
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMOF-derived Ni-based nanocomposites as robust catalysts for chemoselective hydrogenation of functionalized nitro compounds†
Bo Tanga,
Wei-Chao Songa,
En-Cui Yang*a and
Xiao-Jun Zhao *ab
*ab
aKey Laboratory of Inorganic-Organic Hybrid Functional Material Chemistry, Ministry of Education, Tianjin Key Laboratory of Structure and Performance for Functional Molecules, Tianjin Normal University, Tianjin 300387, People's Republic of China. E-mail: encui_yang@163.com; xiaojun_zhao15@163.com
bDepartment of Chemistry, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, People's Republic of China
First published on 5th January 2017
Abstract
Porous graphitic carbon layers encapsulating Ni nanoparticles (Ni@C) were prepared by a facile thermolysis of a Ni-containing metal–organic framework, the structure of which were characterized by power X-ray diffraction (XRD), N2 adsorption–desorption, transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) in detail. The resulting Ni@C nanocomposites served as highly efficient and magnetically recyclable catalysts for the hydrogenation of diverse functionalized nitro compounds to the corresponding anilines under relatively milder conditions. The high catalytic performance and the enhanced stability are ascribed to the synergistic effects and electron transfer between the metallic Ni and graphitic carbon as well as the unique encapsulation structure. The achieved success in the MOF-derived Ni@C nanocomposites may pave the way for designing environmentally benign catalytic hydrogenation processes for industrial applications.
1. Introduction
Functionalized anilines are important feedstocks and essential ingredients for the manufacture of pharmaceuticals, agrochemicals, dyestuffs, functional polymers, fine chemicals, etc.1,2 The method employed generally for the production of anilines is the direct reduction of the corresponding aromatic nitro compounds.3–7 Despite the considerable achievements that have been made, these catalytic systems are unfortunately associated with environmental issues or formation of massive undesired by-products due to utilizing stoichiometric reducing agents, such as NaBH4 or hydrazine hydrate. Alternatively, selective hydrogenation of nitroarenes with hydrogen as the reductant has attracted much attention due to its environmental friendliness, atomic efficiency, and compatibility with industrial processes.Nobel metal catalysts have been proved to be effective for this catalytic hydrogenation process.8–11 However, the presence of other reducible functional groups in the nitroarenes makes the dual requirements of activity and selective reduction of nitro group quite challenging. Additional modification of noble metals with suitable additives was necessary to improve the selectivity, while at the expense of activity.8–11 Furthermore, the use of additives would bring about environmental issues as well as difficult purification. The exploitation of supported Au nanoparticles is a great breakthrough in the field of selective hydrogenation of nitroarenes.12–15 Even so, potential industrial-scale applications are great discounted due to the high cost and scarcity nature of precious metals. Development of heterogeneous catalysts based on earth-abundant metals, i.e. Fe, Co, and Ni, could provide an alternative choice for the reduction of nitroarenes.16–25 In particular, such base metals have the specific advantage in catalyst recovery by an external magnetic field. However, the stability of magnetic nanoparticles is a problem based on the fact that they readily tend to aggregate when being treated without any protection agents, thus losing the unique properties derived from their small size.26 In addition, sintering or leaching of active components is a fatal shortcoming, resulting into irreversible deactivation of base metal catalysts.27,28 Therefore, in view of potential applications in industrial, it is highly desirable and necessary to develop robust magnetic nanomaterials based on earth-abundant metals with high activity and selectivity as well as structure stability and reusability for this transformation.
Recently, metal–organic frameworks (MOFs) have emerged as a promising class of porous materials with very large specific surface area, ultrahigh porosity, and chemical tunability. Because of the distinct advantages and facile synthesis of MOFs, they have been adopted for widespread applications in many fields, including gas storage/separation, luminescence, drug delivery, and catalysis.29–33 Besides, since the first report of MOF-derived nanoporous carbon by Xu et al.,34 many efforts have led to the development of potential MOFs as templates/precursors to construct metal and/or metal nanoparticles embedded in the carbon matrix.35–37 Most notably, the highly ordered metal ions that regularly isolated by organic ligands are imperative in preventing metal aggregation during pyrolysis procedure. And the encapsulation structure appears effective to stabilize the metal nanoparticles, thus suppressing them leaching into the reaction solution. To data, the MOF-derived nanomaterials have displayed great potential in heterogeneous catalysis,38–41 electrochemistry,42,43 and gas adsorption.44,45 Whereas, reports on the employment of MOF-derived metal/carbon magnetic nanomaterials for catalytic hydrogenation reactions are scarce.
On the basis of the good catalytic performance of Ni nanocatalyst in hydrogenation reactions, herein, we report a simple and scalable method to prepare Ni-based hybrids featuring encapsulated Ni nanoparticles within carbon via pyrolysis of Ni-containing MOF. The resultant Ni@C composites serve as high-efficiency and stable catalysts for the hydrogenation of nitro compounds. Near full conversions of various functionalized nitroarenes with excellent selectivity toward corresponding anilines have been achieved under a rather low H2 pressure, which exhibits superior catalytic activities as compared to other Ni-based catalysts, and even noble-catalyzed system. These findings make our catalytic system great potential for large-scale application for the production of anilines from hydrogenation of nitroarenes.
2. Experimental section
2.1 Sample preparation
All starting materials and solvents were purchased from commercial sources and used as received without further purification.Ni-MOF was prepared according to the previous literature.46 In a typical procedure, p-benzenedicarboxylic acid (0.332 g) and NiCl2·6H2O (0.173 g) were dissolved in 5 mL and 10 mL N,N-dimethylformamide (DMF), respectively, under vigorous stirring at room temperature. Afterwards, the DMF solution of NiCl2·6H2O was then slowly added to the other solution with stirring drop by drop. After stirring for 1 h at room temperature, the mixture was transferred into a Teflon-lined stainless steel autoclave and maintained at 120 °C for 16 h. After cooling to room temperature, the resulting green precipitate was collected by centrifugation, washed with DMF and alcohol several times, and then dried at 70 °C in air overnight to derived the Ni-MOF material.
Carbon embedded nickel nanoparticles materials were prepared by direct thermolysis of as-synthesized Ni-MOF. Generally, 200 mg of Ni-MOF was put into a tubular reactor, then sealed and purged with nitrogen gas for 30 min. After air in the tubular reactor was removed, the sample was heated at a heating rate of 10 °C min−1 to the desired target temperature, i.e. 450, 550, 650, and 750 °C, under a continuous nitrogen flow of ca. 40 mL min−1. After being treated for 1 h, the sample was cooled down to room temperature naturally in flowing nitrogen atmosphere. The obtained black powder was denoted as Ni@C-X, where X refers to thermolysis temperature.
For a better understanding of the active sites of as-synthesized Ni@C catalysts, the Ni@C samples after removing the metallic Ni core or carbon shell was prepared, respectively. For this, Ni@C-650 sample was treated with aqua regia to completely remove Ni nanoparticles, and the resultant sample was denoted as Ni@C-650-C. On the other hand, the carbon shell of as-synthesized Ni@C catalyst was eliminated by heating treatment. The Ni@C-650 sample was subjected to heating at 400 °C in air for 1 h and subsequently under 10% H2/Ar atmosphere for another 2 h to derive the final Ni@C-650-Ni sample. Additionally, nickel nanoparticles supported on active carbon material was synthesized via impregnation method. Ni(NO3)2·6H2O was dissolved in a certain amount of ethanol, which was then added dropwise to active carbon and uniformly ground in a quartz mortar to achieve an intimate mixture. The resultant mixture was dried naturally and treated at 400 °C under 10% H2/Ar atmosphere for 2 h to give Ni/AC sample.
