
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclam te1pa for 64Cu PET imaging. Bioconjugation to antibody, radiolabeling and preclinical application in xenografted colorectal cancer†
Mathieu Frindelab,
Patricia Le Saëcb,
Maryline Beylera,
Anne-Sophie Navarrob,
Catherine Saï-Maurelb,
Cyrille Alliotc,
Michel Chérelbd,
Jean-François Gestinb,
Alain Faivre-Chauvet*b and
Raphaël Tripier *a
*a
aUniversité de Brest, UMR-CNRS 6521/SFR148 ScInBioS, UFR Sciences et Techniques, 6 Avenue Victor le Gorgeu, C.S. 93837, 29238 Brest, France. E-mail: raphael.tripier@univ-brest.fr
bCentre de Recherche en Cancérologie Nantes-Angers (CRCNA), Unité INSERM 892 – CNRS 6299, 8 quai Moncousu, BP 70721, 44007 NANTES Cedex, France. E-mail: Alain.Faivre-Chauvet@univ-nantes.fr
cCyclotron ARRONAX, 1 rue Aronnax, CS 10112, 44817 SAINT-HERBLAIN Cedex, France
dInstitut de Cancérologie de l'Ouest, Boulevard Jacques Monod, 44805 SAINT-HERBLAIN Cedex, France
First published on 31st January 2017
Abstract
te1pa is a monopicolinate cyclam previously presented as a better 64Cu chelator than dota, nota and other chelators with an improved biodistribution and in vivo resistance to transchelation. This study aimed to determine whether te1pa could improve the in vivo stability of 64Cu chelation concerning radioimmunoconjugates in order to obtain better contrast in PET imaging. te1pa was activated on its remaining acid function to obtain a N-hydroxysulfosuccinimide ester and was then conjugated to the F6 mouse IgG1a (F6 mAb), directed against CEA (carcinoembryonic antigen), leading to the F6-te1pa immunoconjugate. F6-te1pa was compared to F6–C-dota, i.e. F6 mAb conjugated with a C-functionalized dota which is the only chelator used nowadays in preclinical trials for 64Cu PET imaging. Immunoconjugates were radiolabeled with 64Cu showing an equivalent conjugation rate of 1 ligand per mAb. The study of the complexation kinetics highlighted a relatively fast process and 64Cu–F6-te1pa, exhibiting a specific activity of 69.3 ± 28.9 MBq mg−1, was proved to be inert since only 4.3% of radioactivity was transchelated from the ligand to EDTA (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 equiv., overnight) used as a competitor. All these results are comparable with C-functionalized dota. However, in vivo studies carried out in LS174T tumor-bearing nude mice showed a limited transchelation of superoxide dismutase (SOD) into the liver; 1.6% for 64Cu–F6-te1pa after 24 h post-injection, compared to 4.3% for 64Cu–F6–C-dota. The uptake of 64Cu–F6-te1pa in tumors and radioactivity distribution in organs after 24 and 48 h was satisfactory and equivalent to various standards presented in the literature. Finally, PET-phenotypic images obtained with 64Cu–F6-te1pa at 24 h post-injection showed an excellent contrast between tumors and the healthy tissues around, which agrees well with the results of the biodistribution. The usefulness of te1pa for PET phenotypic imaging using 64Cu has been validated. The synthesis of a bifunctional derivative of te1pa will be the next step of this work to keep the ligand properties intact.
000 equiv., overnight) used as a competitor. All these results are comparable with C-functionalized dota. However, in vivo studies carried out in LS174T tumor-bearing nude mice showed a limited transchelation of superoxide dismutase (SOD) into the liver; 1.6% for 64Cu–F6-te1pa after 24 h post-injection, compared to 4.3% for 64Cu–F6–C-dota. The uptake of 64Cu–F6-te1pa in tumors and radioactivity distribution in organs after 24 and 48 h was satisfactory and equivalent to various standards presented in the literature. Finally, PET-phenotypic images obtained with 64Cu–F6-te1pa at 24 h post-injection showed an excellent contrast between tumors and the healthy tissues around, which agrees well with the results of the biodistribution. The usefulness of te1pa for PET phenotypic imaging using 64Cu has been validated. The synthesis of a bifunctional derivative of te1pa will be the next step of this work to keep the ligand properties intact.
Introduction
Due to its physical properties and its increasing availability, copper-64 (64Cu) represents one of the most promising PET radionuclides after 18-fluorine (18F) and 68-gallium (68Ga). 64Cu is characterized by a longer half-life (12.7 h) compared to 18F (110 min) and 68Ga (68 min) which is particularly interesting for dosimetry calculation, and by β+ and β− emissions (respectively 17.4% and 39.6%) that are adapted to theranostic (ability to perform both therapy and diagnostic) applications.1 Besides, this radionuclide is available as a full GMP (Good Manufacturing Practice) CuCl2 solution for radiopharmaceutical manufacturing and is easily obtained with biomedical cyclotrons by solid 64-nickel (64Ni) target irradiation with protons or deutons.264Cu has already been investigated using several biovectors including peptides and macromolecules in preclinical and clinical studies using various chelating agents for PET imaging studies in oncology3–12 and with the aim to obtain diagnostic companions.13 Copper-64 chloride may also be used as a simple salt to study copper metabolism disorder notably in oncology14,15 and 64Cu-ATSM has been used to visualize tumor hypoxia.16,17 In addition, even if less available than its 64Cu analogue, copper-67 (67Cu) is currently under investigation and presents interesting appropriated properties for therapeutic applications (t1/2 = 62.0 h, β− 100%, Emax = 0.577 MeV). Radiopharmaceuticals based on both copper radiometals are thus attracting the attention of several specialists in nuclear medicine. Consequently, optimized chelation of copper radionuclides is of growing interest.
Chelators suitable for Cu(II) radiopharmaceuticals are generally constituted by N or O donor atoms,18–21 conducting to thermodynamically stable and kinetically inert complexes in order to avoid possible transchelation of the metal by competing with biological ligands or bioreductants, while other preferred properties include good water solubility and fast metal complexation.22,23 The inertness regarding bioreductant systems must particularly be taken in consideration due to the low reduction potential of Cu(II)/Cu(I) (E0: 0.16 V) well adapted to biologic modification of its oxidizing status.24 Then, each copper-complex has its own behavior in reductive media. Among all the chelators presented for 64/67Cu vectorization, dota,20,25 teta,26 te2a,27 cb-te2a28–30 and nota31,32 (see Fig. 1) have received much attention. However, none of them completely fulfills all the criteria for an in vivo application (thermodynamic stability, kinetic inertness, fast complexation process, Cu(II) → Cu(I) stability…) and are still limited to academic researches or early clinical trial phases. Other chelators containing nitrogen atoms as sarcophagines33–35 (Sar ligands, Fig. 1) have also demonstrated their ability to form chelates of interest but still present some real drawbacks. Thus, to our knowledge except two studies involving teta,8,9 dota still remains one of the rare chelators used for clinical trials for 64Cu PET imaging,6,36,37 despite its drawbacks, allowing to continue research in this area.
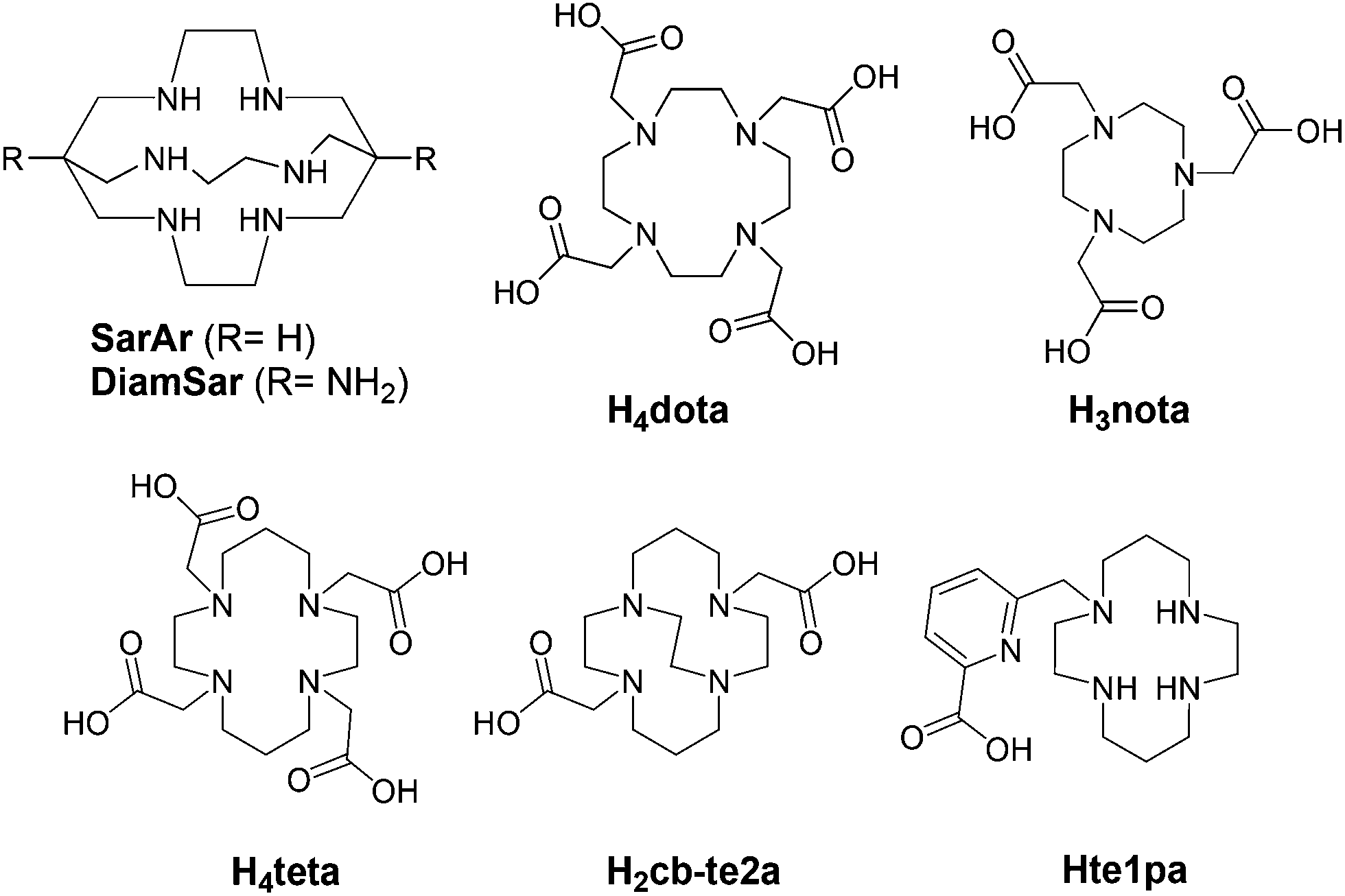 | ||
| Fig. 1 Ligands discussed in this work. | ||
With this in mind we have recently and progressively developed picolinate based copper-chelators that have proved to be very attractive as 64Cu chelators.38–41 Picolinate ligands have indeed already demonstrated their efficiency to form stable and inert complexes with a large panel of cationic ions.42–51 We were the first to report the use of such azamacrocyclic ligands for natCu and 64Cu complexation and radiolabeling including in vitro and in vitro studies.38–40
We previously proved that in the case of [64Cu(te1pa)]+, the carboxylic function of the picolinate pendant did not enter in the coordination scheme of the metal center complex but without any consequence on the overall properties of the chelate. We thus concluded that this function should be ideally used for bioconjugation of the chelate on specific biomolecules to form promising radiopharmaceuticals.
