powder) was found for the first time. In order to fully understand such a staging mechanism, the effects of in situ intercalating agent SDS content, number of milling cycles and incorporation of PP macromolecular chains were investigated. The X-ray diffraction (XRD) and transmission electronic microscopy (TEM) characterizations confirm the successful formation of intercalation/exfoliation structure of LDHs in polymer matrix using S3M technology. The dynamic mechanical properties and crystallization behavior of the nanocomposites prepared were also studied and analyzed.
Experimental
Materials
The isotactic polypropylene (PP), T30S, with diameter of 3–4 mm was supplied by the Dushanzi Petrochemical Co. (Kelamayi, China). MgAl-layered double hydroxides (hydrotalcite) were purchased from Jiangxi Hongyuan Chemical Co., Ltd (China). Sodium dodecyl sulfate (SDS) was obtained from Chemical Reagent Factory of Kelong (Chengdu, China).Equipment
The structure of the self-designed pan type milling equipment was shown in the previous papers,32–34 of which the ingenious design was inspired by the traditional Chinese stone mill. This special pan-mill equipment is designed for pulverization, dispersion, mixing as well as activation. The equipment is operated at room temperature, while the polymer is in solid state, and can exert very strong squeezing force in normal direction and very strong shear force in both radial and tangential directions on the milled materials, functioning like pairs of three-dimensional scissors, and hence has excellent pulverizing, mixing, and activation effect on the polymeric materials. The core parts of the pan-mill equipment are the mill pans, including the stationary pan and the moving pan. They would closely touch when working.Preparation of PP/LDHs co-powders
Mixture of PP, unmodified LDHs and SDS (intercalating agent) at a certain weight ratio was first dispersed in 2000 ml distilled water. The mixture was then fed into the mill pan through the hopper in the center of the milling pan at ambient temperature. The heat produced during milling was removed by cooling water circulation. The milled suspension was discharged from the brim of pans and then collected for next cycle milling. This suspension was dried in an oven at 100 °C for 48 h to obtain the co-powder for the subsequent use. In this paper, the ratio of PP to LDHs was kept constant (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). For the convenience of description, the obtained co-powder was abbreviated as PPLSxp, where LS and x represents LDHs and the percentage content of LDHs in co-powder, respectively. As a contrast, the milled mixture without PP was separated by centrifugation and then washed with distilled water for several times to remove the unreacted SDS monomer prior to drying. The control sample was abbreviated as LDS.
:1). For the convenience of description, the obtained co-powder was abbreviated as PPLSxp, where LS and x represents LDHs and the percentage content of LDHs in co-powder, respectively. As a contrast, the milled mixture without PP was separated by centrifugation and then washed with distilled water for several times to remove the unreacted SDS monomer prior to drying. The control sample was abbreviated as LDS.
Preparation of PP/LDHs nanocomposites
The PPLS25p co-powder with 25 milling cycles as the master batch was diluted using neat PP to obtain various concentration of LDH sample in a twin-screw extruder. The extrudates were pelletised and then dried. The dried pellets were then injection molded into specimen for various measurements. The obtained PP/LDHs sample was named as PP/LSy, where y represents the content of LDHs. The composition of the samples used in this study is summarized in Table 1.| Sample | Component, wt% | Milling cycles | Final content, wt% | |||||
|---|---|---|---|---|---|---|---|---|
| PP | LDHs | SDS | Co-powder | PP | LDHs | SDS | ||
| PP | 100 | — | — | — | — | 100 | — | — |
| LDS | — | 50 | 50 | — | 25/40 | — | — | — |
| PPLS31.3p | 62.5 | 31.3 | 6.2 | — | 25 | — | — | — |
| PPLS25p | 50 | 25 | 25 | — | 15/25 | — | — | — |
| PPLS20p | 40 | 20 | 40 | — | 25 | — | — | — |
| PP/LS0.5 | 98 | — | — | 2 | — | 99 | 0.5 | 0.5 |
| PP/LS1 | 96 | — | — | 4 | — | 98 | 1 | 1 |
| PP/LS2 | 92 | — | — | 8 | — | 96 | 2 | 2 |
| PP/LS3 | 88 | — | — | 12 | — | 94 | 3 | 3 |
Characterization
Results and discussion
Structure of PP/LDHs co-powders
The preparation of PP/LDHs co-powders through wet pan-milling is a key step in the fabrication of PP/LDHs nanocomposites. It is very necessary to characterize the structures of the prepared PP/LDHs co-powders and the mineral raw material LDHs.XRD analysis of PP/LDHs co-powders