2.2 Characterization techniques
Powder X-ray diffraction (XRD) patterns of samples were obtained on a Bruker D8 diffractometer using Cu Kα radiation (λ = 1.54184 Å) from 5° to 80° with a scan speed of 2θ = 6.0° min−1. The TGA curve of Ni-MOF was obtained on DTG-60 with dry sample under an Ar flow with heating rate of 10 °C min−1 from room temperature to 800 °C. The porosity and gas adsorption–desorption isotherms of samples were determined with a Micromeritics ASAP 2020 instrument. Before the adsorption, the samples were degassed at 120 °C for 6 h. The specific area was calculated via the Brunauer–Emmett–Teller (BET) equation, and the NLDFT method with slit/cylindrical pores was applied to the desorption isotherm to obtain the pore size distribution and cumulative pore volumes. The Ni content and in the samples were measured by inductively coupled plasma emission spectrometry (ICP-AES) on a Thermo IRIS Intrepid II XSP atomic emission spectrometer. Element analysis was conducted on a Elementar Vario EL III equipment. TEM images of the samples were acquired on a Tecnai G2 F20 U-TWIN transmission electron microscope at an acceleration voltage of 200 kV. A few drops of the alcohol suspension containing the catalyst samples were placed on a micro grid, followed by evaporation at ambient temperature. X-ray photoelectron spectra (XPS) of the dehydrated samples were recorded on a Kratos Axis Ultra DLD spectrometer with a monochromated Al-Kα X-ray source (hν = 1486.6 eV), hybrid (magnetic/electrostatic) optics and a multi-channel plate and delay line detector (DLD). All spectra were recorded using an aperture slot of 300 × 700 microns. Survey spectra were obtained with a pass energy of 160 eV and high-resolution spectra were obtained with a pass energy of 40 eV. Accurate binding energies (±0.1 eV) were determined with respect to the position of the adventitious C 1s peak at 284.6 eV. CasaXPS software was used for spectra treatment.2.3 Catalytic evaluation
The chemoselective hydrogenation of nitroarenes was performed in a 25 mL stainless-steel autoclave with an external temperature and stirring controller. In a typical experiment, the autoclave was charged with a mixture of nitroarene (0.63 mmol), the green solvent ethanol (10 mL), and 10 mg of catalyst. The reactor was purged with H2 to remove the air for five times, and then sealed tightly and pressured to 0.5 Mpa H2. Afterwards, the autoclave was transferred on a heating panel preheated at 160 °C, and the reactions were started when it was lowered down to the target temperature (140 °C). The stirring speed was kept at 800 rpm and the catalyst was finely pre-ground to avoid either external or internal diffusion limitation. After completion of the reaction, the autoclave was placed into an ice-water bath and cooled to ambient temperature. The remaining H2 was carefully vented, and the content of the reaction mixture was analyzed by a Shimadzu GC 2010 (Agilent HP-5 column, 30 m × 0.25 mm × 0.25 μm; FID detector) using dodecane as internal standard. The GC peaks were identified by comparison with the retention times of the known standard samples, and by means of a Shimadzu GC-MS QP2010 SE equipped with an Agilent HP-5MS column. Carbon balance of over 95% was obtained for all experiments.A hot-filtration test was performed to confirm the heterogeneous nature of the catalyst under the identical conditions employed for the catalytic hydrogenation reaction. After 10 min of reaction time, half of the reaction medium was taken from the reaction solution, the catalyst of which was removed and the liquid was allowed to react in similar ways as the heterogeneous reaction.
To verify the potential recyclability of as-synthesized Ni@C catalysts, six consecutive reactions were performed with the same catalyst material. Between each cycle the catalyst was washed with ethanol for three times, dried at 60 °C, and weighted before subjecting to the next run, in order to guarantee the constant substrate-to-catalyst ratio.
3. Results and discussion
3.1 Synthesis and microstructure of Ni@C composites
In this work, a Ni-MOF with the formula [Ni3(OH)2(C8H4O4)2·(H2O)4]·2H2O was synthesized, which was used as the template/precursor to further prepare the Ni@C materials. The XRD patterns of as-synthesized Ni-MOF (Fig. S1†) matched the published one well, confirming the formation of pure Ni-MOF crystals.46 The conversion of Ni-MOF into Ni@C composites was then realized via direct thermal treatments under a continuous flow of nitrogen. During the course, the ligands of Ni-MOF (p-benzenedicarboxylic acid) were subjected to carbonization, while the Ni2+ were reduced to metallic Ni.47 As the Ni-MOF structure began to decompose when the heating temperature reached above 400 °C, as suggested by the TGA curve (Fig. S2†), the prepared Ni-MOF materials were calcined at four temperatures, i.e. 450, 550, 650, and 750 °C, to derive the final Ni@C-X composites, where X suggests the pyrolysis temperature. As shown in Fig. 1, the XRD patterns of the resultant Ni@C composites exhibited three diffraction peaks at ca. 44.5, 51.8, and 76.4°, separately, which were attributed to metallic Ni with a face-centered cubic (fcc) structure (JCPDS# 87-0712).27 No diffraction peaks characteristic of Ni-MOF could be observed, indicating that the MOF structure was completely collapsed after thermal treatment. The diffraction peaks associated with Ni increased in intensity with calcination at higher temperature, indicating the Ni phase in the Ni@C composites with a higher crystallization degree. In addition, a diffraction peak centered at ca. 25.6° emerged for relatively higher calcination temperature, which could be related to the presence of turbostratic ordering of the carbon atoms in the graphite layers.48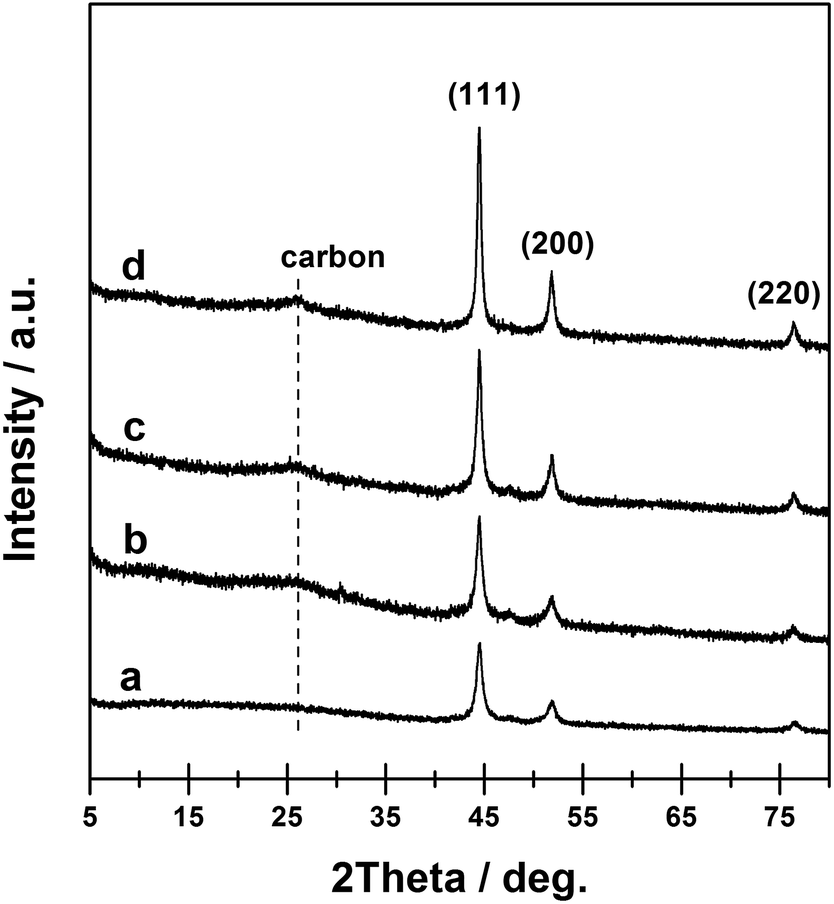 | ||
| Fig. 1 XRD patterns of Ni@C-450 (a), Ni@C-550 (b), Ni@C-660 (c), and Ni@C-760 (d) samples. | ||
The physicochemical properties of Ni@C samples obtained at different pyrolysis temperature were summarized in Table 1. Elemental analyses suggested that all the samples were mainly composed of Ni, C, and H elements, in which the amount of Ni ranged from 31.4 wt% in Ni@C-450 to 49.5 wt% in Ni@C-750, while both the C and H contents showed the opposite trend, decreasing with an increase of pyrolysis temperature. The nitrogen adsorption–desorption isotherms clearly showed the typical microporous structure of Ni@C samples (Fig. S3†). The specific surface areas (SBET) and porosities of Ni@C samples were determined by N2 adsorption–desorption, as shown in Table 1. The BET surface area and pore volume showed obvious enhancement as raising the MOF pyrolysis temperature except that for Ni@C-750, which ranged from 110 m2 g−1 to 160 m2 g−1 and from 0.16 cm3 g−1 to 0.24 cm3 g−1, respectively.