In this work we then present the conjugation reaction of the te1pa chelator with a F6 mouse IgG1a antibody (F6 mAb), directed against CEA (carcinoembryonic antigen), the characterization of the so obtained immunoconjugate, its radiolabeling, in vitro and in vivo behaviors and the first PET phenotypic images in a colorectal human cancer bearing nude mice. Despite the rise of advantageous bifunctional chelators as nota (or noda-GA), a dota derivative was again selected as comparison considering that it represents the actual clinical reference. Additionally, a too large animal study with an exhaustive selection of various chelators is too expensive and ethically questionable.
The choice of the tumor model has been determined by its high percentage human occurrence and its high and regular expression of CEA by tumor cells.52 Moreover, CEA antigens can be easily targeted with different mAbs or derivatives without high nonspecific uptake on healthy tissues.
Results and discussion
Immunoconjugates synthesis
The monoclonal F6 antibody (F6 mAb) is a mouse IgG1a directed against the CEA and more specifically against the CEA epitope Gold 1.53 The first challenge of our study was to manage the activation of the carboxylic function of the picolinate moieties of the te1pa ligand to allow its bioconjugation with this antibody. Such a reaction involving an aromatic carboxylic function has already been performed by Cai et al.54 but is more difficult when the function is linked to a pyridine core. The selected reaction pathway consists in the conventional use of EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) as coupling agent and sulfo-NHS (N-hydroxysulfosuccinimide) to lead to te1pa-SNHS (Scheme 1). To preserve reactivity of the so obtained activated ester, the strategy was to place it directly in reaction with the antibody without further purification. Reaction conditions were inspired by the work of Cai et al. and preliminary works to optimized conjugation yield while lowering formation of aggregates from cross-reactions after incorporation of the mAb in the subsequent conjugation step (see information 1 in ESI:† previous investigations done with the easier accessible hLL2 mAb were used as reference, the F6 mAb being too difficult to obtained in large amount to allow comparable preliminary works). The activation step was performed at 4 °C during 30 minutes at pH 5 with a molar ratio of te1pa:EDC:SNHS of 1:1:1, leading to the corresponding desired activated ester (Scheme 1). This molar ratio was slightly different from those used by Cai et al. (1:1:0.8) but gave equivalent results.
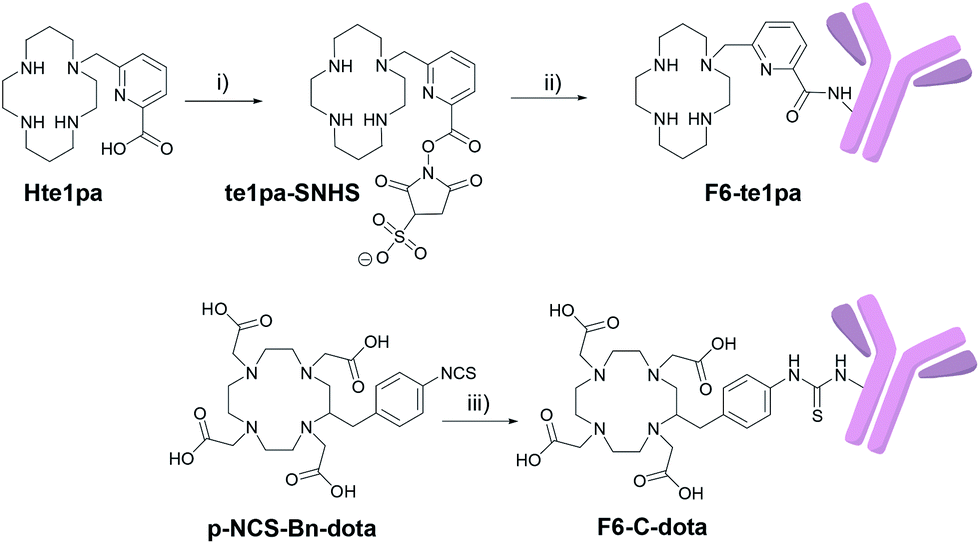 | ||
| Scheme 1 Activation of the carboxylic function of te1pa and conjugation of te1pa-SNHS and p-NCS-Bn-dota with the F6 mAb. (i) MES pH 5 buffer, sulfo-NHS, EDC, 30 min, ice bath; (ii) F6 mAb, carbonate buffer (0.3 M, pH 8.6), 3 h, ice bath; (iii) F6 mAb, carbonate buffer (0.3 M, pH 8.6), 12 h, RT. | ||
The conjugation step was performed from 0 °C to 17 °C by addition of the activated chelator to a solution of F6 mAb at pH 8.6 during 3 hours to obtain the F6-te1pa immunoconjugate. The reaction was performed using 700 equiv. of activated te1pa (theoretical amount by considering a total activation yield) to follow our previously optimized conditions. This leads to the formation of 60% of aggregates (Table 1) eliminated before further experiments by gel filtration (see Experimental section). Compared to the 6 equivalents AmBaSar:cRGD peptide conditions carried out by Cai et al., the chosen conditions clearly underline the weaker reactivity of the carboxylic function of the picolinate. However, the excess of activated te1pa had no impact on the quality of the resulting immunoconjugate.
| F6-te1pa | p-NCS-Bn-dota | |
|---|---|---|
| Equiv. of activated ligand | 700 | 15 |
| % of aggregated after coupling | 60 | 5 |
| Radiolabeled conjugate | 64Cu–F6-te1pa | 64Cu–F6–C-dota |
|---|---|---|
| Ligand per MAbs | 1.05 ± 0.15 | 1.00 ± 0.20 |
| Radiolabeling yield (%) | 93.9 ± 1 | 93.7 ± 2.5 |
| Specific activity (MBq mg−1) | 69.4 ± 27.6 | 69.3 ± 28.9 |
| Immunoreactivity (%) | 85.4 ± 4.6 | 88.1 ± 4.6 |
As already mentioned in our previous work,39 our aim was to compare the results of te1pa with dota, a ligand widely used for 64Cu radiolabeling and especially one of the few to be used for clinical trials. For this study, dota was selected under its C-functionalized form: p-NCS-Bn-dota, which is a 64Cu standard currently used for such experiments. This bifunctional chelating agent analogue of dota presents the advantage to be graftable on biomolecules and to preserve intact the structure of the dota ligand offering thus the best conditions required for a good comparison. Additionally, this compound is commercially available, widely used and its conjugation to mAbs procedure is accessible in literature.
p-NCS-Bn-dota was grafted to the F6 mAb, following the usual protocol used in our laboratory (see Experimental section), leading to 5% of aggregates (Table 1 and Fig. S1 in ESI†). te1pa-SNHS immunoconjugates have shown greater amounts of aggregates in comparison to commercial p-NCS-Bn-dota associated to the same F6 mAb. Purification of the immunoconjugate(s) of interest from antibodies aggregates and from remaining traces of non-conjugated ligands, was performed by size exclusion HPLC. Corresponding chromatograms (immunoconjugate post-conjugation and post-purification) are presented in Fig. S1 (in ESI†) and clearly show that immunoconjugates are finally isolated with less than 5% contaminants. The overall yields of the conjugation/purification procedures were 37% and 90%, respectively for te1pa-SNHS and p-NCS-Bn-dota.
Immunoconjugates characterization
Results are presented in Table 1 and show that the conjugation yields for F6–C-dota and for F6-te1pa are quite equivalent with about 1 accessible ligand grafted per mAb (respectively values of 0.8 to 1.2 and 0.9 to 1.2). This equivalence was a prerequisite for comparing rigorously these 2 immunoconjugates.
