DOI:
10.1039/C6RA25929D
(Paper)
RSC Adv., 2017,
7, 1513-1519
In situ synthesis and self-reinforcement of polymeric composite hydrogel based on particulate macro-RAFT agents†
Received
27th October 2016
, Accepted 30th November 2016
First published on 5th January 2017
Abstract
Excellent mechanical strength is critical for polymer hydrogels to be successfully applied in the field of mechanical devices and tissue engineering scaffolds. Herein, novel nanoparticle-reinforced polyacrylamide-based hydrogel with high mechanical strength was designed and successfully prepared in this work through the RAFT polymerization of acrylamide (AM) and ethylene glycol dimethacrylate (EGDMA) in the presence of particulate macro-RAFT agents in water. Amphiphilic poly(acrylic acid)-b-polystyrene (PAA-b-PS) copolymer was first synthesized via RAFT copolymerization in methanol. Then the vesicles of about 200 nm in size and decorated with RAFT groups were formed by the self-assembly of PAA-b-PS copolymers during the water dialysis, which can be considered as particulate macro-RAFT agents. After the addition and the polymerization of EGDMA and AM monomers in order, hydrogel filled with uniformly distributed PAA-b-PS nanoparticles was in situ formed, showing an extraordinary compression strength and tensile strength up to 7.0 MPa and 2.0 MPa, respectively, as well as an excellent fatigue resistance under loading–unloading. The self-reinforcement effect of the PAA-b-PS nanoparticles should be related to the strong interaction originating from the chemical and hydrogen bonds between the particulate macro-RAFT agents and polyacrylamide chains. This work provides not only a novel method for macro-RAFT agents, but also opens a new feasible strategy to prepare high-performance hydrogels reinforced by polymeric nanoparticles.
1. Introduction
Hydrogels with a three-dimensional network structure composed of hydrophilic polymer chains have attracted considerable attention in various applications including mechanical devices, scaffolds for tissue engineering, carriers for drug delivery, matrices for bioseparation, etc.1–6 Conventional hydrogels are very flexible, having a rather low modulus and compression strength due to their nature specific structure. Therefore, new synthesis and modification methods for hydrogels with high integrated mechanical properties have long been explored with the expansion of hydrogel application fields.7–9 The incorporation of nanoparticles with various structures and morphologies into the polymeric hydrogel matrix can be considered as a facile and effective way to obtain enhanced hydrogels, similar to the particle strengthening effect in polymer bulk matrix.10–14 But this particle strengthening effect in hydrogels greatly depends on the interaction between the filled nanoparticles and hydrogel network. However, most inorganic nanoparticles always have a bad adhesion to hydrogel networks since only weak physical interactions such as van der Waals' force, electrostatic interaction, and hydrogen bonds exist between them.15–19 This will further result in the inhomogeneous distribution of the nanoparticles, as well as the bad structure and mechanical performance stability of hydrogels after receiving frequentative external forces.
Recently, the utilization of polymeric nanoparticles to enhance hydrogels has been extensively studied due to their good compatibility and the easy introduction of myriad active chemical junction sites on polymeric nanoparticles.20–24 For example, Fu et al. prepared Pluronic F127 micelles modified with vinyl functional groups served as multifunctional cross-links for the synthesis of polyacrylamide hydrogel.25 The prepared tough and strengthened composite hydrogels had a tensile strength up to 276 kPa with a fracture strain 2265% and a compressive toughness of 3.5 MPa at 90% strain. However, the post functionalization on the enhanced polymer nanoparticles sometimes is associated with complex or difficult processes. Formation of functionalized polymer nanoparticle direct from the initial synthesis of special-functionalized macromolecular chains is an obviously facile and efficient way. It was found in our previous work26 that RAFT-agent-decorated sulfonated polystyrene (SPS) microspheres can be prepared directly from the radiation RAFT emulsion polymerization of styrene (St) in water with 2-dodecylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (DMP) as the RAFT agent. The DMP decorated SPS microspheres could initiate continuously the RAFT polymerization of acrylic acid (AA) on the SPS microspheres, which means RAFT-agent-decorated microsphere can be considered as a big “living” particulate RAFT agent. This kind of “living” polymer particles evidently can produce strong chemical links with the polymer matrix through the in situ reactions between the active function groups on filled nanoparticles and the polymer chains, which can greatly improve the enhancement effect of the filled polymer nanoparticles.