In order to better understand the structural details of Ni@C composites, TEM and HRTEM characterization were performed on the samples. As observed from the TEM images (Fig. 2), the Ni@C materials were composed of monodisperse metallic Ni nanoparticles throughout the carbon matrix formed from the carbonization of p-benzenedicarboxylic acid ligands. The high dispersion nature of Ni nanoparticles could be ascribed to the regular arrangement of Ni ions and ligands in the Ni-MOF structure. As increasing the pyrolysis temperature, the average size of Ni nanoparticles (Fig. S4†) gradually increased from ca. 7 to ca. 19 nm. Fig. 2e exhibited the typical HRTEM image of an individual Ni nanoparticle in Ni@C-650, which was surrounded tightly by layered graphitic carbon structure with the typical distance value of graphite (0.342 nm). This phenomenon is in accordance with the reported literature works that transition metals could induce catalytic graphitization of carbon,38,39 also as indicated by XRD characterization. The graphite-enwrapped Ni nanoparticles were highly crystallized and exhibited two lattice spacings of 0.204 and 0.176 nm, corresponding to the (111) and (200) planes of the fcc Ni lattice.49 In a recent work, Bao and co-workers reported a hybrid material, characteristic of CoNi alloy nanoparticles totally encapsulated by several layers of graphene, the metal composite of which was not soluble by treating with strong acid solution due to the protection of the carbon layers,50 demonstrating the fact of inaccessibility of the alloy nanoparticles to acid solution. However, in the present study, Ni nanoparticles covered with graphite-like carbon layers could be almost totally dissolved in acid environment (see Experimental section), suggesting that the MOF-derived Ni@C may be not totally covered by carbon layers. As shown in Fig. S5,† it was worth noting that the thin graphitic carbon layers over Ni nanoparticles were not fully closed, and some obvious defects like small channels could be observed. This meant that reactant molecules can have access to the Ni nanoparticles through the carbon layers, where the hydrogenation process occurred.51,52
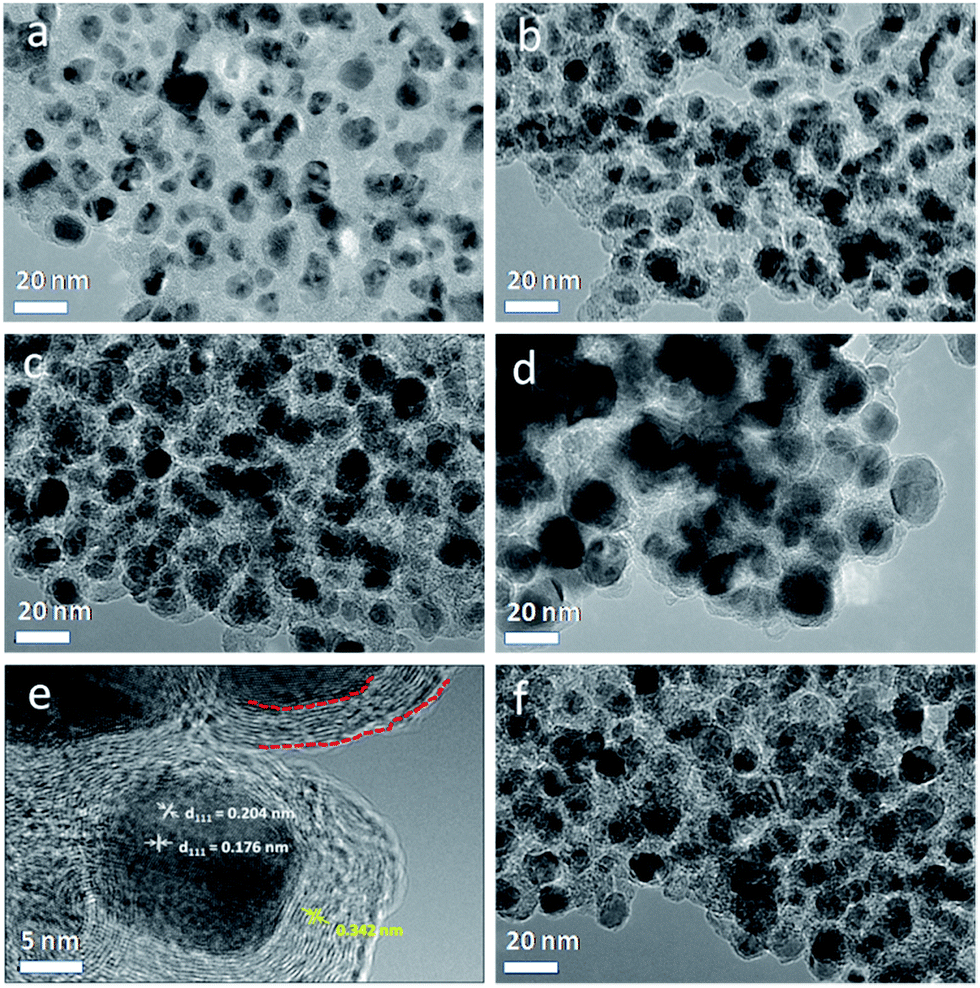 | ||
| Fig. 2 TEM images of Ni@C-450 (a), Ni@C-550 (b), Ni@C-650 (c), Ni@C-750 (d), and Ni@C-650 after five cycles (f). A HRTEM image of an individual Ni nanoparticle in Co@C-650 (e). The dashed red lines in (e) demonstrate graphite carbon layers covering a Ni nanoparticle. | ||
The surface chemical states of Ni@C-650 composite were further confirmed by XPS (Fig. 3). The XPS survey spectrum of Ni@C-650 (Fig. 3a) showed the peaks of three main elements present in the composite (i.e. Ni, C and O). The appearance of the C 1s peak at 284.6 eV was related to the sp2 hybridized graphite-like carbon atom in graphene, and the O 1s peak at 532.0 eV suggested the presence of residual oxygen-containing groups that bounded with carbon atom in graphene.53 In the Ni 2p region of XPS spectra, two peaks with the binding energy values of 870.5 and 853.3 eV, attributed to the spin–orbit splitting of Ni 2p1/2 and Ni 2p3/2, respectively, were observed for Ni@C-650 (Fig. 3d), significantly higher than that of the metallic Ni (Fig. 3b) and Ni/AC (Fig. 3c) references with the typical binding energies of 870.0 and 852.8 eV of metallic Ni.54 This shift to higher values should be ascribed to the charge transfer from the Ni nanoparticles to the surface of graphitic carbon layers, thus leading to an adjustment of electron distribution on the Ni@C composites surface.