Considering the grafting yield, the conjugation of te1pa is comparable or slightly higher than those obtained by Cooper et al. for the conjugation of Sar-CO2H to rituximab, the latter having obtained 0.5 ligands conjugated per mAb after a similar activation of an aromatic carboxylate with EDC and SNHS.55 Regarding the conjugation of p-NCS-Bn-dota, our result is significantly lower than the one obtained by Cooper et al. of 5 ligands conjugated per mAb. This was expected since only 15 eq. of p-NCS-Bn-dota were used to obtain the F6–C-dota while 40 eq. were used with rituximab in Cooper et al. work.
000 equiv. (see Table S3 in ESI†) in 0.1 M ammonium acetate (pH 6.5–7). Results clearly show that 64Cu–F6–C-Bn-dota and 64Cu–F6-te1pa presented the same resistance toward competitive ligand. Respectively, 5.2 and 4.3% of the radioactivity was transchelated from the ligands to EDTA when 50000 equiv. of competitor were introduced in solution in regard to radioimmunoconjugates amounts.It has to be noted that Wu et al. showed that copper-64 complexes of C-functionalized nota (C-nota, 3p-C-ne3ta and 3p-C-nota) were more stable than C-functionalized dota (64Cu–C-dota) when challenged with only 100 equivalents of EDTA.56 Such competition with EDTA is thus able to highlight the differences in stability between copper-64 complexes. It should be noted that the lack of stability of unconjugated 64Cu–C-dota was highlighted at pH 5.5 whereas our competition study was performed at pH 6.5–7. This difference between the two protocols can partially explain the discordant results.
Radiolabeling
Radiolabeling yields, specific activities and immunoreactivities of 64Cu–F6–C-dota and 64Cu–F6-te1pa have been measured and are shown in Table 1 (measurements done after the radiolabeling performed for in vivo experiments; in vitro characterization of radioimmunoconjugates was performed previously and required a separate radiolabeling). Specific activities are given before the adjustment with the unconjugated F6 mAb and are quite equivalent for both radiopharmaceuticals (69.4 ± 27.6 vs. 69.3 ± 28.9 MBq mg−1). It is then possible to obtain immunoconjugates with sufficient specific activity for clinical use despite a relatively low number of chelating agents grafted per antibody.The protocol carried out (see Experimental section) gave equivalent radiolabeling yields (93.7 ± 2.5 vs. 93.9 ± 1) as well as immunoreactivity scores (88.1 ± 4.6 vs. 85.4 ± 4.6) respectively for 64Cu–F6–C-dota and 64Cu–F6-te1pa, ensuring optimal comparison between the two radioimmunoconjugates.
For the PET-phenotypic imaging study, volumic activity was adjusted to 50 MBq mL−1 (instead of 33.3 MBq mL−1 for the biodistribution study) so the injected volume and activity were enough for imaging at 24 h. The two radiolabeled antibodies were injected in the same conditions.
In vivo studies – biodistribution and PET imaging
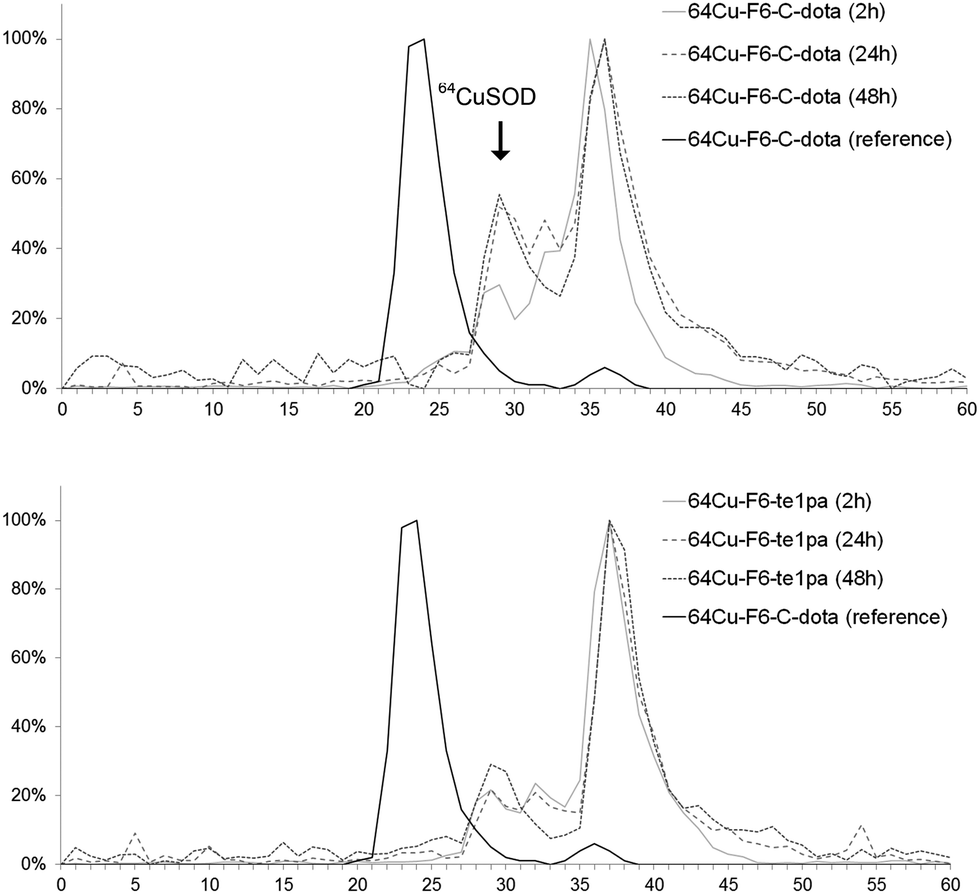 | ||
| Fig. 2 Liver extract chromatograms of sacrified mice at 2 h, 24 h and 48 h post-injection of 64Cu–F6-te1pa and 64Cu–F6–C-dota. The reference is the solution of 64Cu–F6–C-dota post-radiolabeling. | ||
Size exclusion chromatography of hepatic extracts showed a main peak with a retention time (RT) between 35 and 37 min at 2, 24 and 48 h PI in both mice injected with 64Cu–F6-te1pa or 64Cu–F6–C-dota; this peak could reflect the presence of radiolabeled low molecular weight molecules linked with the metabolism of monoclonal antibody, that generates lysyl-aminoacid-dota-64Cu as already mentioned in the literature.58 Another peak was observed at 29 min for all the analyzed samples. It is highly likely that this latter peak corresponds to 64Cu complexed by SOD, considering that the same retention time was observed when injecting bovine SOD previously radiolabeled with copper-64 in vitro. The intensity of the 64Cu–SOD peak and its evolution during time was different depending of the injected radioimmunoconjugate. The intensity of 64Cu-SOD was 30 and 22% after 2 h PI, respectively for 64Cu–F6–C-dota and 64Cu–F6-te1pa. At 24 and 48 h PI, the peak intensity had nearly doubled for 64Cu–F6–C-dota (52% at 24 h and 56% at 48 h) and increased only slightly for 64Cu–F6-te1pa (22% at 24 h and 29% at 48 h). Brought to the injected dose, the amount of 64Cu transchelated to SOD into the liver was equivalent to 4.3% vs. 1.6% at 24 h and 3.8% vs. 2.3% at 48 h respectively for 64Cu–F6–C-dota and 64Cu–F6-te1pa.
These transchelations, although relatively limited, nevertheless prove a better stability for the te1pa derivative when compared to dota derivate.
Regarding chromatographic analyses of the blood plasma (Fig. 3), only one peak was observed for all analyzed samples with a RT between 23 and 25 min. This RT matches with the reference of 64Cu–F6–C-dota (RT of 23–24 min), showing that radiolabeled species present in plasma correspond to radioimmunoconjugates in their intact forms. This was confirmed by the measurement of the immunoreactivity of plasma, which was superior to 80% at 24 and 48 h PI. Almost all plasma activity recorded at these times corresponds to intact radioimmunoconjugates, or at least to species retaining their specific recognition for CEA. Thus, even if transchelation has occurred in liver, this process did not reflect on plasma proteins, notably on ceruloplasmin all over the 48 h studied. This discrepancy between results obtained for liver extract and blood can be explained by the fact that F6 mAb is from murine origins, so it can be recycled in large part by the FcRn receptor in liver capillaries without being metabolized.59 Therefore it can be assumed that only a very small fraction of radioimmunoconjugates will undergo transchelation during the in vivo distribution and elimination follow-up period, taking into account that the half-life of copper-64 is much shorter than the biological half-life of the murine antibody F6.