Inspired by the above concept of “living” polymer nanoparticles, we herein designed and synthesized successfully highly tough and elastic hydrogels taking advantage of the RAFT polymerization of acrylamide (AM) and ethylene glycol dimethacrylate (EGDMA) in the presence of novel macro-RAFT agents with an average size of about 200 nm composed of amphiphilic poly (acrylic acid)-b-polystyrene (PAA-b-PS) copolymer. The uniformly distributed PAA-b-PS nanoparticles in hydrogel acted as multi-arms crosslinkers, which can maintain the structure stability of both the nanoparticles and hydrogel matrix. The resultant nanoparticles reinforced hydrogels showed an extraordinary compressive strength and tensile strength up to 7 MPa and 2.0 MPa, respectively, as well as an excellent fatigue resistance under loading-unloading.
2. Experimental
2.1. Materials
Styrene (St, 99%, Sinopharm Chemical Reagent Co. Ltd) was purified by passing through the alkaline alumina oxide column before use. Acrylic acid (AA, AR, Sinopharm Chemical Reagent Co. Ltd) was distilled under vacuum prior to use. 2,2′-Azobisisobutyronitrile (AIBN, AR, Sinopharm Chemical Reagent Co. Ltd) was recrystallized from ethanol. Ethylene glycol dimethacrylate (EGDMA, 98%, Sigma Aldrich) was used as received. All other analytical pure chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd, and used as received. Deionized water was utilized in all experiments.
2.2. Synthesis of 2-dodecylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (DMP)
The synthesis of DMP was operated according to the previous report.27 Typically, 1-dodecanethiol (20.2 g, 0.10 mol), acetone (58.0 g, 1.0 mol), and tricaprylylmethylammonium chloride (1.0 g, 0.0025 mol) were mixed in a flask under a nitrogen atmosphere in an ice-water bath. Sodium hydroxide solution (50%) (4.5 g) was slowly dropped in over 10 min. After the mixture was stirred mechanically for 20 min, carbon disulfide (7.6 g, 0.10 mol) in acetone (10.0 g) was added dropwise within 30 min. The color of the solution turned gradually red. Then, chloroform (17.8 g, 0.15 mol) was added, followed by the addition of 50% sodium hydroxide solution (40 g) dropwise over 20 min. The mixture was stirred overnight. Finally, 200 mL of water and 80 mL of concentrated HCl (37%) were added in sequence to acidify the aqueous solution, then the generating solid was filtered and re-dispersed into 300 mL of isopropanol. After the insoluble solid was filtered off, the isopropanol solution was concentrated and dried in a vacuum oven at 25 °C for 24 h. The crude product was recrystallized from hexane twice to obtain 13.5 g of the product. The yield of the synthesis of DMP was 36.3%. The 1H NMR of the synthesized product was measured on Bruker AV 300 MHz spectrometer using CDCl3 as the solvent, as shown in Fig. S1.† All the peaks can be assigned as follows: δ: 3.28 (t, 2H, –S–CH2C11H23), 1.73 (s, 6H, –S–C(CH3)2COOH), 1.67 (m, 2H, –S–CH2CH2C10H21), 1.46–1.17 (m, 18H, –CH2CH2C9H18CH3), 0.88 (t, 3H, –C11H22CH3).
2.3. Preparation of poly(acrylic acid)-b-polystyrene (PAA-b-PS) vesicles via reversible addition fragmentation transfer (RAFT) polymerization and self-assembly in water
The RAFT polymerization of amphiphilic PAA-b-PS copolymer can be conducted according to the previous work.28 First, AA (7.2 g, 0.1 mol), DMP (0.364 g, 1 mmol), AIBN (16.4 mg, 0.1 mmol), and tetrahydrofuran (THF, 8 mL) were added into a 25 mL of polymerization tube with a magnetic bar. After degassing through three freeze–evacuate–thaw cycles, the polymerization tube was sealed after being vacuumed and placed into an oil bath at 80 °C. After 2 h, the polymerization was terminated by putting the system into liquid nitrogen. The product, termed PAA-DMP, was purified by precipitating into 100 mL of diethyl ether. The crude product re-dissolved in THF and repeated the above steps, finally PAA-DMP product was dried under vacuum for 24 h at 30 °C. The 1H NMR spectrum of PAA-DMP was measured on Bruker AV 300 MHz spectrometer using DMSO-d6 as the solvent, as shown in Fig. S2.† All the peaks can be assigned as follows: δ: 2.20 (m, 60H, –CHCOOH–), 1.08 (m, 26H, –(CH3)2, –(CH2)10–), 0.85 (m, 3H, –CH3). Based on the above data, the block length of PAA-DMP was calculated to be 60.