Fig. 1(a) shows the XRD pattern of raw material MgAl-LDHs in the 2θ range of 2–70°. The diffraction planes have been indexed on the basis of a hexagonal unit cell with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m rhombohedral symmetry. The three peaks at 2θ = 11.4°, 22.9° and 34.7° corresponding to the typical reflection planes of (003), (006) and (009), respectively, show the existence of a well-defined layered structure in MgAl-LDHs.35 The interlayer spacing of LDHs is found to be 0.77 nm based on (003) reflection plane, which is consistent with the reported value.25,35
m rhombohedral symmetry. The three peaks at 2θ = 11.4°, 22.9° and 34.7° corresponding to the typical reflection planes of (003), (006) and (009), respectively, show the existence of a well-defined layered structure in MgAl-LDHs.35 The interlayer spacing of LDHs is found to be 0.77 nm based on (003) reflection plane, which is consistent with the reported value.25,35
 | ||
| Fig. 1 XRD patterns of (a) pure MgAl-LDHs and (b) PPLSxp with different SDS content at 25 milling cycles: (b-1) PPLS31.3p, (b-2) PPLS25p and (b-3) PPLS20p. ● represents layered structure I; ▼ represents layered structure II. | ||
The XRD patterns of PPLSxp with different SDS content at 25 milling cycles are shown in Fig. 1(b). It can be seen that no new reflection peak appears at the lower angle for PPLS31.3p sample (6.2 wt% SDS loading). However, with increase in SDS loading, interestingly there are several new reflection peaks appearing at the lower angle range in the XRD patterns, which are ascribed to more than three typical order reflection peaks,26 indicating the intercalation of dodecyl sulfate anions and polymer chains into the LDH gallery under the effect of the very strong shear force field induced by pan-milling and formation of two types of layered structures, i.e. layered structure I and layered structure II, respectively. The incorporation of SDS intercalation agent leads to a shift of (003) diffraction plane from 2θ = 11.4° (d003 = 0.77 nm) of pure LDHs to 2θ = 2.35° (d003I = 3.75 nm) of structure I and 2θ = 2.77° (d003II = 3.18 nm) of structure II, respectively. When the SDS loading is further increased to 40 wt% (PPLS20p sample), the involved peak intensity of structure II becomes stronger than that of structure I, but the corresponding basal spacing does not change. This indicates that layered structure I has been transformed to layered structure II with the increase of the SDS content at the same milling cycles and SDS can remarkably promote the anion-exchange intercalation reaction under the effect of shear force field of pan-milling, i.e., the intercalation reaction does not occur until the concentration of SDS increases to a certain value. However, from the X-ray diffraction patterns of various samples (Fig. 1(b)), it can be also seen that the original basal peak of LDHs at 2θ = 11.4° does not disappear, indicating that there are still a fraction of LDH particles with their original structure during pan-milling process. In addition, the content of the original LDH particles is connected with the added SDS content. The theoretical LDHs/SDS ratio used to realize the intercalation of LDHs is about 1:1 according to chemical formula. However, it is obvious that the SDS in the solution is impossible to be completely intercalated into the gallery. On the other hand, the presence of partly unintercalated LDH particles could be explained by the limited pan-milling intercalation reaction and the high affinity of CO32− in the gallery space of LDHs. In order to know how much the CO32− is replaced, element analysis was used to determine the replacement degree of CO32−. The results show that the replacement degree of CO32− of samples PPLS31.3p, PPLS25p and PPLS20p is 0, 73.5% and 86.2%, respectively. It is seen that there is no the intercalation of SDS occurring when SDS content is low (6.2 wt%). However, when increasing the SDS content to 40 wt%, the intercalation of SDS would become very easy and the replacement degree of CO32− even reaches 86.2%. As a result, the proper increase of the SDS content could obviously promote the ion-exchange interaction reaction between SDS and CO32−. This is in agreement with the XRD results (Fig. 1).
Fig. 2 shows the XRD patterns of PPLS25p samples with different milling cycles. It is interestingly seen that when the mixture is milled for only 15 cycles, there are three typical reflection peaks occurring, which could be ascribed to layered structure I. When the number of milling cycles increases to 25, the layered structure II reflection peaks appear, and the intensity of layered structure I reflection peaks is, however, greatly weakened. Above analyses demonstrate that the LDH particles of PPLS25p co-powder experience transformation from structure I to structure II with increment of milling cycles. The reasons will be analyzed subsequently.