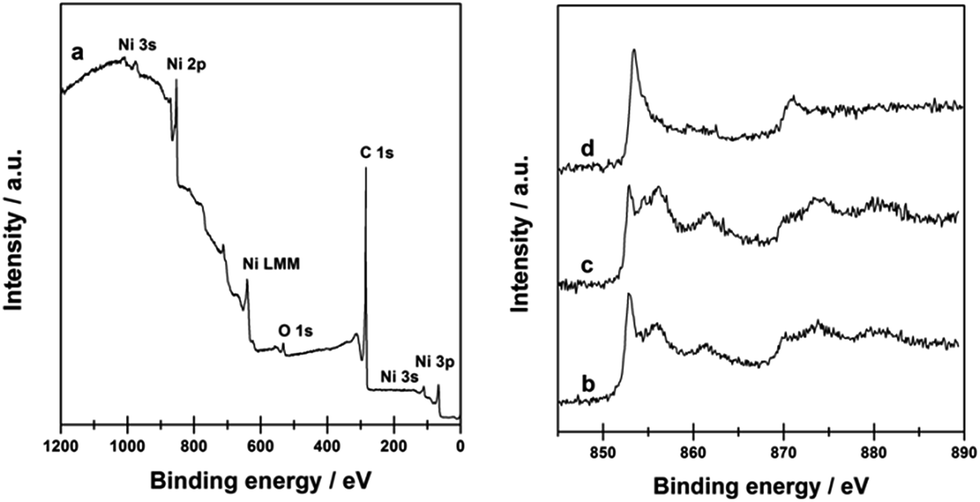 | ||
| Fig. 3 XPS survey spectrum of Ni@C-650 (a); Ni 2p of Ni (b), Ni/AC (c), and Ni@C-650 (d). | ||
3.2 Catalytic properties of Ni@C in the chemoselective hydrogenation of nitroarenes
Generally, non-noble metallic catalysts with higher selectivity require high reaction temperature and higher pressure due to their lower intrinsic activity, which leads to high energy input and difficulties in H2 handling.21,23 In order to explore the catalytic properties of as-obtained Ni@C materials, the hydrogenation of the benchmark substrate o-chloronitrobenzene (o-CNB) was initially investigated, and the similar reaction conditions under which noble metal proceeded efficiently as chemoselective catalysts, i.e. 140 °C and 0.5 Mpa of H2, was selected.8 Pleasingly, the prepared Ni@C materials were highly active in the chemoselective hydrogenation reaction (Table 2, entries 1–4), among which Ni@C-650 exhibited the highest catalytic performance, achieving 99.4% conversion of o-CNB and 94.2% selectivity to o-chloroaniline (o-CAN) in only 40 min. This may be the most promising candidate, to the best of our knowledge, that involves the exceptionally high catalytic performance compared with the hitherto known Ni-based catalysts in the chemoselective hydrogenation of o-CNB reaction based on such low H2 pressure.20,23,27,55 Notably, there was an apparent decline in the catalytic activity of the resultant Ni@C-750 material (entry 4) due to the growth and agglomeration of Ni particles, as suggested by TEM characterization. According to the previous literatures,38,39 transition metals were capable of catalyzing transfer hydrogenation process. However, under our conditions, no obvious conversion of o-CNB could be observed during the whole catalytic test in the absence of H2, ruling out the possibility of hydrogen transfer from solvent, i.e. ethanol, to the reactants over Ni@C-650 material (entry 5). In repeated preparations of Ni@C-650 composite via the facile thermolysis route, the physicochemical properties of the obtained materials were quite similar as each other (not given), and the catalytic performance in the hydrogenation reaction was almost identical (entries 6 and 7). These observations strongly indicated that MOF-based thermolysis strategy was a feasible route to prepare carbon encapsulated metallic Ni nanomaterials with high activity and excellent reproducibility.| Entry | Catalyst | Conversionb (%) | Selectivityb (%) |
|---|---|---|---|
| a Reaction conditions: 0.63 mmol o-chloronitrobenzene, 10 mL C2H5OH, 0.5 Mpa H2, 10 mg catalyst, temperature = 140 °C, reaction time = 40 min.b Experimental accuracy of ±2% from GC analysis.c Experiment under inert N2 atmosphere.d Repeated preparation experiments. | |||
| 1 | Ni@C-450 | 66.2 | 92.5 |
| 2 | Ni@C-550 | 91.8 | 93.8 |
| 3 | Ni@C-650 | 99.4 | 94.2 |
| 4 | Ni@C-750 | 59.5 | 93.6 |
| 5 | Ni@C-650c | <1% | — |
| 6 | Ni@C-650d | 99.1 | 94.0 |
| 7 | Ni@C-650d | 98.5 | 94.3 |
| 8 | Ni(NO3)2 | <1% | — |
| 9 | Ni-MOF | <1% | — |
| 10 | Ni | 30.1 | 85.8 |
| 11 | Ni/AC | 34.3 | 86.0 |
| 12 | Ni@C-650-Ni | 40.8 | 88.7 |
| 13 | Ni@C-650-C | <1% | — |
| 14 | Ni@C-650-C + Ni@C-650-Ni | 36.8 | 88.5 |
| 15 | Ru/C | 100 | 10.5 |
| 16 | Pt/C | 100 | 12.1 |
| 17 | Pd/C | 100 | — |
To gain insight into the unique catalytic characters of the Ni@C materials, a series of control experiments were further performed. Under the identical conditions, no products were observed over homogeneous Ni(NO3)2 and pristine Ni-MOF (entries 8 and 9), suggesting that Ni2+ ions were unable to take part in the hydrogenation process. Commercially available Ni nanoparticles and Ni/AC worked in the hydrogenation of o-CNB but gave poor reactivity (entries 10 and 11), which was inferior to that of prepared Ni@C with respect of both o-CNB conversion and o-CAN selectivity. This distinctive difference could be due to the synergistic effect between highly dispersed Ni and graphitic carbon layers in Ni@C materials. To confirm this hypothesis, we further tested the catalytic efficiency of Ni@C-650 composite after removing the metallic Ni or graphitic carbon layers under the investigated conditions. As expected, Ni@C-650-Ni exhibited a similar low reactivity to those on Ni and Ni/AC (entry 12), while Ni@C-650-C was completely inactive for the reaction (entry 13). On the other hand, by physical mixing of these two separate components, i.e. Ni@C-650-Ni and Ni@C-650-C, a much lower activity than the corresponding Ni@C-650 composite (entry 14) was achieved. Based on these control experiments, it clearly demonstrated the importance of synergic interactions between Ni nanoparticles and graphitic carbon layers in determining the catalytic activities of Ni@C in this hydrogenation reaction. Furthermore, we investigated the catalytic activities of Ru/C, Pt/C, and Pd/C (entries 15–17), and the reaction proceeded unselectively in rather low target product yields due to the formation of undesired dehalogenation by-products severely. This result emphasized the fact that Ni@C exhibited impressive selectively in the hydrogenation of nitroarene compounds having sensitive substituted groups, much better than those of the noble metal catalysts.