Next, the as-prepared PAA-DMP was mixed with St, AIBN, and methanol at different ratios in a 10 mL of polymerization tube. The detailed recipes are listed in Table 1. After being degassed through three cycles of freeze–evacuate–thaw, the polymerization tube was sealed after being vacuumed and placed into an oil bath at 80 °C for 20 h. The reaction was stopped by putting the system in liquid nitrogen. Then let the polymerization tube be exposed to air. The reacted dispersion was divided into two parts. One part was dialyzed in ethanol for 48 h to remove the unreacted monomers, and then dried for 24 h under vacuum at 30 °C. The 1H NMR spectra of the products were measured on Bruker AV 300 MHz spectrometer using the mixture of DMSO-d6 and CDCl3 (DMSO-d6: CDCl3 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the solvent, as shown in Fig. S3.† All the peaks are assigned as follows: δ: 7.01–6.23 (m, 5H, protons of benzene ring of styrene), 2.28 (m, 1H, –CHCOOH– of PAA), 1.50 (m, 3H, –CH2(CH–)– of PS). The length of PS blocks of the obtained product was determined by the above 1H NMR analysis, and the results are listed in Table 1. The other part was dialyzed in water for 48 h, followed by being diluted to a whole volume of 50 mL with water to further use. During the water dialysis process, PAA-b-PS capped with the end groups of DMP formed vesicles in water.
:1 v/v) as the solvent, as shown in Fig. S3.† All the peaks are assigned as follows: δ: 7.01–6.23 (m, 5H, protons of benzene ring of styrene), 2.28 (m, 1H, –CHCOOH– of PAA), 1.50 (m, 3H, –CH2(CH–)– of PS). The length of PS blocks of the obtained product was determined by the above 1H NMR analysis, and the results are listed in Table 1. The other part was dialyzed in water for 48 h, followed by being diluted to a whole volume of 50 mL with water to further use. During the water dialysis process, PAA-b-PS capped with the end groups of DMP formed vesicles in water.
Table 1 The recipes for the synthesis of PAA-b-PS copolymer
| Samples |
PAA-DMP (g) |
St (g) |
nSt/PAA-DMPa |
AIBN (mg) |
nPAA-DMP/AIBNb |
Methanol (g) |
PAAx-b-PSyc |
| The molar ratio of St to PAA-DMP. The molar ratio of PAA-DMP to AIBN. x and y are the average numbers of the AA and St units respectively measured by 1H NMR spectra. |
| Run 1 |
96 |
2.08 |
1000 |
0.33 |
10 |
2 |
PAA60-b-PS127 |
| Run 2 |
96 |
2.08 |
1000 |
0.33 |
10 |
3 |
PAA60-b-PS150 |
| Run 3 |
38.4 |
2.08 |
2500 |
0.13 |
10 |
2 |
PAA60-b-PS180 |
2.4. The removal of the capped DMP end groups on PAA-b-PS
As a control, “dead” PAA-b-PS polymer chains, i.e. without any DMP end groups, were prepared as follows: the aqueous dispersion with a certain concentration of PAA-b-PS capped with DMP end groups was mixed with 50 mg of sodium borohydride after being bubbled with nitrogen 30 min. The system was stirred at room temperature for 24 h. The unreacted sodium borohydride was removed by heating the system to 35 °C under mechanically stirring and standing for 24 h.