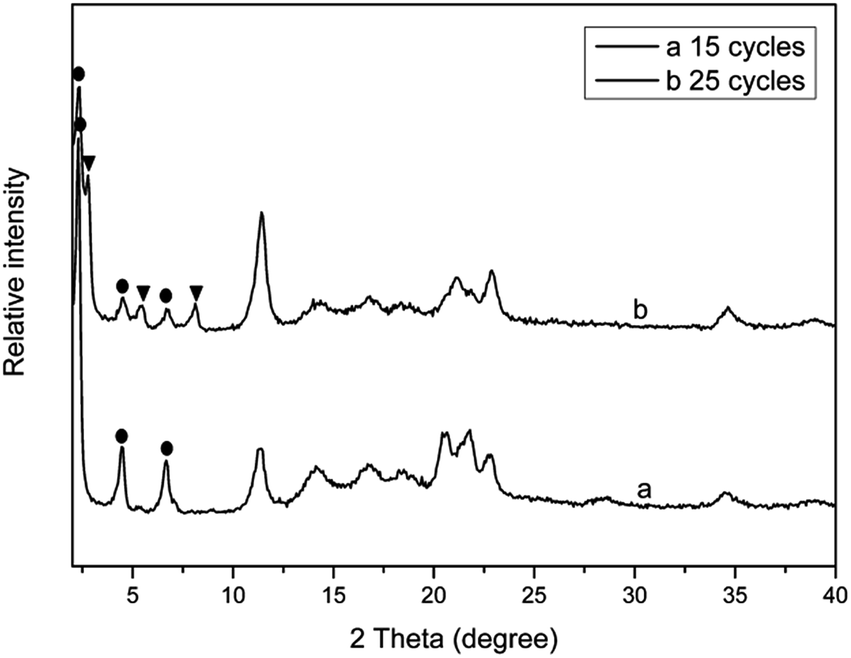 | ||
| Fig. 2 XRD patterns of PPLS25p with different pan-milling cycles. ● represents layered structure I; ▼ represents layered structure II. | ||
Morphology of PP/LDHs co-powders
Fig. 3 shows the SEM images of pure PP and PPLS25p sample at 25 milling cycles. It can be seen that the morphology of pure LDH particles (Fig. 3(a)) shows the presence of thin platelets with irregular hexagonal morphology stacked with each other, differing widely in lateral size. The dimension of PP particles (Fig. 3(b)) is about 10 μm with irregular shape, and the surface of PP particles is covered with a large number of small particles. In addition, some shear induced deformations occur on the surface of PP particles due to the three-dimensional shear force field of pan-milling. Also, it is shown that there is the irregularly lamellar structure of LDH particles instead of hexagonal morphology, which are embedded into PP matrix. This is mainly because under the effects of the very strong shear force and squeeze force fields, the large rigid lamellae of LDHs are first closely combined with the surface and even embedded inside of PP particles during pan-milling. Then, these lamellae would suffer from fragmentation and deformation, simultaneously accompanied with the pulverization of PP particles. | ||
| Fig. 3 SEM images of (a) pure LDHs and (b) PPLS25p at 25 milling cycles. | ||
Structure of LDHs/SDS co-powders
In order to investigate the influence of the matrix polypropylene on pan-milling intercalation reaction, the mixture of LDHs and SDS was milled in the presence of water medium, as shown in Fig. 4. From the obtained XRD patterns (Fig. 4), it can be seen that the reflection peaks are dramatically different from those of samples containing PP. The XRD analysis clearly reveals that both two types of layered structures (I and II) do not exist in the milled mixture of LDHs and SDS (LDS powder). Only a small broad peak at 2θ = 7.7° for reflection plane of (003) appears with a basal spacing of 1.14 nm and this small interlayer space, however, has not been reported by intercalation of SDS into LDH layers.36 So, PP plays an important role in pan-milling intercalation reaction possibly through being as a force carrier. The pan-milling can exert shear force and squeeze force effects on the lamellar LDHs through close combination with PP particles and the intercalation reaction could hardly be realized without PP as carriers.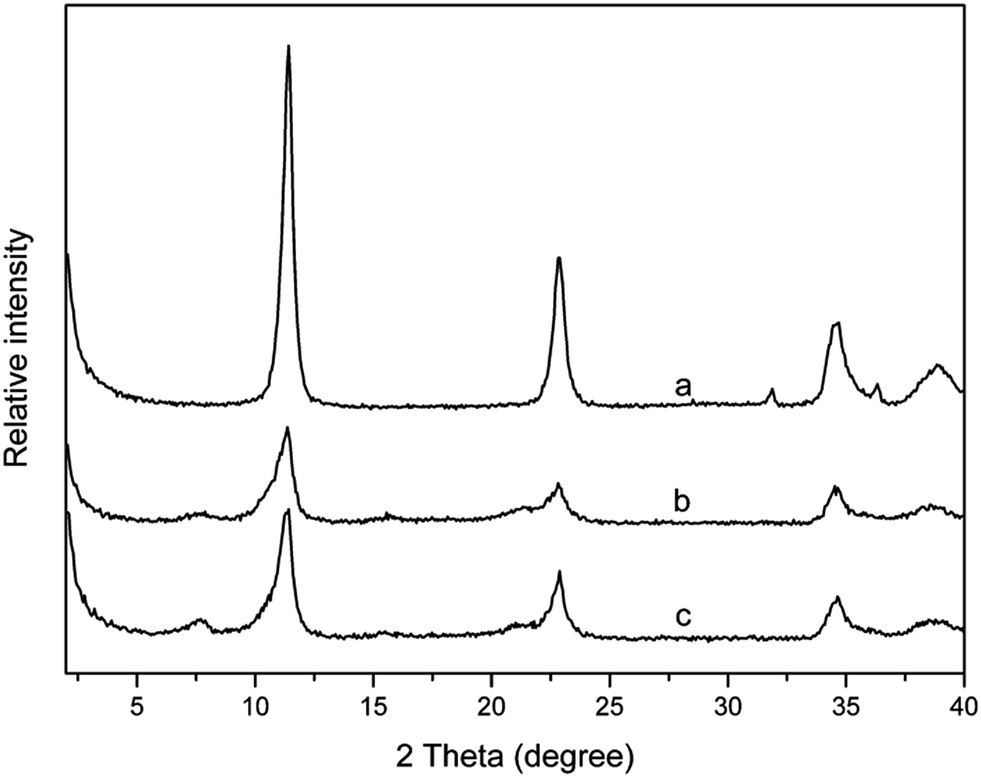 | ||
| Fig. 4 XRD patterns of (a) LDHs, (b) LDS at 25 milling cycles and (c) LDS at 40 milling cycles. | ||
To further confirm the participation of SDS in the intercalation reaction, the corresponding FT-IR spectra of SDS, LDHs and LDS at 25 milling cycles are provided in Fig. 5. For pure LDHs, the broad absorption band around 3507 cm−1 is originated from the OH stretching modes of intra-layer and interlayer water molecules and the shoulder at about 3000 cm−1 is due to hydrogen bonding of interlayer water and carbonate. A peak appearing at 1638 cm−1 is due to the bending mode of interlayer water molecules. In addition, at 1369 cm−1, the band is assigned to the antisymmetric ν3 mode of interlayer carbonate anions. The spectrum of LDS at 25 milling cycles shows new absorption peaks at 2923 and 2854 cm−1, which is associated to the C–H stretching vibrations. These bands are due to the –CH2– and –CH3 groups of SDS anion long chain, and the peak at 1570 cm−1 is due to the deformation vibration of these groups, which is at 1470 cm−1 for pure SDS. The shift is due to the confinement in the gallery of LDH layers. In addition, the bands at 1226 and 1061 cm−1 are attributed to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of sulfate group. The appearance of these new peaks confirms the intercalation of the SDS anions into the gallery of LDHs. However, the characteristic absorption band of the carbonate stretching mode (1369 cm−1) is still strong, indicating that there is still a part of original LDHs which do not interact with dodecyl sulfate anion. This is in accordance with the XRD result.
O stretching vibration of sulfate group. The appearance of these new peaks confirms the intercalation of the SDS anions into the gallery of LDHs. However, the characteristic absorption band of the carbonate stretching mode (1369 cm−1) is still strong, indicating that there is still a part of original LDHs which do not interact with dodecyl sulfate anion. This is in accordance with the XRD result.