Previous literatures indicated that the way that the reactant was adsorbed on the metal catalysts played a key role in determining the chemoselectivity during the hydrogenation of nitroarenes.23,56 As confirmed by the above-mentioned XPS results, a strong Ni–carbon interaction in Ni@C composites formed due to the electron transfer from highly dispersed metallic Ni to the surface of graphitic carbon layers, thus increasing the π-electrons density of the graphite-like carbon support, which could induce an enhanced oriented chemisorption of o-CNB with the nitro group by repulsion of the negative part locating on the chlorine site of o-CNB.23 For this, the hydrogenolysis of the C–Cl bond in o-CNB and o-CAN was greatly inhibited, giving high selectivities to o-CAN on four Ni@C materials obtained in the present work, regardless of the catalytic activities of Ni@C. With respect to Ni/AC, the relative low selectivity to target product should be ascribed to the weak interaction between the Ni and carbon support (as suggested by XPS). Additionally, the reaction conditions were systematically optimized by variation of the temperature and solvent with the results as given in Fig. S6 and Table S1,† respectively. As shown in Table S1,† changing the solvent influenced the activity and chemoselectivity of the hydrogenation reaction dramatically. As one can see, reactions in protonic solvents exhibited better yields as compared to the aprotic ones. Taking THF as an example (entry 2), only 20.8% conversion was achieved among the solvents investigated under the identical conditions. It was noteworthy that a big improvement in conversion from 20.8% to 90.5% was gained when mixing THF with equal volume H2O as solvent (entry 6), suggesting that H2O, as a strong protonic agent, might have a positive effect on the reaction. Whereas, H2O could not promote the reaction to a satisfactory conversion solely, maybe ascribed to the poor solubility of substrate.57 By comparison, C2H5OH seemed to be the optimal solvent with regard to both conversion and chemoselectivity. A comparative catalytic data between the published literatures and the present work was presented in Table 3, which confirms that the MOF-derived Ni@C catalysts investigated in this study are much more efficient in the hydrogenation reaction, with the space-time yield up to 14.0 go-CAN gcat h−1 for Ni@C-650. As can be seen, such a productivity is even superior to some of those achieved with the higher H2 pressure (e.g. 2.0 Mpa).
| Catalyst | H2 pressure (Mpa) | Conv. (%) | Select. (%) | Productivity (go-CAN gcat h−1) | Ref. |
|---|---|---|---|---|---|
| Ni/CNFs | 2.0 | 98 | 97 | 28.2 | 54 |
| Ni/CNFs | 1.0 | 71 | 90 | 3.2 | 54 |
| Ni/C | 0.5 | 100 | 86.6 | 1.7 | 27 |
| Ni/C | 2.0 | 98.9 | 86.0 | 4.3 | 20 |
| Ni-L/P-CNTs | 2.0 | 99.3 | 98.8 | 2.0 | 23 |
| Ni@C-450 | 0.5 | 66.2 | 92.5 | 9.2 | This work |
| Ni@C-550 | 0.5 | 91.8 | 93.8 | 12.9 | This work |
| Ni@C-650 | 0.5 | 99.4 | 94.2 | 14.0 | This work |
| Ni@C-750 | 0.5 | 59.5 | 93.6 | 8.1 | This work |
To verify the true heterogeneity of the Ni@C-650 composite, hot-filtration test was carried out to determine if active components could leach into the reaction mixture and possibly participated in the catalytic reaction. As shown in Fig. 4, removal of the Ni@C-650 leaded to complete termination of the reaction, confirming the truly heterogeneous nature of the hydrogenation process over Ni@C-650. Remarkably, the magnetism of encapsulated Ni nanoparticles was favorable to efficiently separate the Ni@C-650 from the reaction solution by an external magnet (Fig. 4, inset).
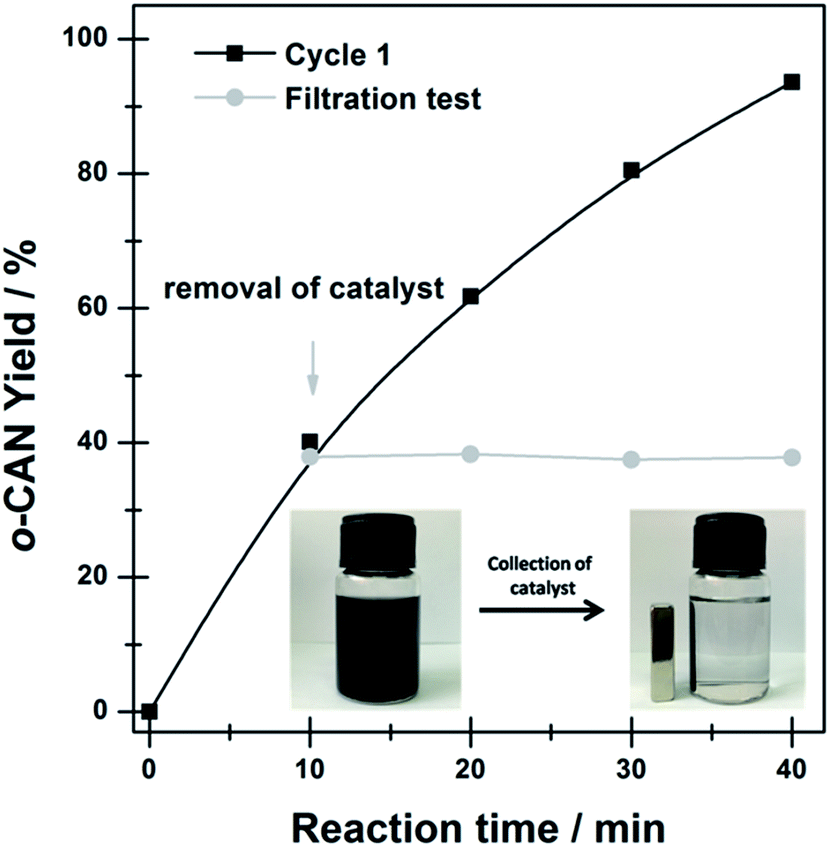 | ||
| Fig. 4 o-CAN yield in the hydrogenation of o-chloronitrobenzene on Ni@C-560, plotted as a function of reaction time. Reaction conditions: 0.63 mmol o-chloronitrobenzene, 10 mL C2H5OH, 0.5 Mpa H2, 10 mg catalyst, temperature = 140 °C. The inset shows the facile magnetic separation of Ni@C-650 catalyst. | ||
In terms of the recycling ability of Ni@C materials, the chemoselective hydrogenation of o-CNB to o-CAN over Ni@C-650 and Ni/AC in successive runs was investigated and the results were shown in Fig. 5. Previous work has revealed that the catalyst used in the hydrogenation of nitroarene was readily subjected to deactivation due to the aggregation of metal particles.27 However, no obvious changes in the catalytic hydrogenation activity for Ni@C-650 in the present study could be observed even after six consecutive cycles. TEM observation clearly demonstrated the maintained nanoparticle size and shape of Ni@C-650 after recycling (Fig. 2f), which should be attributed to the improved resistance of Ni nanoparticles against aggregation with the aid of graphitic carbon layers. In contrast, Ni/AC, characteristic of metal particles deposited on its outer surface, exhibited a remarkable inactivation during the course of the reuse, and obvious Ni particle aggregation occurred (Fig. S7†). In addition, ICP analysis revealed that the reused Ni@C-650 had an identical Ni loading with the fresh one, while the Ni content of Ni/AC declined sharply from 41.5% to 32.7% after six cycles, which, on the other hand, indicated that the coated graphitic carbon layers could stabilize the Ni nanoparticles and prevented them from leaching into the solution. These results strongly emphasized the fact that Ni@C fabricated by the strategy in the present study displayed impressive stability and recyclability, overcoming the poor durability of Ni-based catalysts for the hydrogenation of nitroarenes in liquid phase, thus showing potential application in the chemical industry.