2.5. The preparation of PAA-b-PS nanoparticles reinforced hydrogel (PNR-HG)
Firstly, EGDMA was added to a specific amount of the above prepared aqueous PAA-b-PS dispersion. The dispersion was magnetically stirred for 6 h at room temperature. Then, a certain amount of acryl amide (AM) and ammonium persulphate (APS) were dissolved into the above dispersion. To prepare hydrogel samples for the compression tests, the dispersion was poured into a 10 mL of syringe and placed in an oven of 50 °C for 16 h after being bubbled with nitrogen for 2 min. As to the hydrogel samples for the tensile tests, the dispersion was poured into the cylindrical space with a height of 2 mm fenced by a circular rubber band sandwiched between two glass plates. The whole system was placed in an oven of 50 °C for 16 h after being bubbled with nitrogen for 2 min. Then the formed hydrogel was cut into several strips for the tensile test. As a control, unfilled polymer hydrogel was prepared by the polymerization of AM and EGDMA in water at the similar conditions in the absence of PAA-b-PS copolymers.
2.6. Characterization
The transmission electron microscopy (TEM, Hitachi H-7650, 100 kV) and scanning electron microscopy (SEM, JEOL JSM6700F, 5.0 kV) were used to observe the morphology of vesicles and hydrogels. The number-average diameter of polymer vesicles was measured in the SEM images with at least 100 particles. Mechanical tests of the all samples were carried out on the Electronic Universal Testing machine (WSK-20KB, Intelligent Equipment Co., Ltd.) at room temperature. The compressive test samples were cylindrical hydrogels (14.1 mm in diameter and 10 ± 1 mm in height), and with a 20 kN load cell at a crosshead speed of 5 mm min−1. The compressive stress (σ) and strain (ε) were calculated by the following equations:where r is the radius of the sample, h0 and h are the sample heights before and after compression, respectively. The tensile tests were performed on rectangle shaped hydrogel specimens with a 100 N load cell at a crosshead speed of 5 mm min−1.
3. Results and discussion
3.1. Synthesis of amphiphilic PAA-b-PS copolymers by RAFT polymerization and their self-assembly in water
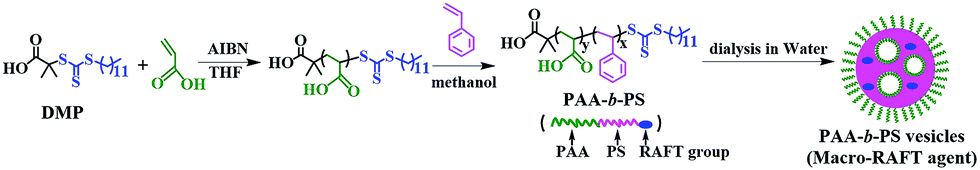
In order to have an effective enhancement on hydrogels, the filled polymer nanoparticles should at least have a hydrophilic surface shell, which will favour to produce a good particle dispersibility and a strong affinity to the hydrogel matrix. But to maintain the morphological stability of the polymer nanoparticles in aqueous environments, hydrophobic polymer cores are also necessary. Therefore, amphiphilic PAA-b-PS copolymers were synthesized via RAFT polymerization method in this work to form the reinforced polymer particles. The synthesis and the formation process of PAA-b-PS vesicles are illustrated in Scheme 1.
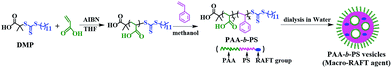 |
| | Scheme 1 The synthesis and the formation process of the PAA-b-PS vesicles as the particulate macro-RAFT agent. | |
PAA with an average of 60 AA units (PAA-DMP) was first obtained by a RAFT polymerization of AA in THF using DMP as the RAFT agent. Then PS chains were continuously grown from the “living” PAA chains in methanol to form amphiphilic PAA-b-PS copolymer chains. The length of PS block increased with the feed molar ratio of St to PAA-DMP (nSt/PAA-DMP) basically at the same molar ratio of PAA-DMP to AIBN, which was confirmed by the 1H NMR spectra displayed in Fig. S3.