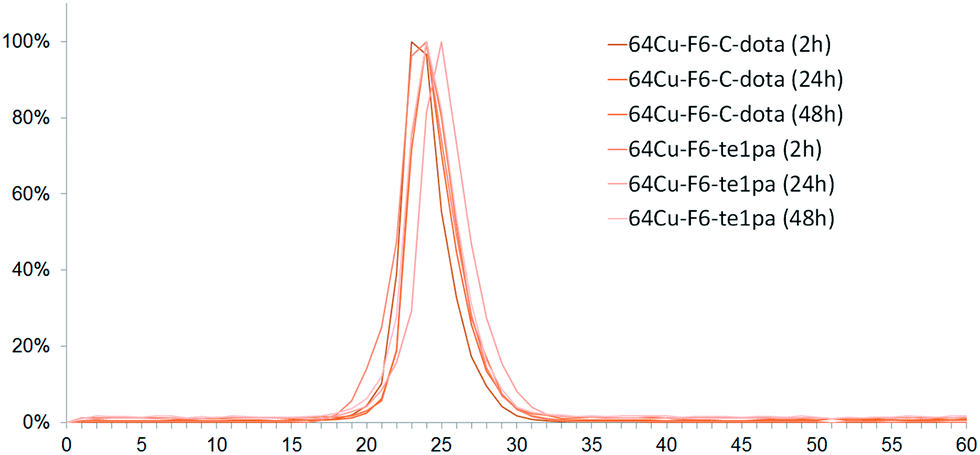 | ||
| Fig. 3 Chromatograms of serum from mice sacrificed at 2 h, 24 h and 48 h post-injection of 64Cu–F6-te1pa and 64Cu–F6–C-dota. | ||
The study of the liver extract nevertheless demonstrates that, following conjugation, 64Cu-te1pa has an excellent stability in comparison to 64Cu-dota regarding transchelation processes. The conjugation pathway that was used for te1pa does not seem to deteriorate the complexation properties of this ligand, which confirms that the carbonyl group of the picolinate does not participate to the coordination of copper and can be used to functionalize the ligand.
| Tissue | 2 h | 24 h | 48 h | |||
|---|---|---|---|---|---|---|
| 64Cu–F6-te1pa | 64Cu–F6-C-dota | 64Cu–F6-te1pa | 64Cu–F6-C-dota | 64Cu–F6-te1pa | 64Cu–F6-C-dota | |
| Tumor | 8.46 ± 1.26 | 6.40 ± 1.37 | 22.01 ± 5.43 | 28.72 ± 7.54 | 29.05 ± 5.92 | 27.89 ± 9.27 |
| Blood | 22.96 ± 3.22 | 21.96 ± 2.62 | 12.94 ± 3.05 | 41.14 ± 1.83 | 13.94 ± 1.23 | 11.62 ± 3.46 |
| Liver | 9.27 ± 1.20 | 8.28 ± 1.18 | 7.41 ± 1.75 | 8.25 ± 1.38 | 8.03 ± 1.13 | 6.83 ± 0.87 |
| Kidney | 5.59 ± 1.09 | 5.22 ± 0.93 | 3.58 ± 0.81 | 4.03 ± 0.46 | 3.73 ± 0.44 | 3.51 ± 0.90 |
| Small intestine | 5.94 ± 1.98 | 4.81 ± 0.86 | 2.69 ± 0.90 | 3.50 ± 0.88 | 3.54 ± 1.16 | 2.95 ± 0.81 |
| Transverse intestine | 5.68 ± 2.07 | 4.04 ± 0.85 | 2.48 ± 0.72 | 2.53 ± 0.30 | 2.73 ± 0.86 | 2.21 ± 0.44 |
| Large intestine | 2.61 ± 2.38 | 2.19 ± 0.41 | 1.80 ± 0.36 | 2.16 ± 0.20 | 2.08 ± 0.46 | 2.01 ± 0.18 |
| Lung | 8.98 ± 1.84 | 10.37 ± 1.81 | 5.80 ± 1.73 | 6.45 ± 1.48 | 6.37 ± 1.39 | 6.00 ± 2.39 |
| Muscle | 0.61 ± 0.09 | 0.58 ± 0.12 | 0.74 ± 0.18 | 0.79 ± 0.09 | 0.85 ± 0.14 | 0.78 ± 0.15 |
| Spleen | 4.94 ± 1.30 | 4.02 ± 0.65 | 4.01 ± 1.24 | 4.87 ± 1.01 | 5.34 ± 1.41 | 4.18 ± 1.93 |
| Skin | 1.42 ± 0.18 | 1.50 ± 0.30 | 3.44 ± 0.74 | 3.60 ± 0.43 | 4.02 ± 0.31 | 3.56 ± 0.95 |
| Brain | 1.24 ± 0.39 | 1.28 ± 0.38 | 0.97 ± 0.23 | 0.89 ± 0.22 | 0.87 ± 0.22 | 0.74 ± 0.23 |
| Heart | 6.05 ± 0.91 | 6.00 ± 0.50 | 4.19 ± 1.00 | 5.02 ± 0.76 | 4.29 ± 0.47 | 4.23 ± 1.05 |
| Bone | 2.30 ± 0.42 | 2.22 ± 0.34 | 1.76 ± 0.48 | 1.89 ± 0.43 | 1.78 ± 0.41 | 1.60 ± 0.44 |
| Stomach | 2.11 ± 0.22 | 1.72 ± 0.40 | 1.98 ± 0.32 | 2.26 ± 0.26 | 2.26 ± 0.53 | 2.11 ± 0.50 |
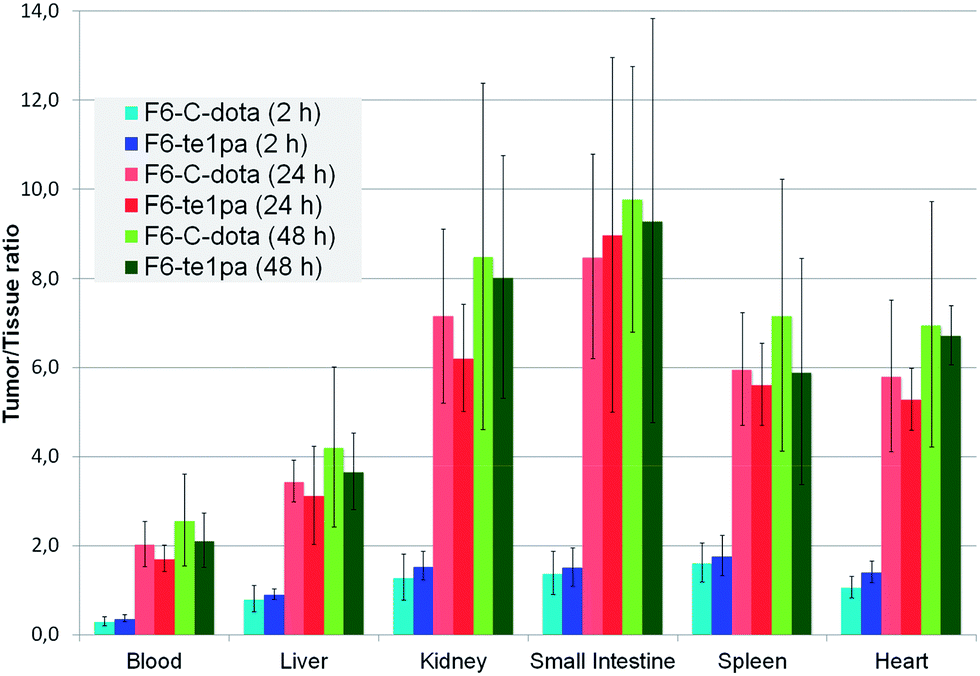 | ||
| Fig. 4 Biodistribution of 64Cu–F6-te1pa and 64Cu–F6–C-dota in nude mice bearing LS174T tumors expressed as a ratio tumor/tissue. | ||
At 2 h PI, the activity is mostly distributed in blood (22 ± 2.6 % ID g−1 for 64Cu–F6–C-dota and 23 ± 3.2 % ID g−1 for 64Cu–F6-te1pa) and in highly vascularized organs like lungs (10.4 ± 1.8 and 9 ± 1.8 % ID g−1). % ID g−1 in the tumor were 6.4 ± 1.4 for 64Cu–F6–C-dota and 8.5 ± 1.3 for 64Cu–F6-te1pa. Tumor/heart ratios were quite low (1.1 ± 0.2 and 1.4 ± 0.2) making difficult to determinate if the radioactivity detected in tumor was due to specific accumulation of the radioimmunoconjugate or to tumor vascularization. At this early time PI, no significant differences were shown between the two radiolabeled antibodies in all studied organs.
At 24 h PI, blood activity decreased by a factor 1.6 and 1.8 (14.1 ± 1.8 and 13 ± 3% ID g−1), while tumor accumulation increased by a factor 4.5 and 2.6 (28.7 ± 7.5 and 22 ± 5.4 % ID g−1) respectively for 64Cu–F6–C-dota and 64Cu–F6-te1pa, in comparison with 2 h PI time. Moreover, a decrease of % ID g−1 was observed for kidney, small intestine, transverse intestine, lung and heart (p < 0.04) for the two radioimmunoconjugates. The uptake in these organs (p > 0.1) and in tumor (p = 0.08) was not statistically different between 64Cu–F6-te1pa and 64Cu–F6–C-dota.
At 48 h PI, no significant differences were observed between 64Cu–F6–C-dota and 64Cu–F6-te1pa for all of the organs and likewise for the tumor (27.9 ± 9.3 and 29.1 ± 5.9 % ID g−1; p = 0.8). No significant difference was observed between 24 and 48 h PI in all organs for the two radioimmunoconjugates. These observations demonstrated the possibility to realize the phenotypic-imaging at 24 h PI, in order to maximize the number of events integrated by the PET camera. The lack of differences between 64Cu–F6–C-dota and 64Cu–F6-te1pa is probably due to the chosen antibody. F6 mAb is indeed a mouse antibody that can be taken in charge by mouse FcRn in our model.59 In these conditions, the mAb metabolism is very low, notably in liver, which can explain the similar uptake in each studied organs for each chosen time point.
The tumor targeting was particularly satisfying for both radioimmunoconjugates as early as 24 h after injection. Such results were similar to those obtained by Bryan et al. in the same animal model with the 64Cu-dota-cT84.66.60 Indeed, in this work the uptake in the tumor was 32 % ID g−1 48 h PI. In terms of tumors/organs ratios, our results were also equivalent to those obtained by Zeglis et al. in a pretargeting approach with a whole antibody modified with a trans cyclooctene derivative and followed by an in vivo coupling reaction with 64Cu-nota modified with a tetrazine.61 Compared to this work, the % ID g−1 was more than 5 times higher in the tumor for 64Cu–F6-te1pa. Compared to pretargeting techniques, the use of a whole antibody is particularly advantageous in the framework of a theranostic approach to maximize targeting and therefore the dose delivered to the tumor. The same immunoconjugate can indeed be radiolabeled with copper-67 to achieve a radioimmunotherapy following PET-phenotypic imaging with copper-64.