 | ||
| Fig. 5 FT-IR spectra of (a) SDS, (b) LDHs and (c) LDS at 25 milling cycles. | ||
Structure and performance of PP/LDHs nanocomposites
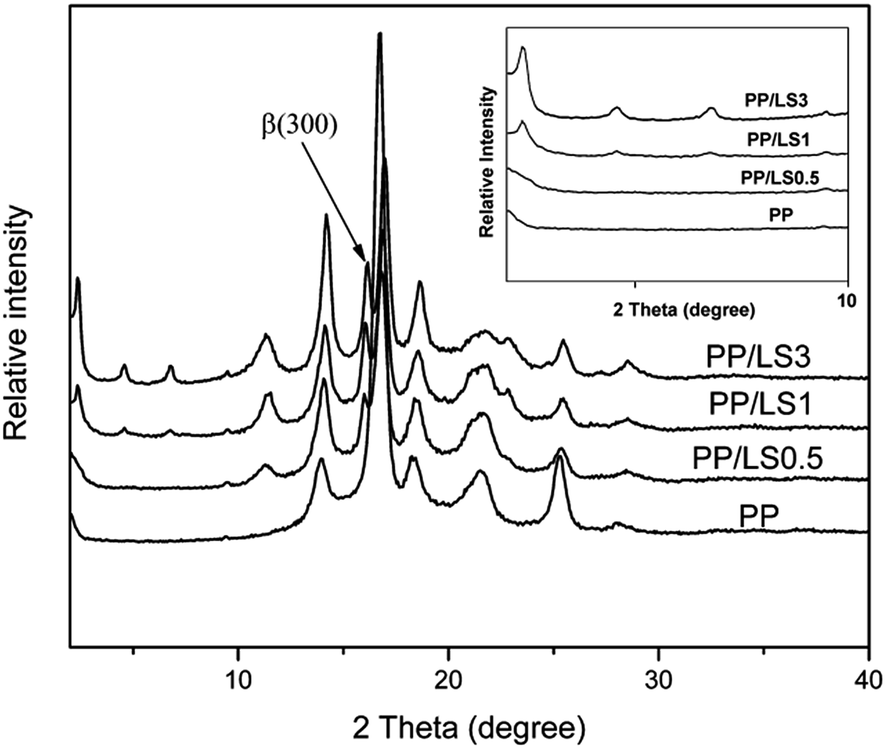 | ||
| Fig. 6 XRD patterns of PP/LDHs nanocomposites with various LDH loading. | ||
In addition, from Fig. 6, it can be also found that the neat PP shows typical α crystal structure according to its featured diffraction peaks. However, for the LDHs filled PP system, there appears a new peak at 2θ = 16.03°, which is the featured diffraction peak of reflection plane (300) of β crystal. Furthermore, with increase of LDH loading, the intensity of (300) peak increases also. Above analyses indicate that the added LDHs could induce the formation of β crystal by acting as a role of β nucleating agent, and, the increase in LDH content would be advantageous to the formation of β crystal.
 | ||
| Fig. 7 TEM micrographs at (a) low and (b) high magnification for PP/LS0.5, and (c) low and (d) high magnification for PP/LS3. | ||
Fig. 8 shows the SEM photos of the fractured surface of PP/LDH (3.0 wt%) (PP/LS3) sample. It is seen that for the PP/LDH (3.0 wt%) nanocomposite prepared through S3M method, the incorporated LDHs fillers are dispersed in PP matrix very homogeneously overall. However, for the conventionally prepared PP/LDH (3.0 wt%) composite (simply melt-compounding without pan-milling treatment), the incorporated LDHs particles are heavily agglomerated. Above results show that pan-milling is effective in realization of the exfoliation and dispersion of LDHs in the non-polar hydrophobic PP matrix.
 | ||
| Fig. 8 The SEM photos of the fractured surface of PP/LDHs (3.0 wt%) sample: (a) S3M method and (b) conventional method. | ||
δ) with the temperature for PP and PP/LDHs nanocomposites. From the Fig. 9(a), it can be seen that the storage modulus (E′) of nanocomposites at low temperature is indeed increased by increasing the LDH loading, indicating that nano-dispersed LDH layers limit the movement of PP macromolecular chain segments. This accordingly enhances the stiffness of PP effectively. However, this increase is less obvious at higher temperature, since the mobility of the PP macromolecules is very high above the glass transition temperature. From the tanδ–T curves (Fig. 9(b)), it can be seen that the broad peaks occurring in the temperature range of 15–40 °C represent the glass transition regions. According to these curves, pure PP exhibits a Tg at 28.5 °C. In the prepared PP/LDHs nanocomposites, by adding LDHs with both 0.5 wt% and 3 wt% content, a slight shift of Tg values to the higher temperature is observed (31.6 °C and 30.2 °C, respectively), due to the strong interaction between the polymeric chains and also the reduction of the macromolecular chain's mobility at the interface around the nanoparticles. Especially, the movement confinement of molecular chains with the LDH nanoparticles in exfoliated single layers (PP/LS0.5) is more obvious than that with the intercalated tactoids (PP/LS3). From Fig. 9(b), it is also seen that the tanδ values of the nanocomposites are higher than those of the pure PP, which is different from the results obtained in most literatures (the addition of filler generally would decrease the tanδ value of polymer matrix near glass transition region because the incorporated fillers would cause the restriction of movement of molecular chains to a certain degree). The reason for this could be explained from the following two aspects: one is the strong internal friction37 possibly present at the LDHs-PP interfaces and the other is possibly the presence of the free unintercalated sodium dodecyl sulfate (SDS) small molecules with a longer alkyl chain in nanocomposite (after pan-milling treatment, the free unintercalated SDS was not washed out). This meanwhile indicates that the obtained PP/LDHs nanocomposites have improved damping properties.