† The PAA-b-PS copolymer chains can be considered as “living” copolymer chains since they are end-capped with the active RAFT groups. After the methanol solution of the “living” PAA-b-PS copolymer was dialyzed with water, the amphiphilic chains would self-assembly and phase separation would take place due to the poor solubility of PS in water. The morphologies of the aggregation of PAA-b-PS copolymer with different PS block length (PAA60-b-PS127, PAA60-b-PS150, and PAA60-b-PS180) in water were fixed by freeze-drying technique and observed by TEM and SEM, as shown in Fig. 1. Hollow or porous particles sized several hundreds of nanometers are displayed, implying PAA-b-PS copolymers formed vesicle-like structure in water. PAA60-b-PS180 particles have a size of about 700 nm, much higher than that of PAA60-b-PS127 (∼240 nm) and PAA60-b-PS150 (∼180 nm). The pores should be left after the evaporation of the inner water phase formed during the phase separation of PS and stabilized by the PAA-b-PS, acting as macromolecular emulsifier, as illustrated in Scheme 1. The PAA-b-PS vesicles can be dispersed very stable in water, no aggregation could be observed even after the dispersions had been standing for 30 d, as shown in Fig. S4.† To achieve the in situ reinforcement of the nanoparticles originated from the self-assembled vesicles on hydrogels, EGDMA and AM monomers were directly added in order in the aqueous dispersion of the PAA-b-PS vesicles. It's well known that AM is a totally water-soluble monomer. In order to let the end group of RAFT agent in the inner hydrophobic PS phase participate the polymerization reaction, we added the hydrophobic and polar monomer EGDMA first in the dispersion. The system stood for 6 h at room temperature to let EGDMA fully swell the vesicles. Then AM monomer and APS were added in the system to start the copolymerization reaction. Evidently, the swelling of vesicles can promote the diffuse of monomer and initiator molecules in the vesicles and enhance the mobility of amphiphilic polymer chains, which favours the copolymerization of AM and EGDMA in vesicles. After the polymerization of EGDMA and AM was initiated by APS at 50 °C, PAA-b-PS nanoparticles reinforced hydrogel (PNR-HG) were one-step fabricated into any shape, as shown in Fig. 2a and b. To make sure whether hydrophobic EGDMA monomers can be permeated into the PAA-b-PS vesicles, EGDMA was mixed with pure water and the aqueous dispersion of PAA-b-PS vesicles, respectively. As shown in Fig. S5,† when EGDMA was dispersed in pure water, large oil droplets can be observed in water under optical microscope (Fig. S5A†). But if EGDMA was dispersed in the aqueous dispersion containing PAA-b-PS vesicles, no large oil droplets could be observed (Fig. S5B†), indicating most of EGDMA will be distributed in the vesicles. At the same time, the size of the vesicles increases obviously after the addition of EGDMA, as detected by DLS (Fig. S5C†). The result indicates that EGDMA monomers can be homogenously distributed into the stable PAA-b-PS vesicles, and then chemically linked on the PAA-b-PS vesicles through RAFT polymerization, similar to the RAFT grafting polymerization of PAA on SPS microspheres capped with the living DMP end groups reported in our previous work. The continuous macroporous structure and nanoparticles originated from the vesicles were uniformly embedded in the gel network (Fig. 2c inset). As a comparison, the SEM image of the freeze-dried PNR-HG in Fig. 2d displays a regular structure of pure PAM hydrogel. Only smooth polymer walls of the macropores can be observed since no particles can be found in the PAM hydrogel matrix.
 |
| | Fig. 1 TEM (a1–c1) and SEM (a2–c2) images of PAA-b-PS nanoparticles after the freeze-drying of the aqueous dispersion of PAA60-b-PS127 (a1, a2), PAA60-b-PS150 (b1, b2), and PAA60-b-PS180 (c1, c2) vesicles. | |
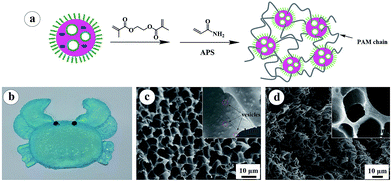 |
| | Fig. 2 (a) The illustration of the in situ synthesis of PAA-b-PS nanoparticles reinforced hydrogel; (b) the appearance of PAA-b-PS nanoparticles reinforced hydrogel synthesized in an crab-shaped mould; (c) SEM image of the freeze-dried PAA-b-PS nanoparticles reinforced hydrogel; (d) SEM image of the freeze-dried pure PAM hydrogel. The insets in (c) and (d) are the corresponding SEM images with a high magnification. | |
3.2. Mechanical performance of PNR-HGs