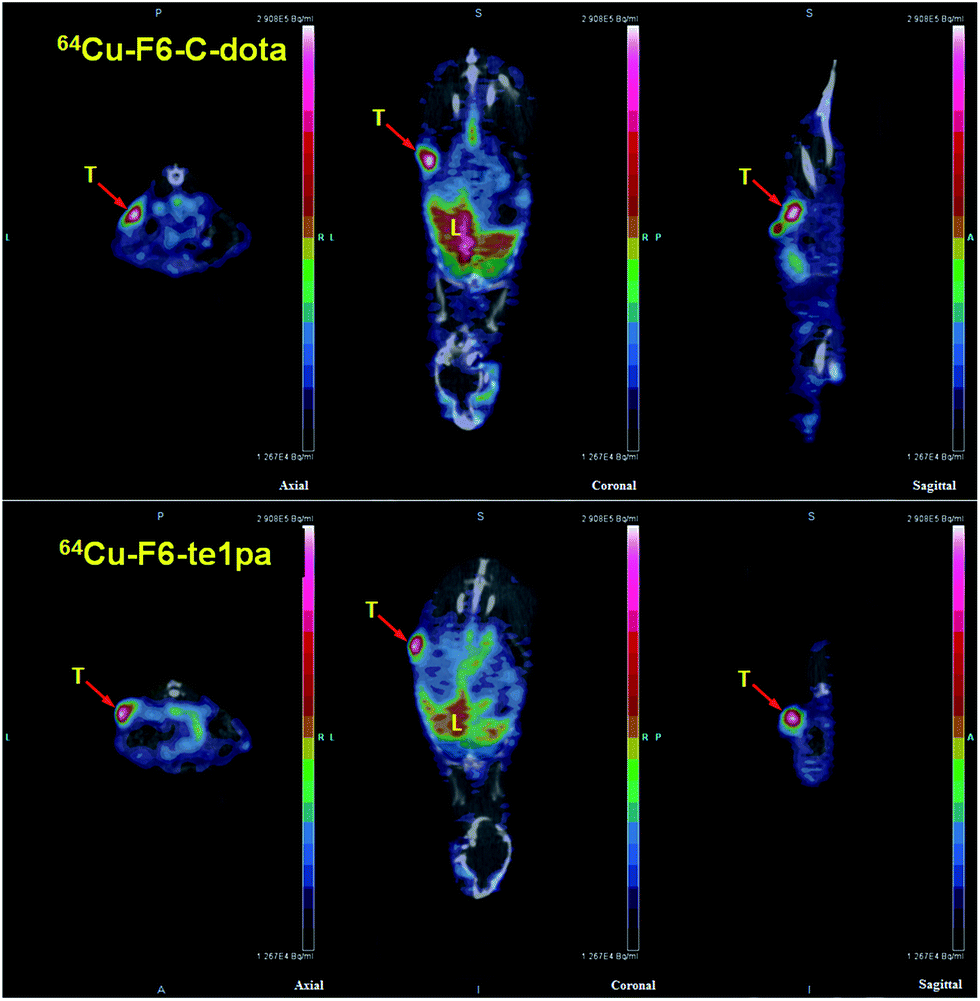 | ||
| Fig. 5 PET-CT images at 24 h post-injection of 64Cu–F6-te1pa and 64Cu–F6–C-dota in nude mice bearing LS174T tumors. T = tumor; L = liver. | ||
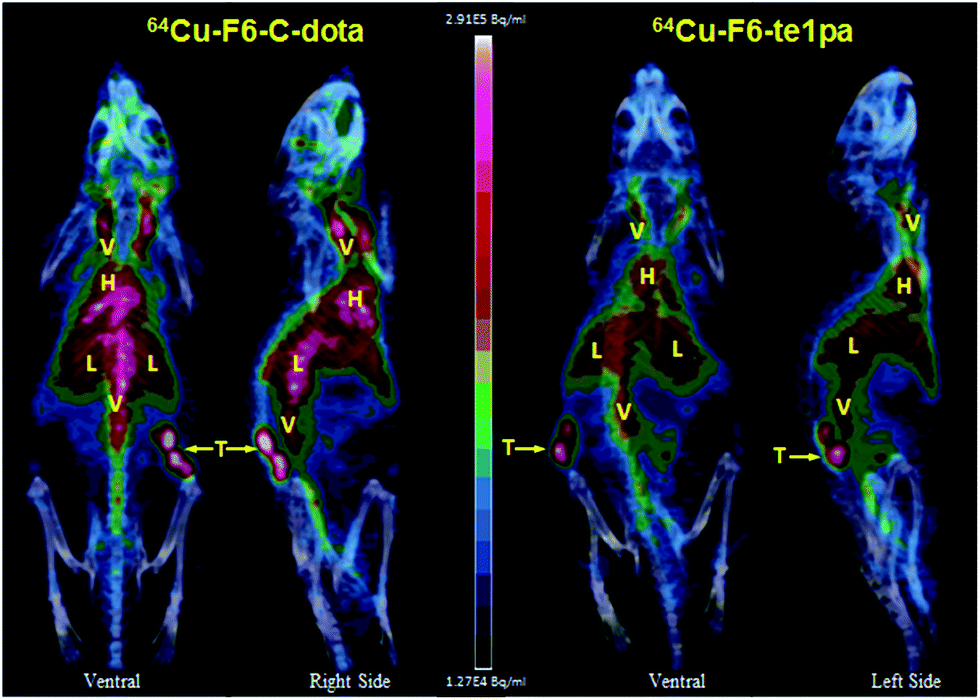 | ||
| Fig. 6 PET – 3D MIP projection at 24 h post-injection of 64Cu–F6-te1pa and 64Cu–F6–C-dota in nude mice bearing LS174T tumors. T = tumor; L = liver; V = vascular; H = heart. | ||
At this stage, a comparison between the bifunctional chelators is tempting. Nevertheless, there is controversy as to the possibility of observing biodistribution discrepancies between copper-64 radioimmunoconjugates of the same mAb labeled through different ligands, when it comes to comparison of macrocyclic ligands. On the one hand, Dearling et al. described very small biodistribution differences between bifunctional derivatives of nota, dota, teta or Sar conjugated to ch14.18 mAb.62 No significant variations were observed between the 24 h and 48 h time points PI, similarly to our results with 64Cu–F6-te1pa vs. 64Cu–F6–C-dota. In the same way, Cooper et al. have compared different macrocyclic and acyclic ligands conjugated to rituximab.55 If no significant difference was observed between macrocyclic derivatives regarding accumulation of radioactivity in the liver and kidneys at 48 h PI, they have however demonstrated greater accumulation in liver and intestine for acyclic ligands derivatives of diethylenetriaminepentaacetic acid (DTPA) in comparison with macrocyclic ligands. On the other hand, Pandya et al. have shown a lower uptake of copper-64 in kidneys and liver when comparing 64Cu-te2a-Bn-NCS-trastuzumab to 64Cu-dota-Bn-NCS-trastuzumab in a murine model of breast cancer.63 In addition, Rogers et al. reported a lower liver and kidney uptake of 64Cu-BAT-2IT-1A3, in rats, compared to SCN-teta, CPTA and PCBA copper-64 radioimmunoconjugates. However, this difference was not significant in the hamster.64
It appears that intrinsic differences in kinetics of macrocyclic or macrobicyclic 64Cu complexes have a low impact on the biodistribution of radioimmunoconjugates based on whole mAb. This can be explained in part by the very slow hepatic metabolism of antibodies due to their active recycling from the liver to the blood through the FcRn receptor in lysosomes.65 Radioactive uptake in liver resulting from transchelation is in fact negligible compared to the intact form of the radioimmunoconjugate which will undergo recycling. The 12.7 h period of copper-64 is not long enough to compare the resistance to transchelation of 64Cu complexes after conjugation to mAbs, which in comparison have a much greater biological half-life. These differences may be observed with copper-67 whose period is closer to the biological half-life of mAbs, which allows to assess the metabolism pathway of copper following the metabolism of the radioimmunoconjugate.66
On the one hand, the choice of an internalizing mAb would be interesting to test the in vivo stability of copper-64 radiolabeling, since it would involve a higher metabolism and also variations of redox intracellular conditions.
In a second hand, a vector with faster metabolism might also allow identification of discrepancies between 64Cu-dota and 64Cu-te1pa. In some particular cases, with smaller vectors and especially with peptides, differences in biodistribution were indeed described, depending on the ligand used to complex 64Cu.10,12,67 Larger molecules such as mAb fragments (minibody, F(ab′)2 etc.) are an attractive alternative to mAbs since their biological half-lives are more adapted to the period of copper-64. These mAb fragments are metabolized faster, which could lead to a larger transchelation process during the time covered by the period of copper-64. As an example, Dearling et al. have shown significant biodistribution differences with ch14.18-ΔCH2, a 120 kDa mAb fragment, radiolabeled through different ligands with 64Cu in a murine model of intrahepatic metastasis.68 This study also highlighted the importance of copper-64 complex overall charge on the uptake of the radioimmunoconjugate in the kidneys. This has been previously demonstrated for non-conjugated 64Cu complexes69 and also for radiolabeled peptides.70 Finally, it should be noted that the conjugation pathway that was used for te1pa in this work induces a +1 modification in the vector overall charge, making it unfavorable for the uptake in liver and kidneys.