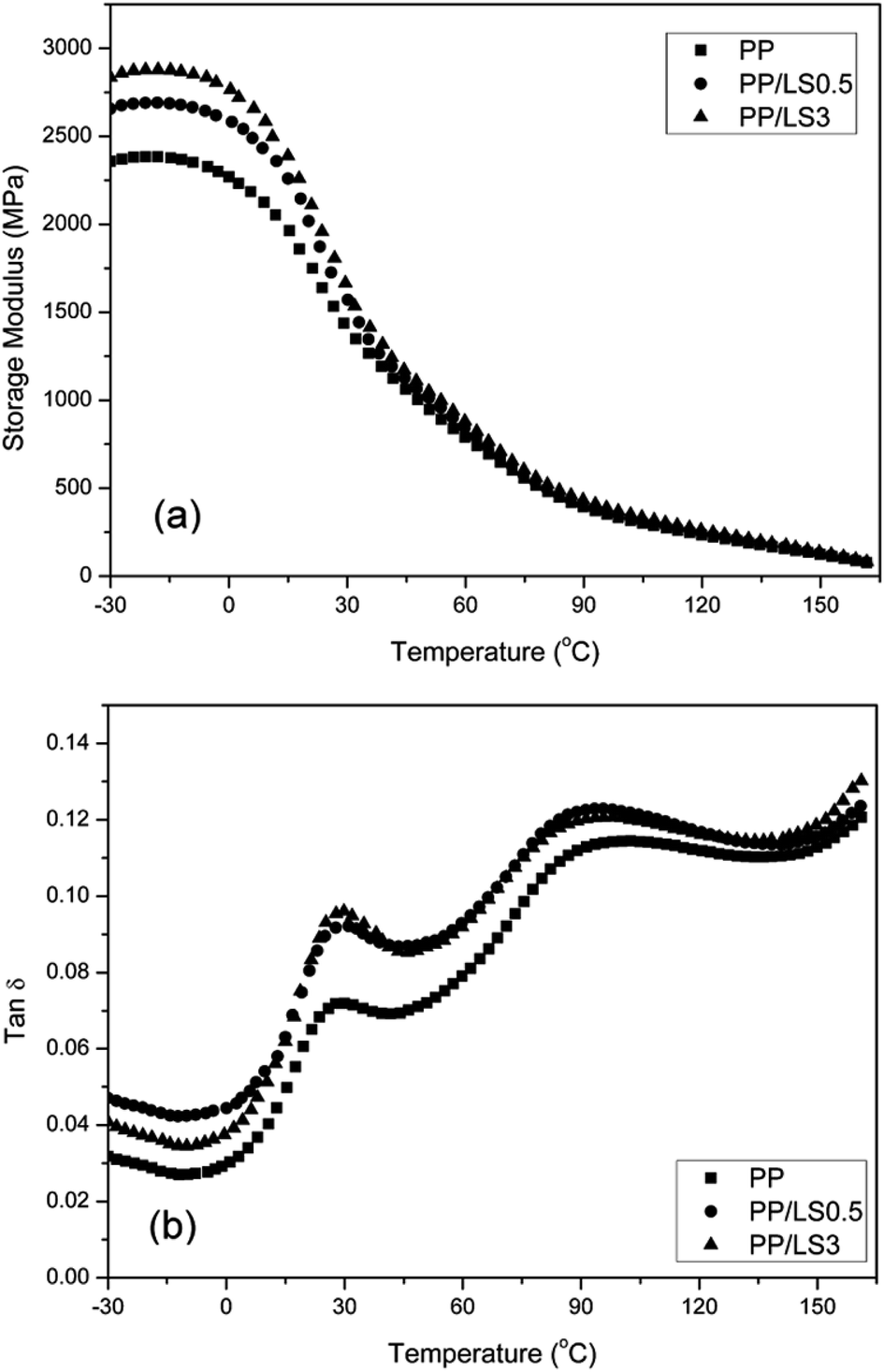 | ||
| Fig. 9 Dynamic mechanical analysis curves of PP and PP/LDHs nanocomposites: (a) storage modulus E′ and (b) tanδ. | ||
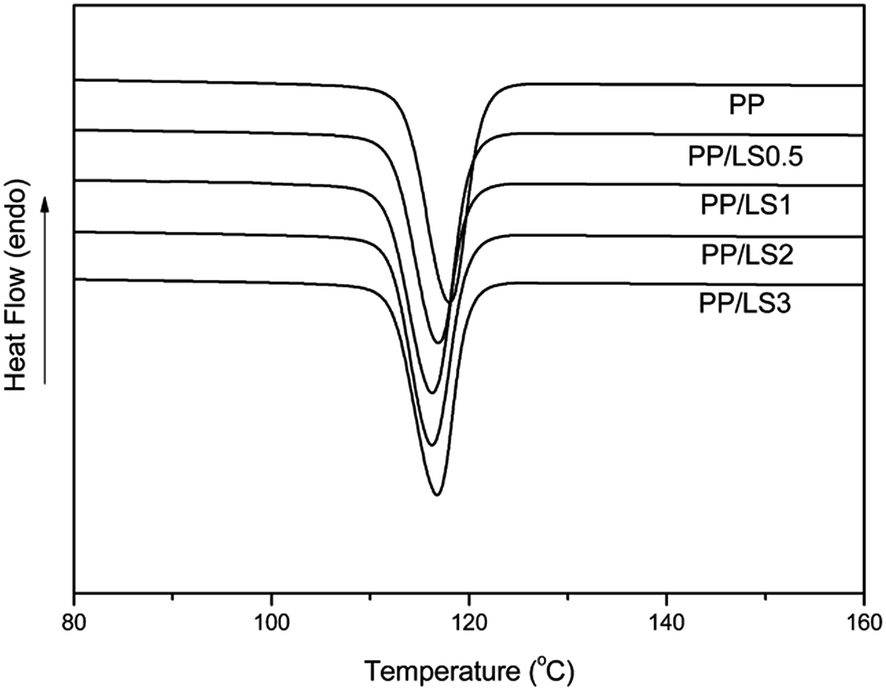 | ||
| Fig. 10 Crystallization of DSC thermograms of pure PP, PP/LS0.5, PP/LS1, PP/LS2 and PP/LS3 samples at cooling rate 10 °C min−1. | ||
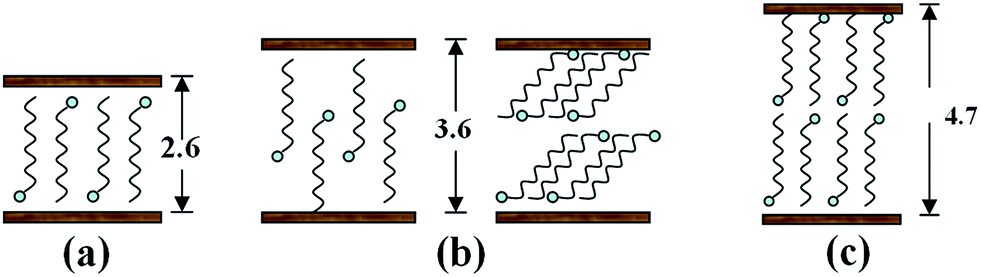 | ||
| Fig. 11 Schematic diagram of the different arrangement of SDS intercalated in LDHs gallery. | ||
During the anion-exchange reaction of LDHs, sometimes there are some well crystallized and incompletely exchanged intermediate products formed, i.e. some interlayer spaces are completely or partially occupied, while the left spaces still remain empty. This phenomenon is called the staging of the exchange reaction, i.e. staging intermediates,38–41 where the involved final product is called the first stage product (the completely anion-exchanged product) with a basal spacing of d; the involved intermediate product is called the second stage intermediate (the partially alternately anion-exchanged product). If the basal spacing of the initial reactant is d0, the basal spacing of the second stage intermediate will be d + d0. This phenomenon can be commonly observed in other layered compound systems such as graphite due to its flexible sheets. However, it is very rare in LDH systems owing to the rigidity of the layer and interlayer attractive forces. O'Hare and the co-workers42 have reported staging phenomenon during the intercalation of organic phosphonates and carboxylates into [LiAl2(OH)6]X·yH2O through an anion exchange reaction, which was also observed in MgAl-LDH system.43 Additionally, Iyi et al.44 have reported the appearance of second staging for MgAl-LDHs intercalated system with an anionic azobenzene derivative (AzAA) through a co-precipitation method.