The compression performance of the PNR-HGs prepared under various compositions have been investigated as shown in Fig. 3. Fig. 3a shows the compressive stress–strain curves of PNR-HGs filled with the PAA-b-PS nanoparticles with different PS block length, compared with that of the unfilled PAM hydrogel. It can be seen that the compressive strengths of all PNR-HGs are much higher than that of the unfilled hydrogel, indicating that the PAA-b-PS nanoparticles do have remarkable reinforcement effect on the hydrogel. It is also noted that the reinforcement effect of PAA-b-PS nanoparticles will be enhanced with the increase of the PS block length under the same particle weight content. Further, the effect of the weight contents of the PAA60-b-PS150 nanoparticles had been investigated as shown in Fig. 3b. When the weight content of PAA60-b-PS150 nanoparticles is lower than 3 × 10−4 g mL−1, a little amount of PAA-b-PS nanoparticles will show a great reinforcement effect on the hydrogel. For example, the compressive strength at 90% strain of PNR-HG reinforced by 2 × 10−4 g mL−1 of PAA60-b-PS150 was raised to 7 MPa, while that of the unfilled hydrogel was only 4 MPa. The compressive stress–strain curve of PNR-HG reinforced by 1.6 × 10−4 g mL−1 of PAA60-b-PS150 is nearly the same with that reinforced by 2 × 10−4 g mL−1 of PAA60-b-PS150, as shown in Fig. S6.† However, the compressive strength of PNR-HGs unexpectedly dropped with the further increase of the weight content of PAA-b-PS nanoparticles. The compressive stress–strain curve of PNR-HG reinforced by 3.2 × 10−4 g mL−1 of PAA60-b-PS150 is similar to that of the unfilled hydrogel. The compressive strength of PNR-HG filled with 5 × 10−4 g mL−1 of PAA60-b-PS150 was still lower than that of PNR-HG filled with 2 × 10−4 g mL−1 of PAA60-b-PS150. This may have relationship with much less amount of monomers swelling in each nanoparticles when the content of nanoparticles is raised but the monomer content is fixed, which will make less polymer chains be linked on the nanoparticles so as to decrease the adhesion between the nanoparticles and hydrogel matrix and result in the deterioration in the compressive strength. The immobilization of hydrogel polymer chains on the PAA-b-PS nanoparticles prepared from RAFT polymerization can be proved by the dramatic decrease of the compressive performance of hydrogel filled with PAA-b-PS nanoparticles without the active DMP end groups, as shown in Fig. 3c, because after the active DMP groups were removed from the PAA-b-PS copolymer chains, “dead” PAA-b-PS nanoparticles would be formed so that the nanoparticles were simply physically blended into the hydrogel. This implies that the PAA-b-PS nanoparticles capped with DMP end groups will take part in the polymerization of AM and EGDMA as particulate macro-RAFT agents, resulting in much more strong chemical connections between the polymer nanoparticles and hydrogel matrix so as to further improve the compression performance of the hydrogel. The existence of EGDMA may have an important contribution on the strength of the PNR-HGs since the bifunctional EGDMA monomers and chains may serve as crosslinkers in the PAM network. As shown in Fig. 4, the hydrogel prepared from an aqueous dispersion containing 20% of AM and 2.25 × 10−2 g mL−1 of PAA60-b-PS150 vesicles is a very sticky fluid, implying that the PAA-b-PS nanoparticles have no reinforcement effect since little hydrophilic AM was distributed in the vesicles. If 10% of monofunctional monomer, e.g. methyl methacrylate (MMA), was added as the comonomer, monolith hydrogel will be obtained. But its strength is evidently lower than that of the hydrogel using the same amount of EGDMA as the comonomer. The result indicates the crosslinking-effect of EGDMA can also make a contribution on the strength of the final PNR-HGs. The effects of the weight contents of monomers, i.e., EGDMA and AM, on the compressive performance of PNR-HGs are also exhibited in Fig. 3d and e, respectively. It is comprehensible that the compressive strength of PNR-HGs increases with the weight content of each monomer because the higher the monomer content, the more crosslinking density and the stronger hydrogen bonding interaction in the gel.