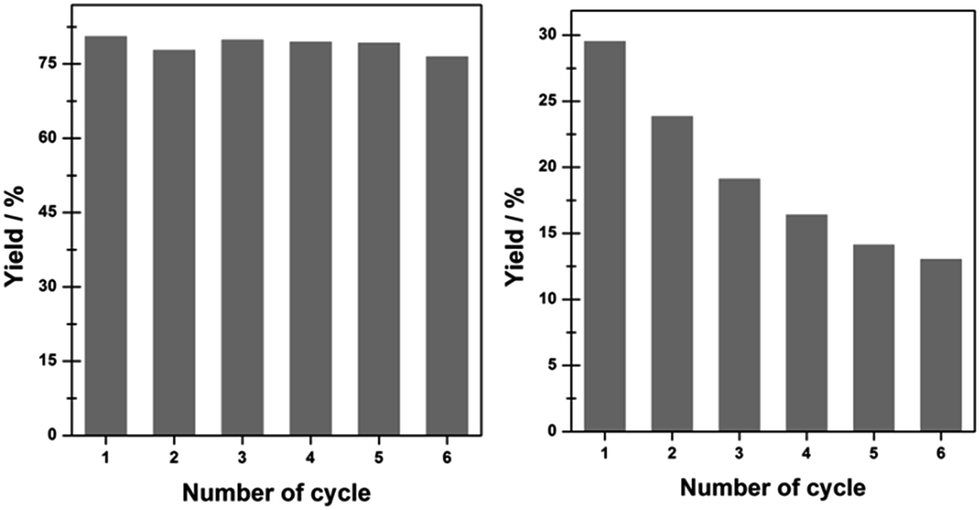 | ||
| Fig. 5 Recycling test for the hydrogenation of o-chloronitrobenzene on Ni@C-560 (left) and Ni/AC (right). Reaction conditions: 0.63 mmol o-chloronitrobenzene, 10 mL C2H5OH, 0.5 Mpa H2, 10 mg catalyst, temperature = 140 °C, reaction time = 30 min. | ||
Inspired by the great success of Ni@C-650 for o-CNB hydrogenation, the general scope of Ni@C-650 in the chemoselective hydrogenation of other functionalized nitro compounds to the corresponding anilines were investigated, as summarized in Table 4. To our delight, the hydrogenation process appeared to be universally valid, and full conversions and essentially perfect chemoselectivity could be achieved. For example, halogen-substituted nitroarenes were efficiently reduced to the corresponding haloaromatic anilines without obvious dehalogenation, and the substituent position did not affect the reactivity significantly (entries 1–6). Meanwhile, the substituted nitroarenes in the presence of other functional groups, i.e. methyl, phenolic hydroxyl, ether and amino, proceeded smoothly with exclusive selectivity in the reduction of nitro group (entries 7–10). Chemoselective hydrogenation of the nitro substrates bearing other easily reducible functional groups is a quite challenging task. Gratifyingly, Ni@C-650 showed outstanding chemoselectivity in the reduction of the nitro group, which was readily transformed to the corresponding anilines selectively, remaining the reducible functional groups such as nitrile, alkene, aldehyde, keto, ester and amide unaffected (entries 11–17). These observations further highlighted the remarkable advantage of as-synthesized Ni@C compared to noble metal-based analogues, with the Ni catalyst exhibiting superior chemoselectivity in the hydrogenation of substituted nitroarenes. In addition, the hydrogenation of heteroaromatic nitro compounds catalyzed by Ni@C-650 was investigated, which also could be hydrogenated smoothly and selectively into the corresponding anilines in almost quantitative yields (entries 18–19). It was noteworthy that aliphatic nitro compounds were also able to be converted into the target aliphatic anilines (entries 20–21), again presenting the superb performance of Ni@C in the hydrogenation of diverse substrates, aromatic, aliphatic as well as heteroaromatic nitro compounds.