Conclusions
The present study has proved the interest to use a te1pa chelator conjugated to F6-mAb in order to improve complexation properties compared to C-functionalization dota derivates. In vitro stability study has shown attractive and encouraging results. Unfortunately, experimental conditions used were not able to demonstrate a better in vivo stability of 64Cu-te1pa immunoconjugate compare to 64Cu-dota immunoconjugate. Nevertheless, the proof of concept for the well-founded of picolinate ligands for copper-64 F6-mAb radiolabeling has been achieved after injection of 64Cu–F6-te1pa radioimmunoconjugates to xenografted nude mice bearing LS174T tumors and the resulting phenotypic-PET images.Even if conjugation of the chelator using the picolinate carboxylic function was expected to not affect the chelating properties of te1pa ligand, we observed a small release of copper from the radiolabeled antibody. This relative instability of the radiotracer could be related to the global charge of the complex consequence of evolution from the acid to amide function during the coupling reaction. To overcome this disadvantage, one of the best ways is to design a C-functionalized te1pa derivative with a conjugable arm like a phenylisothiocyanate.71–73 This functionalization will allow to keep the negative charge of the picolinate at physiological pH and to obtain a neutral effect on the overall charge after conjugation. C-Functionalization of te1pa is also expected to restore the almost instantaneous complexation of copper-64 at RT, to increase the conjugation yield and finally should ensure that the kinetic inertness of the 64Cu complex obtained after conjugation is not impaired. Our efforts therefore are focusing on the synthesis of C-functionalized and bifunctional picolinate chelators for future applications in PET-phenotypic imaging.
Experimental section
Conjugation
Water (18.2 MΩ cm) was obtained from Milli-Q® Gradient system (Millipore). S-2-(4-Isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane tetraacetic acid (p-NCS-Bn-dota) was purchased from Macrocyclics USA.
Copper-64 dichloride in 0.1 M hydrochloric acid was obtained from ARRONAX cyclotron (Saint-Herblain, France). Radionuclidic purity was determined by gamma spectroscopy using a DSPEC-JR-2.0 type 98-24B HPGE detector (AMETEK) and purity on metals was assessed by ICP-OES with an iCAP 6500 DUO (Thermo Fisher Scientific).
F6 mAb – a mouse IgG1a directed against Gold-1 epitope of carcinoembryonic antigen (CEA) – was kindly provided by Immunotech (Marseille, France).
The te1pa activation reaction – to obtain a N-hydroxysulfosuccinimide intermediate compound – and coupling reactions were carried out in glass hemolysis tubes with bar magnets. All these materials have been treated prior to use by being dipped overnight in a 10 mM EDTA solution (pH 4.5) and were then rinsed several times with water.
:1:1. The reaction mixture was kept in an ice bath and gently stirred during 30 min. Following this reaction, the te1pa N-hydroxysulfosuccinimide intermediate (te1pa-SNHS) was not isolated; 302 μL of the reaction mixture was immediately added to 1.55 mL (41.4 nmol; 4 mg mL−1) of F6 mAb in coupling buffer (carbonate, 0.3 M, pH 8.6). The resulting F6 mAb:te1pa-SNHS molar ratio is 1:700. The reaction mixture was stirred during 3 h, initially in an ice bath then allowing the temperature to return gradually to room temperature.The purification, i.e. the separation of the immunoconjugate(s) of interest, antibodies aggregates and also remaining traces of non-conjugated ligand, was performed by size exclusion HPLC. The system is composed of a 500 μL peek tubing loop, a Sephadex G200 column (GE Healthcare Life Science), an Eckert&Ziegler HPLC module, Knauer K120 pumps, Knauer degasser and smartline UV 2520 set at 227 nm and an Eckert&Ziegler shielded gamma-detector. The system is controlled using the Modular-Lab (Eckert&Ziegler) software.
The purification was carried out by elution in isocratic mode with phosphate buffer 0.1 M (pH 7) at a flow rate of 0.5 mL min−1 for a run of 35 min. The UV-vis spectrophotometric was taken into account to collect manually the fraction corresponding to the peak of immunoconjugate. The latter was concentrated by ultrafiltration and the exact concentration of the solution was measured by UV-vis spectrophotometry and exact volume was determined by weighing.
Characterization of the immunoconjugates
In an Eppendorf tube, 5 μL of 64CuCl2 solution was added to 1 M ammonium acetate (volume sufficient to obtain a final reaction volume of 200 μL and a pH of 6.5–7). The mixture was vortexed before adding the calculated volume of immunoconjugate to obtain the appropriate ratio. This volume is variable from one 64CuCl2 production to another, depending on the contamination by cold copper.
The reaction mixture was incubated for 30 min at 40 °C. Then, a fraction was collected to carry out the radiolabeling yield monitoring. The solution was deposited on an ITLC-SG plate (Agilent Technologies) and the TLC was developed using 0.1 M citrate buffer pH 5. Then, the plate was dried and put exposing for 5 min on a storage phosphor plate followed by development using a Cyclone® Plus (Perkin Elmer) and Optiquant® software to obtain a radio-chromatogram. The chromatographic conditions used allow to separate the copper-64 non chelated (Rf. 1.0) and the radioimmunoconjugate (Rf. 0). For each radiolabeling, the labeling yield was multiplied by the ratio copper-to-immunoconjugate and the results were averaged to obtain the number of ligands conjugated per antibody.
:3 (1.5) copper-to-immunoconjugate ratio according to the protocol described previously. The radiolabeling solution was divided into 4 aliquots and then supplemented with a volume of a 0.1 M pH 7.5 EDTA solution corresponding to 2000, 10000, 25000 and 50000 molar equivalents of EDTA relative to the immunoconjugate. The mixture was stirred at RT during one night. TLC controls were performed before and after supplementation of EDTA according to the protocol described previously.:1 copper:immunoconjugate ratio, each vial being associated with a single control time. Half of the vials were placed at 40 °C while the other half was stirred at RT.Evaluations of immunoreactivity were made in PBS/BSA 0.5% to promote the interaction between antigens and antibodies.
1.5 pmol of radioimmunoconjugate was introduced in the 44.12.13-coated tube. This volume was calculated taking into account the specific activity and activity concentration of the labeling solution. The tube was stirred for 1 hour, then the solution was removed and introduced in a conventional tube. The 44.12.13-coated tube was rinsed with 500 μL PBS and the rinsing volume has been reserved. The coated tube, the tube containing the supernatant and that containing the rinsing liquid were counted using NaI(Tl) spectrometer (Wallac 1480-Wizard® 3; Perkin Elmer).
The test result is obtained by calculating the percentage for the number of CPM (counts per minute) of the 44.12.13-coated tube compared to the total activity.
Radiolabeling
For the radiolabeling of F6-te1pa, 62 μL of 64CuCl2 solution (volume activity: 0.98 MBq μL−1; specific activity: 25.1 GBq μmol−1) were first added to 162 μL of 0.2 M ammonium acetate and mixed. Then, 100 μL of 5.5 mg mL−1 F6-te1pa solution were added to the mixture, in slight excess (1.5 equiv.) relative to the total amount of copper dosed in 64CuCl2. The reaction mixture was heated at 40 °C for 25 min, then 8 μL of 10 mM EDTA solution were added to stop the reaction and complex free 64Cu(II). After mixing, the radiolabeling yield was assessed by ITLC-SG in 0.1 M (pH 5) citrate buffer, according to the previously described protocol. Finally, the specific activity of the radiolabeled immunoconjugate was adjusted to 50 MBq mg−1 by adding 220 μL of 3 mg mL−1 F6 mAb in PBS. Volume activity was also adjusted to 33.3 MBq mL−1 by adding 1272 μL of phosphate buffer 0.15 M.In the same way, radiolabeling of F6–C-dota was achieved with 137 μL of 0.2 M ammonium acetate, 62 μL of 64CuCl2, 75 μL of 7.3 mg mL−1 F6-NCS-Bn-dota, 8 μL of 10 mM EDTA, 220 μL of 3 mg mL−1 F6 mAb and 1322 μL of phosphate buffer 0.15 M.
In vivo studies – biodistribution and PET imaging
The other chemical products correspond to those previously described for conjugation and radiolabeling. NUDE female NMRI mice were obtained from Harlan CPB then hosted in our animal facility (IRS-UN, Nantes) under standard conditions and received a standard diet ad libitum. LS174T cells (ATCC® CL-188™) were purchased from American Type Culture Collection (Rockville, MD).
The animal experimentation protocol was conducted in accordance with French regulations and was approved by the ethics committee on animal testing in the Region Pays de la Loire (Protocol CEEA.2012.171) and by the Ministry for higher education and research (Ministère de l'enseignement supérieur et de la recherche – No. 00143.02).
The injection of the radioimmunoconjugate was carried out 2 to 3 weeks after transplantation when the tumor volume was greater than 30 mm.3 A total of 40 mice was necessary to achieve biodistribution study (36 mice) and PET imaging study (4 mice).
The number of counts in the tissue samples were decay-corrected and calibrated, by comparison with aliquots (n = 3) of a dilution of the injected dose, which were counted at the same time. The activity in the selected tissues and organs was expressed as the percentage of the injected dose per gram of organ (% ID g−1). Results were also expressed as tumor-to-organ ratio by dividing the % ID g−1 of the tumor by the % ID g−1 contained in the organ concerned.
A total of 18 mice were sacrificed for each radioimmunoconjugate: 5 mice at 2 h, 7 mice at 24 h and 6 mice at 48 h after injection.