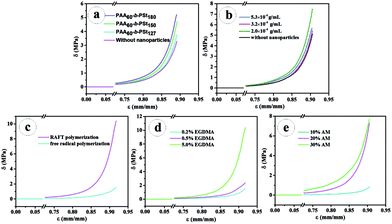 |
| | Fig. 3 The compressive stress–strain curves of unfilled PAM hydrogel and PNR-HGs filled with PAA-b-PS nanoparticles with different PS block length (the nanoparticle weight content in all PNR-HGs was 2 × 10−4 g mL−1) (a); the compressive stress–strain curves of PNR-HGs reinforced by different weight contents of the PAA60-b-PS150 nanoparticles (b); the compressive stress–strain curves of PNR-HGs filled with PAA60-b-PS150 nanoparticles prepared via RAFT polymerization and classic free radical polymerization (c); the compressive stress–strain curves of PNR-HGs reinforced by the PAA60-b-PS150 nanoparticles with different weight contents of EGDMA (d) and AM (e). | |
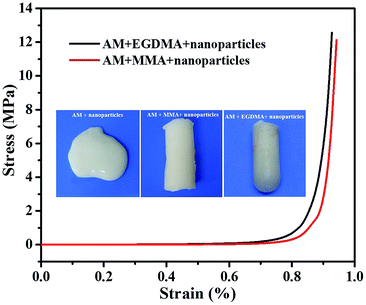 |
| | Fig. 4 The compressive stress–strain curves and the digital photos (inset images) of PNR-HGs prepared with different comonomers at the presence of 2.25 × 10−2 g mL−1 of PAA60-b-PS150 vesicles. The weight contents of AM and comonomer are 20% and 10%, respectively. | |
To investigate the fatigue resistance of the prepared PNR-HG, consecutive cyclic compression tests were conducted at a strain of 0.8 and resulted in almost overlapping hysteresis loops, as shown in Fig. 5a. This indicated that no significant irrecoverable damage has been occurred in the hydrogel. The compression stress–time curves also demonstrated the excellent fatigue resistance of the hydrogel under the loading–unloading as shown in Fig. 5b. It is intuitively obvious in Fig. 5c that the PNR-HG quickly recovered to its original shape upon the removal of the external compression force, indicating the hydrogel with excellent elasticity and fatigue resistance in each compressive cycle.
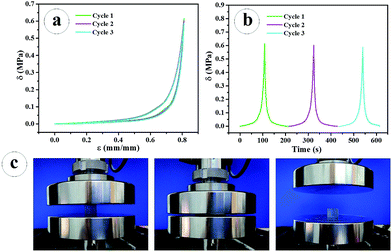 |
| | Fig. 5 Cyclic compressive stress–strain (a); stress–time curves at a strain of 80% (b); the compression-recovery process recorded by a digital camera (c). | |
Tensile strength of the PNR-HG was measured using rectangle shaped hydrogel samples prepared at a condition of 20% AM, 5% EGDMA, and 2 × 10−4 g mL−1 PAA60-b-PS150 nanoparticles. As shown in Fig. 6a, the PNR-HG showed a tensile fracture stress of 2 MPa at 225% strain, and it can lift a 100 g weight (Fig. 6a, inset). Cyclic tensile tests at different stains and 100% strain were also investigated respectively. The hysteresis increased with the extension, but the hysteresis is very slight in every loading-unloading cycle, indicating that the hydrogel has an excellent recovery ability after the remove of the tensile stress (Fig. 6b). When the cyclic tensile tests were conducted at a 100% strain, the loading-unloading curves completely overlapped, as shown in Fig. 6c. These results suggest that the PNR-HG possesses a high stiffness and tensile resilience.
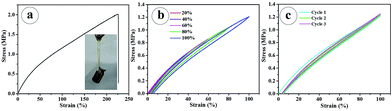 |
| | Fig. 6 Tensile stress–strain curve of PAA60-b-PSt150 nanoparticles reinforced hydrogel (a). Inset: the photography of hydrogel lifting a 100 g weight; the cyclic tensile curves of hydrogel at different strain (b) and at 100% strain (c). | |
4. Conclusion
In summary, we prepared novel PAA-b-PS vesicles through RAFT copolymerization of AA and St with DMP in methanol, followed by the self-assembly of PAA-b-PS molecular chains during the water dialysis. The active DMP end group decorated PAA-b-PS vesicles can act as the particulate macro-RAFT agents to initiate the “living” polymerization of monomers swollen in the vesicles. Therefore, after mixing the aqueous dispersion of PAA-b-PS vesicles with the comonomers (AM and EGDMA), PAM based hydrogels with high mechanical performance were then successfully prepared via the copolymerization of monomers. Different from the conventional physical blending with polymer nanoparticles, the particulate macro-RAFT agents filled in hydrogels can link polymer chains of hydrogel matrix through the initiation of RAFT polymerization of monomers in the nanoparticles so as to produce strong adhesions between nanoparticles and hydrogel matrix and achieve the self-reinforcement effect of the hydrogels. The prepared hydrogels show the extraordinary compression strength up to 7 MPa at 90% strain and high tensile strength up to 2.0 MPa, as well as excellent fatigue resistance under loading–unloading. This work opens a new direction to the synthesis of high-performance hydrogels taking advantage of the self-reinforcement effect of active polymer nanoparticles.