As a consequence, by combing the literatures with our experimental results, the pan-milling assisted staging anion-exchange intercalation reaction mechanism is proposed here. Fig. 10 shows the schematic diagram of the staging process for anion-exchange intercalation of dodecyl sulfate anions and PP chains into the layer spaces of MgAl-LDHs during pan-milling. Because during pan-milling, the PP is introduced and the d value would change accordingly, the d1 and d2 were used in Fig. 12. It can be seen that the reaction does not proceed directly from the host to the first stage product in one-step process, but instead proceeds via second stage intermediate in two-step process. When the PP/LDHs/SDS suspensions suffer wet-milling, the second stage intermediate corresponding to layered structure I is firstly formed, which has interlayer spaces alternately occupied by dodecyl sulfate and carbonate anions. The first stage product with interlayer spaces all occupied by dodecyl sulfate anion (formed by the further intercalation in the incompletely intercalated layered structure I products), corresponding to layered structure II, is then produced. Therefore, the staging can well explain the conversion from layered structure I to layered structure II with increase in milling cycles and SDS loading.
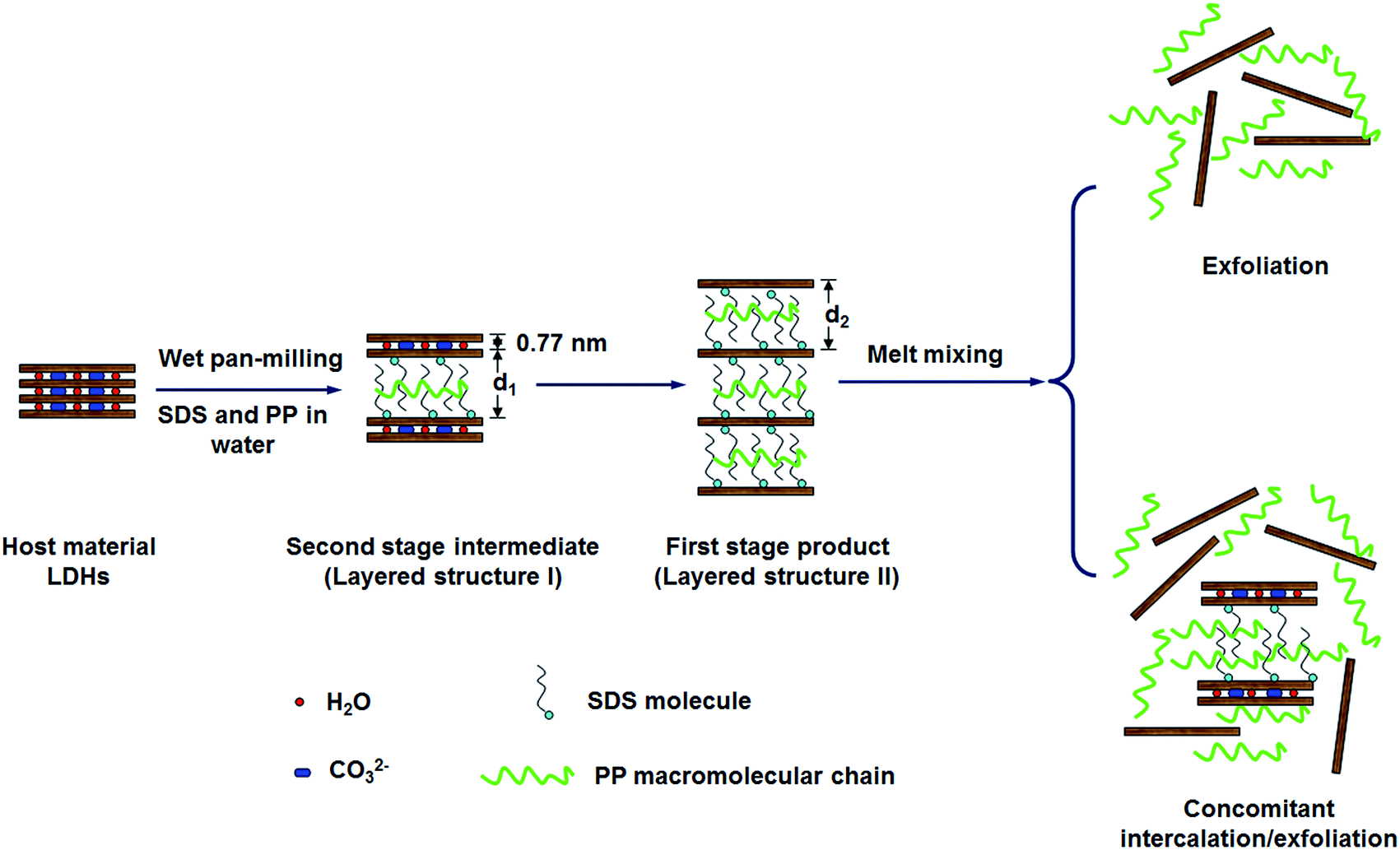 | ||
| Fig. 12 Schematic diagram illustrating the formation of the second stage intermediate for intercalation of dodecyl sulfate anion and PP macromolecular chain into LDH gallery through wet pan-milling. | ||
Combined with XRD results, the basal spacing of first stage product is d2 = 3.18 nm, while the d value (3.75 nm) of the second stage intermediate is the sum of d1 and the basal spacing of original LDHs (d0 = 0.77 nm). So, the calculated d1 value (d1 = 3.75 − 0.77 = 2.98 nm) is larger than ∼2.6 nm.36 This indicates that there must be occurrence of co-intercalation of PP macromolecular chains into LDH interlayer space under the strong three dimensional shear force field of pan milling. Meanwhile, the further intercalation of PP macromolecular chains makes d2 > d1. This simultaneously reveals the formation of a second-staging phase with the 2.98 nm and 0.77 nm phases stacked alternatively. The host, second stage intermediate and first stage products may co-exist in co-powder according to the related XRD results. The ratio between the three components above mentioned may vary with SDS content and milling cycles. When SDS content is low (e.g. PPLS31.3p), no anion-exchange intercalation reaction would occur due to the too low concentration or the diffusion constraint. However, when SDS content becomes high (e.g. PPLS20p), the second stage intermediates (layered structure I) have most been converted into the completely intercalated first stage products (layered structure II).