| Entry | Substrate | Product | Time (min) | Conv.b (%) | Sel.b (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.63 mmol nitroarene, 10 mL C2H5OH, 0.5 Mpa H2, 10 mg catalyst, temperature = 140 °C.b Experimental accuracy of ±2% from GC analysis. | |||||
| 1 |  |
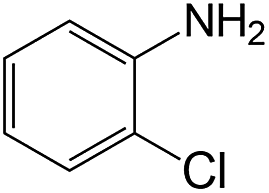 |
40 | >99 | 95.0 |
| 2 | 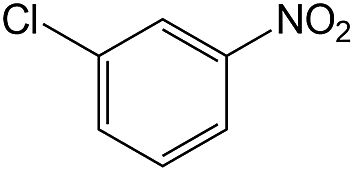 |
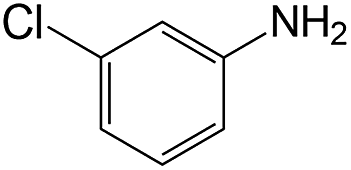 |
40 | >99 | 93.1 |
| 3 |  |
 |
40 | >99 | 94.3 |
| 4 |  |
 |
40 | >99 | 94.8 |
| 5 |  |
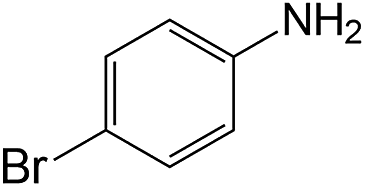 |
60 | 98.5 | 93.8 |
| 6 | 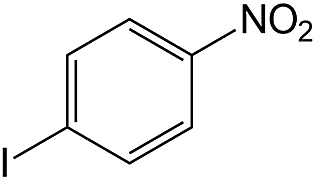 |
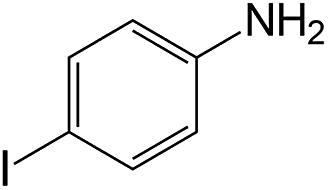 |
60 | 93 | 90.3 |
| 7 | 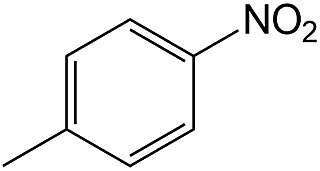 |
 |
50 | 98.3 | >99 |
| 8 |  |
 |
30 | 96.0 | >99 |
| 9 |  |
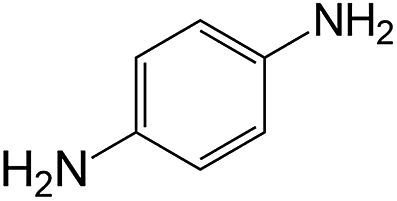 |
50 | >99 | >99 |
| 10 |  |
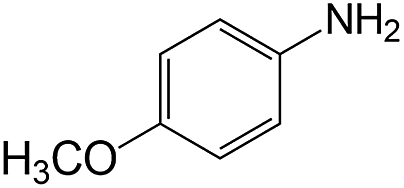 |
30 | 97.4 | 98.4 |
| 11 |  |
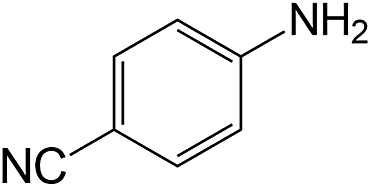 |
40 | >99 | 98.7 |
| 12 | 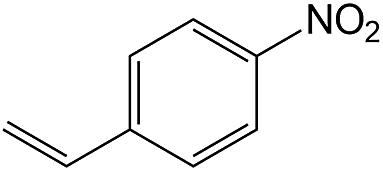 |
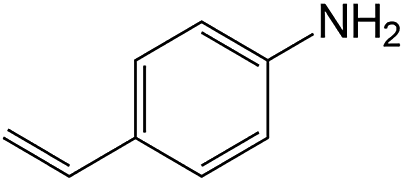 |
40 | >99 | 95.3 |
| 13 | 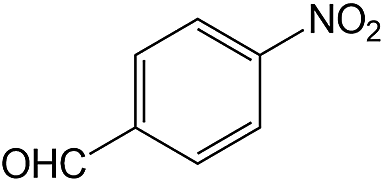 |
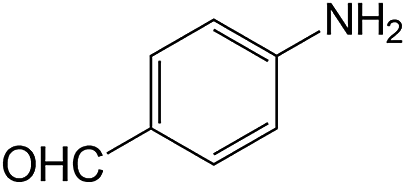 |
40 | >99 | >99 |
| 14 | 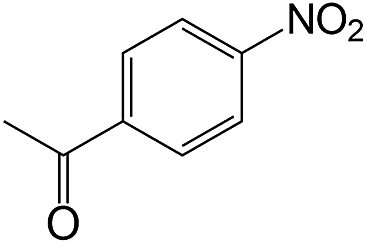 |
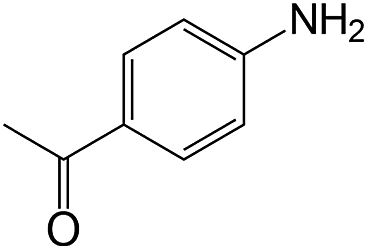 |
40 | >99 | 98.8 |
| 15 | 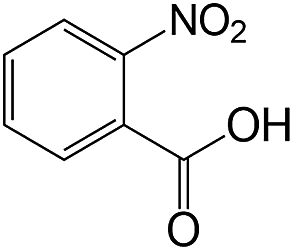 |
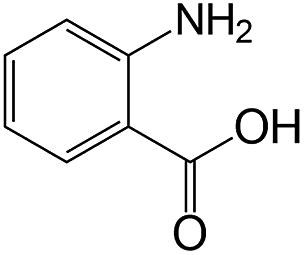 |
30 | >99 | 98.0 |
| 16 | 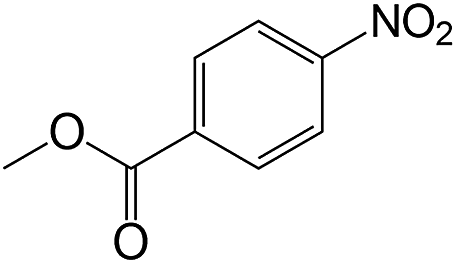 |
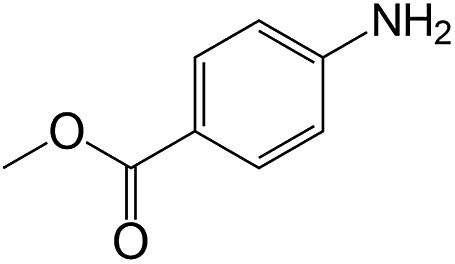 |
40 | >99 | >99 |
| 17 | 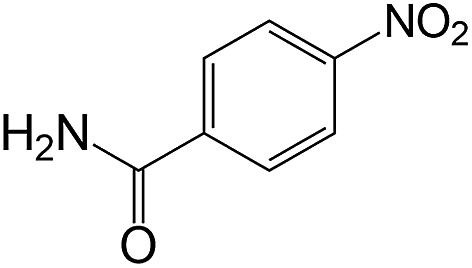 |
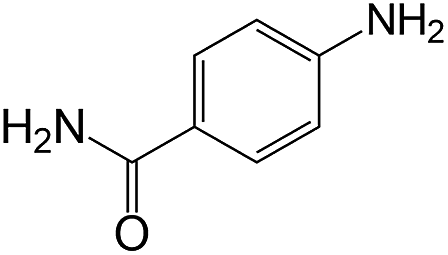 |
50 | >99 | >99 |
| 18 | 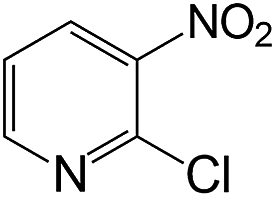 |
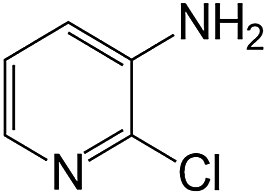 |
40 | >99 | 98.5 |
| 19 | 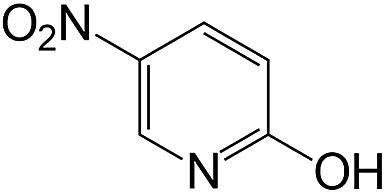 |
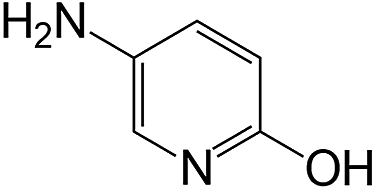 |
40 | >99 | >99 |
| 20 | CH3NO2 | CH3NH2 | 60 | 75.3 | >99 |
| 21 | CH3CH2NO2 | CH3CH2NH2 | 80 | 61.5 | >99 |
4. Conclusions
In summary, we presented highly efficient and magnetically recyclable Ni-based hybrids for selective hydrogenation of functionalized nitro compounds. The Ni@C composites were prepared by facile thermolysis of a Ni-containing MOF, characteristic of highly dispersed Ni nanoparticles within the porous graphitic carbon layers. Such materials exhibited outstanding catalytic performance in the hydrogenation of a variety of substituted aromatic, heteroaromatic and aliphatic nitroarene compounds, affording their corresponding anilines with remarkably high chemoselectivity. The hot-filtration and recycling experiments revealed the true heterogeneity and perfect stability of Ni@C materials during the liquid hydrogenation process. The superior performance could be ascribed to the following aspects. (1) The large surface area and defects surrounded carbon layers allows the easily approached catalytically active sites. (2) The synergistic effect and electron transfer between Ni nanoparticles and graphitic carbon layers enables the superior activity and chemoselectivity. (3) The unique encapsulation structure played a very important role in stabilizing metal nanoparticles, protecting them from growing larger and suppressing leaching of active sites. The achieved success in the MOF-derived metal nanoparticles embedding within graphitic carbon layers would provide an alternative methodology for the production of anilines from selective hydrogenation of nitroarenes.Acknowledgements
This work was financially supported by the NSFC (Grants 21531005) and the 973 program (2014CB845601). The support from the doctoral program foundation of Tianjin Normal University (52XB1509) and the program for Innovative Research Team in University of Tianjin (TD12-5038) were also acknowledged.Notes and references
- R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS.