In order to accurately estimate the theoretical amount of radioimmunoconjugate contained in plasma, 200 μL of plasma were counted following a standard tube containing one hundredth of the activity injected per mouse. The number of counts measured for the plasma sample was divided by the number of counts for the standard. Taking into account the theoretical activity of the standard, the activity of the plasma sample was obtained (in Bq). The theoretical amount of radioimmunoconjugate contained in plasma was obtained by dividing the activity of the plasma by the specific activity of the radioimmunoconjugate decay-corrected.
:35 ethanol/0.1 M ammonium acetate added with protease inhibitors (5 μg mL−1 aprotinin, 5 μg mL−1 leupeptin, 4 mM AEBSF) using a PTFE – inox pellet piston and a 15 mL Falcon tube (Fisher Scientific) as a tissue grinder. Centrifugation at 8600g and 4 °C during 30 min was conducted with a Sigma 2-16PK (Fisher Bioblock Scientific) centrifuge. The supernatant was separated from the pellet and both were counted. The supernatant was filtered (0.45 μm) and 200 μL were analyzed using the following HPLC system. Also, the plasma from blood pool was also analyzed with the same HPLC system.HPLC system consists on a Waters 600 controller module, Empower pro 1154 software, Waters 600 quaternary pump, Waters 487 UV detector and Gilson FC203B fraction collector. Radioactive species were separated using a gel-filtration column (GE Healthcare Life Science Superose 12 10/300 GL) eluted isocratically with 20 mM HEPES buffer and 150 mM NaCl (pH 7.3) during 60 min, flow rate: 0.5 mL min−1. The calibration of gel-filtration column was conducted using three standards with molecular weight of 32.6 kDa, 66 kDa and 150 kDa, corresponding to molecular weight of respectively SOD, BSA and a standard IgG. The collected fractions were counted using a Wallac 1480-Wizard® 3 gamma counter (Perkin Elmer). No discrimination between small size molecules with molecular weight bellow 10 kDa can be achieved with this column (they consequently present the same retention time of 36 min in the experimental conditions).
Acknowledgements
The authors are grateful to Ferid Haddad, Thomas Carlier, Nicolas Chouin, Julie Rousseau, Aurore Rauscher and Sébastien Gouard. They also acknowledge the Ministère de l'Enseignement Supérieur et de la Recherche, the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale and the Conseil General du Finistère. The authors thank the support of the LABEX IRON (ANR-LABX-0018-01) and Equipex ArronaxPlus (ANR-11-EQPX-0004) operated by the French National Research Agency (ANR) within the program “Investissements d'Avenir”.Notes and references
- C. Qin, H. Liu, K. Chen, X. Hu, X. Ma, X. Lan, Y. Zhang and Z. Cheng, J. Nucl. Med., 2014, 55, 812–817 CrossRef CAS PubMed.
- C. Alliot, N. Audouin, J. Barbet, A.-C. Bonraisin, V. Bossé, C. Bourdeau, M. Bourgeois, C. Duchemin, A. Guertin, F. Haddad, S. Huclier-Markai, R. Kerdjoudj, J. Laizé, V. Métivier, N. Michel, M. Mokili, M. Pageau and A. Vidal, Front. Med., 2015, 2, 31 Search PubMed.
- Z. Miao, G. Ren, H. Liu, L. Jiang and Z. Cheng, Bioconjugate Chem., 2010, 21, 947–954 CrossRef CAS PubMed.
- D. T. T. Yapp, C. L. Ferreira, R. K. Gill, E. Boros, M. Q. Wong, D. Mandel, P. Jurek and G. E. Kiefer, Mol. Imaging, 2013, 12, 263–272 CAS.
- A. L. Vavere, E. R. Butch, J. L. J. Dearling, A. B. Packard, F. Navid, B. L. Shulkin, R. C. Barfield and S. E. Snyder, J. Nucl. Med., 2012, 53, 1772–1778 CrossRef CAS PubMed.
- A. Pfeifer, U. Knigge, J. Mortensen, P. Oturai, A. K. Berthelsen, A. Loft, T. Binderup, P. Rasmussen, D. Elema, T. L. Klausen, S. Holm, E. von Benzon, L. Højgaard and A. Kjaer, J. Nucl. Med., 2012, 53, 1207–1215 CrossRef CAS PubMed.
- Y. Zhang, H. Hong, J. W. Engle, J. Bean, Y. Yang, B. R. Leigh, T. E. Barnhart and W. Cai, PLoS One, 2011, 6, e28005 CAS.
- C. J. Anderson, F. Dehdashti, P. D. Cutler, S. W. Schwarz, R. Laforest, L. A. Bass, J. S. Lewis and D. W. McCarthy, J. Nucl. Med., 2001, 42, 213–221 CAS.
- G. W. Philpott, S. W. Schwarz, C. J. Anderson, F. Dehdashti, J. M. Connett, K. R. Zinn, C. F. Meares, P. D. Cutler, M. J. Welch and B. A. Siegel, J. Nucl. Med., 1995, 36, 1818–1824 CAS.
- S. Roosenburg, P. Laverman, L. Joosten, M. S. Cooper, P. K. Kolenc-Peitl, J. M. Foster, C. Hudson, J. Leyton, J. Burnet, W. J. G. Oyen, P. J. Blower, S. J. Mather, O. C. Boerman and J. K. Sosabowski, Mol. Pharm., 2014, 11, 3930–3937 CrossRef CAS PubMed.
- D. Liu, D. Overbey, L. D. Watkinson, C. J. Smith, S. Daibes-Figueroa, T. J. Hoffman, L. R. Forte, W. A. Volkert and M. F. Giblin, Bioconjugate Chem., 2010, 21, 1171–1176 CrossRef CAS PubMed.
- H. Cai, Z. Li, C.-W. Huang, A. H. Shahinian, H. Wang, R. Park and P. S. Conti, Bioconjugate Chem., 2010, 21, 1417–1424 CrossRef CAS PubMed.
- F. Kraeber-Bodéré, C. Rousseau, C. Bodet-Milin, C. Mathieu, F. Guérard, E. Frampas, T. Carlier, N. Chouin, F. Haddad, J.-F. Chatal, A. Faivre-Chauvet, M. Chérel and J. Barbet, Int. J. Mol. Sci., 2015, 16, 3932–3954 CrossRef PubMed.
- P. Panichelli, C. Villano, A. Cistaro, A. Bruno, F. Barbato, A. Piccardo and A. Duatti, Cancer Biother. Radiopharm., 2016, 31, 159–167 CrossRef CAS PubMed.
- H. Zhang, H. Cai, X. Lu, O. Muzik and F. Peng, Acad. Radiol., 2011, 18, 1561–1568 CrossRef PubMed.
- J. S. Lewis, R. Laforest, F. Dehdashti, P. W. Grigsby, M. J. Welch and B. A. Siegel, J. Nucl. Med., 2008, 49, 1177–1182 CrossRef PubMed.
- I. Grassi, C. Nanni, G. Cicoria, C. Blasi, F. Bunkheila, E. Lopci, P. M. Colletti, D. Rubello and S. Fanti, Clin. Nucl. Med., 2014, 39, e59–63 CrossRef PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Curr. Pharm. Des., 2007, 13, 3–16 CrossRef CAS PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- M. Shokeen and C. J. Anderson, Acc. Chem. Res., 2009, 42, 832–841 CrossRef CAS PubMed.
- C. J. Anderson, T. J. Wadas, E. H. Wong and G. R. Weisman, Chin. J. Nucl. Med. Mol. Imaging, 2008, 52, 185–192 CAS.
- R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS.
- J. D. Silversides, R. Smith and S. J. Archibald, Dalton Trans., 2011, 40, 6289–6297 RSC.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 2005, 4829–4833 CrossRef.
- G. J. Stasiuk and N. J. Long, Chem. Commun., 2013, 49, 2732–2746 RSC.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 2005, 4829–4833 CrossRef.
- D. N. Pandya, J. Y. Kim, J. C. Park, H. Lee, P. B. Phapale, W. Kwak, T. H. Choi, G. J. Cheon, Y.-R. Yoon and J. Yoo, Chem. Commun., 2010, 46, 3517–3519 RSC.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K.-C. Lam and I. A. Guzei, et al., J. Am. Chem. Soc., 2000, 122, 10561–10572 CrossRef CAS.
- R. Ferdani, D. J. Stigers, A. L. Fiamengo, L. Wei, B. T. Y. Li, J. A. Golen, A. L. Rheingold, G. R. Weisman, E. H. Wong and C. J. Anderson, Dalton Trans., 2012, 41, 1938–1950 RSC.
- P. Comba, M. Kubeil, J. Pietzsch, H. Rudolf, H. Stephan and K. Zarschler, Inorg. Chem., 2014, 53, 6698–6707 CrossRef CAS PubMed.
- A. Bevilacqua, R. I. Gelb, W. B. Hebard and L. J. Zompa, Inorg. Chem., 1987, 26, 2699–2706 CrossRef CAS.
- P. Chaudhuri and K. Wieghardt, Prog. Inorg. Chem., 1987, 35, 329–436 CrossRef CAS.
- L. R. Gahan and J. M. Harrowfield, Polyhedron, 2015, 94, 1–51 CrossRef CAS.
- A. M. Sargeson, Coord. Chem. Rev., 1996, 151, 89–114 CrossRef CAS.
- N. Di Bartolo, A. M. Sargeson and S. V. Smith, Org. Biomol. Chem., 2006, 4, 3350–3357 CAS.