The previous TEM results show that when the co-powder is diluted with neat PP through melt mixing, the exfoliation of LDH nanosheets can be observed at low loading, while at high loading both intercalated and exfoliated structures can be found. In general, the layers with larger spacing should be more easily exfoliated. However, from the staging process, the first stage product (layered structure II) with one phase can be more easily further intercalated and exfoliated under strong shear condition in melt-compounding process than the second stage intermediate (layered structure I) containing vertical DS−-LDH phase and CO32−-LDH phase at high LDH loading. This further verifies the rationality of the staging anion-exchange intercalation reaction we proposed.
The intercalation of organic anions into hydroxides layers is a heterogeneous process, and therefore the reaction can be considered from the kinetics and thermodynamics. For the intercalation reaction of layered material, there are three principal steps requiring energy:41 first, the guest anion must diffuse through the reaction solvent to reach the host. Second, once the guest has reached the host, the layers must be prised apart in order to allow the guest to enter the interlayers. Finally, the guests must move through the host to completely fill the interlayer space. Staging is a way where the reaction barrier can be reduced, which localizes the organic anions in certain interlayer regions so as to reduce the activation energy. Because of the high affinity of carbonate anions, the activation energy of the traditional anion-exchange reaction for CO32−-LDHs is so high that the intercalation reaction cannot take place. In this study, however, the anion-exchange reaction can be priorly achieved under the very strong three-dimensional shear force field of pan-milling, which can provide external energy to the system. Why does the reaction not proceed directly from the host to the first stage product, but in a two step-process via a second-stage intermediate? It is mainly because the energy provided by pan-milling is not enough for the host to overcome the barrier of the first stage product, but only can overcome the barrier of the second stage intermediate. Obviously, the energy provided by three-dimensional shear force field, which is required for forcible delamination of the layers, plays the most important part in facilitating organic anions entry, as mentioned before. However, the transfer of the energy externally provided could be realized only through using PP as a force carrier. As a consequence, it is easy to understand that during pan-milling, if there is absence of PP particles, the anion-exchange intercalation reaction system cannot overcome any of the barriers to form the corresponding products. Therefore, the staging in wet pan-milling process can be well explained from kinetics and thermodynamics view.
By now, it is for the first time that CO32−-LDHs is successfully modified with aqueous SDS via anion-exchange reaction under the effect of pan milling, simultaneously achieving the blending with PP. The staging phenomenon and incomplete intercalation reaction in this paper further show the high affinity of carbonate anions in hydrotalcite, which is difficult to be exchanged with other inorganic or organic anions.
Conclusions
Wet pan-milling method is utilized to successfully prepare PP/LDHs nanocomposites without prior organic modification of LDHs. The organic modification of LDHs with SDS modifier and their compounding with PP are finished just in one pan-milling process, greatly simplifying the preparation procedure of PP/LDHs nanocomposites. XRD study shows that there are two types of layered structures (I and II) formed during pan-milling and presence of transformation from structure I to II when increasing both milling cycles and SDS content. In addition, during pan-milling process, LDH particles with hexagonal platelets are found to be embedded in the surface of PP particles and suffer from co-pulverization with PP, which plays an important role in anion-exchange intercalation reaction as stress carriers. Staging is used to illustrate the anion-exchange intercalation and the related structure change from kinetics and thermodynamics view, especially transformation from second stage intermediate corresponding to layered structure I with larger basal spacing (d003I = 3.75 nm) to first stage product corresponding to layered structure II with smaller basal spacing (d003II = 3.18 nm). There are the exfoliated structures at low LDH loading and the concomitant intercalated/exfoliated structures at high LDH loading in PP/LDHs nanocomposites prepared by further melt mixing of PP/LDHs co-powders with PP without use of compatibilizer. The intercalated and exfoliated LDH layers are found to be homogeneously dispersed in PP matrix.In addition, DMA analysis shows that the Tg and storage modulus (E′) of the prepared nanocomposites slightly enhance due to the restriction of PP macromolecular chains segments in the presence of nano LDH particles. However, the crystallization behavior analysis reveals that LDH particles are found to hinder the nucleation of PP macromolecular chains and delay the crystallization process. This study provides a novel technique for preparation of PP/LDHs nanocomposites.
Acknowledgements
This work is supported by National Natural Science Foundation of China (51421061), the Program of Innovative Research Team for Young Scientists of Sichuan Province (2016TD0010) and the Project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (sklpme2016-2-01).Notes and references
- Y. Zhao, N. Li, F. Z. Yuan, H. D. Zhang and S. J. Xia, J. Appl. Polym. Sci., 2016, 133(39), 43988 CrossRef.
- F. Rafiee, M. Otadia, V. Goodarzib, H. A. Khonakdar, S. H. Jafaric, E. Mardania and U. Reuterd, Composites, Part B, 2016, 103, 122–130 CrossRef CAS.
- X. Wang, E. N. Kalali and D.-Y. Wang, ACS Sustainable Chem. Eng., 2015, 3(12), 3281–3290 CrossRef CAS.
- L. Z. Qiu, W. Chen and B. J. Qu, Polymer, 2006, 47, 922–930 CrossRef CAS.
- S. S. Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef CAS.
- S. P. Newman and W. Jones, New J. Chem., 1998, 22, 105–115 RSC.