- H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed.
- R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A. E. Surkus, M. M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972–10974 RSC.
- M. Shalom, V. Molinari, D. Esposito, G. Clavel, D. Ressnig, C. Giordano and M. Antonietti, Adv. Mater., 2014, 26, 1272–1276 CrossRef CAS PubMed.
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. J. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 6799–6802 CrossRef CAS PubMed.
- Z. Zhao, H. Yang, Y. Li and X. Guo, Green Chem., 2014, 16, 1274–1281 RSC.
- A. Corma, P. Serna, P. Concepción and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS PubMed.
- A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed.
- E. Boymans, S. Boland, P. T. Witte, C. Müller and D. Vogt, ChemCatChem, 2013, 5, 431 CrossRef CAS.
- H. U. Blaser, U. Siegrist, H. Steiner and M. Studer, in Aromatic Nitro Compounds: Fine Chemicals through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley-VCH, Weinheim, 2001 Search PubMed.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- M. Boronat, P. Concepcion, A. Corma, S. Gonzalez, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS PubMed.
- L. He, L. C. Wang, H. Sun, J. Ni, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2009, 48, 9538–9541 CrossRef CAS PubMed.
- X. Liu, H. Q. Li, S. Ye, Y. M. Liu, H. Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 7624–7628 CrossRef CAS PubMed.
- R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Junge, J. Radnik, J. Rabeah, H. M. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhofer, M. M. Pohl, J. Radnik, A. E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Bruckner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS PubMed.
- D. Cantillo, M. Baghbanzadeh and C. O. Kappe, Angew. Chem., Int. Ed., 2012, 51, 10190–10193 CrossRef CAS PubMed.
- P. Zhang, C. Yu, X. M. Fan, X. N. Wang, Z. Ling, Z. H. Wang and J. S. Qiu, Phys. Chem. Chem. Phys., 2015, 17, 145–150 RSC.
- Z. Z. Wei, J. Wang, S. J. Mao, D. F. Su, H. Y. Jin, Y. H. Wang, F. Xu, H. R. Li and Y. Wang, ACS Catal., 2015, 5, 4783–4789 CrossRef CAS.
- Y. G. Wu, M. Wen, Q. S. Wu and H. Fang, J. Phys. Chem. C, 2014, 118, 6307–6313 CAS.
- J. Wang, G. L. Fan and F. Li, Catal. Sci. Technol., 2013, 3, 982–991 CAS.
- Y. Li, Y. X. Zhou, X. Ma and H. L. Jiang, Chem. Commun., 2016, 52, 4199–4202 RSC.
- X. Wang and Y. W. Li, J. Mol. Catal. A: Chem., 2016, 420, 56–65 CrossRef CAS.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- P. Zhang, Z. B. Zhao, B. Dyatkin, C. Liu and J. S. Qiu, Green Chem., 2016, 18, 3594–3599 RSC.
- J. Lee, D. H. K. Jackson, T. Li, R. E. Winans, J. A. Dumesic, T. F. Kuech and G. W. Huber, Energy Environ. Sci., 2014, 7, 1657–1660 CAS.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- M. O'Keefe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- S. S. Chen, M. Chen, S. Takamizawa, P. Wang, G. C. Lv and W. Y. Sun, Chem. Commun., 2011, 47, 4902–4904 RSC.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750–5765 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- W. Chaikittisilp, K. Ariga and Y. Yamauchi, J. Mater. Chem. A, 2013, 1, 14–19 CAS.
- H. J. Lee, W. Cho, E. Lim and M. Oh, Chem. Commun., 2014, 50, 5476–5479 RSC.
- P. Zhang, F. Sun, Z. H. Xiang, Z. G. Shen, J. Yun and D. P. Cao, Energy Environ. Sci., 2014, 7, 442–450 CAS.
- K. Shen, X. D. Chen, J. Y. Chen and Y. W. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS.
- W. Zhong, H. L. Liu, C. H. Bai, S. J. Liao and Y. W. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS.
- T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan, A. Chojecki, A. C. J. Koeken, M. Ruitenbeek, G. Meima, H. U. Islam, G. Sankar, M. Makkee, F. Kapteijn and J. Gascon, ACS Catal., 2016, 6, 3236–3247 CrossRef CAS.
- F. Zhang, C. Chen, W. M. Xiao, L. Xu and N. Zhang, Catal. Commun., 2012, 26, 25–29 CrossRef CAS.
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394–7397 CrossRef CAS PubMed.
- A. J. Amali, J. K. Sun and Q. Xu, Chem. Commun., 2014, 50, 1519–1522 RSC.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed.
- Y. Jiao, J. Pei, C. S. Yan, D. H. Chen, Y. Y. Hu and G. Chen, J. Mater. Chem. A, 2016, 4, 13344–13351 CAS.
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC.
- Y. F. Yang, L. T. Jia, B. Hou, D. B. Li, J. G. Wang and Y. H. Sun, J. Phys. Chem. C, 2014, 118, 268–277 CAS.
- W. L. Zhen, B. Li, G. X. Lu and J. T. Ma, Chem. Commun., 2015, 51, 1728–1731 RSC.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- L. Liu, P. Concepción and A. Corma, J. Catal., 2016, 340, 1–9 CrossRef CAS.
- Z. Wei, Y. Chen, J. Wang, D. Su, M. Tang, S. Mao and Y. Wang, ACS Catal., 2016, 6, 5816–5822 CrossRef CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- S. Oswald and W. Brückner, Surf. Interface Anal., 2004, 36, 17–22 CrossRef CAS.
- C. Wang, J. S. Qiu, C. H. Liang, L. Xing and X. M. Yang, Catal. Commun., 2008, 9, 1749–1753 CrossRef CAS.
- M. Boronat, P. Concepcion, A. Corma, S. Gonzalez, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS PubMed.
- R. J. Rahaim and R. E. Maleczka, Org. Lett., 2005, 7, 5087–5090 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26699a |
| This journal is © The Royal Society of Chemistry 2017 |