- C. B. Johnbeck, U. Knigge, A. Loft, A. K. Berthelsen, J. Mortensen, P. Oturai, S. Langer, D. Elema and A. Kjaer, J. Nucl. Med., 2016 DOI:jnumed.116.180430.
- C. Malmberg, R. S. Ripa, C. B. Johnbeck, U. Knigge, S. W. Langer, J. Mortensen, P. Oturai, A. Loft, A. M. Hag and A. Kjær, J. Nucl. Med., 2015, 56, 1895–1900 CrossRef CAS PubMed.
- L. M. P. Lima, D. Esteban-Gómez, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2012, 51, 6916–6927 CrossRef CAS PubMed.
- M. Frindel, N. Camus, A. Rauscher, M. Bourgeois, C. Alliot, L. Barré, J.-F. Gestin, R. Tripier and A. Faivre-Chauvet, Nucl. Med. Biol., 2014, 41, e49–57 CrossRef CAS PubMed.
- L. M. P. Lima, Z. Halime, R. Marion, N. Camus, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2014, 53, 5269–5279 CrossRef CAS PubMed.
- M. Roger, L. M. P. Lima, M. Frindel, C. Platas-Iglesias, J.-F. Gestin, R. Delgado, V. Patinec and R. Tripier, Inorg. Chem., 2013, 52, 5246–5259 CrossRef CAS PubMed.
- A. Rodríguez-Rodríguez, M. Regueiro-Figueroa, D. Esteban-Gómez, R. Tripier, G. Tircsó, F. K. Kálmán, A. C. Bényei, I. Tóth, A. de Blas and T. Rodríguez-Blas, et al., Inorg. Chem., 2016, 55, 2227–2239 CrossRef PubMed.
- A. Rodríguez-Rodríguez, D. Esteban-Gómez, R. Tripier, G. Tircsó, Z. Garda, I. Tóth, A. de Blas, T. Rodríguez-Blas and C. Platas-Iglesias, J. Am. Chem. Soc., 2014, 136, 17954–17957 CrossRef PubMed.
- R. Tripier and Z. Halime, World Patent, WO 2015/071334 A1, 2015.
- A. Rodríguez-Rodríguez, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, M. Botta, R. Tripier and C. Platas-Iglesias, Inorg. Chem., 2012, 51, 13419–13429 CrossRef PubMed.
- A. Rodríguez-Rodríguez, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, M. Fekete, M. Botta, R. Tripier and C. Platas-Iglesias, Inorg. Chem., 2012, 51, 2509–2521 CrossRef PubMed.
- L. M. Lima, M. Beyler, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2015, 54, 7045–7057 CrossRef CAS PubMed.
- A. Rodríguez-Rodríguez, Z. Garda, E. Ruscsák, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, L. M. Lima, M. Beyler, R. Tripier and G. Tircsó, et al., Dalton Trans., 2015, 44, 5017–5031 RSC.
- L. M. Lima, M. Beyler, F. Oukhatar, P. Le Saec, A. Faivre-Chauvet, C. Platas-Iglesias, R. Delgado and R. Tripier, Chem. Commun., 2014, 50, 12371–12374 RSC.
- R. Tripier, C. Platas-Iglesias, L. M. Lima and M. Beyler, Word Patent, WO 2014/195416 A1, 2014.
- R. Tripier, C. Platas-iglesias, L. Lima and M. Beyler, Chelates of Lead(II) and Bismuth(III) Based on Trans-Di-N-Picolinate Tetraazacycloalkanes, 2016 Search PubMed.
- S. Benchimol, A. Fuks, S. Jothy, N. Beauchemin, K. Shirota and C. P. Stanners, Cell, 1989, 57, 327–334 CrossRef CAS PubMed.
- M. Ychou, A. Pelegrin, P. Faurous, B. Robert, J. C. Saccavini, D. Guerreau, J. F. Rossi, M. Fabbro, F. Buchegger, J. P. Mach and J. C. Artus, Int. J. Cancer, 1998, 75, 615–619 CrossRef CAS PubMed.
- H. Cai, J. Fissekis and P. S. Conti, Dalton Trans., 2009, 5395–5400 RSC.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS PubMed.
- N. Wu, C. S. Kang, I. Sin, S. Ren, D. Liu, V. C. Ruthengael, M. R. Lewis and H.-S. Chong, J. Biol. Inorg Chem., 2015, 21, 177–184 CrossRef PubMed.
- S. D. Voss, S. V. Smith, N. DiBartolo, L. J. McIntosh, E. M. Cyr, A. A. Bonab, J. L. J. Dearling, E. A. Carter, A. J. Fischman, S. T. Treves, S. D. Gillies, A. M. Sargeson, J. S. Huston and A. B. Packard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17489–17493 CrossRef CAS PubMed.
- F.-T. Lee, A. Rigopoulos, C. Hall, K. Clarke, S. H. Cody, F. E. Smyth, Z. Liu, M. W. Brechbiel, N. Hanai, E. C. Nice, B. Catimel, A. W. Burgess, S. Welt, G. Ritter, L. J. Old and A. M. Scott, Cancer Res., 2001, 61, 4474–4482 CAS.
- R. J. Ober, C. G. Radu, V. Ghetie and E. S. Ward, Int. Immunol., 2001, 13, 1551–1559 CrossRef CAS PubMed.
- J. N. Bryan, F. Jia, H. Mohsin, G. Sivaguru, W. H. Miller, C. J. Anderson, C. J. Henry and M. R. Lewis, Nucl. Med. Biol., 2005, 32, 851–858 CrossRef CAS PubMed.
- B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D. Carlin, P. Zanzonico, R. Weissleder and J. S. Lewis, J. Nucl. Med., 2013, 54, 1389–1396 CrossRef CAS PubMed.
- J. L. J. Dearling, S. D. Voss, P. Dunning, E. Snay, F. Fahey, S. V. Smith, J. S. Huston, C. F. Meares, S. T. Treves and A. B. Packard, Nucl. Med. Biol., 2011, 38, 29–38 CrossRef CAS PubMed.
- D. N. Pandya, N. Bhatt, A. V. Dale, J. Y. Kim, H. Lee, Y. S. Ha, J.-E. Lee, G. I. An and J. Yoo, Bioconjugate Chem., 2013, 24, 1356–1366 CrossRef CAS PubMed.
- B. E. Rogers, C. J. Anderson, J. M. Connett, L. W. Guo, W. B. Edwards, E. L. C. Sherman, K. R. Zinn and M. J. Welch, Bioconjugate Chem., 1996, 7, 511–522 CrossRef CAS PubMed.
- V. Ghetie and E. S. Ward, Annu. Rev. Immunol., 2000, 18, 739–766 CrossRef CAS PubMed.
- G. L. Denardo, S. J. Denardo, D. L. Kukis, R. T. O'Donnell, S. Shen, D. S. Goldstein, L. A. Kroger, Q. Salako, D. A. Denardo, G. R. Mirick, L. F. Mausner, S. C. Srivastava and C. F. Meares, Anticancer Res., 1998, 18, 2779–2788 CAS.
- J. C. Garrison, T. L. Rold, G. L. Sieckman, S. D. Figueroa, W. A. Volkert, S. S. Jurisson and T. J. Hoffman, J. Nucl. Med., 2007, 48, 1327–1337 CrossRef CAS PubMed.
- J. L. J. Dearling, B. M. Paterson, V. Akurathi, S. Betanzos-Lara, S. T. Treves, S. D. Voss, J. M. White, J. S. Huston, S. V. Smith, P. S. Donnelly and A. B. Packard, Bioconjugate Chem., 2015, 26, 707–717 CrossRef CAS PubMed.
- T. M. Jones-Wilson, K. A. Deal, C. J. Anderson, D. W. McCarthy, Z. Kovacs, R. J. Motekaitis, A. D. Sherry, A. E. Martell and M. J. Welch, Nucl. Med. Biol., 1998, 25, 523–530 CrossRef CAS PubMed.
- M. Fani, L. Del Pozzo, K. Abiraj, R. Mansi, M. L. Tamma, R. Cescato, B. Waser, W. A. Weber, J. C. Reubi and H. R. Maecke, J. Nucl. Med., 2011, 52, 1110–1118 CrossRef CAS PubMed.
- N. Camus, Z. Halime, N. le Bris, H. Bernard, M. Beyler, C. Platas-Iglesias and R. Tripier, RSC Adv., 2015, 5, 85898–85910 RSC.
- Z. Halime, M. Frindel, N. Camus, P.-Y. Orain, M. Lacombe, M. Chérel, J.-F. Gestin, A. Faivre-Chauvet and R. Tripier, Org. Biomol. Chem., 2015, 13, 11302–11314 CAS.
- N. Camus, Z. Halime, N. Le Bris, H. Bernard, C. Platas-Iglesias and R. Tripier, J. Org. Chem., 2014, 79, 1885–1899 CrossRef CAS PubMed.
- L. A. Bass, M. Wang, M. J. Welch and C. J. Anderson, Bioconjugate Chem., 2000, 11, 527–532 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of te1pa conjugation on HLL2 mAb; radiolabelling of each immunoconjugate with 64Cu; competition experiments with EDTA; HPLC chromatograms of immunoconjugates and radioimmunoconjugates; kinetic process of radiolabelling of F6-te1pa with 64Cu; biodistribution of 64Cu–F6-te1pa and 64Cu–F6–C-dota in nude mice bearing LS174T tumors expressed as a ratio tumor/tissue; biodistribution of 64Cu–F6-te1pa and 64Cu–F6–C-dota in nude mice bearing LS174T tumors expressed in % ID/g ± SD. See DOI: 10.1039/c6ra26003a |
| This journal is © The Royal Society of Chemistry 2017 |