Acknowledgements
This work was supported by the PetroChina Innovation Foundation (2014D-5006-0201), the National Natural Science Foundation of China (No. 51473152 and 51573174), and the Fundamental Research Funds for the Central Universities (WK2060200012, WK3450000001).
Notes and references
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. V. Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020–6041 CrossRef CAS PubMed.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- Y. Shi, C. B. Ma, L. L. Peng and G. H. Yu, Adv. Funct. Mater., 2015, 25, 1219–1225 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, Adv. Mater., 2007, 19, 1622–1626 CrossRef CAS.
- Y. T. Wu, Z. Zhou, Q. Q. Fan, L. Chen and M. F. Zhu, J. Mater. Chem., 2009, 19, 7340 RSC.
- J. Yang, X. P. Wang and X. M. Xie, Soft Matter, 2012, 8, 1058–1063 RSC.
- Y. Tan, K. Xu, P. X. Wang, W. B. Li, S. M. Sun and L. S. Dong, Soft Matter, 2010, 6, 1467–1471 RSC.
- D. Yang, X. W. Peng, L. X. Zhong, X. F. Cao, W. Chen, S. Wang, C. F. Liu and R. C. Sun, RSC Adv., 2015, 5, 13878–13885 RSC.
- E. A. Appel, M. W. Tibbitt, J. M. Greer, O. S. Fenton, K. Kreuels, D. G. Anderson and R. Langer, ACS Macro Lett., 2015, 4, 848–852 CrossRef CAS.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS PubMed.
- W. C. Lin, A. Marcellan, D. Hourdet and C. Creton, Soft Matter, 2011, 7, 6578 RSC.
- J. Yang, L. H. Deng, C. R. Han, J. F. Duan, M. G. Ma, X. M. Zhang, F. Xu and R. C. Sun, Soft Matter, 2013, 9, 1220–1230 RSC.
- G. R. Gao, Y. Xiao, Q. Wang and J. Fu, RSC Adv., 2016, 6, 37974–37981 RSC.
- Y. N. Sun, S. Liu, G. L. Du, G. R. Gao and J. Fu, Chem. Commun., 2015, 51, 8512–8515 RSC.
- Y. Tan, K. Xu, P. X. Wang, W. B. Li, S. M. Sun and L. S. Dong, Soft Matter, 2010, 6, 1467 RSC.
- L. W. Xia, X. J. Ju, J. J. Liu, R. Xie and L. Y. Chu, J. Colloid Interface Sci., 2010, 349, 106–113 CrossRef CAS PubMed.
- K. Xu, Y. Tan, Q. Chen, H. Y. An, W. B. Li, L. S. Dong and P. X. Wang, J. Colloid Interface Sci., 2010, 345, 360–368 CrossRef CAS PubMed.
- S. Sharifi, S. B. G. Blanquer, T. G. V. Kooten and D. W. Grijpma, Acta Biomater., 2012, 8, 4233–4243 CrossRef CAS PubMed.
- Y. N. Sun, G. R. Gao, G. L. Du, Y. J. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS.
- H. Q. Weng, H. H. Huang, Y. S. Wang, M. Z. Wang and X. W. Ge, Polym. Chem., 2015, 6, 7717–7725 RSC.
- Y. Z. You and D. Oupicky, Biomacromolecules, 2007, 8, 98–105 CrossRef CAS PubMed.
- W. D. He, X. L. Sun, W. M. Wan and C. Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25929d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Xuewu Ge*a
*a and
Xuewu Ge*a