- M. V. Reddy, N. T. K. Lien, G. C. S. Reddy, K. T. Lim and Y. T. Jeong, Green Chem., 2016, 18, 4228–4239 RSC.
- N. Chubar and M. Szlachta, Chem. Eng. J., 2015, 279, 885–896 CrossRef CAS.
- Y. S. Gao, J. W. Wu, Q. Wang, C. A. Wilkie and D. O'Hare, J. Mater. Chem. A, 2014, 2(29), 10996–11016 CAS.
- S. S. D. Richardson-Chong, R. Patel and G. R. Williams, Ind. Eng. Chem. Res., 2012, 51(7), 2913–2921 CrossRef CAS.
- M. Kotal and A. K. Bhowmick, Prog. Polym. Sci., 2015, 51, 127–187 CrossRef CAS.
- Q. Wang, J. W. Wu, Y. S. Gao, Z. Zhang, J. Y. Wang, X. Zhang, X. R. Yan, A. Umar, Z. H. Guob and D. O'Hare, RSC Adv., 2013, 3, 26017–26024 RSC.
- J. Y. Wang, W. L. Bao, A. Umar, Q. Wang, D. O'Hare and Y. L. Wan, J. Biomed. Nanotechnol., 2016, 12(5), 922–933 CrossRef CAS PubMed.
- W. Cui, Q. Jiao, Y. Zhao, H. Li, H. Liu and M. Zhou, eXPRESS Polym. Lett., 2012, 6(6), 485–493 CrossRef CAS.
- Z. Q. Hu and G. M. Chen, J. Mater. Chem. A, 2014, 2(33), 13593–13601 CAS.
- H. D. Peng, W. C. Tjiu, L. Shen, S. Huang, C. B. He and T. X. Liu, Compos. Sci. Technol., 2009, 69, 991–996 CrossRef CAS.
- Y. H. Wang, Q. R. Li, L. F. Bo, X. P. Wang, T. Zhang, S. Li, P. Ren and G. Y. Wei, Chem. Eng. J., 2016, 284, 989–994 CrossRef CAS.
- S. Zhang, X. S. Liu, X. Y. Gu, P. Jiang and J. Sun, Polym. Compos., 2015, 36(12), 2230–2237 CrossRef CAS.
- C. Nyambo, E. Kandare and C. A. Wilkie, Polym. Degrad. Stab., 2009, 94, 513–520 CrossRef CAS.
- F. Gu, X. Cheng, S. F. Wang, X. Wang and P. S. Lee, Small, 2015, 11(17), 2044–2050 CrossRef CAS PubMed.
- R. Z. Ma, Z. P. Liu, L. Li, N. Iyi and T. Sasaki, J. Mater. Chem., 2006, 16, 3809–3813 RSC.
- U. Costantino, M. Nocchetti, M. Sisani and R. Vivani, Z. Kristallogr., 2009, 224, 273–281 CrossRef CAS.
- N. Iyi, T. Matsumoto, Y. Kaneko and K. Kitamura, Chem. Mater., 2004, 16, 2926–2932 CrossRef CAS.
- N. Iyi and T. Sasaki, Appl. Clay Sci., 2008, 42, 246–251 CrossRef CAS.
- M. Ardanuy, J. I. Velasco, M. L. Maspoch, L. Haurie and A. I. Fernández, J. Appl. Polym. Sci., 2009, 113, 950–958 CrossRef CAS.
- S. P. Lonkar and R. P. Singh, Thermochim. Acta, 2009, 491, 63–70 CrossRef CAS.
- S. P. Lonkar, S. Morlat-Therias, N. Caperaa, F. Leroux, J. L. Gardette and R. P. Singh, Polymer, 2009, 50, 1505–1515 CrossRef CAS.
- M. Zhang, P. Ding and B. J. Qu, Polym. Compos., 2009, 30, 1000–1006 CrossRef CAS.
- W. G. Shao, Q. Wang and K. S. Li, Polym. Eng. Sci., 2005, 45, 451–457 CAS.
- W. G. Shao and Q. Wang, J. Appl. Polym. Sci., 2006, 101, 1806–1809 CrossRef CAS.
- G. L. Wang, Y. H. Chen and Q. Wang, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 807–817 CrossRef CAS.
- X. Xu and Q. Wang, CN Pat., ZL95111258.9, 1995.
- X. Xu, Q. Wang, X. G. Kong, X. D. Zhang and J. G. Huang, Plast., Rubber Compos., 1996, 25, 152–158 CAS.
- Q. Wang, J. Z. Cao, J. G. Huang and X. Xu, Polym. Eng. Sci., 1996, 37, 1091–1101 Search PubMed.
- H. Acharya, S. K. Srivastava and A. K. Bhowmick, Compos. Sci. Technol., 2007, 67, 2807–2816 CrossRef CAS.
- A. Clearfield, M. Kieke, J. Kwan, J. L. Colon and R. C. Wang, J. Inclusion Phenom. Macrocyclic Chem., 1991, 11, 361–378 CrossRef CAS.
- K. Kumar, P. K. Ghosh and A. Kumar, Composites, Part B, 2016, 97, 353–360 CrossRef CAS.
- A. M. Fogg, J. S. Dunn and D. O'Hare, Chem. Mater., 1998, 10, 356–360 CrossRef CAS.
- M. Kaneyoshi and W. Jones, Chem. Phys. Lett., 1998, 296, 183–187 CrossRef CAS.
- N. Iyi, K. Fujii, K. Okamoto and T. Sasaki, Appl. Clay Sci., 2007, 35, 218–227 CrossRef CAS.
- G. R. Williams, A. M. Fogg, J. Sloan, C. Taviot-Guého and D. O'Hare, Dalton Trans., 2007, 3499–3506 RSC.
- G. R. Williams and D. O'Hare, Chem. Mater., 2005, 17, 2632–2640 CrossRef CAS.
- S. L. Ma, C. H. Fan, L. Du, G. L. Huang, X. J. Yang, W. P. Tang, Y. Makita and K. Ooi, Chem. Mater., 2009, 21, 3602–3610 CrossRef CAS.
- N. Iyi, K. Kurashima and T. Fujita, Chem. Mater., 2002, 14, 583–589 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |